

Abstract
Animal models of neuromyelitis optica (NMO) are needed for elucidation of disease mechanisms and for testing new therapeutics. Prior animal models of NMO involved administration of human anti-aquaporin-4 immunoglobulin G antibody (NMO-IgG) to rats with pre-existing neuroinflammation, or to naïve mice supplemented with human complement. We report here the development of NMO pathology following passive transfer of NMO-IgG to naïve rats. A single intracerebral infusion of NMO-IgG to adult Lewis rats produced robust lesions around the needle track in 100% of rats; at 5 days there was marked loss of aquaporin-4 (AQP4), glial fibrillary acidic protein (GFAP) and myelin, granulocyte and macrophage infiltration, vasculocentric complement deposition, blood-brain barrier disruption, microglial activation and neuron death. Remarkably, a distinct ‘penumbra’ was seen around lesions, with loss of AQP4 but not of GFAP or myelin. No lesions or penumbra were seen in rats receiving control IgG. The size of the main lesion with loss of myelin was greatly reduced in rats made complement-deficient by cobra venom factor or administered NMO-IgG lacking complement-dependent cytotoxicity (CDC) effector function. However, the penumbra was seen under these conditions, suggesting a complement-independent pathogenesis mechanism. The penumbra was absent with NMO-IgG lacking both CDC and antibody-dependent cellular cytotoxicity (ADCC) effector functions. Finally, lesion size was significantly reduced after macrophage depletion with clodronate liposomes. These results: (i) establish a robust, passive-transfer model of NMO in rats that does not require pre-existing neuroinflammation or complement administration; (ii) implicate ADCC as responsible for a unique type of pathology also seen in human NMO; and (iii) support a pathogenic role of macrophages in NMO.
Keywords: NMO, aquaporin-4, astrocyte, complement, neuroinflammation
Introduction
Neuromyelitis optica (NMO) is an inflammatory demyelinating disease of the central nervous system that can produce significant motor and visual impairment [11,12,45]. Immunoglobulin G autoantibodies against aquaporin-4 (AQP4) are found in the serum of most NMO patients [13,19]. AQP4 is a water channel expressed in astrocytes throughout the central nervous system, as well as in a subset of skeletal muscle cells and of epithelial cells in kidney, lung and the gastrointestinal tract [8,9,25,27]. It is thought that anti-AQP4 autoantibodies (NMO-IgG) cause NMO pathology by binding to AQP4 on astrocytes, which results in complement-dependent cytotoxicity (CDC) and downstream inflammation and blood-brain barrier disruption, leading to secondary oligodendrocyte injury, demyelination and neuronal injury [17,23,26]. The major pathological features of NMO lesions in the human central nervous system include inflammation with marked granulocyte and macrophage infiltration, loss of AQP4 and glial fibrillary acidic protein (GFAP), vasculocentric deposition of activated complement, and demyelination [32].
Animal models of NMO that closely recapitulate human NMO pathology are needed to investigate NMO pathogenesis mechanisms, such as the role of specific effector leukocytes, and to test novel therapeutics, such as monoclonal antibody blockers of NMO-IgG binding to AQP4 [40] and enzymatic NMO-IgG inactivation [38,39]. Early work showed that intraperitoneal injection of IgG purified from NMO patient serum exacerbated CNS inflammation and produced perivascular astrocyte depletion and complement deposition in rats with experimental autoimmune encephalomyelitis (EAE) [15] or in rats pre-treated with complete Freund's adjuvant [16]. Though these studies provided evidence that NMO-IgG can worsen pathology, the pre-existing inflammation in these models precluded unambiguous interpretation, as EAE involves a targeted response of myelin-sensitized T-cells, very different from the AQP4-targeted humoral response in NMO.
Mouse models involving intracerebral injection or infusion of NMO-IgG and human complement have generated lesions with pathology similar to that of human NMO [34], and have been used to address complement- and cell-mediated pathogenesis mechanisms, including the role of neutrophils and eosinophils [35,48]. However, the mouse models required administration of human complement directly into brain. Attempts to develop mouse models of NMO based on peripheral NMO-IgG administration were not successful, despite efficient access of serum NMO-IgG to brain tissue in circumventricular organs [30]. In addition to the known weak activity of the mouse complement system [3], we found that mouse serum strongly inhibits the classical complement pathway, preventing complement-dependent cytotoxicity (unpublished results). As complement activation through the classical pathway is central in NMO pathogenesis, mice thus have limited utility as models of NMO.
Building on methods developed in mice, here we report robust NMO pathology in rat brain following intracerebral injection of NMO-IgG, without complement supplementation and without preexisting T cell-related neuroinflammation. The model was characterized and applied to investigate the role of macrophages in NMO pathology. In addition, an unexpected pathological feature, a penumbra with selective AQP4 loss, was identified and characterized, and evidence was obtained supporting a novel role for antibody-dependent cellular cytotoxicity (ADCC) in NMO pathology.
Materials and Methods
Rats
Lewis rats were purchased from Charles River Lab (Wilmington, MA). Experiments were done using weight-matched rats (200–300 g), age 8 to 12-weeks. Protocols were approved by the University of California San Francisco Committee on Animal Research.
Antibodies and sera
Recombinant monoclonal NMO antibody rAb-53 (referred to as NMO-IgG) was generated from a clonally expanded plasma blast population from cerebrospinal fluid of an NMO patient, as described and characterized previously [2,6]. NMO serum was obtained from seropositive individuals who met the revised diagnostic criteria for clinical disease [46]. Non-NMO (seronegative) human serum was used as control. In some studies IgG was purified from NMO or control serum using a Protein A-resin (GenScript, Piscataway, NY) and concentrated using Amicon Ultra Centrifugal Filter Units (Millipore, Billerica, MA).
Cell culture
Chinese hamster ovary (CHO) cells stably expressing human M23-AQP4 were generated as described [28] and cultured at 37 °C in 5% CO2 95% air in F-12 Ham's Nutrient Mixture medium supplemented with 10% fetal bovine serum, 200 μg/ml geneticin (selection marker), 100 U/ml penicillin and 100 μg/ml streptomycin. Primary astrocyte cultures were generated from cortex of newborn rats, as described [1,4]. After 8-10 days in culture, cells were treated for two weeks with 0.25 mM dibutyryl cAMP (Sigma-Aldrich, St. Louis, MO) to induce differentiation. Greater than 95% of cells were positive for the astrocyte marker GFAP.
NMO-IgG binding and cytotoxicity assays
Cells were grown on glass coverslips for 24 h. After blocking with 1% bovine serum albumin in phosphate-buffered saline (PBS), cells were incubated with NMO-IgG or purified IgG from NMO serum for 1 h at room temperature. Cells were washed with PBS and incubated with AlexaFluor 488 goat anti-human IgG secondary antibody (1:200; Invitrogen, Carlsbad, CA). For AQP4 immunostaining, cells were fixed in 4% paraformaldehyde (PFA) and permeabilized with 0.2% Triton-X. Rabbit anti-AQP4 antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) was added followed by Alexa Fluor-555 goat anti-rabbit IgG secondary antibody (1:200, Invitrogen), as described [6]. For assay of CDC, cells were incubated for 60 min at 28 °C with NMO-IgG or NMO serum with 5% human or rat complement (Innovative Research, Novi, MI). Cytotoxicity was measured by the Alamar Blue assay (Invitrogen).
Quantification of cell surface AQP4
Rat astrocyte cultures and AQP4-transfected cells were grown on coverglasses until confluence and incubated for specified times with 100 μg/ml rAb-53 at 37 °C. Cells were then washed extensively in cold PBS and blocked for 20 min in 1% BSA at 4 °C. Remaining AQP4 at the cell surface was labeled with 100 μg/ml rAb-53 at 4 °C for 1 h. Subsequently, rAb-53 was labeled by incubation for 1 h at 4 °C with goat anti-human IgG-conjugated Alexa Fluor 555 (1:200, Invitrogen), and the plasma membrane was stained using a fluorescent lectin, wheat germ agglutinin (WGA)-conjugated Alexa Fluor 488 (1:400; Invitrogen). Cells were then washed in cold PBS and fixed for 15 min in 4% paraformaldehyde (PFA). Surface AQP4 was quantified as the (background-subtracted) ratio of red (surface AQP4) to green (plasma membrane) fluorescence using ImageJ, as described [29].
Intracerebral injection protocols
Adult rats (200–300 g) were anesthetized with intraperitoneal ketamine (75-100 mg/kg) and xylazine (5-10 mg/kg) and mounted in a stereotaxic frame. Following a midline scalp incision, a burr hole of diameter 1 mm was made in the skull 3.5 mm to the right of the bregma. A 30-gauge needle attached to 50-μl gas-tight glass syringe (Hamilton, Reno, NV) was inserted 5 mm deep to infuse 10 μg NMO-IgG (in a total of 21 rats) or control IgG (21 rats) in a total volume of 10 μl (at 2 μl/min). In some experiments, purified IgG from NMO or control serum (1 mg) was injected in a total volume of 30 μl (3 rats per group). After 5 days, rats were anesthetized and perfused through the left cardiac ventricle with 100 ml PBS and then 25 ml of PBS containing 4% PFA.
For macrophage depletion experiments, clodronate-liposomes were used as previously described [22,41]. Briefly, clodronate or control (PBS) liposomes (2 ml) (3 rats per group) were injected intraperitoneally twice, at 48 h before and 48 h after intracerebral injection of NMO-IgG. In some studies rat complement was depleted by intraperitoneal injection of cobra venom factor (350 U/kg; Quidel Corporation, Santa Clara, CA) 24 h before and 48 h after intracerebral injection of NMO-IgG (3 rats) [20,42].
Immunofluorescence
Brains were post-fixed for 2 h in 4% PFA. Five micrometer-thick paraffin sections were immunostained at room temperature for 1 h with antibodies against rat AQP4 (1:200, Santa Cruz Biotechnology), GFAP (1:100, Millipore), myelin basic protein (MBP) (1:200, Santa Cruz Biotechnology), ionized calcium-binding adaptor molecule-1 (Iba1; 1:1,000; Wako), albumin (1:200, Santa Cruz Biotechnology), Ly-6G (1:100, Santa Cruz Biotechnology), C5b-9 (1:50, Hycult Biotech), CD45 (1:10, BD Biosciences), CD163 (1:50, Bio-Rad Laboratories), neurofilament (1:200, Millipore), iNOS (1:100, BD Biosciences), or arginase-1 (1:50, Santa Cruz Biotechnology) followed by the appropriate fluorescent secondary antibody (1:200, Invitrogen) or biotinylated secondary antibody (1:500, Vector Laboratories). Tissue sections were examined with a Leica (Wetzlar, Germany) DM 4000 B microscope at 25× magnification. AQP4, GFAP and MBP immunonegative areas were defined by hand and quantified using ImageJ. Data are presented as area (mm2) of immunonegative area.
Fluoro Jade-C staining
Coronal brain sections through the injection tract were deparaffinized, rehydrated, incubated in 0.06% potassium permanganate solution for 15 min, water-rinsed and transferred for 30 min to a 0.0001% solution of Fluoro Jade-C (FluoroJade) (Chemicon) dissolved in 0.1% acetic acid. Slides were then rinsed with distilled water, air-dried at 25 °C and coverslipped with VectaMount mounting medium (Vector Laboratories) [34].
Statistical analysis
Comparisons between two groups were performed using an unpaired t-test. P < 0.05 was considered statistically significant. Values are presented as mean ± S.E.
Results
NMO-IgG binds to AQP4 in rat astrocytes and causes CDC with rat complement
Many of the studies were done using a human recombinant monoclonal NMO antibody, rAb-53 (referred to as NMO-IgG), which binds tightly to human and mouse AQP4 and produces CDC [6,28]. Well-differentiated primary astrocyte cultures expressing high levels of AQP4 and GFAP were generated from brain cortex of neonatal rats (Fig. 1a, top). Fig. 1a (bottom) shows that NMO-IgG, as well as purified IgG from NMO patient serum (visualized with a green fluorescent secondary antibody that recognizes human IgG), binds to rat AQP4 (visualized using a primary anti-C-terminus AQP4 antibody and red fluorescent secondary antibody) on the primary astrocyte cultures. Binding of the recombinant and serum NMO-IgG to rat AQP4 produced CDC in the presence of rat complement, as shown using an Alamar Blue cytotoxicity assay (Fig. 1b).
Figure 1. NMO-IgG binds to AQP4 in rat astrocytes and produces CDC in the presence of rat complement.
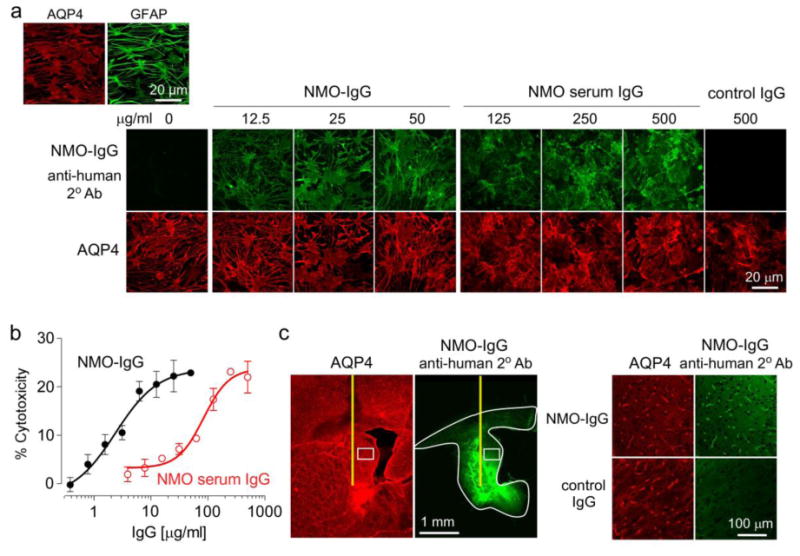
a. (Top left) AQP4 and GFAP immunofluorescence in well-differentiated primary cultures of rat astrocytes. (Bottom) Binding of NMO-IgG (recombinant human antibody rAb-53) or control IgG purified from NMO patient serum (each stained green with anti-human secondary antibody) to AQP4 (red). b. CDC (by Alamar blue assay) in rat astrocyte cultures following 3-h incubation with 5% rat complement and NMO-IgG or IgG purified from NMO patient serum (S.E., n=6). c. (Left) Immumofluorescence of brain sections at 3 h after intracerebral injection of 10 μg NMO-IgG showing area of antibody diffusion (white line). (Right) Expanded micrograph of boxed region along with data for control IgG.
The binding and distribution of NMO-IgG in rat brain was determined following intracerebral administration 5 mm beneath the brain surface using a small needle. At 3 h after injection, NMO-IgG was seen in an ∼6.2 mm2 brain area around its injection site, representing ∼20% of the area of a brain hemi-slice (Fig. 1c, left). At high magnification the distribution of NMO-IgG overlapped with that of AQP4, which was concentrated at the end-feet of astrocytes around the blood capillaries (Fig. 1c, right). No AQP4 binding was seen of a control, isotype-matched human antibody (control IgG).
Intracerebral injection of NMO-IgG in rats produces NMO pathology
Rats were administered NMO-IgG (or control IgG) by a single intracerebral injection that involved insertion of a 30-gauge needle 5 mm deep into brain and infusion of 10 μg NMO-IgG in a 10 μL volume over 5 min. Fig. 2a (left) shows immunofluorescence for AQP4, GFAP and myelin basic protein (MBP) at 5 days after antibody injection. A NMO-like lesion was seen with loss of AQP4, GFAP and MBP immunofluorescence around the site of antibody injection in rats receiving NMO-IgG, but not control IgG. Interestingly, a large area (referred to as ‘penumbra’) was observed around the lesion in the NMO-IgG-injected mice, characterized by decreased AQP4 but intact GFAP and myelin staining (Fig. 2a, left). The lesion and penumbra areas were well-demarcated as seen at high magnification (Fig. 2a, center). Fig. 2a (right) summarizes lesion and penumbra size from a series of rats. The lesion observed in rats administered NMO-IgG was a typical demyelinating lesion with loss of myelin but preserved axons as seen by intact neurofilament staining (Fig. 2b).
Figure 2. Intracerebral injection of NMO-IgG in rats produces NMO pathology.
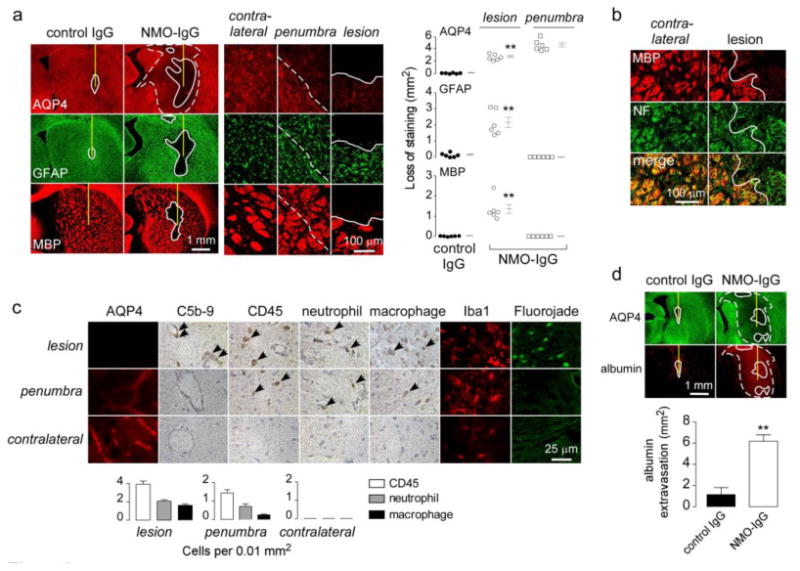
a. Brains were injected with 10 μg NMO-IgG or control IgG. (Left) AQP4, GFAP and MBP immunofluorescence at 5 days after injection. In this and subsequent figures the needle tract shown as a yellow line, white line demarcates region with loss of immunofluorescence, and white dashed line demarcates the penumbra (reduced AQP4 but normal GFAP and MBP immunofluorescence). (Middle) Higher magnification of indicated regions. (Right) Summary of lesion areas showing data for individual rats (S.E., n=6, ** P < 0.01). b. MBP and neurofilament [5] immunofluorescence. White line demarcates region with loss of immunofluorescence. c. (Left) Immunostaining for AQP4, activated complement (C5b-9), leukocytes (CD45), neutrophils (Ly6-G), macrophages (CD163), microglia (Iba1), and degenerating neurons (Fluorojade) in indicated regions. (Right) Number of infiltrating leukocytes per 0.01 mm2 (S.E., n=3). d. (Top) Albumin immunofluorescence. (Bottom) Area of albumin extravasation (S.E., n=3, ** P < 0.01).
Fig. 2c shows immunostaining for additional markers in the lesion, penumbra and contralateral hemisphere of NMO-IgG-injected rats. The NMO-like lesion with near complete loss of AQP4 showed vasculocentric deposition of activated complement (C5b-9), leukocyte infiltration (CD45), consisting mainly of neutrophils (Ly-6G) and macrophages (CD163), microglial activation (Iba1), and neuronal degeneration (FluoroJade). The contralateral hemisphere (and rats receiving control antibody, not shown) did not show these changes. Complement deposition and neuronal degeneration were absent in the penumbra, though there was mild inflammation (mainly neutrophils and macrophages). Fig. 2d shows albumin immunofluorescence in the brain as a measure of blood-brain barrier integrity. Rats receiving NMO-IgG showed marked albumin extravasation in the lesion as well as in the penumbra.
To characterize the kinetics over which NMO pathology develops, rats receiving NMO-IgG or control antibody were sacrificed at 1 day after antibody injection. Fig. 3a shows loss of AQP4 and GFAP immunofluorescence following NMO-IgG administration, indicating early astrocyte damage with marked disruption of the blood-brain barrier seen by albumin extravasation. However, little or no myelin loss was seen at 1 day after NMO-IgG administration. Complement deposition and inflammation were also seen at 1 day (Fig. 3b).
Figure 3. Characterization of early pathology and pathology produced by intracerebral injection of NMO patient serum.
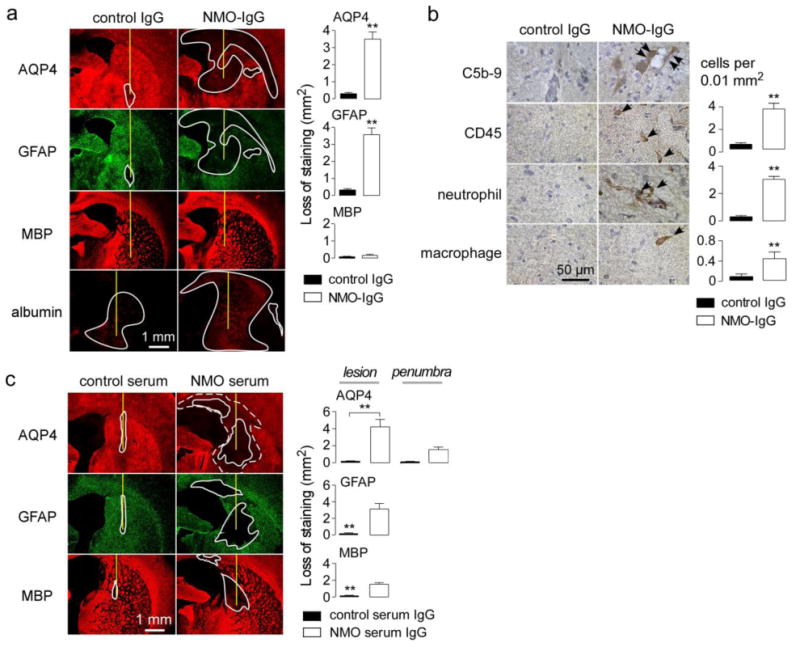
a. Brains were injected with 10 μg NMO-IgG or control IgG. (Left) AQP4, GFAP, MBP and albumin immunofluorescence at 1 day after injection. Needle track shown as yellow line and lesion demarcated by white line. (Right) Summary of lesion areas (S.E., n=3, ** P < 0.01). b. (Left) Immunohistochemistry in lesions from A. (Right) Number of infiltrating leukocytes per 0.01 mm2 (S.E., n=3). c. Brains were injected with 1 mg of purified IgG from NMO patient serum or control serum. (Left) AQP4, GFAP and MBP immunofluorescence at 5 days after injection. Summary of lesion areas (S.E., n=3, ** P < 0.01).
The characteristic NMO pathology produced by the recombinant NMO-IgG was also seen following intracerebral injection of IgG purified from NMO patient serum. Fig. 3c (left) shows marked loss of AQP4, GFAP and MBP immunofluorescence at 5 days after injection of 1 mg IgG purified from NMO patient serum, which was not seen in rats receiving the same amount of IgG from control (non-NMO) serum. A penumbra with decreased AQP4 immunoreactivity was seen as well. Because only a small fraction of serum IgG is NMO-IgG (AQP4-reactive), and because polyclonal NMO-IgGs bind less tightly to AQP4 than rAb-53 (which was selected for tight binding), substantially more serum IgG than rAb-53 was needed to produce comparable NMO pathology (as quantified in Fig. 3c, right).
NMO lesion formation is complement-dependent
The involvement of rat complement in our model was tested using a mutated NMO-IgG in which complement effector function was abolished, and, in separate studies, using cobra venom toxin to inactivate rat complement. The rAb-53 mutant S239D/A330L/I332E (NMO-IgGCDC-) was shown previously to lack CDC effector function, without impairment of ADCC effector function or AQP4 binding [40]. Rats were administered NMO-IgG by intracerebral injection in one hemisphere, with the same quantity of NMO-IgGCDC- in the contralateral hemiphere. Fig. 4a shows remarkably reduced lesion size (as seen by loss of AQP4, GFAP and myelin) in the brain hemisphere receiving the antibody lacking CDC effector function. Interestingly, the penumbra with decreased AQP4 immunoreactivity was observed in the hemisphere injected with NMO-IgGCDC-.
Figure 4. Generation of NMO lesions in rats is complement-dependent.
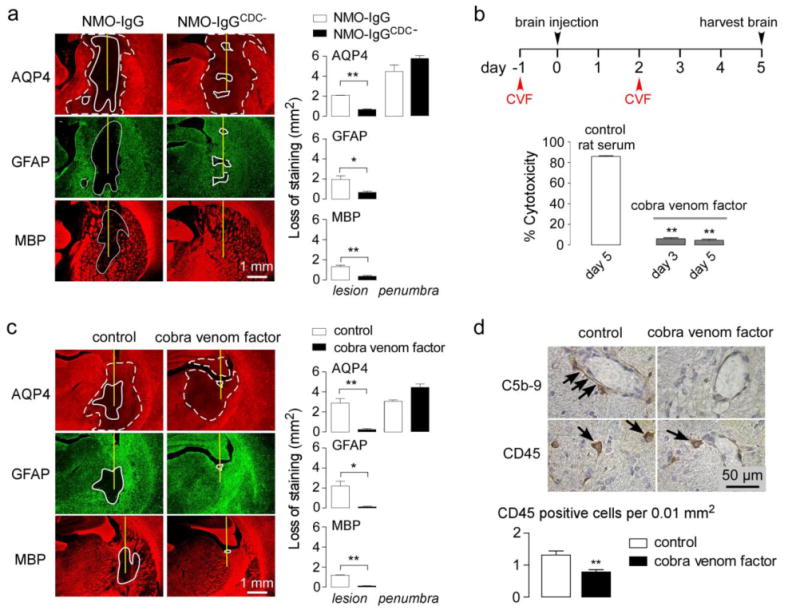
a. Brains were injected with 10 μg of NMO-IgG or NMO-IgGCDC- (lacking CDC effector function). (Left) AQP4, GFAP and MBP immunofluorescence at 5 days after injection. (Right) Summary of lesion areas (S.E., n=3, ** P < 0.01, * P<0.05). b. Cytotoxicity (by Alamar blue assay) in AQP4-expressing CHO cells incubated with 5 μg/ml NMO-IgG and 5% serum from control and cobra venom factor-treated rats (S.E., n=3, ** P< 0.01). c. (Left) AQP4, GFAP and MBP immunofluorescence at 5 days after injection of 10 μg NMO-IgG in control and cobra venom factor-treated rats. (Right) Summary of lesion areas (S.E., n=3, ** P < 0.01, * P<0.05). d. (Top) C5b-9 and CD45 immunohistochemistry. (Bottom) Number of CD45-positive cells per 0.01 mm2 (S.E., n=3, ** P< 0.01).
In another set of studies rat complement was inactivated by peripheral administration of cobra venom factor, a well-established approach to investigate the involvement of complement in disease pathogenesis [20,42]. Rats were injected with cobra venom factor at days -1 and 2, as diagrammed in Fig. 4b (top), and NMO-IgG was administered at day 0 by intracerebral injection. Assay of complement activity in rat serum (by CDC in NMO-IgG-treated AQP4-expressing cells) showed near zero complement activity at days 3 and 5 (Fig. 4B, bottom). Fig. 4c shows remarkably reduced lesion size at 5 days after NMO-IgG administration of NMO-IgG in the cobra venom toxin-treated rats compared with control rats. The penumbra area was seen in rats treated with cobra venom toxin. Fig. 4d shows near absence of activated complement (C5b-9) deposition in the penumbra of the cobra venom-treated rats, though leukocyte infiltration (CD45) was seen.
These data support the conclusion that complement is required for development of a full lesion but not for the penumbra.
Absence of a penumbra in mice administered NMO-IgG lacking CDC and ADCC effector functions
The pathogenic mechanisms involved in the penumbra were further investigated. Upon binding to its target, an antibody can mediate several effector functions, including target internalization, CDC and ADCC. The results above showed that administration of a mutant NMO-IgG lacking CDC effector function produced a penumbra. We also tested NMO-IgG made deficient in both CDC and ADCC effector functions by enzymatic deglysosylation using endoglycosidase S, as previously reported [39]. Binding of the deglycosylated antibody (NMO-IgGGL-) was similar to that of control NMO-IgG (Fig. 5a, top). NMO-IgGGL- did not produce CDC (Fig. 5a, bottom) or ADCC (not shown). Intracerebral injection of NMO-IgGGL- produced very minimal pathology around the needle tract compared to NMO-IgG, and, importantly, no penumbra was seen (Fig. 5b). To confirm these findings by an independent approach, we administered a mutant rAb-53 (NMO-IgGCDC-/ADCC-, ref [40]) lacking both CDC and ADCC effector functions. Injection of NMO-IgGCDC-/ADCC- in rat brain did not produce penumbra pathology (Fig. 5b). These results implicate the involvement of ADCC mediated in generation of the penumbra lesion.
Figure 5. Mechanisms of penumbra pathology.
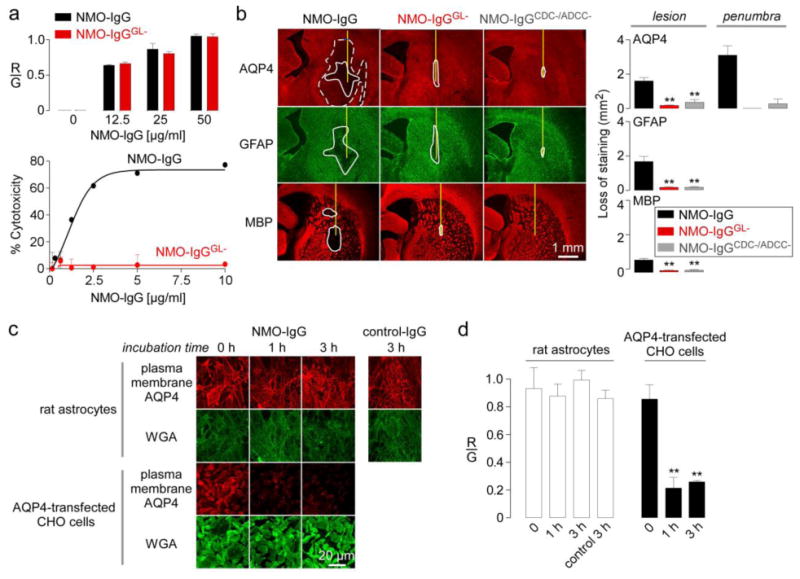
a. (Top) Binding of control and deglycosylated NMO-IgG (NMO-IgGGL-) to AQP4-expressing CHO cells showing red-to-green fluorescence ratio (R/G) at indicated NMO-IgG concentrations. (Bottom) Cytotoxicity (by Alamar blue assay) following incubation for 1 h with NMO-IgG or NMO-IgGGL- and 5% complement (S.E., n=3). b. AQP4, GFAP and MBP immunofluorescence at 5 days after injection with NMO-IgG, NMO-IgGGL- or NMO-IgGCDC-/ADCC-. (Right) Summary of lesion areas (S.E., n=3, ** P < 0.01). c. Internalization of AQP4 in rat primary astrocyte cultures. Cells were incubated for 1 or 3 h at 37 °C with 100 μg/ml NMO-IgG or control IgG. Surface AQP4 immunofluorescence shown in red, with plasma membrane marker (WGA) in green. AQP4 internalization in AQP4-expressing CHO cells shown as positive control. d. R/G ratios from experiments as in c (S.E., n=3, ** P < 0.01).
Similar pathology was reported in human NMO, with loss of AQP4 but intact GFAP and myelin immunofluorescence [24], in which the authors proposed a mechanism involving AQP4 internalization in response to NMO-IgG binding. We previously demonstrated that NMO-IgG binding did not cause internalization of AQP4 in primary cultures of mouse astrocytes and in mouse brain in vivo [29]. Studies were done here in rat astrocytes. Evidence against significant AQP4 internalization in rat astrocytes is the finding above (Fig. 5b) that intracerebral administration of NMO-IgGGL- or NMO-IgGCDC-/ADCC-, each of which bound as strongly to AQP4 as the original antibody, did not alter AQP4 expression. Further, surface AQP4 in rat primary astrocyte cultures was quantified in response to NMO-IgG incubation (Fig. 5c), as done previously in other culture models [29]. In this assay, cells were incubated for indicated times with NMO-IgG or a control IgG at 37 °C, washed, and cooled to 4 °C to inhibit internalization. Surface AQP4 on live cells was then labeled with NMO-IgG and a fluorescent secondary antibody (red), and plasma membrane was labeled with fluorescently labeled wheat germ agglutinin (WGA, green). Surface AQP4 was quantified by the ratio of red-to-green fluorescence. Fig. 5c shows similar surface AQP4 in control and NMO-IgG-treated cultures. Results are quantified in Fig. 5d. As a positive control for internalization, CHO cells stably expressing the M23 isoform of human AQP4 and incubated with NMO-IgG showed marked reduction in cell surface AQP4 (Figs. 5c, d).
Macrophage depletion in rats reduces NMO pathology
Human NMO lesions are characterized by prominent infiltration of granulocytes (neutrophils and eosinophils) and macrophages. Prior studies have implicated the involvement of neutrophils and eosinophils in NMO pathogenesis by ADCC and complement-dependent cell-mediated cytotoxicity (CDCC) mechanisms [18,35,48]. Like granulocytes, macrophages can produce ADCC and CDCC [37]. We showed previously that macrophages exacerbated NMO pathology in spinal cord slice cultures exposed to low concentrations of NMO-IgG and complement [47].
Here, to investigate the role of macrophages in vivo, NMO pathology was studied in rats following intraperitoneal injection of clodronate-containing liposomes, an established approach to deplete monocytes and macrophages [41]. Fig. 6a (left) shows a ∼ 4-fold reduction in serum monocyte count at 3 days following the second clodronate liposome injection. Neutrophil and eosinophil counts did not change significantly. Fig. 6a (right) shows depletion of peripheral macrophages as assessed by Iba1 immunofluorescence in liver.
Figure 6. Pathogenic role of macrophages in NMO.
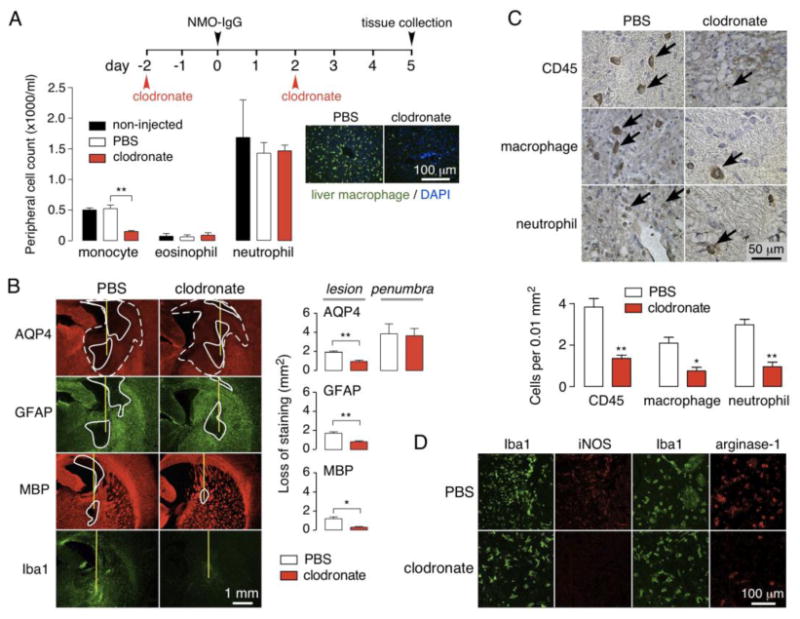
a. Macrophages were depleted by intraperitoneal injection of clodronate liposomes. (Left) Peripheral monocyte, eosinophil and neutrophil counts at day 5 in control (non-injected), and PBS-liposome and clodronate-liposome injected rats (S.E., n=3, ** P < 0.01). (Right) Liver sections immunostained for macrophages (green) with DAPI (blue) counterstain. b. AQP4, GFAP and MBP immunofluorescence at 5 days after intracerebral injection of 10 μg NMO-IgG in rat treated with control or clodronate liposomes. (Right) Summary of lesion areas (S.E., n=3, ** P < 0.01, * P < 0.05). c. (Top) Immunohistochemistry showing infiltrating leukocytes (arrows) in lesions. (Bottom) Number of CD45, CD163 and Ly-6G positive cells per 0.01 mm2 (S.E., n=3, ** P < 0.01, * P < 0.05). d. Iba1, M1 (M1 mac) and M2 (M2 mac) macrophage staining in rats treated with PBS- or clodronate-liposomes.
Fig. 6b shows reduced NMO pathology in the monocyte/macrophage-depleted rats compared to control rats at 5 days after intracerebral injection of NMO-IgG. The size of the main lesion, with loss of AQP4, GFAP and myelin, was reduced in the macrophage-depleted rats, whereas the penumbra remained. Leukocyte infiltration in the lesion was reduced in clodronate liposome-treated rats compared to control rats (Fig. 6c). Both M1 and M2 macrophages were present in the lesion at 5 days (Fig. 6d), with a predominance of the M2 sub-type as seen by co-staining with the macrophage markers Iba1, iNOS (M1) or arginase-1 (M2). Treatment with clodronate liposomes reduced both sub-types in the lesion.
Discussion
The major pathological features of human NMO, including loss of AQP4, GFAP and myelin, perivascular deposition of activated complement, blood-brain barrier disruption, and inflammation with granulocyte and macrophage infiltration and microglial activation, were produced in rat brain by passive transfer of NMO-IgG, without the need for pre-existing neuroinflammation or administration of human complement. In contrast to prior mouse models of NMO that require complement administration, NMO pathology in rats utilizes endogenous complement, as shown by the protective effect of complement inactivation by cobra venom toxin and of a CDC-disrupting NMO-IgG mutation. The robust and reproducible pathology in this model make it suitable for investigation of NMO-IgG-dependent pathogenesis mechanisms, such as the role of specific effector cells or cytokines, and for in vivo testing of potential therapeutics, such as small molecules or biologics targeting AQP4, NMO-IgG or complement. As the study design here was focused on pathogenic mechanisms triggered by NMO-IgG binding to AQP4, this model does not address the immune mechanisms triggering NMO-IgG generation or its access into the CNS, nor does it address the preferential localization of NMO pathology in spinal cord and optic nerve. Rat NMO models based on peripheral administration of NMO-IgG may address some of these questions.
An unanticipated and interesting finding was the presence of a penumbra around the central lesion, which, as seen in the central lesion, showed reduced AQP4 expression, but, in contrast to the central lesion did not show loss of GFAP or myelin, or complement deposition. Inactivation of rat complement by cobra venom factor or neutralization of antibody CDC effector function by NMO-IgG mutation produced selective AQP4 loss resembling that of the penumbra. However, complete neutralization of antibody CDC and ADCC effector functions by endoglycosidase S digestion or antibody mutation prevented all pathology, including the complement-independent lesion with selective AQP4 loss. These findings suggest that the penumbra lesion is dependent on the ADCC but not the CDC effector function of NMO-IgG.
The molecular-level mechanisms for selective loss of AQP4 and its relationship to ADCC are unclear. There is conflicting evidence that NMO-IgG-induced AQP4 endocytosis may be responsible for AQP4 loss in astrocytes [10]. However, work from our lab did not find significant NMO-IgG-induced AQP4 endocytosis and loss in mouse astrocyte cultures or live mice [29], and, here, in rat astrocyte cultures. Also, the absence of reduced AQP4 following administration of NMO-IgGs lacking CDC and ADCC effector functions argues against an endocytosis mechanism as accounting for the selective loss of AQP4. We previously reported that a pure ADCC mechanism produced by intracerebral injection of NMO-IgG and natural-killer cells in mice produced lesions with loss of AQP4 but not myelin [31]. Perhaps certain inflammatory mediators produced by infiltrating granulocytes or macrophages trigger accelerated degradation of AQP4 transcript or protein in astrocytes.
The penumbra lesion seen in our rat model likely has relevance to human NMO pathology and pathogenesis mechanisms. A recent study described multiple distinct pathological lesions in NMO patients, one of which (‘type 4 pathology’, ref. [24]) is a lesion with selective loss of AQP4 but not of GFAP or myelin. The lesions showed mild-to-moderate perivascular inflammatory infiltration but no complement deposition, as in the penumbra pathology here. With regard to NMO pathogenesis, the penumbra lesion supports a growing body of evidence implicating the involvement of ADCC in NMO. In addition to showing that a pure ADCC mechanism can produce NMO-like pathology as mentioned above [31], there is evidence for involvement of neutrophils and eosinophils in NMO pathogenesis as discussed just below. Also, we recently found that mice administered a mutated NMO-IgG lacking ADCC effector function but with enhanced CDC effector function, together with human complement, developed only minimal NMO pathology (Ratelade et al., unpublished results). We conclude that ADCC is an underappreciated mechanism in NMO pathogenesis and suggest ADCC as a potential target for development of a new class of NMO therapeutics.
The heterogeneous responses of astrocytes within the lesion may also play a role in NMO pathogenesis. After the initial attack by NMO-IgG and complement, astrocytes at the lesion core are injured by a CDC mechanism, which promotes inflammatory cell infiltration, oligodendrocyte damage and myelin loss. This mechanism is consistent with transgenic loss-of-function studies in which ablation of proliferating, reactive astrocyte produces marked tissue invasion by inflammatory cells, including macrophages and neutrophils, in both EAE and spinal cord injury models [5,7]. In the penumbra, GFAP-positive reactive astrocytes were present, myelin was preserved, and a lesser inflammatory reaction was seen. Perhaps these reactive astrocytes form a barrier-like structure surrounding the lesion that restricts leukocyte influx into CNS [43,44].
The rat model of NMO established here was used to investigate the role of macrophages in NMO pathogenesis. Granulocytes and macrophages are the major leukocyte types found in human NMO lesions, with few lymphocytes, cytotoxic T cells and natural killer cells [21,23,32,33]. Prior studies in mice have implicated the involvement of granulocytes, both neutrophils and eosinophils, in NMO pathogenesis. In a mouse model involving intracerebral injection of NMO-IgG and complement, NMO lesion size was reduced in mice made neutropenic with anti-neutrophil antibody, or made hypoeosinophilic with anti-IL-5 antibody or by genetic mutation of Gata1 promoter [35,48]. NMO lesion size was also reduced by inhibition of neutrophil elastase and by small-molecule inhibitors of eosinophil degranulation. NMO lesion size was increased in mice made neutrophilic by gm-GSF [35] and in hypereosinophilic IL-5 Tg mice [48]. In contrast, NMO lesions were not reduced in T-cell deficient (nude) mice [36]. These results suggested the potential utility of granulocyte-targeted inhibitors as adjunctive therapy in NMO. Here, we found that depletion of monocytes and macrophages (both M1 and M2 sub-types) in rats by clodronate liposomes reduced the severity of NMO pathology produced by intracerebral injection of NMO-IgG. Macrophages can exacerbate astrocyte damage by several mechanisms, including generation of oxidative metabolites (such as nitric oxide and superoxide) and of proinflammatory cytokines, as well as by phagocytosis, which can be enhanced by binding of complement components to receptors on macrophage [14,37].
In conclusion, our results establish a novel passive-transfer model of NMO in rats that does not require complement administration or pre-existing inflammation. The model was applied to establish a pathogenic role of macrophages in NMO, and the model produced an unexpected pathological lesion with relevance to human NMO.
Acknowledgments
This work was supported by grants EY13574, EB00415, DK35124, HL73856, DK86125 and DK72517 from the National Institutes of Health, and a grant from the Guthy-Jackson Charitable Foundation. We thank Dr. Jeffrey Bennett (Univ. Colorado Denver, Aurora, CO) for providing recombinant monoclonal NMO antibody and for Accelerated Cure (Waltham, MA) for providing human NMO sera.
Abbreviations
- ADCC
antibody-dependent cellular cytotoxicity
- AQP4
aquaporin-4
- CDC
complement-dependent cytotoxicity
- CDCC
complement-dependent cell-mediated cytotoxicity
- EAE
experimental autoimmune encephalomyelitis
- GFAP
glial fibrillary acidic protein
- MBP
myelin basic protein
- NMO
neuromyelitis optica
- NMO-IgG
neuromyelitis optica immunoglobulin G antibody
- WGA
wheat germ agglutinin
References
- 1.Aoyama M, Kakita H, Kato S, Tomita M, Asai K. Region-specific expression of a water channel protein, aquaporin 4, on brain astrocytes. J Neurosci Res. 2012;90(12):2272–2280. doi: 10.1002/jnr.23117. [DOI] [PubMed] [Google Scholar]
- 2.Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B. Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol. 2009;66(5):617–629. doi: 10.1002/ana.21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergman I, Basse PH, Barmada MA, Griffin JA, Cheung NK. Comparison of in vitro antibody-targeted cytotoxicity using mouse, rat and human effectors. Cancer Immunol Immunother. 2000;49(4-5):259–266. doi: 10.1007/s002620000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bodega G, Suarez I, Lopez-Fernandez LA, Almonacid L, Zaballos A, Fernandez B. Possible implication of ciliary neurotrophic factor (CNTF) and beta-synuclein in the ammonia effect on cultured rat astroglial cells: a study using DNA and protein microarrays. Neurochem Int. 2006;48(8):729–738. doi: 10.1016/j.neuint.2005.12.014. doi:S0197-0186(06)00004-0. [DOI] [PubMed] [Google Scholar]
- 5.Bush TG, Puvanachandra N, Horner CH, Polito A, Ostenfeld T, Svendsen CN, Mucke L, Johnson MH, Sofroniew MV. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23(2):297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 6.Crane JM, Lam C, Rossi A, Gupta T, Bennett JL, Verkman AS. Binding affinity and specificity of neuromyelitis optica autoantibodies to aquaporin-4 M1/M23 isoforms and orthogonal arrays. J Biol Chem. 2011;286(18):16516–16524. doi: 10.1074/jbc.M111.227298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Faulkner JR, Herrmann JE, Woo MJ, Tansey KE, Doan NB, Sofroniew MV. Reactive astrocytes protect tissue and preserve function after spinal cord injury. J Neurosci. 2004;24(9):2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frigeri A, Gropper MA, Turck CW, Verkman AS. Immunolocalization of the mercurial-insensitive water channel and glycerol intrinsic protein in epithelial cell plasma membranes. Proc Natl Acad Sci U S A. 1995;92(10):4328–4331. doi: 10.1073/pnas.92.10.4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frigeri A, Gropper MA, Umenishi F, Kawashima M, Brown D, Verkman AS. Localization of MIWC and GLIP water channel homologs in neuromuscular, epithelial and glandular tissues. J Cell Sci. 1995;108(Pt 9):2993–3002. doi: 10.1242/jcs.108.9.2993. [DOI] [PubMed] [Google Scholar]
- 10.Hinson SR, Pittock SJ, Lucchinetti CF, Roemer SF, Fryer JP, Kryzer TJ, Lennon VA. Pathogenic potential of IgG binding to water channel extracellular domain in neuromyelitis optica. Neurology. 2007;69(24):2221–2231. doi: 10.1212/01.WNL.0000289761.64862.ce. doi:01.WNL.0000289761.64862.ce. [DOI] [PubMed] [Google Scholar]
- 11.Jacob A, McKeon A, Nakashima I, Sato DK, Elsone L, Fujihara K, de Seze J. Current concept of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84(8):922–930. doi: 10.1136/jnnp-2012-302310. [DOI] [PubMed] [Google Scholar]
- 12.Jarius S, Paul F, Franciotta D, Waters P, Zipp F, Hohlfeld R, Vincent A, Wildemann B. Mechanisms of disease: aquaporin-4 antibodies in neuromyelitis optica. Nat Clin Pract Neurol. 2008;4(4):202–214. doi: 10.1038/ncpneuro0764. [DOI] [PubMed] [Google Scholar]
- 13.Jarius S, Wildemann B. AQP4 antibodies in neuromyelitis optica: diagnostic and pathogenetic relevance. Nat Rev Neurol. 2010;6(7):383–392. doi: 10.1038/nrneurol.2010.72. [DOI] [PubMed] [Google Scholar]
- 14.Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29(43):13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S. Neuromyelitis optica: Passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun. 2009;386(4):623–627. doi: 10.1016/j.bbrc.2009.06.085. [DOI] [PubMed] [Google Scholar]
- 16.Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S. Anti-aquaporin-4 antibody induces astrocytic cytotoxicity in the absence of CNS antigen-specific T cells. Biochem Biophys Res Commun. 2010;394(1):205–210. doi: 10.1016/j.bbrc.2010.02.157. [DOI] [PubMed] [Google Scholar]
- 17.Kira J. Autoimmunity in neuromyelitis optica and opticospinal multiple sclerosis: astrocytopathy as a common denominator in demyelinating disorders. J Neurol Sci. 2011;311(1-2):69–77. doi: 10.1016/j.jns.2011.08.043. [DOI] [PubMed] [Google Scholar]
- 18.Klos A, Tenner AJ, Johswich KO, Ager RR, Reis ES, Kohl J. The role of the anaphylatoxins in health and disease. Mol Immunol. 2009;46(14):2753–2766. doi: 10.1016/j.molimm.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med. 2005;202(4):473–477. doi: 10.1084/jem.20050304. doi:jem.20050304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levin ME, Jin JG, Ji RR, Tong J, Pomonis JD, Lavery DJ, Miller SW, Chiang LW. Complement activation in the peripheral nervous system following the spinal nerve ligation model of neuropathic pain. Pain. 2008;137(1):182–201. doi: 10.1016/j.pain.2007.11.005. doi:S0304-3959(07)00672-0. [DOI] [PubMed] [Google Scholar]
- 21.Lucchinetti CF, Mandler RN, McGavern D, Bruck W, Gleich G, Ransohoff RM, Trebst C, Weinshenker B, Wingerchuk D, Parisi JE, Lassmann H. A role for humoral mechanisms in the pathogenesis of Devic's neuromyelitis optica. Brain. 2002;125(Pt 7):1450–1461. doi: 10.1093/brain/awf151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, Park JK, Beck FX, Muller DN, Derer W, Goss J, Ziomber A, Dietsch P, Wagner H, van Rooijen N, Kurtz A, Hilgers KF, Alitalo K, Eckardt KU, Luft FC, Kerjaschki D, Titze J. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–552. doi: 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 23.Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y. Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain. 2007;130(Pt 5):1224–1234. doi: 10.1093/brain/awm047. doi:awm047. [DOI] [PubMed] [Google Scholar]
- 24.Misu T, Hoftberger R, Fujihara K, Wimmer I, Takai Y, Nishiyama S, Nakashima I, Konno H, Bradl M, Garzuly F, Itoyama Y, Aoki M, Lassmann H. Presence of six different lesion types suggests diverse mechanisms of tissue injury in neuromyelitis optica. Acta Neuropathol. 2013;125(6):815–827. doi: 10.1007/s00401-013-1116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17(1):171–180. doi: 10.1523/JNEUROSCI.17-01-00171.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Papadopoulos MC, Verkman AS. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012;11(6):535–544. doi: 10.1016/S1474-4422(12)70133-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papadopoulos MC, Verkman AS. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14(4):265–277. doi: 10.1038/nrn3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Phuan PW, Ratelade J, Rossi A, Tradtrantip L, Verkman AS. Complement-dependent cytotoxicity in neuromyelitis optica requires aquaporin-4 protein assembly in orthogonal arrays. J Biol Chem. 2012;287(17):13829–13839. doi: 10.1074/jbc.M112.344325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ratelade J, Bennett JL, Verkman AS. Evidence against cellular internalization in vivo of NMO-IgG, aquaporin-4, and excitatory amino acid transporter 2 in neuromyelitis optica. J Biol Chem. 2011;286(52):45156–45164. doi: 10.1074/jbc.M111.297275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ratelade J, Bennett JL, Verkman AS. Intravenous neuromyelitis optica autoantibody in mice targets aquaporin-4 in peripheral organs and area postrema. PLoS One. 2011;6(11):e27412. doi: 10.1371/journal.pone.0027412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ratelade J, Zhang H, Saadoun S, Bennett JL, Papadopoulos MC, Verkman AS. Neuromyelitis optica IgG and natural killer cells produce NMO lesions in mice without myelin loss. Acta Neuropathol. 2012;123(6):861–872. doi: 10.1007/s00401-012-0986-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF. Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain. 2007;130(Pt 5):1194–1205. doi: 10.1093/brain/awl371. doi:awl371. [DOI] [PubMed] [Google Scholar]
- 33.Saadoun S, Bridges LR, Verkman AS, Papadopoulos MC. Paucity of natural killer and cytotoxic T cells in human neuromyelitis optica lesions. Neuroreport. 2012;23(18):1044–1047. doi: 10.1097/WNR.0b013e32835ab480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133(Pt 2):349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saadoun S, Waters P, MacDonald C, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. Neutrophil protease inhibition reduces neuromyelitis optica-immunoglobulin G-induced damage in mouse brain. Ann Neurol. 2012;71(3):323–333. doi: 10.1002/ana.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saadoun S, Waters P, Macdonald C, Bridges LR, Bell BA, Vincent A, Verkman AS, Papadopoulos MC. T cell deficiency does not reduce lesions in mice produced by intracerebral injection of NMO-IgG and complement. J Neuroimmunol. 2011;235(1-2):27–32. doi: 10.1016/j.jneuroim.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Schlesinger LS, Horwitz MA. Phagocytosis of Mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1 (CD35), CR3 (CD11b/CD18), and CR4 (CD11c/CD18) and IFN-gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J Immunol. 1991;147(6):1983–1994. [PubMed] [Google Scholar]
- 38.Tradtrantip L, Asavapanumas N, Verkman AS. Therapeutic cleavage of anti-aquaporin-4 autoantibody in neuromyelitis optica by an IgG-selective proteinase. Mol Pharmacol. 2013;83(6):1268–1275. doi: 10.1124/mol.113.086470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tradtrantip L, Ratelade J, Zhang H, Verkman AS. Enzymatic deglycosylation converts pathogenic neuromyelitis optica anti-aquaporin-4 immunoglobulin G into therapeutic antibody. Ann Neurol. 2013;73(1):77–85. doi: 10.1002/ana.23741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol. 2012;71(3):314–322. doi: 10.1002/ana.22657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Meth. 1994;174(1-2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 42.Vogel CW, Fritzinger DC. Cobra venom factor: Structure, function, and humanization for therapeutic complement depletion. Toxicon. 2010;56(7):1198–1222. doi: 10.1016/j.toxicon.2010.04.007. [DOI] [PubMed] [Google Scholar]
- 43.Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci. 2009;29(37):11511–11522. doi: 10.1523/JNEUROSCI.1514-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV. Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci. 2013;33(31):12870–12886. doi: 10.1523/JNEUROSCI.2121-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol. 2007;6(9):805–815. doi: 10.1016/S1474-4422(07)70216-8. doi:S1474-4422(07)70216-8. [DOI] [PubMed] [Google Scholar]
- 46.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology. 2006;66(10):1485–1489. doi: 10.1212/01.wnl.0000216139.44259.74. [DOI] [PubMed] [Google Scholar]
- 47.Zhang H, Bennett JL, Verkman AS. Ex vivo spinal cord slice model of neuromyelitis optica reveals novel immunopathogenic mechanisms. Ann Neurol. 2011;70(6):943–954. doi: 10.1002/ana.22551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang H, Verkman AS. Eosinophil pathogenicity mechanisms and therapeutics in neuromyelitis optica. J Clin Invest. 2013;123(5):2306–2316. doi: 10.1172/JCI67554. [DOI] [PMC free article] [PubMed] [Google Scholar]