

Abstract
A decline in skeletal muscle mass and function with aging is well recognized, but remains poorly characterized at the molecular level. Here, we report for the first time a genome-wide study of DNA methylation dynamics in skeletal muscle of healthy male individuals during normal human aging. We predominantly observed hypermethylation throughout the genome within the aged group as compared to the young subjects. Differentially methylated CpG (dmCpG) nucleotides tend to arise intragenically and are underrepresented in promoters and are overrepresented in the middle and 3′ end of genes. The intragenic methylation changes are overrepresented in genes that guide the formation of the junction of the motor neuron and myofibers. We report a low level of correlation of gene expression from previous studies of aged muscle with our current analysis of DNA methylation status. For those genes that had both changes in methylation and gene expression with age, we observed a reverse correlation, with the exception of intragenic hypermethylated genes that were correlated with an increased gene expression. We suggest that a minimal number of dmCpG sites or select sites are required to be altered in order to correlate with gene expression changes. Finally, we identified 500 dmCpG sites that perform well in discriminating young from old samples. Our findings highlight epigenetic links between aging postmitotic skeletal muscle and DNA methylation.
Keywords: DNA methylation, skeletal muscle, human aging, epigenome, genomics, postmitotic
Introduction
Aging is accompanied by a reduction in muscle mass and function, which is commonly referred to as sarcopenia and leads to decreased mobility (Marzetti et al., 2009), with an estimated cost to the US economy of some 18.5 billion a year (Janssen et al., 2004). Although numerous different biochemical pathways have been linked with sarcopenia, the mechanisms that drive the development of this important age-related disorder remain to be uncovered. There have been several studies focused at the molecular level that have reported changes in skeletal muscle with age and exercise. However, these studies have primarily focused on the quantitation of RNA abundance (Melov et al., 2007; Drummond et al., 2011) or have not stringently controlled for confounding effects such as activity level, comorbidities, medications, and timing of the last exercise bout. In addition to measuring gene expression, the epigenetic modification of DNA cytosine plays important roles in many processes including differentiation and cellular senescence (Berdasco & Esteller, 2011). Aging cells, different tissue types as well as a variety of human diseases possess their own distinct DNA methylation profiles (Fernandez et al., 2012). Dysregulation of DNA methylation has also been reported with age-related disorders such as Alzheimer’s disease (Irier & Jin, 2012) and cancer (Lopez-Serra & Esteller, 2012).
Changes in DNA methylation with aging have also been reported in a select few tissues of human beings, as well as in a number of other species (Calvanese et al., 2009). One of the initial studies reported a decline in methylation in a number of aged mouse tissues (Wilson et al., 1987). Another early study observed promoter hypermethylation with age in human colorectal mucosa and argued that aging is a major contributing factor to hypermethylation in cancer of this tissue (Ahuja et al., 1998). A longitudinal study of DNA methylation in human blood cells reported that DNA methylation changes with age (Bjornsson et al., 2008). A recent report compared global hypomethylation of DNA from blood in centenarians with middle-aged individuals and newborns (Heyn et al., 2012). However, in general, molecular studies on aged tissues from well-characterized cohorts absent of potentially confounding age-related disorders are relatively uncommon. The majority of DNA methylation studies on aging have been carried out on blood comprised of multiple cell types. No studies have as of yet examined postmitotic skeletal muscle for epigenetic changes between young and old individuals. A minor surgical procedure is required to obtain such tissues, contributing to the difficulty in carrying out such studies.
Here, we report for the first time a genome-wide study of DNA methylation dynamics in a postmitotic tissue – skeletal muscle taken from healthy older adults compared to young adults under controlled conditions at over 480 000 sites throughout the genome. We report a predominant pattern of hypermethylation in the DNA of aging skeletal muscle. Surprisingly, the locations of these dmCpG sites are underrepresented in promoter regions and are overrepresented in the middle and 3′ ends of genes. These epigenetic changes could be very significant to the biology of aging muscle as recent studies have suggested that age-related loss of muscle mass is not due to the loss of muscle cells, but a reduction in type II fiber size (Nilwik et al., 2013). In comparing our data with prior gene expression studies, we did not observe a general correlation of DNA methylation with differentially expressed genes in aged human skeletal muscle. Finally, using a novel approach to identify dmCpGs associated with aging, we have identified over 500 methylated sites that separate younger subjects from older subjects in this study.
Results
Distribution of differentially methylated CpG sites
We have studied DNA methylation dynamics in skeletal muscle taken from 24 healthy older male adults (age range: 68–89 years) compared to 24 young male adults (age range: 18–27 years) under controlled conditions at 485 577 sites throughout the genome (Illumina 450 K methylation arrays). We found an overall global trend of genomic hypermethylation in human skeletal muscle between the young and old groups (Fig. 1). We identified 5963 CpG sites that are differentially methylated between the two groups (dmCpG). Of these, 92% (5518 dmCpG sites) were hypermethylated in the older subjects, while the remaining 8% were hypomethylated (Table S1, Supporting Information). We observed that the distribution of dmCpG sites was relatively even over all chromosomes, with exceptions of a significant overrepresentation of dmCpGs in chromosomes 16, 17 and a relative underrepresentation in the X chromosome (Table S2, Supporting Information). For both hyper- and hypomethylated dmCpG sites, there was a significant enrichment within a gene and there was an underrepresentation outside of genes (Fig. 2A, Table S2). In order to further refine positional information about dmCpGs with age relative to gene structure, we segmented genes into the following structural regions: the 5′ region, the ‘central’ region, the 3′ region, and regions around the transcription start, and end sites (TSS and TES). DmCpG sites within genes could be divided into two broad regions – those that were underrepresented near the 5′ regions of genes and those that were enriched in the central and 3′ regions (Fig. 2B, Table S2).
Figure 1.
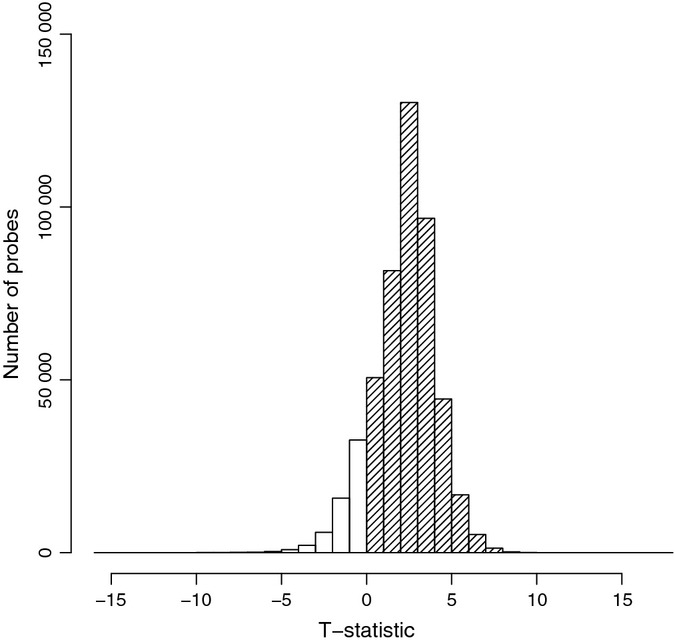
Changes in methylation status with age. Distribution of the t-statistic shows a significant hypermethylation of DNA derived from skeletal muscle of older versus younger adults (distribution is shifted to the right). Y-axis: number of CpG probes; x-axis: t-statistic. Filled columns are hypermethylated with age, and uncolored columns hypomethylated with age.
Figure 2.
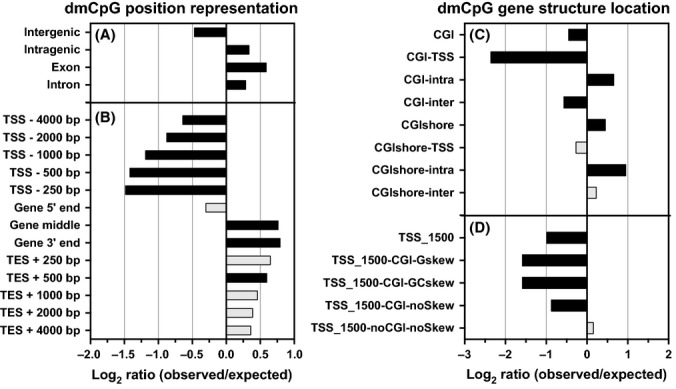
Distribution of dmCpG. Log(2) ratio of observed [fraction of dmCpG from all CpG sites on the microarrays (aCpG) in a region] to expected (fraction of dmCpG from aCpG in genome) for a region. Black color indicates a significant overrepresentation (P < 0.05) of dmCpG. The positional categories of dmCpG contain the location of dmCpG sites in relation to specific sequence features in the genome. The number following TSS/TES refers to the number of nucleotides upstream and downstream of TSS/TES. A detailed regional description and the breakdown of the hyper- and hypomethylated site observed/expected ratios can be found in Table S12 (Supporting Information) and Table S2.
CpG islands (CGI) are present in 60% of gene promoters in the 450 K chip, and methylation deregulation in CGI overlapping the promoter has often been linked with cancer (Draht et al., 2012). Other relevant features of note in the methylated DNA of cancer are increases in the inter- and intragenic CGI (Deaton et al., 2011) and CGI shores (Irizarry et al., 2009). In order to examine whether a similar phenomenon arises in aged human skeletal muscle, we tested the association between methylation and age in CGI and CGI shores (2000 base pairs upstream and downstream of a CGI) that overlap with promoters, and intra-/intergenic regions. We determined that in aging human skeletal muscle, there is an overall enrichment of dmCpG sites in CGI shores and there is a decrease in CGI, except for CGI within a gene (Fig. 2C, Table S2). All data including methylation values/subject, and methylated vs. unmethylated probe data, are deposited in GEO and are accessible via the GEO number GSE50498.
Recently, it was determined that an R-loop (a three-stranded nucleic acid structure formed due to biased strand distribution of guanines and cytosines, or GC-skew) negatively correlates with the corresponding promoter methylation level (Ginno et al., 2012). For each promoter region (−1500 +1500 bp around TSS), we defined those that have or do not have CGI or GC-skew, or both. We identified that there was an underrepresentation of dmCpG sites in all promoters, unless it is a promoter with no CGI and nonskew (Fig. 2D, Table S2). Ginno et al. reported that promoters with CGI and positive GC-skew are the most protected from methylation, while as promoters with weak CGI and weak GC-skew showed little to no protection. Concordant with these results, we report that less protected promoters in DNA of aged skeletal muscle tend to acquire more methylation than the younger tissue.
As a final analysis of the positional information of the dmCpG sites within aged skeletal muscle, we performed an analysis for possible enrichment of differential methylation of transcriptional regulator-binding sites. Using the ENCODE ChIP-Seq Significance Tool, we were able to probe existing datasets for enriched transcription factors within our gene list (Auerbach et al., 2013). We used all skeletal muscle cell lines (Hsmm, Hsmmt) available within the ENCODE project and then analyzed gene sets from our data that have at least 2, 4, or 8 dmCpG within their gene boundaries. In all three cases, the only significantly enriched motif (P < 0.01) was the transcription factor CTCF (Table S3, Supporting Information).
Genes with altered methylation status within aged muscle tissue
We identified 2114 genes with at least one dmCpG site located intragenically (Table S4, Supporting Information) in DNA from aged skeletal muscle. The location of these dmCpG sites may be significant, as methylation changes in aged individuals may affect gene function (Maunakea et al., 2010) and consequently muscle function. We observed that the number of dmCpG sites for a gene was not dependent on the total number of CpG sites present on the microarray chip for the same gene (Pearson’s correlation = 0.55, and Fig. S1, Supporting Information). One example of a gene with dmCpG that is affected by aging is the tubulin-folding cofactor D (TBCD) gene (Fig. 3). Tubulin-folding cofactor D has the highest number of intragenic dmCpG sites (46 distinct sites, which is 13.2% of the total number of CpG sites present for the gene). Of these, 45 of them were hypermethylated in the aged group compared to the young muscle tissue, a significant enrichment in the older age group. As an important component of microtubules, TBCD plays a key role in all cell types, and disruption of microtubule pathways was previously reported to affect motor neuron function (Molon et al., 2004). Another interesting example of dmCpG within the older age group is the gene for pericentrin, with three dmCpG sites. We report that all three of these sites were hypomethylated with age (4.1% of total), and expression of this gene has also been reported to be increased with age (Melov et al., 2007). Like TBCD, the pericentrin protein is a part of the cytoskeleton, and it plays an important role in microtubule assembly and reorganization during muscle differentiation (Bugnard et al., 2005).
Figure 3.

Schematic distribution of CpG islands (CGI), GC-skew, and CpG sites within TBCD gene. This figure was generated using the Genome Browser (Kent et al., 2002). GC-skew track: Gskew on positive strand marked with green; Gskew on negative strand marked with brown. Methylation (meth) track: CpG sites that are present on the arrays are colored black; dmCpG sites are colored red.
Ontology and pathway enrichment analysis
In order to better understand the biological significance of dmCpG changes in aged skeletal muscle, we applied STOP, an ontology enrichment tool (Wittkop et al., 2013), to genes with two or more intragenic dmCpG sites. Only one enriched term was found from cell type ontology analysis, the term ‘muscle cell’ (P = 0.0004). This suggests that the dmCpG sites we identified within the aged group are relevant to muscle tissue. There were 54 enriched terms with a P < 0.01 (Table S5, Supporting Information). This list includes muscle-related terms (two terms) and signal transduction/intracellular transport (25 terms), such as axon (P = 2.05E-07) and axon guidance (P = 2.11E-07), which may be related to muscle innervation and aging. A decline of function in the neuromuscular junction has long been thought to contribute to the decline of muscle mass with age (Bütikofer et al., 2011). Actin cytoskeleton (P = 3.89E-05), cytoskeleton (P = 0.0003), and cytoskeleton organization (P = 0.001) are all linked to cytoskeleton function, and the latter plays an important role in proper muscle contractile function (Berthier & Blaineau, 1997). Other significantly identified terms could be put together into the cell adhesion/motility group (six terms), including plasma membrane (P = 3.23E-08), homophilic cell adhesion (P = 8.43E-08), and cell adhesion (P = 1.70E-05), with cell adhesion being associated with fiber degeneration (Campbell, 1995). Additionally, we observed several terms that could be gathered into a growth and differentiation group (six terms), ion binding (two terms), broad cellular function (11 terms), and two terms that stand separately.
We further expanded our analysis by examining the overrepresentation of canonical genetic pathways specific to muscle tissue using ingenuity pathway analysis (www.ingenuity.com) (Table S6, Supporting Information). This analysis further expanded on the STOP ontology evaluation by identifying ‘axon guidance signaling’ as the top pathway within the dmCpG dataset (P = 6.16E-10) with 50 of 216 pathway members (23.1%) differentially methylated in at least one intragenic site (Table S7 and Fig. S2, Supporting Information).
A recent report utilized genes identified as having dmCpGs with age to examine stem cell differentiation pathways (West et al., 2013). We compared our results from postmitotic skeletal muscle to their findings of the best age predictor in whole blood – the UTF1 gene, which is also hypermethylated with age. We did not find any dmCpG sites within or around this gene in DNA obtained from skeletal muscle. We also did not observe dmCpG sites in/around the other age predictors WNT3A, SFRP1, FZD2, or FZD9 previously reported for whole blood. Another study of DNA methylation of whole blood and aging found a hypermethylation of bivalent chromatin domains with increasing age (Rakyan et al., 2010). The comparison of the sites from that study and our current study of skeletal muscle found that there was a significant overlap in genes that contain both dmCpG sites in bivalent chromatin domains (hypergeometric test, P = 7E-33).
Comparison of DNA methylation state with gene expression
Next, we compared the differentially methylated sites identified in DNA from aged skeletal muscle with a previously reported study on gene expression using a similar experimental design, which was focused on healthy older adults absent age-related disease (Melov et al., 2007). We could not carry out gene expression and genome-wide methylation on all samples, as the amount of material obtained via needle biopsy was limited at the time we carried out the study. We wished to determine whether there was any relationship between gene expression in healthy aged skeletal muscle and the level of DNA methylation. We identified suites of genes that have a minimum number of differentially methylated CpG sites (1, 2, 4, 8, and 16) within specific regions, for example intragenic, 5′ end, or CGI-TSS. If a gene has only hypermethylated or hypomethylated sites, it was designated as such. We then calculated the number of genes from either hypo- or hypermethylated groups that overlap with genes that were previously reported to be overexpressed or underexpressed within aged muscle tissue, as well as genes that did not change in expression between young and old subjects. We determined that the majority of genes with altered methylation in DNA from healthy aged human skeletal muscle are characterized by unchanged gene expression within aged tissue (Table S8, Supporting Information).
We then focused only on genes that were significantly altered in the aged group as compared to the young group, and calculated how changes in gene expression correlate with changes in DNA methylation. We found that a majority of hypomethylated genes had an increase in gene expression with age, while hypermethylated genes displayed a reduction in gene expression (Fig. 4). Only for dmCpG sites in the 3′ ends of genes did we observe a positive concordance between hypermethylation and up-regulation of gene expression, which has also been reported by other groups in differing biological contexts (Hellman & Chess, 2007; Feng et al., 2010). These correlations were especially strong with increasing numbers of dmCpG sites within a gene or gene region. These results suggest that a minimal number of dmCpG sites or select sites are required to be altered in order to inversely modulate the gene expression in aging skeletal muscle. Similar results were also reported for different tissues in another methylation study (Weber et al., 2007).
Figure 4.
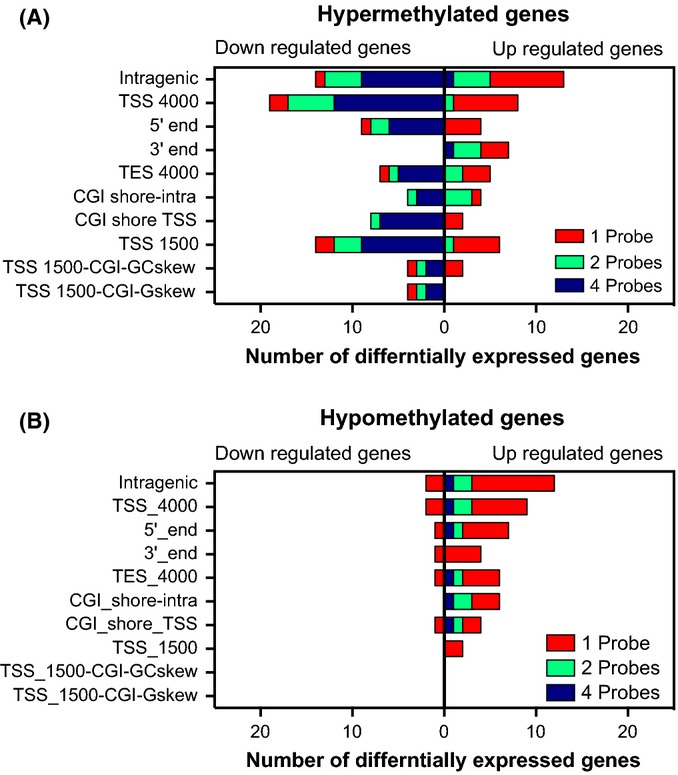
Correspondence of hyper- and hypo-dmCpG in genes with changes in gene expression. Number of genes (x-axis) that have a minimal number of hypermethylated (A) or hypomethylated dmCpG (B). The data are displayed as a stacked bar chart for one probe (red), two probes (green), or four probes (blue) for each genome position category. The expression change in aged tissue is indicated as bars. Left of axis is down-regulated and bars to the right are up-regulated. Detailed description of genome position categories is described in Table S12.
Differential methylation sites with age
From all dmCpG in DNA from aged skeletal muscle, there are 500 high probability predicting CpG sites (Table S9, Supporting Information) and 21 CpG sites that are universally changed in either the young or old age groups’ CpG predictors (i.e., either all increased in all old samples, and all decreased in all young samples, or the inverse) (Fig. 5). We used a procedure in which cross-validation was used to estimate the misclassification rate. It also incorporates simple stepwise constant functions as candidates and will result in no misclassified samples (see methods for a more detailed description). A majority of these sites are increased in methylation within the aged tissue, although some do decrease. Any reasonable prediction procedure that looks for simple cutoffs using even one probe comparing young samples to old samples would have found probes that completely cross-validated (unbiased) in terms of classifying young from old in 21 of the sites, and nearly completely separated young from old for at least 500. Three probes of these 21 sites are excellent at distinguishing young from old, and nine of 500 of these excellent/good predictors were also determined as age-predicting markers by a recent study on DNA methylation in blood (Hannum et al., 2012) (Table S10, Supporting Information). However, we stress that such sites would need to be validated in a completely independent population in order to verify their biological significance, although it remains statistically highly unlikely that these 21 probes are differentially methylated between the two age groups by chance alone.
Figure 5.
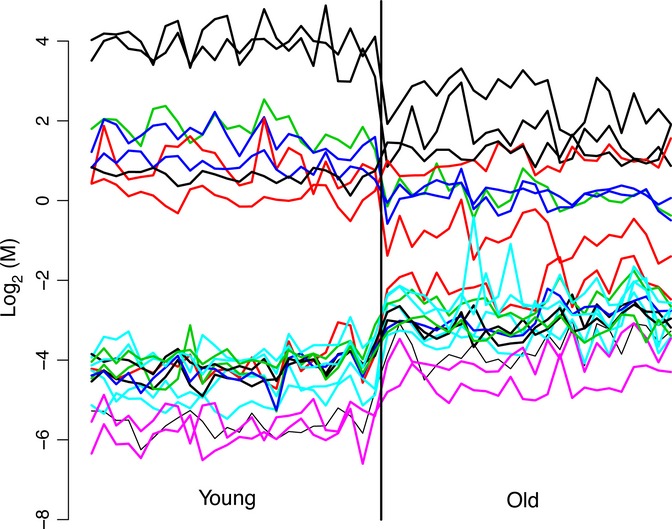
Methylation level of 21 CpG probes with M-values ranked by age. Individual methylation levels of 21 CpG sites that distinguish young from old subjects.
Discussion
We report for the first time a genome-wide survey of DNA methylation status of skeletal muscle DNA from healthy younger versus older male individuals. We determined that differentially methylated CpG nucleotides predominantly arise intragenically and are underrepresented in promoters and are overrepresented in the middle and 3′ end of genes. We also identified a number of sites that discriminated with high confidence the younger subjects from older subjects.
Historically, it has long been known that DNA methylation changes with age to varying extents, dependent upon the sites being assayed within the genome, species, tissue, or cell (Calvanese et al., 2009). Several recent studies have reported genome-wide increases in DNA hypermethylation with age in cells from whole blood or skin (Grönniger et al., 2010; Rakyan et al., 2010; Bell et al., 2012), saliva (Bocklandt et al., 2011), and brain (Hernandez et al., 2011). Other studies have reported substantial decreases in DNA methylation with age in blood (Teschendorff et al., 2010; Heyn et al., 2012). These genome-wide studies have used a variety of approaches to cover the genome to a greater or lesser extent, and true genome-wide coverage at the level of the single base is fast approaching. We have compared the dmCpG sites we identified in postmitotic human skeletal muscle to the reported dmCpG sites from six other studies of different tissues. We did not find any common dmCpG site with age in skeletal muscle (this study) and brain (Hernandez et al., 2011). With four of these (Rakyan et al., 2010; Teschendorff et al., 2010; Bocklandt et al., 2011; Bell et al., 2012), there were only 2–6 dmCpG sites in common (Table S11, Supporting Information). We identified 141 concordant dmCpG sites between aged skeletal muscle and blood (Heyn et al., 2012) (Fig. S11, Supporting Information). The technology for identifying dmCpG sites has rapidly evolved over the last few years, progressing from relatively low-throughput assessment of single sites of interest to more than 450 thousand (450 K) sites throughout the genome. We and others (Heyn et al., 2012) employed the 450 K Illumina array technology to identify differentially methylated sites with age, while other studies (Rakyan et al., 2010; Teschendorff et al., 2010; Bocklandt et al., 2011; Bell et al., 2012) have used the smaller 27 K Illumina platform, surveying < 6% of the total sites present on the 450 K chip. The difference in the number of methylation sites being surveyed between the 27 K and 450 K formats could be a major contributory factor with regard to a lack of concordance between studies, because only three CpG sites from 141 dmCpGs we identified were also present on the smaller format 27 K Illumina array chip. Interestingly, one CpG site was differentially methylated with age in three studies (including this study), another two CpG sites in four studies, and one CpG in five studies. Each of the methylation sites of interest is located intragenically close to the 5′ end of four genes. The genes containing these CpG sites are CELF6, NHLRC1, CECR6, and INSM2. There is little functional information known about CECR6 or INSM2; however, NHLRC1 encodes the subunit E3 ubiquitin ligase and is associated with Lafora epilepsy (Salar et al., 2012). CELF6 is a member of CELF protein family, which carries an RNA-binding domain.
For genes with two or more intragenic dmCpG sites identified in DNA from aged skeletal muscle, we have identified several associated gene ontology terms and pathways which were enriched within the aged group versus the young subjects, including ‘axon guidance signaling’. Differential methylation changes in this pathway were used as a focus to identify how epigenetic changes during aging could potentially relate to the well-known loss of skeletal muscle function with increasing age. Motor unit loss (denervation) occurs with aging and is a major contributory factor in sarcopenia (Doherty et al., 1993). Therefore, regulation and signaling at the interphase between the nervous system and the musculature is of critical importance during aging to guide the motor neuron to the muscle fiber. Within the ‘axon guidance pathway’ ontology, the gene with the highest number of intragenic dmCpG sites was the NFATC1 (nuclear factor of activated T cells, cytoplasmic 1 gene). This gene contained 17 dmCpG sites in aged skeletal muscle: 16 hypermethylated and one hypomethylated (Table S7). NFATC1 has an established role in transcriptional regulation of skeletal muscle in direct response to electrical stimulation via calcium/calmodulin signaling (McCullagh et al., 2004; Rana et al., 2008). NFATC1 regulates both signaling at the neuromuscular junction and acts as a nuclear transcriptional regulator of muscle fibers relating information about the contractile electrical impulses entering the cell via motor neurons (Salanova et al., 2011). Methylation changes in the DNA for this gene and others within the axon guidance signaling pathway may alter transcriptional events leading to the loss of proper reinnervation at the neuromuscular junction during muscle turnover. The loss of this plasticity mechanism has been shown to be a critical step preceding muscle wasting in both animal models and humans (Lauretani et al., 2006; Chai et al., 2011; Jang & Van Remmen, 2011). Therefore, the intragenic differential methylation data we identified in DNA from aged skeletal muscle provide potential candidate genes for investigating the role of denervation of the neuromuscular junction loss prior to age-related muscle loss and sarcopenia. This provides a potential functional linkage between epigenetic changes in aged muscle tissue and loss of muscle mass.
In aging human skeletal muscle, we identified a strong preference for the dmCpG sites to localize within a gene, and in the central and 3′ end regions of genes, but surprisingly no preference for the promoter. Some studies have suggested that intragenic DNA methylation could regulate alternative splicing (Sati et al., 2012) and may be involved in the regulation of alternative splicing in differentiation and cancer (Irizarry et al., 2009). These sites may be also important for gene regulation as they can potentially modulate alternative promoter activity (Maunakea et al., 2010). We also identified similar results within CGI, where dmCpGs are overrepresented in intragenic CGI and underrepresented in CGI that overlap the transcriptional start site. Another possible reason for the preferential distribution of dmCpG within DNA of aging skeletal muscle may relate to a potential protective mechanism, where some areas of the genome are more vulnerable to differential methylation than others. Partial evidence for this hypothesis is provided by the observation that promoters without CGI were overrepresented with dmCpG compared to the CGI promoters. It has been previously reported that weak CGI promoters tend to be de novo methylated preferentially in somatic cells during differentiation (Weber et al., 2007). Promoters with CGI and positive GC-skew are more protected from methylation and de novo methylation; CGI and GC-skew promoters, however, are less protected from methylation; and finally, promoters with weak CGI and week GC-skew showed little to no protection (Ginno et al., 2012). The overall conclusions of these prior studies on targeted protection are concordant with the observations we report here on DNA from aging human skeletal muscle, where we found less than expected dmCpG events in CGI-positive GC-skew promoters compared to promoters without CGI and CG-skew.
A potential issue with the biopsy procedure used in our study is tissue heterogeneity between biopsies, which may impact the resultant molecular signatures. There are other cell types in muscle that might contaminate muscle-specific methylation signatures. Such sample variation may influence the results. However, it was previously demonstrated that there is remarkably little technical variation between discrete biopsies sampled from the right and left leg of the same individual with regard to fiber type differences, gene expression as assessed by microarray, fiber size, or even strength (Tarnopolsky et al., 2007).
In summary, we describe for the first time a genome-wide DNA methylation survey of a postmitotic tissue between young and old healthy human skeletal muscle. We also determined that there were several hundred robust dmCpGs that discriminated younger males from older males. Ontology analysis of the terms associated with dmCpGs provided several interesting hints of a potential role for epigenetic changes in the neuromuscular junction during aging. Resistance exercise can reverse specific aspects of the gene expression changes in aging to more youthful levels (Melov et al., 2007). Acute endurance exercise has also been shown to decrease promoter methylation in human skeletal muscle (Barrès et al., 2012). Collectively, this prior work in combination with the data we report here shows a dynamic inter-relationship between DNA methylation, gene expression, age, and exercise. With exercise, gene expression and DNA methylation can both be ‘reversed’, emphasizing the importance of determining the mechanisms of the beneficial effects of exercise. Future studies will be needed to determine whether resistance exercise alters DNA methylation and can reverse the age-specific methylation profiles we report here.
Experimental procedures
All methods and experimental procedures are available in online supplement.
Acknowledgments
This work was supported in part by NIH R01 LM009722 (SDM), NIH U54-HG004028 (SDM), NIH UL1DE019608 supporting the Interdisciplinary Research Consortium on Geroscience (SM), the Glenn Foundation for Medical Research (SM), and the Buck Trust. Grant support for Dr. Tarnopolsky for this work was supported by CIHR – Institute of Aging. We would like to thank Brittany Garret for technical support and Thomas Down and Vardhman Rakyan for kindly sharing the list of genes with bivalent chromatin domains.
Author contributions
SM, AZ, MFF, and MT conceived and carried out the experiments. AH, AZ, JMF, and SM analyzed the data. CK, SDM, and DO provided material and logistic support. All authors edited the manuscript.
Funding
No funding information provided.
Conflict of interest
The authors declare that they have no competing interests.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
Fig. S1 Numbers of CpGs present on array in a gene and dmCpG site for same gene.
Fig. S2 Ingenuity canonical axon guidance pathway.
{kind=link}
Fig. S3 dmCpG sites that are common for this study and Heyn et al.
Table S1 List of differentially methylated CpG probes with age.
Table S2 Distribution of dmCpG.
Table S3 ENCODE ChIP-Seq significance tool results.
Table S4 Genes with at least one intragenic dmCpG site.
Table S5 Ontology enrichment analysis.
Table S6 Muscle-specific canonical pathway analysis.
Table S7 Differential methylation of axon guidance genes.
Table S8 Correspondence of intragenic differential methylation to gene expression.
Table S9 List of CpG sites that discriminate young from old samples.
Table S10 List of overlapping CpG predictors of biological age with Hannum et al. study.
Table S11 Number of overlapping dmCpG sites with other studies.
Table S12 Description of genomic regions.
Table S13 Description of human subjects.
Table S14 Illumina 450 K data quality control report.
References
- Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. [PubMed] [Google Scholar]
- Auerbach RK, Chen B, Butte AJ. Relating genes to function: identifying enriched transcription factors using the ENCODE ChIP-Seq significance tool. Bioinformatics. 2013;29:1922–1924. doi: 10.1093/bioinformatics/btt316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrès R, Yan J, Egan B, Treebak JT, Rasmussen M, Fritz T, Caidahl K, Krook A, O’Gorman DJ, Zierath JR. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012;15:405–411. doi: 10.1016/j.cmet.2012.01.001. [DOI] [PubMed] [Google Scholar]
- Bell JT, Tsai PC, Yang TP, Pidsley R, Nisbet J, Glass D, Mangino M, Zhai G, Zhang F, Valdes A, Shin SY, Dempster EL, Murray RM, Grundberg E, Hedman AK, Nica A, Small KS, Dermitzakis ET, McCarthy MI, Mill J, Spector TD, Deloukas P. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012;8:e1002629. doi: 10.1371/journal.pgen.1002629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berdasco M, Esteller M. DNA methylation in stem cell renewal and multipotency. Stem Cell Res. Ther. 2011;2:42. doi: 10.1186/scrt83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthier C, Blaineau S. Supramolecular organization of the subsarcolemmal cytoskeleton of adult skeletal muscle fibers. A review. Biol. Cell. 1997;89:413–434. doi: 10.1016/s0248-4900(97)89313-6. [DOI] [PubMed] [Google Scholar]
- Bjornsson HT, Sigurdsson MI, Fallin MD, Irizarry RA, Aspelund T, Cui H, Yu W, Rongione MA, Ekström TJ, Harris TB, Launer LJ, Eiriksdottir G, Leppert MF, Sapienza C, Gudnason V, Feinberg AP. Intra-individual change over time in DNA methylation with familial clustering. JAMA. 2008;299:2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocklandt S, Lin W, Sehl ME, Sánchez FJ, Sinsheimer JS, Horvath S, Vilain E. Epigenetic predictor of age. PLoS One. 2011;6:e14821. doi: 10.1371/journal.pone.0014821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugnard E, Zaal KJ, Ralston E. Reorganization of microtubule nucleation during muscle differentiation. Cell Motil. Cytoskeleton. 2005;60:1–13. doi: 10.1002/cm.20042. [DOI] [PubMed] [Google Scholar]
- Bütikofer L, Zurlinden A, Bolliger MF, Kunz B, Sonderegger P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011;25:4378–4393. doi: 10.1096/fj.11-191262. [DOI] [PubMed] [Google Scholar]
- Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Res. Rev. 2009;8:268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Campbell KP. Three muscular dystrophies: review loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. doi: 10.1016/0092-8674(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Chai RJ, Vukovic J, Dunlop S, Grounds MD, Shavlakadze T. Striking denervation of neuromuscular junctions without lumbar motoneuron loss in geriatric mouse muscle. PLoS One. 2011;6:e28090. doi: 10.1371/journal.pone.0028090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deaton AM, Webb S, Kerr AR, Illingworth RS, Guy J, Andrews R, Bird A. Cell type-specific DNA methylation at intragenic CpG islands in the immune system. Genome Res. 2011;21:1074–1086. doi: 10.1101/gr.118703.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty TJ, Vandervoort AA, Taylor AW, Brown WF. Effects of motor unit losses on strength in older men and women. J. Appl. Physiol. 1993;74:868–874. doi: 10.1152/jappl.1993.74.2.868. [DOI] [PubMed] [Google Scholar]
- Draht MX, Riedl RR, Niessen H, Carvalho B, Meijer GA, Herman JG, van Engeland M, Melotte V, Smits KM. Promoter CpG island methylation markers in colorectal cancer: the road ahead. Epigenomics. 2012;4:179–194. doi: 10.2217/epi.12.9. [DOI] [PubMed] [Google Scholar]
- Drummond MJ, McCarthy JJ, Sinha M, Spratt HM, Volpi E, Esser KA, Rasmussen BB. Aging and microRNA expression in human skeletal muscle: a microarray and bioinformatics analysis. Physiol. Genomics. 2011;43:595–603. doi: 10.1152/physiolgenomics.00148.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Cokus SJ, Zhang X, Chen PY, Bostick M, Goll MG, Hetzel J, Jain J, Strauss SH, Halpern ME, Ukomadu C, Sadler KC, Pradhan S, Pellegrini M, Jacobsen SE. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl Acad. Sci U S A. 2010;107:8689–8694. doi: 10.1073/pnas.1002720107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC, Galm O, Ferrer I, Sanchez-Cespedes M, Villanueva A, Carmona J, Sanchez-Mut JV, Berdasco M, Moreno V, Capella G, Monk D, Ballestar E, Ropero S, Martinez R, Sanchez-Carbayo M, Prosper F, Agirre X, Fraga MF, Graña O, Perez-Jurado L, Mora J, Puig S, Prat J, Badimon L, Puca AA, Meltzer SJ, Lengauer T, Bridgewater J, Bock C, Esteller M. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012;22:407–419. doi: 10.1101/gr.119867.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012;45:814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönniger E, Weber B, Heil O, Peters N, Stäb F, Wenck H, Korn B, Winnefeld M, Lyko F. Aging and chronic sun exposure cause distinct epigenetic changes in human skin. PLoS Genet. 2010;6:e1000971. doi: 10.1371/journal.pgen.1000971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, Deconde R, Chen M, Rajapakse I, Friend S, Ideker T, Zhang K. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell. 2012;49:359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- Hernandez DG, Nalls MA, Gibbs JR, Arepalli S, van der Brug M, Chong S, Moore M, Longo DL, Cookson MR, Traynor BJ, Singleton AB. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Li N, Ferreira HJ, Moran S, Pisano DG, Gomez A, Diez J, Sanchez-Mut JV, Setien F, Carmona FJ, Puca AA, Sayols S, Pujana MA, Serra-Musach J, Iglesias-Platas I, Formiga F, Fernandez AF, Fraga MF, Heath SC, Valencia A, Gut IG, Wang J, Esteller M. Distinct DNA methylomes of newborns and centenarians. Proc. Natl Acad. Sci. U S A. 2012;109:10522–10527. doi: 10.1073/pnas.1120658109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irier HA, Jin P. Dynamics of DNA methylation in aging and Alzheimer’s disease. DNA Cell Biol. 2012;(Suppl. 1):S42–S48. doi: 10.1089/dna.2011.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang YC, Van Remmen H. Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 2011;46:193–198. doi: 10.1016/j.exger.2010.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen I, Baumgartner RN, Ross R, Rosenberg IH, Roubenoff R. Skeletal muscle cutpoints associated with elevated physical disability risk in older men and women. Am. J. Epidemiol. 2004;159:413–421. doi: 10.1093/aje/kwh058. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauretani F, Bandinelli S, Bartali B, Di Iorio A, Giacomini V, Corsi AM, Guralnik JM, Ferrucci L. Axonal degeneration affects muscle density in older men and women. Neurobiol. Aging. 2006;27:1145–1154. doi: 10.1016/j.neurobiolaging.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Serra P, Esteller M. DNA methylation-associated silencing of tumor-suppressor microRNAs in cancer. Oncogene. 2012;31:1609–1622. doi: 10.1038/onc.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzetti E, Lees HA, Wohlgemuth SE, Leeuwenburgh C. Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. BioFactors. 2009;35:28–35. doi: 10.1002/biof.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T, Costello JF. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullagh KJ, Calabria E, Pallafacchina G, Ciciliot S, Serrano AL, Argentini C, Kalhovde JM, Lømo T, Schiaffino S. NFAT is a nerve activity sensor in skeletal muscle and controls activity-dependent myosin switching. Proc. Natl Acad. Sci U S A. 2004;101:10590–10595. doi: 10.1073/pnas.0308035101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Tarnopolsky MA, Beckman K, Felkey K, Hubbard A. Resistance exercise reverses aging in human skeletal muscle. PLoS One. 2007;2:e465. doi: 10.1371/journal.pone.0000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molon A, Di Giovanni S, Chen YW, Clarkson PM, Angelini C, Pegoraro E, Hoffman EP. Large-scale disruption of microtubule pathways in morphologically normal human spastin muscle. Neurology. 2004;62:1097–1104. doi: 10.1212/01.wnl.0000118204.90814.5a. [DOI] [PubMed] [Google Scholar]
- Nilwik R, Snijders T, Leenders M, Groen BB, van Kranenburg J, Verdijk LB, van Loon LJ. The decline in skeletal muscle mass with aging is mainly attributed to a reduction in type II muscle fiber size. Exp. Gerontol. 2013;48:492–498. doi: 10.1016/j.exger.2013.02.012. [DOI] [PubMed] [Google Scholar]
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010;20:434–439. doi: 10.1101/gr.103101.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana ZA, Gundersen K, Buonanno A. Activity-dependent repression of muscle genes by NFAT. Proc. Natl Acad. Sci U S A. 2008;105:5921–5926. doi: 10.1073/pnas.0801330105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salanova M, Bortoloso E, Schiffl G, Gutsmann M, Belavy DL, Felsenberg D, Furlan S, Volpe P, Blottner D. Expression and regulation of Homer in human skeletal muscle during neuromuscular junction adaptation to disuse and exercise. FASEB J. 2011;25:4312–4325. doi: 10.1096/fj.11-186049. [DOI] [PubMed] [Google Scholar]
- Salar S, Yeni N, Gündüz A, Güler A, Gökçay A, Velioğlu S, Gündoğdu A, Hande Çağlayan S. Four novel and two recurrent NHLRC1 (EPM2B) and EPM2A gene mutations leading to Lafora disease in six Turkish families. Epilepsy Res. 2012;98:273–276. doi: 10.1016/j.eplepsyres.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Sati S, Tanwar VS, Kumar KA, Patowary A, Jain V, Ghosh S, Ahmad S, Singh M, Reddy SU, Chandak GR, Raghunath M, Sivasubbu S, Chakraborty K, Scaria V, Sengupta S. High resolution methylome map of rat indicates role of intragenic DNA methylation in identification of coding region. PLoS One. 2012;7:e31621. doi: 10.1371/journal.pone.0031621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarnopolsky M, Phillips S, Parise G, Varbanov A, Demuth J, Stevens P, Qu A, Wang F, Isfort R. Gene expression, fiber type, and strength are similar between left and right legs in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2007;62:1088–1095. doi: 10.1093/gerona/62.10.1088. [DOI] [PubMed] [Google Scholar]
- Teschendorff AE, Menon U, Gentry-Maharaj A, Ramus SJ, Weisenberger DJ, Shen H, Campan M, Noushmehr H, Bell CG, Maxwell AP, Savage DA, Mueller-Holzner E, Marth C, Kocjan G, Gayther SA, Jones A, Beck S, Wagner W, Laird PW, Jacobs IJ, Widschwendter M. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010;20:440–446. doi: 10.1101/gr.103606.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Hellmann I, Stadler MB, Ramos L, Pääbo S, Rebhan M, Schübeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- West J, Beck S, Wang X, Teschendorff AE. An integrative network algorithm identifies age-associated differential methylation interactome hotspots targeting stem-cell differentiation pathways. Sci. Rep. 2013;3:1630. doi: 10.1038/srep01630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J. Biol. Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- Wittkop T, TerAvest E, Evani US, Fleisch KM, Berman AE, Powell C, Shah NH, Mooney SD. STOP using just GO: a multi-ontology hypothesis generation tool for high throughput experimentation. BMC Bioinformatics. 2013;14:53. doi: 10.1186/1471-2105-14-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Numbers of CpGs present on array in a gene and dmCpG site for same gene.
Fig. S2 Ingenuity canonical axon guidance pathway.
Fig. S3 dmCpG sites that are common for this study and Heyn et al.
Table S1 List of differentially methylated CpG probes with age.
Table S2 Distribution of dmCpG.
Table S3 ENCODE ChIP-Seq significance tool results.
Table S4 Genes with at least one intragenic dmCpG site.
Table S5 Ontology enrichment analysis.
Table S6 Muscle-specific canonical pathway analysis.
Table S7 Differential methylation of axon guidance genes.
Table S8 Correspondence of intragenic differential methylation to gene expression.
Table S9 List of CpG sites that discriminate young from old samples.
Table S10 List of overlapping CpG predictors of biological age with Hannum et al. study.
Table S11 Number of overlapping dmCpG sites with other studies.
Table S12 Description of genomic regions.
Table S13 Description of human subjects.
Table S14 Illumina 450 K data quality control report.