

Abstract
We present the case of a 53‐year‐old Caucasian male smoker with remote history of left lower extremity deep venous thrombosis (DVT) and a strong family history of thrombosis, who presented to the Center for Wound Healing at MedStar Georgetown University Hospital with spontaneous left leg ulceration. Prothrombotic evaluation showed homozygosity for the factor V Leiden (FVL) mutation. Therapeutic anticoagulation was commenced with warfarin (Coumadin®) and the patient underwent successful debridement and Apligraf® followed by split‐thickness skin graft (STSG) of two wounds. He had an uneventful postoperative course and on the 27th postoperative day the grafts were 95% intact. However, by postoperative day 41 there was 10% graft loss, and over the subsequent 2 weeks both grafts necrosed. On further questioning, it transpired that the patient had discontinued his warfarin on postoperative day 37 because he thought that it was no longer necessary. The patient is enrolled in the Wound Etiology and Healing (WE‐HEAL) study, and at the time of the original graft, residual skin fragments from the STSG were transplanted onto a nude mouse for development of an animal model of wound healing. The mouse graft was successful and was harvested at postoperative day 87 for pathological examination. We review the mechanisms by which prothrombotic states, particularly FVL mutation, can contribute to skin graft failure and delayed wound healing. This case highlights the importance of considering prothrombotic conditions in patients with spontaneous leg ulcerations and the impact of therapeutic anticoagulation on healing. It further allows us to demonstrate the efficacy of the animal model in which residual fragments of STSG tissue are utilised for transplant onto nude mice for manipulation in the laboratory.
Keywords: Animal model, Factor V Leiden, Hypercoagulable state, Leg ulcer, Prothrombotic state
Case history
A 53‐year‐old Caucasian male presented to the Center for Wound Healing at MedStar Georgetown University Hospital (MGUH) with a 4‐month history of spontaneous and painful left leg ulceration. About 4 months prior to the current presentation, the patient had developed an unusual sore on the left medial ankle, which he thought was a spider bite. It was painful but eventually healed spontaneously. Then, 3–4 weeks later he developed two lesions on the anterior shin and lateral side of the left ankle, which began as small dark necrotic areas and over a period of a few weeks broke down. He was evaluated at an outside wound centre and underwent bedside debridement in the clinic. The wound did not heal and he subsequently presented to our facility.
Past medical history was notable for a previous left lower extremity deep venous thrombosis (DVT) at the time of an arthroscopic knee surgery, 14 years prior to the current presentation. He had a strong family history of thrombosis with his mother and sister also suffering deep venous thrombi. At the time of the initial thrombosis, he was evaluated by a haematologist and found to have homozygous factor V Leiden (FVL) R506Q mutation. He was anticoagulated for approximately 6 months but then remained well without anticoagulation until the current presentation. He is a lifelong smoker with a 24 pack‐year exposure history.
Physical examination
Physical examination at the initial presentation to MGUH showed a healthy appearing Caucasian male in no acute distress. His body mass index was 24·64. Blood pressure was 154/84 and he reported a visual analogue pain score of 8/10. Cardiovascular, respiratory and abdominal examinations were unremarkable. Examination of the lower extremities showed bilateral lower extremity swelling, worse on the left, with three ulcerations of the left lower extremity extending to subcutaneous tissue, measuring 9·3 × 6·3 cm (58·59 cm2), 1·2 × 1·4 cm (1·68 cm2) and 3·2 × 7·1 cm (22·72 cm2), respectively (Figure 1A). He was also noted to have an erythematous tender cord on the right thigh. Peripheral pulses were normal.
Figure 1.
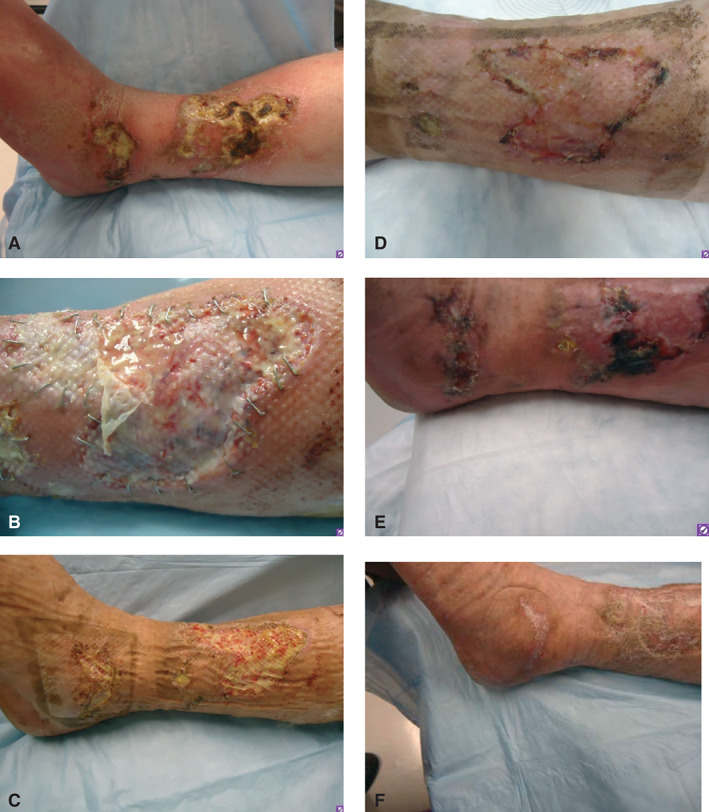
Photographs of patient' wounds on the leg. (A) At presentation; (B) after initial debridement and Apligraf placement; (C) prior to split‐thickness skin graft (STSG) demonstrating healthy wound bed; (D) at postoperative day 25 after STSG; (E) postoperative day 52 demonstrating necrosis of the STSG; and (F) improvement of wounds following reinstitution of anticoagulation therapy.
Laboratory data
Laboratory evaluation at the time of presentation is tabulated in Table 1. Autoimmune screen was unremarkable, but prothrombotic work up was notable for the presence of homozygous FVL mutation, and heterozygosity for the PAI‐1 and MTHFR mutations.
Table 1.
Haematological parameters and autoimmune profile at presentation
| Patient result | Normal range | |
|---|---|---|
| White blood cell count | 10·6k/µl | 4·0–10·8k/µl |
| Differential | 57% neutrophils, 33% lymphocytes, 10% monocytes | |
| Haemoglobin | 14·3 g/dl | 12·5–16·5 g/dl |
| Haematocrit | 43·0% | 41–53% |
| Platelet count | 348k/µl | 145–400k/µl |
| Sodium | 138 mmol/l | 137–145 mmol/l |
| Potassium | 3·7 mmol/l | 3·5–5·1 mmol/l |
| Blood urea nitrogen (BUN) | 18 mg/dl | 9–20 mg/dl |
| Creatinine | 0·9 mg/dl | 0·66–1·50 mg/dl |
| Alkaline phosphatase | 89 U/l | 38–126 U/l |
| Aspartate aminotransferase (AST) | 11 U/l | 3–34 U/l |
| Alanine aminotransferase (ALT) | 25 U/l | 15–41 U/l |
| Antinuclear antibody (ANA) immunofluorescence | Negative | Negative |
| Double‐stranded DNA antibodies | Negative | Negative |
| Antineutrophil cytoplasmic antibodies (ANCA) | Negative | Negative |
| Sjogren's antibodies (SSA and SSB) | Negative | Negative |
| Sm antibody | Negative | Negative |
| Ribonucleoprotein (RNP) antibody | Negative | Negative |
| Thyroid stimulating hormone | 0·863 IU/ml | 0·40–4·0 IU/ml |
| Uric acid | 5·7 mg/dl | 3·5–7·2 mg/dl |
| Haemoglobin A1c | 5·9% | 4·2–5·6% |
| Beta‐2 glycoprotein I antibodies IgG | <9 U/ml | <20·0 U/ml negative |
| Beta‐2 glycoprotein I antibodies IgA | <9 U/ml | <20·0 U/ml negative |
| Beta‐2 glycoprotein I antibodies IgM | <9 U/ml | <20·0 U/ml negative |
| Anticardiolipin antibodies IgG | <9 U/ml | <15 U/ml negative |
| Anticardiolipin antibodies IgA | <9 U/ml | <12 U/ml negative |
| Anticardiolipin antibodies IgM | <9 U/ml | <13 U/ml negative |
| Lupus anticoagulant ratio | 1·2 | 1·2–1·5 Weakly positive, 1·5–2·0 moderately positive, >2·0 strongly positive |
| Sedimentation rate | 20 mm/hour | 0–16 mm/hour |
| C‐reactive protein | 6·23 mg/l | 0·00–3·00 mg/l |
| Human immunodeficiency virus 1 and 2 | Negative | Negative |
| Hepatitis B surface antibody | Negative | Negative |
| Hepatitis C antibody | Negative | Negative |
| Plasminogen activator inhibitor mutation | 4G/5G heterozygous | 5G/5G |
| Factor V Leiden (R506Q) mutation | R506Q homozygous | Negative |
| Methyl‐tetrahydrofolate reductase (MTHFR) C677T mutation | C677T heterozygous | Negative |
| Prothrombin gene (G20210A) mutation | Negative | Negative |
| Protein S activity | 102% | 60–145% |
| Protein C activity | 149% | 74–151% |
| Antithrombin III activity | 61% | 75–135% |
| Plasma homocysteine | 13·3 µmol/l | 0–15·0 µmol/l |
Clinical course
Venous ultrasound showed acute thrombosis of the greater saphenous vein in the mid‐thigh on the right leg, and chronic thrombosis of the mid femoral, distal femoral and popliteal veins on the left leg. Lower extremity arterial Doppler results were within normal limits with ankle‐brachial pressure indices of 1·28 on the right and 0·95 on the left. He was commenced on therapeutic anticoagulation with enoxaparin (Lovenox®, Sanofi, Bridgewater, NJ). He underwent surgical debridement and Apligraf® (Organogenesis Inc., Canton, MA) placement and was transitioned to warfarin (Coumadin®, Bristol‐Myers Squibb, New York, NY) postoperatively. Pathology from the initial debridement showed skin with ulceration, inflammatory granulation tissue and dermal fibrosis (Figure 2). He demonstrated good improvement with the Apligraf® (Figure 1B,C) and 6 weeks later he was again transitioned to enoxaparin for split‐thickness skin grafting (STSG) of the remaining two wounds. Postoperatively, he was transitioned back to warfarin and the wounds demonstrated 95% graft take at the 25th postoperative day (Figure 1D). However, by postoperative day 41 there was 10% graft loss and over the subsequent 2 weeks both grafts necrosed (Figure 1E). On further questioning, it transpired that the patient had discontinued his warfarin from postoperative day 37 because he thought that it was no longer necessary. The patient was reinitiated on enoxaparin and transitioned to warfarin. His wounds slowly healed by secondary intention and at the last visit the wounds were completely healed (Figures 1F and 3).
Figure 2.
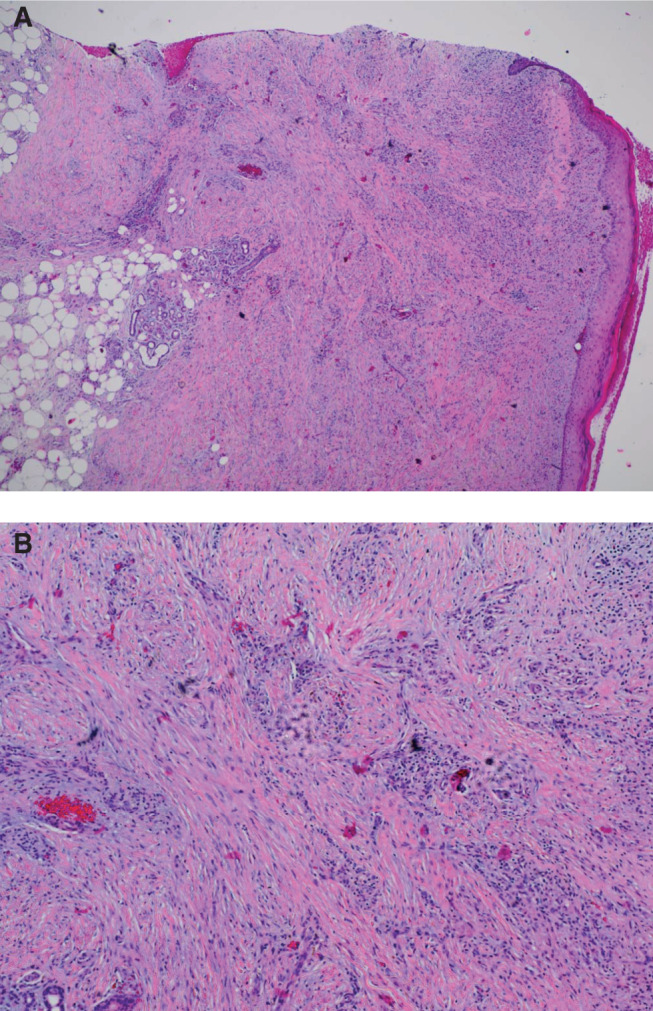
Haematoxylin and eosin‐stained histopathology specimen from wound biopsy specimen taken at wound debridement demonstrating skin ulceration with inflammatory granulation tissue and dermal fibrosis (A: ×4 and B: ×10).
Figure 3.
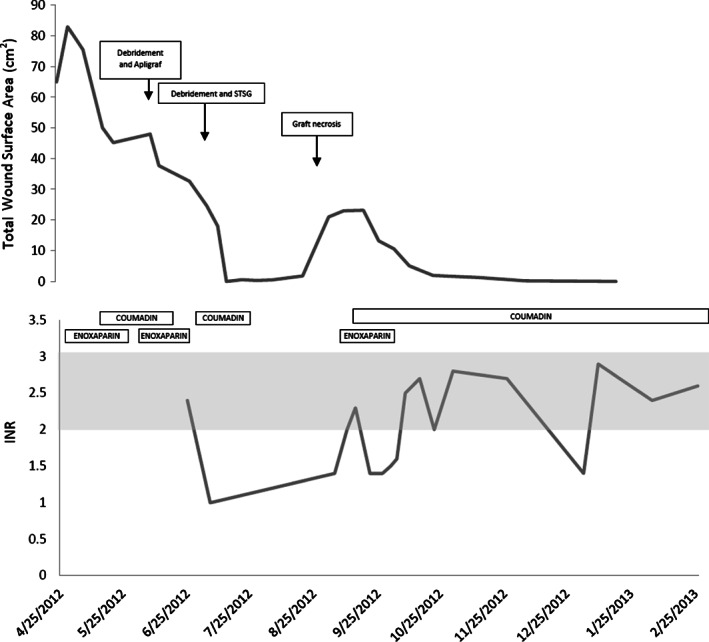
Graphs demonstrating timeline of total wound surface area (top panel) with surgical interventions superimposed and anticoagulation therapy superimposed on laboratory results from international normalised ratio (INR) testing (bottom panel, therapeutic INR 2–3 shown in shaded area). The covariation of wound size and graft necrosis with therapeutic anticoagulation is clearly demonstrated.
Correlative mouse model
This patient is also enrolled in the Wound Etiology and Healing (WE‐HEAL) study, an IRB‐approved specimen and data biorepository (IRB 2011‐055 and NCT 01352078) in which patients can elect to donate residual tissue from STSG and wound debridement for use in research. Under a protocol approved by the Georgetown University Animal Care and Use Committee (GUACUC 2011‐050) samples collected through the WE‐HEAL study may also be used for research to develop a correlative animal model for studying human wound healing. Currently available mouse models for studying human wound healing are limited because wound contraction contributes to wound shrinkage independent of epithelialisation. We have developed a model in which human skin, either collected from elective abdominoplasty or from residual skin discarded after STSG harvest, is transplanted onto an athymic nude (nu/nu) mouse and allowed to engraft for several months prior to wounding experiments. As a result of enrollment in the WE‐HEAL study, this patient's residual STSG was transplanted onto a nude mouse, coincidentally providing a correlative animal model.
Method of mouse grafting
In this case, an athymic nude (nu/nu) mouse (Harlan Laboratories) that had been allowed to acclimatise for 14 days in the Georgetown University Division of Comparative Medicine (DCM) rodent barrier facility was used for transplantation. The mouse was anaesthetised using inhalation of 1–3% isoflurane in oxygen and the skin was sterilised using povidone iodine and isopropyl alcohol. Full‐thickness skin was removed from two 1·5‐cm diameter graft beds on either side of the mouse flank and the residual 1‐cm diameter human STSG skin was placed on the wound bed. The graft was secured using steri‐strips and dressed using a Telfa non‐adherent dressing (Tyco Healthcare, Mansfield, MA) and a compressive wrap (Coban, 3M Health Care, Neuss, Germany). Postoperatively, the mouse was housed in an individual cage to minimise disturbance of the xenograft. Dressings were changed on postoperative days 7 and 14, at which time the steri‐strips were removed.
Results of mouse xenograft
Engraftment of the human STSG on the mouse was successful (Figure 4A) and in contrast to the graft on the patient that underwent necrosis on postoperative days 41–52, the graft on the mouse remained intact at 87 days postoperatively. Histopathology of the STSG prior to xenografting and the STSG at the time of harvest are shown in Figure 4B–D.
Figure 4.
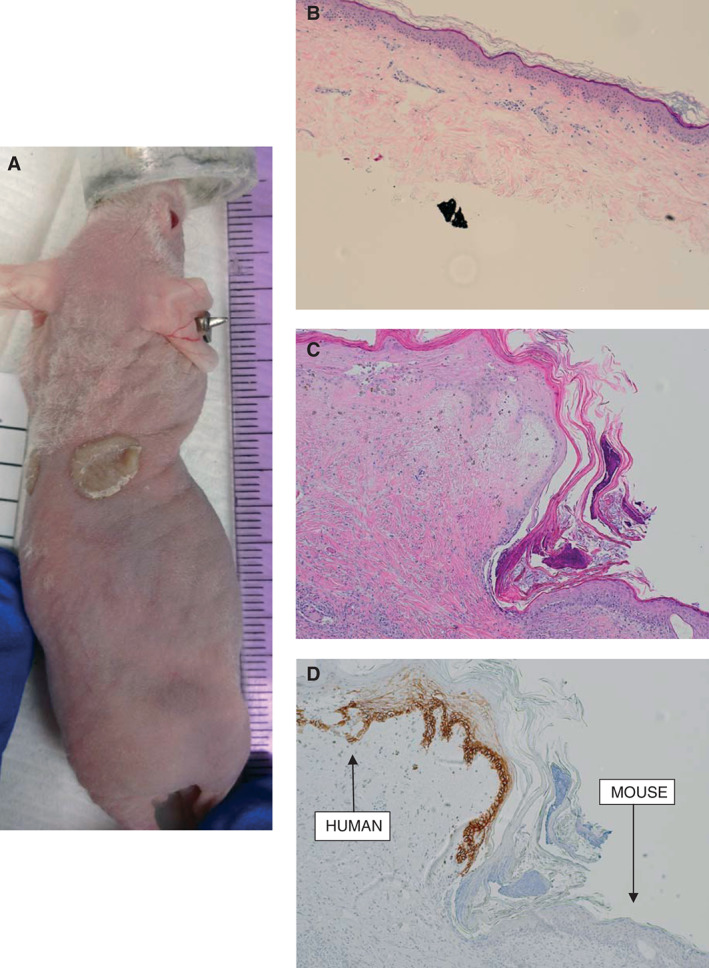
Correlative mouse xenograft model. (A) Photograph showing successful engraftment of patient skin onto nude mouse at day 31. (B) Haematoxylin and eosin staining of xenografted tissue prior to transplantation (×4). (C) Haematoxylin and eosin staining of xenograft after harvest at 87 days post‐transplant (×10). (D) Staining for human major histocompatability complex‐1 (MHC‐1) in the harvested graft clearly demonstrating viable epithelial human tissue engrafted adjacent to mouse tissue (×10).
Discussion
Chronic leg ulcers that have failed to heal after 3 months of appropriate wound care affect approximately 6·5 million people in the USA with a prevalence of 1% and costs estimated at $25 billion per year 1. In addition to the financial costs, these wounds significantly impact mortality 2 and cause considerable pain, affecting patient's psychosocial well‐being and quality of life 3, 4.
Inherited prothrombotic states are well recognised to be associated with venous leg ulceration and non‐healing lower extremity wounds 5, 6, 7, 8, with 41% prevalence of thrombophilia in a population of patients presenting to a hospital‐based leg ulcer clinic 8. The FVL mutation is highly prevalent in patients with post‐thrombotic venous leg ulcers, with various rates reported in the literature including 23% 9, 36% 10 and 41% 11, depending on the racial and geographic region being studied. Data additionally show an increased incidence of prothrombotic states including FVL mutation in patients presenting with vasculitic leg ulcers 12 and livedoid vasculopathy 13, 14, 15, suggesting a synergy between vascular wall abnormalities and prothrombotic states contributing to tissue injury 16.
Mechanisms of thrombosis and delayed healing in patients with FVL mutation
FVL mutation is common, accounting for 40–50% of cases of inherited thrombophilia 17. The prevalence of the FVL mutation is approximately 5·3% in Caucasians, 2·2% in Hispanic Americans and 1·2% in African Americans 18 and approximately 1% of patients with FVL mutation are homozygous for the mutation.
The mechanisms by which FVL mutation contributes to hypercoagulability can be understood by reviewing the role of factor V in the coagulation cascade. Circulating factor V is an inactive molecule that is activated by thrombin to become factor Va. Factor Va serves as a cofactor in the conversion of prothrombin to thrombin. Factor Va is then inactivated by enzymatic cleavage of its heavy chain in a two‐step process by activated protein C. Cleavage of the protein at Arg506 results in exposure of cleavage sites, facilitating subsequent cleavage at Arg306 and Arg 679. The FVL mutation involves replacement of guanine by adenine at nucleotide 1691 (G1691) resulting in replacement of arginine at position 506 in factor V by glutamine. This renders factor Va resistant to cleavage at position 506 by activated protein C, and thus delays inactivation. Delayed inactivation of factor Va results in more factor Va within the prothrombinase complex, increasing thrombin generation and thereby activating coagulation. Additionally, normal factor V cleaved at position 506 synergistically acts as a cofactor with protein S, supporting the role of activated protein C in the degradation of factor VIIIa and factor Va; in the absence of this cleavage product the anticoagulant effect of activated protein C is reduced.
The major clinical manifestation of FVL is venous thromboembolic disease. The prevalence of FVL heterozygosity in patients with an initial confirmed DVT or pulmonary embolism is 12% 19, and in otherwise healthy men older than 60 years who develop a DVT the prevalence of FVL is 25·8%.
Synergistic effect of FVL with other prothrombotic states
FVL mutation may be seen in association with other prothrombotic defects including prothrombin gene mutation and deficiencies of proteins C and S and antithrombin III. Carriers of two defects have a higher risk of thrombosis than those with one; for example, FVL heterozygotes have an odds ratio of thrombosis of 4·9, prothrombin gene mutation heterozygotes have an odds ratio of 3·8, but individuals heterozygous for both mutations have an odds ratio of 20·0 for thrombosis 20.
Smoking and venous thromboembolic disease
The association between smoking and venous thromboembolic disease is unclear; some studies have shown increased risk of thromboembolism in smokers 21, 22, whereas others suggest only a modest increased risk 23. No studies have investigated the associations between prothrombotic states, smoking and STSG failure. However, it is possible that the combined effect of the two prothrombotic states with cigarette smoking contributed to thrombosis and graft loss in this case.
Warfarin‐associated skin necrosis
Warfarin‐induced skin necrosis has been reported in association with FVL mutation 24, 25. This syndrome is caused by a transient prothrombotic state resulting from the acute drop in protein C activity, and increased generation of thrombin during the early phases of warfarin therapy 26. In the case reported here, necrosis of the STSG coincided with cessation of warfarin therapy, suggesting that the primary hypercoagulable state was more likely to be the cause of the skin necrosis. However, it is essential that in patients with primary hypercoagulable states, treatment is bridged with another agent during the initiation of warfarin therapy.
Utility of the human–mouse xenograft model for studying wound healing
Mouse models of human skin healing are limited for many reasons, particularly because wound contraction contributes to surface area shrinkage independent of epithelialisation 27, 28, 29. To overcome this challenge, we developed a model for studying human wound healing in vivo using normal human skin engrafted onto athymic nude mice. Taking this model one step further, to try to develop a correlative model for patients with delayed wound healing, we have been performing xenografts with residual STSG from patients enrolled in the WE‐HEAL study. Our hypothesis was that we would see correlation between human and mouse graft failures in certain disease states. However, what was striking in this case was that the STSG failed in the patient but was maintained in the mouse. This confirms our suspicion that in patients with coagulopathy, activation of the coagulation cascade in the circulation, as opposed to primary skin pathology, contributes to the graft loss.
Conclusions
A thorough preoperative history and examination should be completed in all patients with delayed wound healing, particularly patients undergoing STSG, to identify prior thrombotic events. Also, haematological evaluation should be completed prior to operative intervention, when indicated.
We report a novel mouse model in which residual human skin from STSGs was successfully transplanted onto an athymic nude mouse. This model will be a useful tool for studying not only patients with intrinsic skin diseases but also patients in whom serological factors (such as abnormalities of the coagulation cascade, or autoimmune diseases) play a role in the disease pathogenesis.
Disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources, the National Cancer Institute or the National Institutes of Health.
Acknowledgements
VKS is currently supported by award numbers KL2RR031974 and UL1TR000101 (previously UL1RR031975) from the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through the Clinical and Translational Science Awards Program (CTSA). SM, VKS, CEA, ET and AW are supported by award R01NR013888 from the National Institute of Nursing Research. These studies were conducted in part at the Lombardi Comprehensive Cancer Center Histopathology and Tissue Shared Resource, which is supported in part by the National Cancer Institute grant P30CA051008.
References
- 1. Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 2009;17:763–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Escandon J, Vivas AC, Tang J, Rowland KJ, Kirsner RS. High mortality in patients with chronic wounds. Wound Repair Regen 2011;19:526–8. [DOI] [PubMed] [Google Scholar]
- 3. Price P, Harding K. The impact of foot complications on health‐related quality of life in patients with diabetes. J Cutan Med Surg 2000;4:45–50. [DOI] [PubMed] [Google Scholar]
- 4. Price P, Harding K. Cardiff wound impact schedule: the development of a condition‐specific questionnaire to assess health‐related quality of life in patients with chronic wounds of the lower limb. Int Wound J 2004;1:10–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wiszniewski A, Bykowska K, Bilski R, Jaśkowiak W, Proniewski J. Prevalence rate for inherited thrombophilia in patients with chronic and recurrent venous leg ulceration. Wound Repair Regen 2011;19:552–8. [DOI] [PubMed] [Google Scholar]
- 6. Calistru A, Baudrier T, Goncalves L, Azevedo F. Thrombophilia in venous leg ulcers: a comparative study in early and later onset. Indian J Dermatol Venereol Leprol 2012;78:406. [DOI] [PubMed] [Google Scholar]
- 7. Bradbury A, MacKenzie R, Burns P, Fegan C. Thrombophilia and chronic venous ulceration. Eur J Vasc Endovasc Surg 2002;24:97–104. [DOI] [PubMed] [Google Scholar]
- 8. MacKenzie RK, Ludlam CA, Ruckley CV, Allan PL, Burns P, Bradbury AW. The prevalence of thrombophilia in patients with chronic venous leg ulceration. J Vasc Surg 2002;35:718–22. [DOI] [PubMed] [Google Scholar]
- 9. Maessen‐Visch MB, Hamulyak K, Tazelaar DJ, Crombag NHCMN, Neumann HAM. The prevalence of Factor V Leiden mutation in patients with leg ulcers and venous insufficiency. Arch Dermatol 1999;135:41–4. [DOI] [PubMed] [Google Scholar]
- 10. Gaber Y, Siemens HJ, Schmeller W. Resistance to activated protein C due to factor V Leiden mutation: high prevalence in patients with post‐thrombotic leg ulcers. Br J Dermatol 2001;144:546–8. [DOI] [PubMed] [Google Scholar]
- 11. Hafner J, Kühne A, Schär B. Factor V Leiden mutation in postthrombotic and non‐postthrombotic venous ulcers. Arch Dermatol 2001;137:599–603. [PubMed] [Google Scholar]
- 12. Mekkes JR, Loots MA, van der Wal AC, Bos JD. Increased incidence of hypercoagulability in patients with leg ulcers caused by leukocytoclastic vasculitis. J Am Acad Dermatol 2004;50:104–7. [DOI] [PubMed] [Google Scholar]
- 13. Hairston BR, Davis M, Pittelkow MR, Ahmed I. Livedoid vasculopathy: further evidence for procoagulant pathogenesis. Arch Dermatol 2006;142:1413–8. [DOI] [PubMed] [Google Scholar]
- 14. Calamia K, Balabanova M, Perniciaro C, Walsh J. Livedo (livedoid) vasculitis and the factor V Leiden mutation: additional evidence for abnormal coagulation. J Am Acad Dermatol 2002;46:133–7. [DOI] [PubMed] [Google Scholar]
- 15. Biedermann T, Flaig MJ, Sander CA. Livedoid vasculopathy in a patient with factor V mutation (Leiden). J Cutan Pathol 2000;27:410–2. [DOI] [PubMed] [Google Scholar]
- 16. Shanmugam V, Steen V, Cupps T. Lower extremity ulcers in connective tissue disease. Isr Med Assoc J 2008;10:534–6. [PubMed] [Google Scholar]
- 17. Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature 1994;369:64–7. [DOI] [PubMed] [Google Scholar]
- 18. Ridker P, Miletich J, Hennekens C, Buring J. Ethnic distribution of factor V Leiden in 4047 men and women: implications for venous thromboembolism screening. JAMA 1997;277:1305–7. [PubMed] [Google Scholar]
- 19. Ridker PM, Hennekens CH, Lindpaintner K, Stampfer MJ, Eisenberg PR, Miletich JP. Mutation in the gene coding for coagulation Factor V and the risk of myocardial infarction, stroke, and venous thrombosis in apparently healthy men. N Engl J Med 1995;332:912–7. [DOI] [PubMed] [Google Scholar]
- 20. Emmerich J, Rosendaal FR, Cattaneo M, Margaglione M, De Stefano V, Cumming T, Arruda V, Hillarp A, Reny JL. Combined effect of factor V Leiden and prothrombin 20210A on the risk of venous thromboembolism‐‐pooled analysis of 8 case–control studies including 2310 cases and 3204 controls. Study group for pooled‐analysis in venous thromboembolism. Thromb Haemost 2001;86:809–16. [PubMed] [Google Scholar]
- 21. Holst AG, Jensen G, Prescott E. Risk factors for venous thromboembolism: results from the Copenhagen City Heart Study. Circulation 2010;121:1896–903. [DOI] [PubMed] [Google Scholar]
- 22. Pomp ER, Rosendaal FR, Doggen CJM. Smoking increases the risk of venous thrombosis and acts synergistically with oral contraceptive use. Am J Hematol 2008;83:97–102. [DOI] [PubMed] [Google Scholar]
- 23. Blondon M, Wiggins K, McKnight B, Psaty BM, Rice KM, Heckbert SR, Smith NL. The association of smoking with venous thrombosis in women. A population‐based, case–control study. Thromb Haemost 2013;109:891–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Makris M, Bardhan G, Preston F. Warfarin induced skin necrosis associated with activated protein C resistance. Thromb Haemost 1996;75:523–4. [PubMed] [Google Scholar]
- 25. McGehee WG, Klotz TA, Epstein DJ, Rapaport SI. Coumarin necrosis associated with hereditary protein C deficiency. Ann Intern Med 1984;101:59–60. [DOI] [PubMed] [Google Scholar]
- 26. Vigano D'Angelo S, Comp PC, Esmon CT, D'Angelo A. Relationship between protein C antigen and anticoagulant activity during oral anticoagulation and in selected disease states. J Clin Invest 1986;77:416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X, Ge J, Tredget EE, Wu Y. The mouse excisional wound splinting model, including applications for stem cell transplantation. Nat Protoc 2013;8:302–9. [DOI] [PubMed] [Google Scholar]
- 28. Wong V, Sorkin M, Glotzbach J, Longaker M, Gurtner G. Surgical approaches to create murine models of human wound healing. J Biomed Biotechnol 2011;2011:969618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ansell DM, Holden KA, Hardman MJ. Animal models of wound repair: are they cutting it? Exp Dermatol 2012;21:581–5. [DOI] [PubMed] [Google Scholar]