

Abstract
Objective
We examined whether deficiency of Shp2 signaling in forebrain neurons alters metabolic and cardiovascular regulation under various conditions and if it attenuates the anorexic and cardiovascular effects of leptin. We also tested whether forebrain Shp2 deficiency alters blood pressure (BP) and heart rate (HR) responses to acute stress.
Design
Forebrain Shp2-/- mice were generated by crossing Shp2flox/flox mice with CamKIIα-cre mice. At 22 to 24 weeks of age, mice were instrumented for telemetry for measurement of BP, HR and body temperature (BT). Oxygen consumption (VO2), energy expenditure and motor activity were monitored by indirect calorimetry.
Results
Shp2/CamKIIα-cre mice were heavier (46±3 vs 32±1 g), hyperglycemic, hyperleptinemic, hyperinsulinemic, and hyperphagic compared to Shp2flox/flox control mice. Shp2/CamKIIα-cre mice exhibited reduced food intake responses to fasting/refeeding and impaired regulation of BT when exposed to 15°C and 30°C ambient temperatures. Despite being obese and having many features of metabolic syndrome, Shp2/CamKIIα-cre mice had similar daily average BP and HR compared to Shp2flox/flox mice (112±2 vs 113±1 mmHg and 595±34 vs 650±40 bpm), but exhibited increased BP and HR responses to cold exposure and acute air-jet stress test. Leptin's ability to reduce food intake and to raise BP were markedly attenuated in Shp2/CamKIIα-cre mice.
Conclusion
These results suggest that forebrain Shp2 signaling regulates food intake, appetite responses to caloric deprivation, and thermogenic control of body temperature during variations in ambient temperature. Deficiency of Shp2 signaling in the forebrain is associated with augmented cardiovascular responses to cold and acute stress but attenuated BP responses to leptin.
Keywords: Energy expenditure, food intake, blood pressure, heart rate, acute stress, thermoneutrality
Introduction
Src homology 2 containing phosphatase (Shp2), a non-transmembrane protein tyrosine phosphatase with two Src homology domains, has been implicated as a signaling pathway for a variety of growth factors and cytokines. Shp2 signaling in the central nervous system (CNS) has been shown to contribute to the regulation of body weight. Zheng et al. (1) found that mice with Shp2 deficiency in forebrain neurons develop obesity and some of the characteristics of metabolic syndrome including hyperglycemia, hyperinsulinemia and insulin resistance, suggesting that Shp2 may play an important role in the regulation of energy balance and metabolism. Although these observations suggest a potential role for Shp2 signaling in the CNS in regulating body weight and metabolism, the mechanisms by which defective forebrain Shp 2 signaling causes obesity are still unclear and could be related to increased food intake, increased food efficiency, or altered metabolic rate. Also, the role of Shp signaling in controlling appetite, metabolic rate and body weight responses to changes in ambient temperature have not, to our knowledge, been previously reported.
Since Shp2 is one of the three primary pathways for leptin receptor (LRb) signaling in the CNS, mainly by eliciting downstream mitogen-activated protein kinase (MAPK) (2-5), Shp2 deficiency may cause obesity, in part, by impairing the effects of leptin, a peptide hormone secreted by adipocytes, which normally reduces appetite and increases metabolic rate (6-10). CNS Shp2 signaling may also contribute to the cardiovascular effects of leptin. We previously provided evidence that leptin may play a key role in linking excess weight gain with increased sympathetic nervous system (SNS) activity and elevated blood pressure in obesity (8, 11,12). However, the role of CNS Shp2 signaling in chronic cardiovascular regulation has not, to our knowledge, been previously reported.
In the present study, we used a genetic approach to produce Shp2 deficiency specifically in forebrain neurons and determined if the resulting early-onset obesity is associated with increased food efficiency, altered appetite and body weight responses to changes in ambient temperature, and if these mice have altered food responses to refeeding after prolonged fasting. We also determined if the obese phenotype in this model is accompanied by increases in BP and heart rate (HR) and altered cardiovascular responses to acute stress. In addition, we examined if Shp2 deficiency in forebrain neurons attenuates the effects of leptin on food intake and BP regulation.
Our data suggest that Shp2 deficiency in forebrain neurons causes early onset obesity due mainly to mild hyperphagia and increased food efficiency rather than reduced energy expenditure. Our results also indicate that obesity in this model is not accompanied by increased BP, measured 24 hours/day, and that these mice also exhibited attenuated BP response to chronic leptin infusion, suggesting that functional Shp2 in the forebrain may be essential for obesity and hyperleptinemia to cause hypertension. Forebrain Shp2 deletion, however, was also associated with enhanced cardiovascular responses to acute stress.
Methods
The experimental procedures and protocols follow the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Mississippi Medical Center.
Animals
Male 24- to 25-week-old Shp2flox/flox control mice (n=22) and Shp2/CamkIIα-cre mice (n=25) with forebrain deficiency of Shp2 signaling were used in these studies. Shp2/CamkIIα-cre mice were generated by crossing CamkIIα-Cre mice (generously provided by Dr. Ionanis Dragatsis, University of Tennessee Health Sciences Center) with Shp2flox/flox mice (generously provided by Dr. Gen-Sheng Feng, Burnham Institute). Homozygous mice for the Shp2flox allele that also expressed Cre-recombinase were used as Shp2/CamkIIα-cre mice (Fig. 1A). Homozygous mice for the Shp2flox allele that did not express Cre-recombinase were used as controls. Both strains were on the C57BL/6 background.
Figure 1.
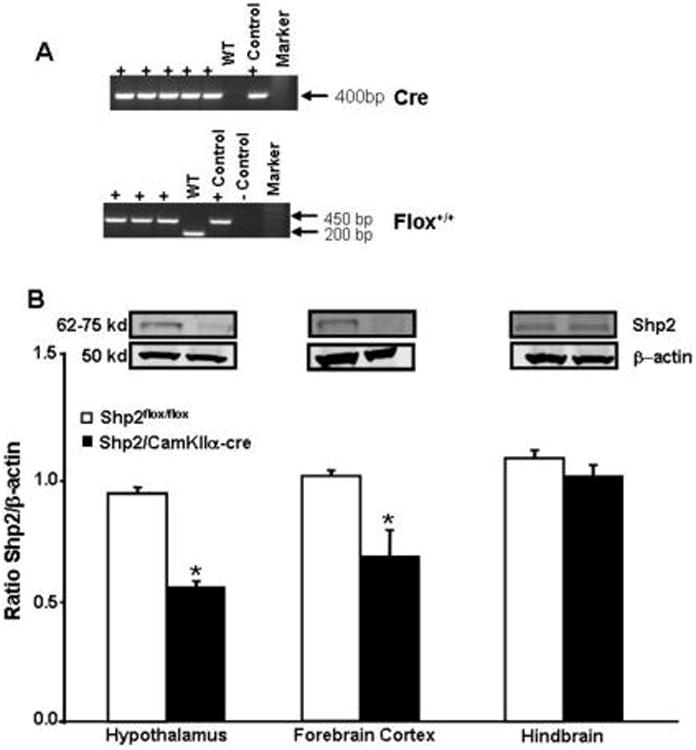
(A) Polymerase chain reaction (PCR) gels for Cre-recombinase (Top panel) and Shp2flox/flox gene from tail snip samples with analysis indicating presence of Cre-recombinase and Shp2flox/flox. Positive (+) indicates presence of Cre (loxP sequence). The WT is C57 negative control for Cre whereas “+ positive” is a positive Cre control sample that is added to the gel. The presence of floxed Shp2 gene product, the + sign indicates homozygote and + positive” is a positive floxed control sample that is added to the gel and negative (-) control” is just negative (no bands) control samples containing or not the specific gene product of the floxed Shp2 gene. The WT sample represents a sample from a regular C57 mouse containing only the normal gene product of the Shp2 gene product, and (B) Bottom panel, western blot data showing the ratio between Shp2 protein expression by beta-actin from hypothalamus, forebrain cortex and hindbrain of Shp2flox/flox and Shp2/CamkIIα-cre mice. * p<0.05 compared to Shp2flox/flox mice (n=5)
Specificity of Cre expression in forebrain neurons in Shp2/CamkIIα-cre mice compared to control mice has been detailed elsewhere (1). We observed a 50% reduction in Shp2 protein content in hypothalamus and forebrain cortex whereas no change in Shp2 protein content was found in the brainstem of Shp2/CamkIIα-cre mice when compared to Shp2flox/flox mice (Fig. 1B).
Body weight and food intake measurements from weaning to adulthood
After weaning, the mice were single-housed in a controlled temperature room (23 to 24°C) with 12-hr lights on/off cycle (lights on at 6:00 am). Food and water were offered ad libitum, daily food intake was measured between 8:30-9:30 am, and body weight was measured twice a week until 23 weeks of age. The cumulative food intake was calculated as the sum of food consumed from 6 to 13 weeks of age and net cumulative food intake was calculated as the sum of the daily difference in food intake after a change in ambient temperature minus the average food intake at 23°C. The food efficiency was calculated as the ratio of the cumulative calories consumed divided by body weight gain from 6 to 23 weeks of age.
Telemetry measurement of blood pressure (BP) and heart rate (HR) and venous catheter implantation
Under isoflurane anesthesia, Shp2flox/flox and Shp2/CamkIIα-cre mice, at 24 weeks of age were implanted with telemetric pressure transmitter probes inserted into the left carotid artery for determination of mean arterial pressure (MAP) and HR 24-hr day using computerized methods for data collection as previously described (11,12). Daily MAP and HR were obtained from the average of 24 hours of recording using a sampling rate of 1000 Hz with duration of 10 seconds for every 10–minute period. The mice were allowed to recover for 8 to 10 days after the surgery before 4 consecutive days of measurements were taken.
In additional groups of Shp2flox/flox (n=5) and Shp2/CamkIIα-cre mice (n=6) we also implanted a venous catheter into the jugular vein and tunneled it subcutaneously, exteriorizing it between the scapulae, passing through a spring and connecting to a mouse swivel mounted on the top of the metabolic cage. The venous catheter was connected through a sterile filter to a syringe pump for continuous 24-hr/day saline or leptin infusion. After 8 to 10 days to recover from surgery and 5 days of stable baseline control measurements leptin infusion experiments were begun.
Acute air-jet stress test
To determine whether deletion of Shp2 signaling in forebrain neurons alters the MAP and HR responses to an acute stress, 2 days after stable baseline measurements of MAP and HR were obtained (described above) the mice were placed in a special cage for air-jet stress testing. Briefly, after a 2-hour period for acclimatization to the cage, MAP and HR were continuously measured, using telemetry, for 30 minutes before the air-jet stress test was applied. The air-jet stress test consisted of 2-second pulses of air jet delivered every 5 seconds during 5 consecutive minutes aimed at the forehead of the mice at an approximate distance of 5 cm using a 14 gauge needle opening at the front of the tube connected to compressed air. After the 5-minute air-jet stress, MAP and HR were measured for an additional 30-minute recovery period. BP and HR responses during the air-jet stress and recovery period following the air-jet stress were calculated as the changes compared to baseline period (average of the last 10 minutes of baseline measurements before air-jet stress was initiated). We also calculated the area under curves (AUC) for MAP and HR during the air-jet stress and recovery periods using the following parameters: average change in MAP for each minute during the 5-minute air-jet stress test and for each 5 minutes during the 30-minute recovery period.
Oxygen consumption (VO2), motor activity, body temperature (BT), and food intake response to fasting/refeeding at different ambient temperatures
In separate experiments, Shp2flox/flox (n=10) and Shp2/CamkIIα-cre mice (n=15) at 25 weeks of age were placed individually in metabolic cages in a temperature adjustable system (CLAMS, Columbus Inst., Columbus OH) equipped with oxygen sensors to measure VO2 and receivers to measure BT. Ambulatory spontaneous motor activity was determined using infrared light beams mounted in the cages in X, Y and Z axes. BT was measured using telemetry probes (Respironics Mini Emitter 4000, Bend, OR) implanted intraperitonealy 8 to 10 days prior to the beginning of the experiment. VO2 was measured using Zirconia oxygen sensors. VO2 and BT were measured for 2 minutes at every 10-minute interval continuously 24-hr/day. After the mice were acclimatized to the new environment for 3 days, VO2, BT and motor activity were measured for 5 consecutive days at the control temperature of 23°C, and then the mice were kept at 15 or 30°C for an additional 5-day period.
To determine whether refeeding responses after prolonged fasting were affected by Shp2 deletion in forebrain neurons the mice were subjected to a 24-hr fasting followed by a 3-day period where food was available ad libitum. This fasting/refeeding protocol was performed in different groups of mice at 15, 23 and 30°C.
Food intake and blood pressure responses to leptin
To determine the role of Shp2 in forebrain neurons in mediating the acute effects of leptin on appetite, food intake was measured 24-hr after an intraperitoneal injection of leptin (10 mg/kg) or saline vehicle (0.3 mL) at 5:00 PM in additional groups of non-instrumented Shp2flox/flox (n=6) and Shp2/CamkIIα-cre (n=5) mice at 22 weeks of age.
We also studied the effects of leptin on regulation of blood pressure, measured 25 hrs/day by telemetry, in mice with Shp2 deficiency. In these experiments, control measurements were made for 5 days during i.v. isotonic saline vehicle infusion. Then leptin was added to the saline vehicle and infused i.v. at a rate of 2 μg/kg/min for 7 days in Shp2flox/flox and Shp2/CamKα-cre mice. Leptin infusion was then stopped and post-treatment measurements were made for 5 days while only saline vehicle was infused.
Western Blotting
Hypothalamus, brainstem and forebrain cortex were homogenized in lysis buffer (KPO4, pH 7.4), sonicated and cleared by centrifugation (3.500 × g, 5 min at 4°C). After the supernatant protein concentration was determined using the Bradford method (Bio-rad, Hercules, CA), 50 μg of protein were separated in a 4-15% precast linear gradient polyacrylamide gel (Bio-Rad, CA). After being transferred to nitrocellulose membranes, blots were rinsed in PBS and blocked in Odyssey blocking buffer (LI-COR, Lincoln, NE) for 1 h at room temperature, and incubated with mouse monoclonal anti-Shp2 antibody (1:2,500; BD Transduction Laboratories, USA) overnight at 4°C. The membranes were probed with rabbit anti-β-actin (1:5,000; Abcam) as a loading control. Membranes were incubated with IR700-conjugated donkey anti-mouse IgG and IR800-conjugated donkey anti-rabbit (1:2,000; Rockland Immunologicals). Antibody labeling was visualized using the Odyssey Infrared Scanner (LI-COR) for simultaneous detection of two fluorprobes. Fluorescence intensity analyses were performed using Odyssey software (LI-COR). Shp2 protein level was normalized to β-actin.
Analytical Methods
Plasma leptin and insulin levels were measured by ELISA kit (R&D Systems and Crystal Chem Inc., respectively), and plasma glucose concentrations were determined using the glucose oxidation method (Beckman glucose analyzer 2).
Statistical Analysis
The results are expressed as mean ± SEM. The data obtained were analyzed by paired t test or 1-way ANOVA with repeated measures followed by Dunnett's post hoc test for comparison between control and experimental values within each group when appropriate. Different groups were compared by unpaired t test or 2-way ANOVA followed by Bonferroni's post hoc test when appropriate. Statistical significance was accepted at a level of P<0.05.
Results
Shp2 deficiency in forebrain neurons causes early-onset obesity associated with hyperphagia, increased food efficiency and features of the metabolic syndrome
Shp2/CamkIIα-cre mice were significantly heavier (∼60%) than Shp2flox/flox control mice as early as 6 weeks of age and this difference persisted until 23 weeks of age when experiments were initiated (Fig. 2A). No differences in body length (nasal-anal distance) were observed between groups (10.3±0.2 vs. 10.8±0.3 cm). The increased body weight in Shp2/CamkIIα-cre mice was associated with slightly higher food intake compared to lean control mice (Fig. 2B) which was confirmed when total cumulative food intake was measured during the 18 weeks of observation (Fig. 2C). In addition, Shp2/CamkIIα-cre mice also exhibited increased food efficiency from 6 to 23 weeks of age compared to control mice (Fig. 2D). Although cumulative food intake was significantly higher in Shp2/CamkIIα-cre mice, their hyperphagia was modest with only small increases in food intake which were not statistically significant during some weeks from weaning until experiments began. For instance, food intake was not significantly different between groups at 23 weeks of age (Fig. 2B) while at 25 weeks of age Shp2/CamkIIα-cre mice ate significantly more than control mice (Fig. 5F). However, average food intake from weaning to 23 weeks of age was significantly increased in Shp2/CamkIIα-cre mice compared to control Shp2flox/flox mice, as reflected by the cumulative food intake.
Figure 2.
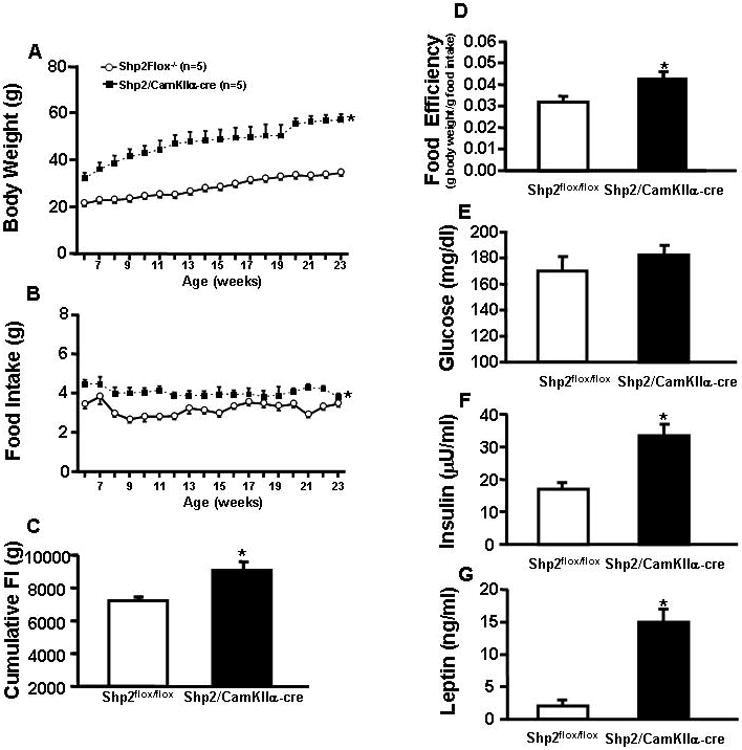
(A) Average weekly body weight; (B) food intake from 6 to 23 weeks; (C) cumulative food intake from 6 to 23 weeks; (D) food efficiency; (E) plasma glucose; (F) plasma leptin and (G) plasma insulin of Shp2flox/flox and Shp2/CamkIIα-cre mice. * p<0.05 compared to Shp2flox/flox mice.
Figure 5.
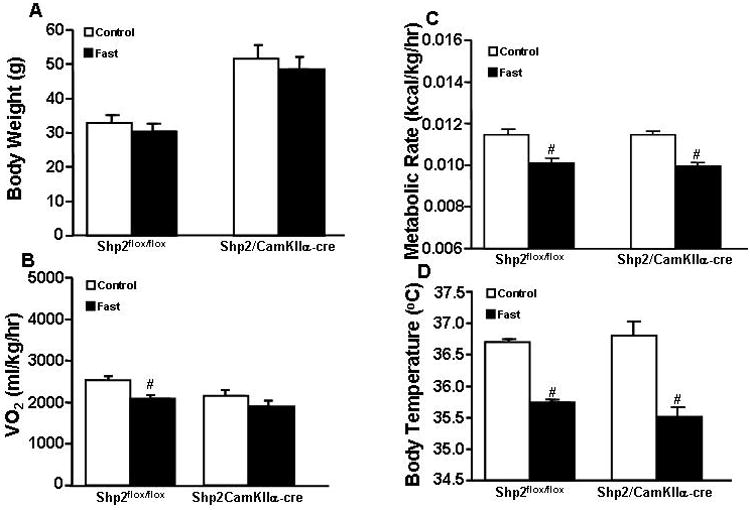
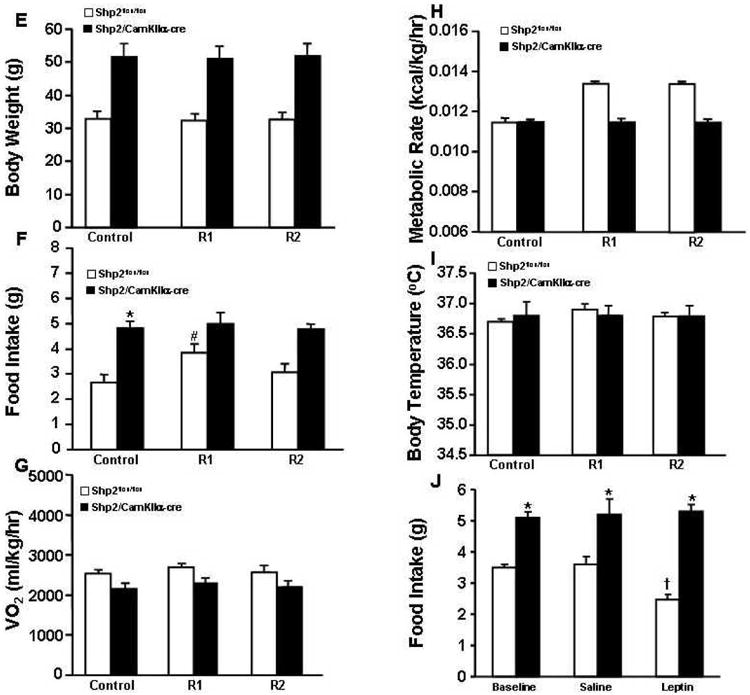
(A) Body weight; (B) oxygen consumption (VO2); (C) metabolic rate; and (D) body temperature responses to 24-hour fast; (E) Body weight; (F) food intake; (G) oxygen consumption (VO2); (H) metabolic rate and (I) body temperature during refeeding after 24-hour fast in Shp2flox/flox and Shp2/CamkIIα-cre mice at 23°C. # p<0.05 compared to control period.
At 25 weeks of age, Shp2/CamkIIα-cre mice were hyperleptinemic, hyperinsulinemic and had slightly, but not significantly, higher plasma glucose levels (Fig. 2E-G).
Food intake responses to changes in ambient temperature in Shp2flox/flox and Shp2/CamKIIα-cre mice
At 25 weeks of age, Shp2/CamkIIα-cre and Shp2flox/flox mice were exposed for 5 days to cold (15°C) or warm ambient temperatures (thermoneutral zone of 30°C) in order to examine if Shp2 deletion in forebrain neurons alters appetite and body weight responses to changes in ambient temperature. No significant changes in body weight were observed in either group when ambient temperature was changed from 23°C to 30°C (Fig. 3A). However, body weight was significantly increased in Shp2/CamkIIα-cre but not in Shp2flox/flox mice when ambient temperature was changed from 23°C to 15°C for 5 days (Fig. 4A) Food intake was markedly impacted by ambient temperature in Shp2/CamkIIα-cre mice while only modest changes were observed in control mice (Fig. 3B and 4B). For instance, raising ambient temperature from 23°C to 30°C reduced food intake by 40 and 18% in Shp2/CamkIIα-cre and Shp2flox/flox mice, respectively (Fig. 3B), leading to a greater negative net change in cumulative food intake in Shp2/CamkIIα-cre mice during the 5-day exposure period (Fig. 3C). Lowering ambient temperature from 23°C to 15°C increased food intake in Shp2/CamkIIα-cre mice by 30% compared to only 16% increase in control mice (Fig. 4B), causing a greater positive change in net cumulative food intake in Shp2/CamkIIα-cre mice (Fig. 4C). These results indicate that Shp2 deletion in forebrain neurons enhances appetite sensitivity to variations in ambient temperature.
Figure 3.
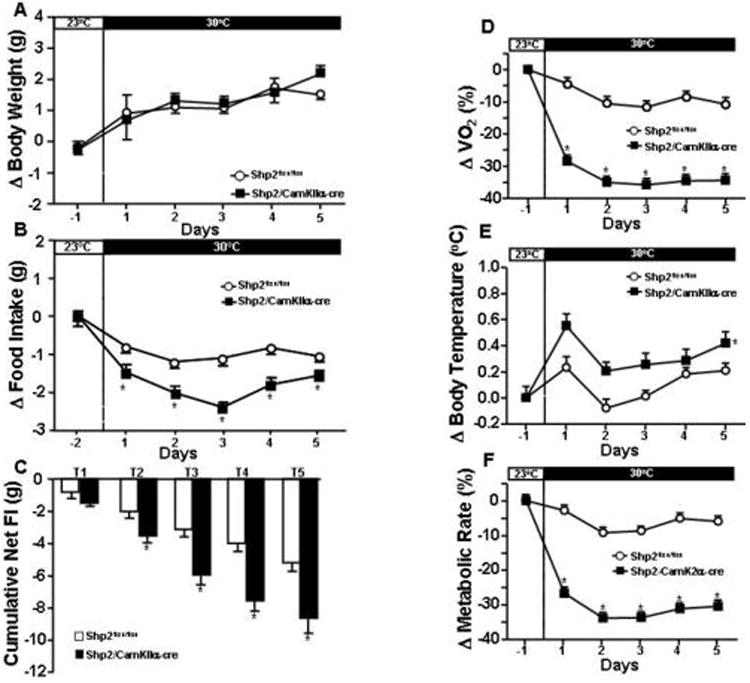
(A) change (Δ) in body weight; (B) change (Δ) food intake and (C) cumulative net food intake (FI); (D) percentage changes in oxygen consumption (VO2); (E) change (Δ) in body temperature and (F) percentage changes in metabolic rate at 15°C in Shp2flox/flox and Shp2/CamkIIα-cre mice. * p<0.05 compared to Shp2flox/flox mice.
Figure 4.

(A) change (Δ) in body weight; (B) change (Δ) food intake and (C) cumulative net food intake (FI); (D) percentage changes in oxygen consumption (VO2); (E) change (Δ) in body temperature; (F) percentage changes in metabolic rate at 30°C (thermoneutral zone) and (I) response to acute leptin injection (10 mg/kg, IP) on food intake in Shp2flox/flox and Shp2/CamkIIα-cre mice. * p<0.05 compared to Shp2flox/flox mice; # p<0.05 compared to 23°C; † p<0.05 compared to baseline.
Oxygen consumption (VO2), motor activity and body temperature (BT) responses to changes in ambient temperature in Shp2flox/flox and Shp2/CamKIIα-cre mice
At baseline, Shp2/CamkIIα-cre mice showed slightly reduced VO2 (2036±341 vs 2414±171 ml/kg/hr) and similar metabolic rate (0.011±0.002 vs 0.012±0.004 kcal/kg/hr) compared to Shp2flox/flox mice. BT was not significantly different between groups at baseline (36.8±0.1 vs 36.6±0.1 °C). To determine whether Shp2/CamkIIα-cre mice exhibit greater sensitivity of energy balance to the effects of changes in ambient temperature we also measured metabolic rate, BT, and VO2 at 15 and 30°C. Exposure to thermoneutral temperature (30°C) caused greater reductions in VO2 and metabolic rate in Shp2/CamkIIα-cre mice than in control mice (Fig. 3D and F), while BT also increased to a greater extent (∼0.5°C) in these mice compared to controls (∼0.2°C, Fig. 3E). At 15°C, however, the marked increases in VO2 and metabolic rate were similar in both groups (Fig. 4D and F), while BT decreased more in Shp2/CamkIIα-cre mice than in controls (Fig. 4E). No significant changes were observed in motor activity in either group during exposure to 15 or 30°C, compared to 23°C (Suppl. Fig. S1A and S1B). Collectively, these data demonstrate that deletion of Shp2 in forebrain neurons impairs regulation of body temperature and energy balance during variations in ambient temperature, especially at thermoneutral temperatures.
Food intake, VO2, metabolic rate, motor activity and BT responses to fasting and refeeding and food intake responses to acute leptin injection
Prolonged (24-hr) fasting significantly reduced VO2, metabolic rate and BT in Shp2/CamkIIα-cre and Shp2flox/flox control mice at the control ambient temperature of 23°C (Fig. 5B-D). The reduction in VO2, however, did not reach significance in Shp2/CamkIIα-cre mice (Fig. 5B). Although there was a tendency for reduced body weight in both groups the changes were not significant (Fig. 5A). In Shp2flox/flox mice refeeding was associated with a significant increase in food intake that returned to control values 48 hrs after normal access to feed was restored (Fig. 5F); in Shp2/CamkIIα-cre mice no initial hyperphagia was observed during refeeding after fasting (Fig. 5F). No significant changes were observed in body weight, VO2, metabolic rate or BT during refeeding for groups (Fig. 5E, 5G-I). Acute leptin injection reduced 24-hr food intake in Shp2flox/flox mice by ∼32% when compared with saline vehicle injection, whereas no differences were observed between leptin and vehicle injection in Shp2/CamkIIα-cre mice (Fig. 5J). These results indicate that Shp2 signaling in forebrain neurons contributes to the acute effects of leptin to reduce appetite.
At 15°C or thermoneutrality, fasting caused similar changes in body weight, VO2, metabolic rate and BT in Shp2/CamkIIα-cre and Shp2flox/flox mice as observed at 23°C (Fig. S2A-H). Similar responses to refeeding were also observed at 15°C or thermoneutral temperatures (Fig. S3A-J).
Baseline MAP and HR, and cardiovascular responses to fasting/refeeding, acute air-jet stress, and to chronic leptin infusion
To test the hypothesis that Shp2 signaling in forebrain neurons is also important for the regulation of cardiovascular function, we measured BP and HR 24-hr/day using telemetry. In addition, we examined the cardiovascular response of Shp2/CamkIIα-cre and Shp2flox/flox control mice to acute stress. We found that although baseline BP (112±2 vs 113±1 mmHg) and HR (595±34 vs 650±40 bpm) for Shp2/CamkIIα-cre and Shp2flox/flox mice, respectively, were similar (Fig. 6A and 6B), the BP and HR responses to acute air-jet stress were much more pronounced in Shp2/CamkIIα-cre when compared to control mice. For instance, we observed a 25% and 110% greater rise in BP and HR, respectively, in Shp2/CamkIIα-cre mice during the 5-minute air-jet stress period (Fig. 6C and 6D). Moreover, BP and HR quickly returned to baseline values in Shp2flox/flox control mice during the 30-minute recovery period that followed the exposure to the air-jet, whereas in Shp2/CamkIIα-cre mice BP and HR remained elevated (Fig. 6C and 6D).
Figure 6.


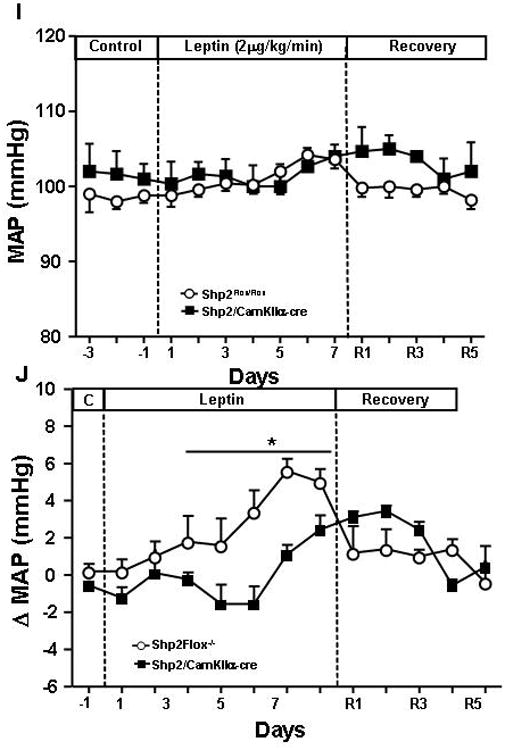
(A) Baseline mean arterial pressure (MAP); (B) heart rate (HR); (C) area under curve (AUC) for change (Δ) in blood pressure; and (D) changes in HR during air-jet stress and recovery period in Shp2flox/flox and Shp2/CamKα-cre mice; (E) change (Δ) in MAP, and (F) change (Δ) in HR at 15°C and thermoneutrality (30°C) compared to 23°C; (G) MAP and (H) HR responses to fasting and refeeding; (I) response to chronic leptin infusion (2 μg/kg/min, i.v.) for 7 days on MAP and (J) delta MAP values in Shp2flox/flox and Shp2/CamKα-cre mice. * p<0.05 compared to Shp2flox/flox mice. # p<0.05 compared to control period.
To determine whether Shp2/CamkIIα-cre mice exhibit greater sensitivity to the effects of changes in ambient temperature and prolonged fasting (24hr) and refeeding on cardiovascular function, we measured MAP and HR at 15°C and thermoneutrality (30°C). Exposure to cold temperature (15°C) caused greater increases in MAP and HR in Shp2/CamkIIα-cre mice than in control mice (Fig. 6E and 6F), while no significant changes were observed at thermoneutral temperature (30°C) between groups (Fig. 6E and 6F).
Prolonged fasting significantly reduced MAP and HR in Shp2/CamkIIα-cre and Shp2flox/flox control mice at the control ambient temperature of 23°C, at 15°C and thermoneutrality (Fig. 6G and 6H). Refeeding returned MAP and HR all the way back to control values (Fig. 6G and 6H). These results indicate that Shp2 signaling in forebrain neurons also plays an important role in modulating cardiovascular response to cold and to acute stress.
In Shp2flox/flox control mice chronic leptin infusion caused a gradual and significant elevation in MAP while in Shp2/CamkIIα-cre mice the effects of leptin on MAP were attenuated (Fig. 6I and 6J). This suggests that Shp2 signaling in forebrain neurons also contributes, at least in part, to the effects of leptin on BP regulation.
Discussion
Our studies indicate that Shp2 signaling in forebrain neurons contributes importantly to regulation of body weight, energy expenditure, and cardiovascular function. In addition, we found that Shp2 deficiency in forebrain neurons impairs regulation of body temperature during changes in ambient temperature. We also found that exposure to cold temperature caused greater increases in MAP and HR in Shp2/CamkIIα-cre mice than in control mice. In addition, the acute effect of leptin on food intake was completely abolished and the chronic BP response to leptin was attenuated in mice with Shp2 deficiency in forebrain neurons. To our knowledge the role of CNS Shp2 signaling in controlling metabolic and cardiovascular function under normal conditions, during acute and chronic leptin infusion, and during stress stimuli (e.g. fluctuations in ambient temperature, prolonged fasting, air jet stress, etc.) have not been previously reported. Here we show that Shp2 deficiency specifically in forebrain neurons enhances the blood pressure and heart rate responses to cold and acute air-jet stress, modulates food intake responses to caloric deprivation, and impairs body temperature regulation during exposure to cold and warm temperatures. We also show that Shp2 deficiency attenuates the effects of leptin to reduce food intake and increase BP.
Mice with Shp2 deficiency in forebrain neurons were heavier after weaning compared to lean control mice, suggesting that Shp2 signaling in these neurons plays an important role in regulating body weight early in life. Our results also confirm previous reports (1) that mice with forebrain Shp2 deficiency exhibit many metabolic abnormalities including insulin resistance and high plasma leptin levels (1,13). However, despite early-onset obesity and metabolic abnormalities these mice had similar BP and HR compared to lean control littermates, suggesting that activation of Shp2 signaling in these neurons may be important in linking obesity with elevation in BP and HR.
Shp2 –MAPK activation is one of the key pathways for LRb signaling in the CNS (2-5) and previous studies from our lab and others have provided evidence that leptin-induced SNS activation may play a key role in obesity-induced hypertension. For example, chronic leptin infusion at rates that raise blood leptin to levels comparable to those found in obesity increase BP and HR. Also, experimental animals and humans with leptin deficiency (14-18) or leptin receptor deletion in the entire CNS (15) have severe obesity but normal or reduced BP and SNS activity (14,15) suggesting that a functional leptin system is necessary for obesity to increase SNS activity and BP. Moreover, we showed that leptin-induced activation of proopiomelanocortin (POMC) neurons is required for obesity and the accompanying hyperleptinemia to increase SNS activity and raise BP and HR (11). Thus, our current observations suggest that Shp2 signaling in forebrain neurons may be a key intracellular pathway for BP and HR responses to obesity, as well as playing an important role in regulating energy balance. Shp2 deficiency in forebrain neurons is also associated with resistance to the acute effects of leptin to reduce appetite and with attenuated BP response to chronic hyperleptinemia. Determining whether these effects are mediated specifically by Shp2 signaling in POMC neurons or in other forebrain neurons will require further investigation. Since in this model Shp2 was probably deleted not only in LRb-containing neurons but also in non LRb-containing neurons it is possible that some of the phenotypes observed in these mice are caused by alterations in other signaling pathways besides leptin signaling.
Although Shp2/CamkIIα-cre mice were not hypertensive despite marked early-onset obesity, these mice showed exacerbated BP and HR responses to cold and acute air-jet stress. The mechanisms leading to augmented cardiovascular responses to this pressor stimulus, however, are still unclear. One possibility is that Shp2 signaling in forebrain neurons modulates SNS responses to external stress stimuli and defective Shp2 levels in these neurons cause greater variations in SNS activity and BP during stress. Another possibility is that obesity may exacerbate cardiovascular responses to stress through mechanisms that are independent of Shp signaling. Separating the effects of obesity from defective Shp2 signaling in the forebrain is challenging since dietary models of obesity, for example, add other variables such as increased BP; also prevention of obesity by pair feeding young Shp2/CamkIIα-cre mice adds the chronic stress of food deprivation in animals whose brains are reset for hyperphagia. Supporting the idea that defective forebrain Shp2 signaling greatly enhances cardiovascular responses to acute stress are our previous findings that mice with obesity comparable (or even more severe) to that found in Shp2 deficient mice have attenuated rather than enhanced responses to air jet stress, the same stimulus as used in the present study (11).
Our results indicate that deletion of forebrain Shp2 signaling leads to obesity mainly because of increased food intake and food efficiency from 7 to 23 weeks of age. Zhang et al (1) observed no differences in food consumption in young mice from postnatal days 23 to 32 and concluded that obesity was due to decreased energy expenditure, although this was not directly assessed (1). In the present study, we found that at 23 weeks of age these mice did exhibit significant differences in food intake, but not in oxygen consumption or motor activity compared to control mice. These findings suggest that Shp2 deficiency in forebrain neurons causes obesity mainly by increasing food intake and food efficiency rather than decreasing energy expenditure. These findings are consistent with the possibility that CNS Shp2 signaling may play an important role in regulating body weight by altering food intake rather than by altering energy expenditure.
In response to changes in ambient temperature we found significant differences in body temperature and food intake responses in Shp2/CamKIIα-cre and control mice. For instance, we found that upon exposure to thermoneutrality Shp2/CamKIIα-cre mice had more pronounced reduction in food intake leading to greater negative net cumulative food intake than observed in control mice. At 15°C Shp2/CamKIIα-cre mice exhibited more pronounced increases in food intake resulting in higher positive net cumulative food intake compared to control mice. These results suggest that Shp2 signaling in forebrain neurons plays a role in regulating food intake responses to changes in ambient temperature.
Another important observation from our study is that Shp2/CamKIIα-cre mice exhibited greater changes in body temperature during variations in ambient temperature which were not associated with changes in physical activity. Thus, our data suggest an important role for Shp2 signaling in forebrain neurons in regulation of body temperature and food intake in response to changes in metabolic demand caused by varying ambient temperature.
Caloric deprivation causes increased appetite, reduced metabolic rate and decreased sympathetic nervous system activity to the heart, kidneys and brown adipose tissue, and decreased heart rate and blood pressure (21-23). In the present study we examined whether Shp2 signaling in forebrain neurons plays an important role in mediating the appetite and metabolic and cardiovascular responses to caloric deprivation. We found that mice with Shp2 deficiency in forebrain neurons had impaired food intake responses to refeeding after 24 hours of fasting at 15°C and thermoneutrality, despite significant reductions in body temperature and heat production. We also found that exposure to cold temperature caused greater increases in MAP and HR in Shp2/CamkIIα-cre mice than in control mice; however the BP and HR responses to refeeding were similar between Shp2/CamKIIα-cre and control mice. Moreover, chronic leptin infusion at a rate to raise plasma leptin levels to those found in severe obesity caused a gradual increase in MAP in control mice, and this increase in MAP was attenuated in mice with Shp2 deficiency in forebrain neurons. These mice were also resistant to the acute anorexic effects of leptin. These observations suggest that Shp2 signaling in forebrain neurons is important for leptin-mediated decreases in appetite, and to a lesser extent in mediating the actions of leptin on BP regulation. Taken together, our findings suggest that functional Shp2 signaling in forebrain neurons is required for normal metabolic responses to caloric deprivation. Although the precise mechanisms are still unknown, Zhang et al. (1) previously showed that mice with Shp2 deficiency in forebrain neurons do not exhibit the normal increase in hypothalamic neuropeptide Y (NPY) levels, an orexigenic factor important in regulating feeding behavior, after a 20-hr fast. Whether changes in NPY levels alone can fully explain the lack of reactive hyperphagia to prolonged fasting in Shp2/CamkIIα-cre mice has not been determined.
In summary, we demonstrated that Shp2 deficiency in forebrain neurons leads to early onset obesity associated with mild hyperphagia and increased food efficiency, metabolic abnormalities and impaired body temperature regulation during changes in ambient temperature. We also found that Shp2 signaling is involved in the food intake responses to refeeding and to leptin injection, and that forebrain Shp2 plays an important role in mediating the cardiovascular response to hyperleptinemia, to cold exposure, and to an acute air-jet stress Since obesity results from an imbalance between energy intake and expenditure, understanding the mechanisms that underlie regulation of energy expenditure and appetite in response to external stimuli will be important for the development of novel treatment strategies.
Supplementary Material
Acknowledgments
This research was supported by the National Heart, Lung and Blood Institute Grant PO1HL-51971, NIGMS P20GM104357, and by a Scientist Development Grant from the American Heart Association to JM do Carmo.
Footnotes
Disclosures: None.
Conflict of Interest: None.
Supplementary information is available at the journal's website.
References
- 1.Zhang EE, Chapeau E, Hagihara K, Feng GS. Neuronal Shp2 tyrosine phosphatase controls energy balance and metabolism. PNAS. 2004;101:16064–16069. doi: 10.1073/pnas.0405041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG., Jr Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem. 2002;277:41547–55. doi: 10.1074/jbc.M205148200. [DOI] [PubMed] [Google Scholar]
- 3.Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, et al. Divergent roles of SHP-3 in ERK activation by leptin receptors. J Biol Chem. 2001;276:4747–55. doi: 10.1074/jbc.M007439200. [DOI] [PubMed] [Google Scholar]
- 4.East JB, Royer AR, Middlemas DS. The protein tyrosine phosphatase, Shp2, is required for the complete activation of the RAS/MAPK pathway by the brain-derived neurotrophic factor. J Neurochem. 2006;97:834–45. doi: 10.1111/j.1471-4159.2006.03789.x. [DOI] [PubMed] [Google Scholar]
- 5.Myers MP, Anderson JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, et al. TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem. 2001;276:47771–4. doi: 10.1074/jbc.C100583200. [DOI] [PubMed] [Google Scholar]
- 6.Lou PH, Yang G, Huang L, Cui Y, Pourbahrami T, Radda GK, et al. Reduced body weight nad increased energy expenditure in transgenic mice over-expressing soluble leptin receptor. PLoS One. 2010;20:e11669. doi: 10.1371/journal.pone.0011669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singh A, Wirtz M, Parker N, Hogan M, Strahler J, Michailidis G, et al. Leptin-mediated changes in hepatic mitochondrial metabolism, structure, and protein levels. Proc Natl Acad Sci USA. 2009;106:13100–5. doi: 10.1073/pnas.0903723106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shek EW, Brands MW, Hall JE. Chronic leptin infusion increases arterial pressure. Hypertension. 1998;31:409–414. doi: 10.1161/01.hyp.31.1.409. [DOI] [PubMed] [Google Scholar]
- 9.Hall JE, da Silva AA, do Carmo JM, Dubinion J, Hamza S, Munusamy S, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. J Biol Chem. 2010;285:17271–6. doi: 10.1074/jbc.R110.113175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.da Silva AA, do Carmo JM, Dubinion J, Hall JE. The role of leptin and central nervous system melanocortins in obesity hypertension. Curr Opion Nephrol Hypertens. 2013;22:135–40. doi: 10.1097/MNH.0b013e32835d0c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.do Carmo JM, da Silva AA, Cai Z, Lin S, Dubinion JH, Hall JE. Control of blood pressure, appetite, and glucose by leptin in mice lacking leptin receptors in proopiomelanocortin neurons. Hypertension. 2011;57:918–26. doi: 10.1161/HYPERTENSIONAHA.110.161349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tallam LS, da Silva AA, Hall JE. Melanocortin-4 receptor mediates chronic cardiovascular and metabolic actions of leptin. Hypertension. 2006;48:58–64. doi: 10.1161/01.HYP.0000227966.36744.d9. [DOI] [PubMed] [Google Scholar]
- 13.Krajewska M, Banares S, Zhang EE, Huang X, Scadeng M, Jhala US, et al. Development of diabesity in mice with neuronal deletion of Shp2 tyrosine phosphatase. Am J Pathol. 2008;172:1312–24. doi: 10.2353/ajpath.2008.070594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mark AL, Shafer RA, Correia ML, Morgan DA, Sigmund CD, Haynes WG. Contrasting blood pressure effects of obesity in leptin deficient ob/ob mice and agouti yellow obese mice. J Hypertens. 1999;17:1949–53. doi: 10.1097/00004872-199917121-00026. [DOI] [PubMed] [Google Scholar]
- 15.do Carmo JM, Bassi M, da Silva AA, Hall JE. Control of cardiovascular, metabolic and respiratory functions during prolonged obesity in leptin-deficient and diet-induced obese mice. Hypertension. 2008;15:109. [Google Scholar]
- 16.Ozata M, Ozdemir IC, Licinio J. Human leptin deficiency caused by a missense mutation: multiple endocrine defects, decreased sympathetic tone, and immune system dysfunction indicate new targets for leptin action, greater central than peripheral resistance to the effects of leptin, and spontaneous correction of leptin-mediated defects. J Clin Endocrinol Metab. 1999;10:3686–95. doi: 10.1210/jcem.84.10.5999. [DOI] [PubMed] [Google Scholar]
- 18.Farooqi S, O'Rahilly S. Genetics of obesity in humans. Endocr Rev. 2006;27:710–8. doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 19.Giri H, Muthuramu I, Dhar M, Rathnakumark K, Ram U, Dixit M. Protein tyrosine phosphatase SHP2 mediates chronic insulin-induced endothelial inflammation. Arterioscler Tromb Vasc Biol. 2012;32:1943–50. doi: 10.1161/ATVBAHA.111.239251. [DOI] [PubMed] [Google Scholar]
- 20.He Z, Zhang SS, Meng Q, Li S, Zhu HH, Raquil MA, et al. Shp2 controls female body weight and energy balance by integrating leptin and estrogen signals. Mol Cell Biol. 2012;32:1867–78. doi: 10.1128/MCB.06712-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams TD, Chambers JB, Henderson RP, Rashotte ME, Overton JM. Cardiovascular responses to caloric restriction and thermoneutrality in C57BL/6J mice. Am J Physiol Regul Integr Comp Physiol. 2002;282:R1459–67. doi: 10.1152/ajpregu.00612.2001. [DOI] [PubMed] [Google Scholar]
- 22.Muralidhara DV, Shetty PS. Cold-induced thermogenesis in rats nutritionally deprived early in life. Indian J Physiol Pharmacol. 1987;31:149–58. [PubMed] [Google Scholar]
- 23.Overton JM, Williams TD, Chambers JB, Rashotte ME. Central leptin attenuates the cardiovascular and metabolic effects of fasting in rats. Hypertension. 2001;37:663–9. doi: 10.1161/01.hyp.37.2.663. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.