

Summary
Intracellular bacterial pathogens often subvert apoptosis signaling to regulate survival of their host cell, allowing propagation of the bacterial population. Coxiella burnetii, the intracellular agent of human Q fever, inhibits host cell apoptosis through several mechanisms, including prevention of mitochondrial cytochrome c release, triggering of an anti-apoptotic transcriptional program, and activation of pro-survival kinases. To control host cell survival, C. burnetii delivers effector proteins to the eukaryotic cytosol using a specialized Dot/Icm type IV secretion system (T4SS). Effectors are predicted to regulate activity of pro-survival host signaling proteins, such as Akt and cAMP-dependent protein kinase (PKA), to control infection. Here, we show that host PKA activity is required for C. burnetii inhibition of macrophage apoptosis. PKA is activated during infection and inhibits activity of the pro-apoptotic protein Bad via phosphorylation. Bad is also phosphorylated at an Akt-specific residue, indicating C. burnetii uses two kinases to fully inactivate Bad. Additionally, Bad and the tethering protein 14-3-3β co-localize at the C. burnetii parasitophorous vacuole (PV) membrane during infection, an event predicted to alter Bad promotion of apoptosis. Inhibiting PKA activity prevents Bad recruitment to the PV, but the protein is retained at the membrane during induction of apoptosis. Finally, PKA regulatory subunit I (RI) traffics to the PV membrane in a T4SS-dependent manner, suggesting a C. burnetii effector(s) regulates PKA-dependent activities. This study is the first to demonstrate subversion of host PKA activity by an intracellular bacterial pathogen to prevent apoptosis and survive within macrophages.
Introduction
Coxiella burnetii is the intracellular bacterial agent of human Q fever, a debilitating flu-like illness that can progress to endocarditis in immunocompromised individuals (Raoult et al., 2005). With an infectious dose approaching one organism and the potential for aerosolization, C. burnetii is a category B select agent with potential for illegitimate use (Control, 2002). There is currently no Q fever vaccine approved for civilian use in the U. S., and the rise of Q fever cases worldwide over the last decade suggests C. burnetii is an emerging pathogen (Control, 2002, Enserink, 2010). Following inhalation by a human host, C. burnetii enters macrophages by phagocytosis, traffics through the phagolysosomal maturation pathway, and ultimately resides within an acidic, lysosome- like parasitophorous vacuole (PV) required for bacterial survival (Hackstadt et al., 1981, Graham et al., 2012, Beron et al., 2002, Howe et al., 2000, Voth et al., 2007a). During infection, C. burnetii uses a Dot/Icm type IV secretion system (T4SS) to deliver bacterial proteins termed effectors to the host cytoplasm where they control PV generation and host cell survival (Beare et al., 2011, Carey et al., 2011, Chen et al., 2010). T4SS effectors are also predicted to regulate host kinase signaling, and we recently identified several host kinases required for PV formation, including calmodulin kinase II, myosin light chain kinase, protein kinase C, and cAMP-dependent protein kinase (PKA) (Hussain et al., 2010). In addition to promoting PV formation, we predict these and other signaling cascades are required for additional critical aspects of C. burnetii infection, including prevention of host cell death.
Eukaryotic cells often respond to intracellular pathogen invasion by committing a form of cell death termed apoptosis as part of the intrinsic immune defense (Lamkanfi et al., 2010). Apoptosis allows pathogen clearance in the absence of inflammation (Gallucci et al., 1999) and apoptotic body engulfment by dendritic cells activates the adaptive immune response (Elliott et al., 2010). To counter apoptosis, intracellular pathogens either induce premature cell death to allow spread to bystander cells or inhibit death to maintain a viable replicative niche. For example, Yersinia pestis induces apoptosis by secreting YopJ to block MAPKK signaling (Monack et al., 1997, Orth et al., 1999). In contrast, Legionella pneumophila inhibits apoptosis by several mechanisms using T4SS effectors such as SdhA, which inhibits activation of caspases that typically promote DNA damage and apoptosis (Laguna et al., 2006). Additionally, L. pneumophila LnaB activates anti-apoptotic NF-κB signaling (Losick et al., 2010) and SidF inhibits apoptosis by binding to BNIP3 and Bcl-rambo, preventing their pro-apoptotic activity (Banga et al., 2007). Mycobacterium tuberculosis shrewdly inhibits apoptosis at early times post-infection to allow replication, then later induces death to escape from macrophages (Loeuillet et al., 2006, Behar et al., 2010). C. burnetii is no exception among intracellular pathogens, using secreted effectors and host signaling to prevent macrophage apoptosis by multiple mechanisms (Voth et al., 2007b, Luhrmann et al., 2007, Beare et al., 2011, Voth et al., 2009a).
C. burnetii inhibits both intrinsic, mitochondrial-dependent and extrinsic, death receptor-mediated apoptosis (Voth et al., 2007b, Luhrmann et al., 2007). Infected cells display reduced caspase processing under apoptosis-inducing conditions, and production of anti-apoptotic A1/Bfl-1 and c-IAP2 increases during intracellular growth (Voth et al., 2007b). While mechanisms of extrinsic apoptosis inhibition by C. burnetii have not been elucidated, the pathogen prevents intrinsic apoptosis by activating pro-survival Erk1/2 and Akt signaling and inhibiting cytochrome c release (Voth et al., 2009a, Luhrmann et al., 2007). Recent studies demonstrated that C. burnetii effector proteins participate in this process (Beare et al., 2011), as AnkG antagonizes cell death by preventing p32 activity (Luhrmann et al., 2010) and CaeB stabilizes mitochondrial membrane potential (Klingenbeck et al., 2012). Collectively, these studies indicate C. burnetii uses many methods to ensure host cell survival, making anti-apoptotic activity a hallmark of infection.
The balance of pro- and anti-apoptotic mitochondrial proteins is critical in controlling intrinsic apoptosis. A subset of these proteins, including Bak and Bax, promotes apoptosis through perturbation of mitochondrial membrane integrity and cytochrome c release (Shimizu et al., 2000). The Bcl-2 protein family promotes survival by inhibiting pro-apoptotic protein activity (Vaux et al., 1988, Willis et al., 2007, Suzuki et al., 2000, Letai et al., 2002, Chen et al., 2005, Kuwana et al., 2005, Kim et al., 2006). A third group of proteins known as the pro-apoptotic BH3-only proteins, include Bad, Bid, and Bim, and form dimers with Bcl-2 proteins, inhibiting pro-survival effects (Yang et al., 1995, Youle et al., 2008). Phosphorylation by Akt and PKA results in Bad binding to the adaptor protein 14-3-3, inhibiting Bad interaction with Bcl-2 survival proteins (Datta et al., 2000). In the absence of pro-survival kinase activity, Bad binds to Bcl-2 proteins, promoting disruption of the mitochondrial membrane (Zha et al., 1997). Loss of mitochondrial membrane potential results in cytochrome c release and formation of the apoptosome, ultimately leading to caspase activation required for DNA damage and death (Cain et al., 2002).
We recently discovered that Bad phosphorylation increases substantially during C. burnetii infection (MacDonald et al., 2012), suggesting the pathogen regulates Bad activity to influence host cell survival. In the current study, we found that PKA activity is required for C. burnetii anti-apoptotic activity, and Bad phosphorylation increases during infection at Akt- and PKA-specific residues, indicating both kinases are required for prevention of apoptosis. Interestingly, Bad localizes to the PV membrane in a PKA-dependent manner. We also discovered that PKA regulatory subunit I (RI) localizes to the PV membrane in a T4SS-dependent fashion. These results suggest a scaffolding complex present at the PV membrane controls apoptotic signaling during C. burnetii infection. Collectively, we have uncovered a unique mechanism for intracellular pathogen prevention of macrophage apoptosis using host PKA signaling.
Results
PKA activity is required for C. burnetii-mediated apoptosis prevention
To test the hypothesis that PKA regulates apoptosis during C. burnetii infection, we assessed nuclear fragmentation, indicative of apoptosis, in infected cells treated with PKA inhibitors. THP-1 macrophage-like cells were infected with avirulent Nine Mile II (NMII) C. burnetii for 48 hours, then treated with the pharmacologic inhibitor H-89 to prevent PKA activity (Aronoff et al., 2005). Twenty-four hours after H-89 addition, cells were incubated with the apoptosis inducer staurosporine (Zhu et al., 1995). As evidenced by apoptotic nuclei, increased death was observed in infected cell cultures treated with H-89 and staurosporine, with PKA-inhibited cells showing ~ 35% death similar to staurosporine-treated uninfected cells (~ 37%), and non-inhibited, infected cells showing ~ 5% death (Fig. 1A). These results were confirmed with a second PKA-specific inhibitor, Rp-cAMPS (Bell et al., 1994) (Fig. 1B), where ~40% of inhibited cells contained fragmented nuclei following staurosporine treatment, indicating PKA is required for C. burnetii prevention of apoptosis. Importantly, neither inhibitor alone induced substantial levels of apoptosis in infected cells (~ 2–3%).
Fig. 1.

PKA is required for C. burnetii anti-apoptotic activity. THP-1 cells were infected with NMII C. burnetii for 48 h, then treated with H-89 (A) or Rp-cAMPS (Rp; B) where indicated. At 72 hpi, indicated cells were treated with staurosporine (Sts; 750 nM) to induce intrinsic apoptosis. At 4 h post-treatment, cells were processed for fluorescence microscopy. DNA was stained with DAPI and bacteria were detected with a C. burnetii-specific antibody. The percentage of cells containing fragmented nuclei indicative of apoptosis was quantified from 950 cells repeated in triplicate. n.s. = not significant and ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. PKA inhibition by H-89 or Rp-cAMPS antagonizes C. burnetii anti-apoptotic activity. (C) Cells infected with C. burnetii expressing CyaA-CpeE were treated with H-89 at 48 hpi, then cAMP levels indicative of effector secretion were determined at 72 hpi. Cells infected with C. burnetii expressing CyaA alone served as a negative control. Results are representative of two independent infections, ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard error from the mean. Following H-89 treatment for 24 h, significant levels of CyaA-CpeE, although lower than untreated cells, remain present in the host cell cytosol.
To determine if H-89 treatment prevents accumulation of cytosolic T4SS effectors in addition to preventing PKA activity at the time of apoptosis analysis (72 hpi), we used a strain expressing a known T4SS effector (CpeE) fused to an adenylate cyclase reporter of translocation (Voth et al., 2011). Cells infected with C. burnetii expressing CyaA-CpeE were treated with H-89 at 48 hpi, then cAMP levels indicative of effector presence in the host cytosol were determined at 72 hpi. As shown in Fig. 1C, cAMP levels decreased following H-89 treatment as a result of slowed PV expansion and fewer organisms present compared to untreated cells. However, cAMP levels triggered by CyaA-CpeE translocation into H-89-treated cells remained significantly elevated compared to cells infected with C. burnetii expressing CyaA alone. Therefore, T4SS effectors were still present in the cytoplasm of H-89-treated, C. burnetii-infected cells when apoptosis was assessed, indicating H-89 does not abolish effector secretion to prevent anti-apoptotic activity.
We next examined poly (ADP-ribose) polymerase (PARP) processing as a reliable indicator of apoptosis in infected cells treated with H-89 and staurosporine (Voth et al., 2007b). Active PARP normally repairs damaged DNA, and processing by caspases inactivates the protein (Jin et al., 2005). As shown in Fig. 2, and similar to fragmented nuclei results, significantly more cleaved PARP-positive cells (~ 35%) were present in infected, staurosporine-treated cell cultures incubated with H-89 than infected cells treated with staurosporine alone and similar to uninfected, staurosporine-treated cells (~ 27%) in the presence or absence of H-89 (data not shown). A corresponding increase in PARP cleavage was detected by immunoblot in infected cells treated with staurosporine and H-89. Together, these results indicate PKA inhibition impairs C. burnetii’s capacity to prevent PARP cleavage and protect its host cell from apoptotic death.
Fig. 2.

Increased PARP processing is observed in infected, PKA-inhibited cell cultures following induction of apoptosis. THP-1 cells were infected with NMII C. burnetii for 48 hpi, then treated with H-89 where indicated. At 72 hpi, indicated cells were treated with staurosporine (Sts) for 4 h, then processed for fluorescence microscopy or lysates harvested for immunoblot analysis. DNA (blue) was stained with DAPI, cleaved PARP (green) labeled using an anti-cleaved PARP antibody, and bacteria (red) were detected with a C. burnetii-specific antibody. The percentage of cleaved PARP-positive cells was quantified from 950 cells repeated in triplicate and densitometry values are provided as percent above uninfected levels under the blot. * indicates p<0.05 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. Bar, 20 μM. Cellular lysates were assessed by immunoblot using antibody directed against cleaved PARP. Blots are representative of three independent experiments and conditions are described above the blots. PKA inhibition increases PARP cleavage in C. burnetii-infected cells treated with staurosporine.
Phosphorylated Bad levels increase during C. burnetii infection
Bad is a pro-apoptotic protein regulated by phosphorylation at amino acid residues Ser136 and Ser155 by the growth stimulatory kinases including Akt and PKA (Datta et al., 2000). Because Bad phosphorylation at the PKA-specific residue Ser155 increases during infection (MacDonald et al., 2012), we sought to determine the timing of phosphorylation at this residue and the possibility of Akt-specific Ser136 phosphorylation during C. burnetii intracellular growth. THP-1 cells were infected with NMII C. burnetii and lysates harvested from 12 – 72 hpi and probed for phosphorylation of Bad at both serine residues. Increased Ser136 phosphorylation was observed as early as 12 hpi, while Ser155 phosphorylation levels increased between 24 and 48 hpi (Fig. 3A and B). Total Bad levels remained constant throughout the infection time course. Akt activity is required for C. burnetii prevention of apoptosis, and is activated between 2 and 6 hpi [25], while PKA activation is observed from 24–96 hpi (MacDonald et al., 2012, Voth et al., 2009a). Densitometry analysis confirmed that the timing of increased Bad phosphorylation corresponds to kinase activation, suggesting Akt is activated first during infection and phosphorylates Bad at Ser136, followed by PKA activation and Bad Ser155 phosphorylation (Fig. 3B). We next investigated Bad phosphorylation in C. burnetii-infected cells lacking PKA activity. As shown in Figure 3, increased Ser155 phosphorylation was abrogated at 72 hpi in the presence of H-89 (Fig. 3A), while Ser136 phosphorylation levels remained elevated under the same conditions, supporting a model in which C. burnetii activates both Akt and PKA to completely abrogate Bad activity. Interestingly, H-89 treatment triggered increased Ser136 phosphorylation suggesting Akt activity may increase in PKA-inhibited cells.
Fig. 3.

Bad phosphorylation at Ser136 and Ser155 increases during infection. (A) THP-1 cells were infected with NMII C. burnetii and treated where indicated with H-89. Lysates were harvested at the indicated times post-infection and assessed by immunoblot using antibody directed against total or phosphorylated Bad. C = control, uninfected cells, C + H = control, uninfected cells treated with H-89, 72 + H = infected cells treated with H-89 at 48 hpi and harvested at 72 hpi, p = phosphorylated. Blots are representative of three independent experiments and the arrow denotes phosphorylated Bad. (B) Densitometry analysis of Ser136 and Ser155 phosphorylation was performed and values were calculated as percent above levels in uninfected cells (C). n.s. = not significant and ** indicates p<0.01 compared to control levels as analyzed by the Student’s t test, and error bars represent standard deviation from the mean from at least three representative blots. Bad phosphorylation at Ser136 increases by 12 hpi, while Ser155 phosphorylation increases between 24–48 hpi. Importantly, total levels of Bad remain constant throughout infection. Increased Ser155, but not Ser136, phosphorylation levels are abrogated by H-89 treatment.
PKA activity is required for C. burnetii prevention of primary human alveolar macrophage apoptosis
We recently demonstrated that primary human alveolar macrophages (hAMs) are a human disease-relevant platform to investigate C. burnetii-regulated signaling required for infection (Graham et al., 2012), and PKA is activated by virulent C. burnetii during growth in primary hAMs (MacDonald et al., 2012). To determine if virulent C. burnetii modulates PKA-dependent pro-survival signaling, we infected primary hAMs with virulent Nine Mile I (NMI) C. burnetii and assessed the impact of PKA inhibition on apoptosis. At 72 hpi, H-89 was added to indicated cell cultures and at 96 hpi, staurosporine was added and cells were processed for fluorescence microscopy to quantify fragmented nuclei. Results from hAMs infected with virulent C. burnetii mirrored those seen for avirulent NMII infection of THP-1 cells, as ~ 37% of infected hAMs treated with staurosporine and H-89 were apoptotic compared to ~ 10% of infected hAMs only treated with staurosporine (Fig. 4A). In addition, lysates were harvested at the indicated time points and Bad Ser155 phosphorylation assessed. Similar to THP-1 results, Bad phosphorylation at Ser155 increased above uninfected cell levels from 24 – 96 hpi (Fig. 4B). This increase was also PKA-dependent (data not shown). These results indicate virulent C. burnetii uses PKA to inactivate Bad and prevent apoptosis of primary hAMs, the pathogen’s in vivo target cell.
Fig. 4.

Apoptosis increases in PKA-inhibited, infected primary hAMs. (A) Primary hAMs were infected with virulent NMI C. burnetii and treated with H-89 where indicated at 72 hpi. At 96 hpi, indicated cells were treated with staurosporine (Sts) for 4 hours, then processed for fluorescence microscopy. Experiments were performed in triplicate and fragmented nuclei quantified from 950 cells. n.s. = not significant, * indicates p<0.05, and ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. PKA inhibition antagonizes virulent C. burnetii anti-apoptotic activity in primary hAMs. (B) Lysates from infected hAMs were harvested at the indicated times and assessed by immunoblot using antibody directed against total or phosphorylated Bad. C = control, uninfected cells. Blots are representative of three independent experiments. Increased Bad phosphorylation is observed from 24–96 hpi similar to NMII-infected THP-1 cells.
Bad and the adaptor protein 14-3-3β localize to the C. burnetii PV membrane
Intracellular pathogens often sequester host proteins to their replication vacuole to alter protein activity and promote infection (Verbeke et al., 2006). Because C. burnetii recruits host proteins such as Beclin-1 and Bcl-2 (Vazquez et al., 2009) to the PV membrane and Bad localization to mitochondria is required for pro-apoptotic activity, we assessed whether Bad traffics to the PV membrane during infection. Because native Bad could not be detected by antibody-based methods, we generated a green fluorescent protein (GFP)-Bad fusion construct and transfected HeLa cells with GFP-Bad or GFP alone. HeLa cells were used for these experiments because GFP-Bad expression was highly toxic to THP-1 cells (data not shown) and C. burnetii efficiently prevents HeLa cell apoptosis (Luhrmann et al., 2007). At 72 hpi, cells were processed for fluorescence microscopy using an antibody to detect the PV membrane marker LAMP-1 (Heinzen et al., 1996). GFP-Bad localized to ~ 89% of PV compared to 15% that co-localized with GFP alone (Fig. 5A). As shown in a representative intensity plot, Bad co-localization with LAMP-1 was also observed (Fig. 5B), confirming recruitment of the protein to the PV membrane. Because phosphorylation of Bad promotes binding to 14-3-3 proteins (Datta et al., 2000, Datta et al., 1997), we also examined localization of 14-3-3β. Similar to Bad trafficking, 14-3-3β was also present at the PV membrane as evidenced by fluorescence overlap with LAMP-1 (Fig. 5B), suggesting 14-3-3β mediates Bad sequestration away from mitochondria during infection. Importantly, Bad mis-localization was not simply due to mitochondrial association with the PV, as the mitochondrial protein CoxIV did not localize to the PV membrane (Fig. 5C).
Fig. 5.

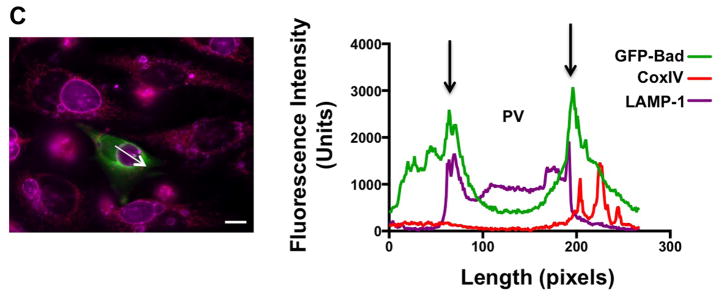
Bad and 14-3-3β traffic to the PV membrane during infection. HeLa cells were infected with NMII C. burnetii for 56 h, then transfected with GFP alone or GFP-Bad constructs. (A) At 72 hpi, cells were processed for fluorescence microscopy and the percentage of PV positive for membrane-localized GFP was quantified from at least 100 cells. Experiments were performed in triplicate. ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. Bar, 10 μM. GFP-Bad is present on ~ 90% of PV at 72 hpi. (B) Co-localization of GFP-Bad, LAMP- 1, and the Bad tethering protein 14-3-3β was assessed using antibodies, and intensity profiles of representative PV were acquired to analyze fluorescence overlap (region indicated by arrow in the micrograph). Arrows in the intensity profile indicate overlap of GFP-Bad (green), LAMP-1 (violet), and 14-3-3β (red) surrounding the PV. Data is representative of three independent experiments. Bar, 10 μM. (C) Co-localization of GFP-Bad with the mitochondrial protein CoxIV (red) was assessed using antibodies and intensity profiles of representative PV were acquired to assess fluorescence overlap. Bar, 10 μM. GFP-Bad and 14-3-3β traffic to the PV membrane during infection as evidenced by co-localization with LAMP-1, while CoxIV does not overlap with LAMP1 at the PV membrane.
Because Bad is recruited to the PV membrane, we predicted C. burnetii sequesters the protein in this location upon sensing an apoptotic stimulus. To determine if Bad is retained at the PV membrane under apoptotic conditions, we treated infected, GFP-Bad-expressing HeLa cells with staurosporine and assessed Bad trafficking. Apoptosis was induced for 3 h in HeLa cells because this condition yields fragmented nuclei counts comparable to THP-1 cells (data not shown). As shown in Fig. 6, Bad remained at the PV membrane in ~ 90% of infected cells following staurosporine treatment similar to untreated cells. However, when H-89 was added at the time of transfection, Bad was present primarily in the cytoplasm of ~ 70% of infected cells (Fig. 6), suggesting dephosphorylation of Bad prevents recruitment to the PV. Together, these results suggest C. burnetii actively sequesters Bad away from mitochondria during apoptosis in a PKA-directed manner.
Fig. 6.

PKA inhibition, but not apoptosis induction, prevents Bad localization to the PV membrane. HeLa cells were infected with NMII C. burnetii for 56 h, then transfected with GFP alone or GFP-Bad constructs. Indicated cells were incubated with H-89 for 12 h. At 70 hpi, indicated cells were treated with staurosporine (Sts) for 3 h, then harvested for fluorescence microscopy. Arrowheads indicate apoptotic nuclei, asterisks and boxes denote PV, and the percentage of PV positive for membrane-localized GFP was quantified from 950 cells. * indicates p<0.05 and ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. Graphs and images are representative of three independent experiments. Bar, 10 μM. GFP-Bad is retained at the PV membrane during induction of apoptosis by staurosporine. In contrast, inhibition of PKA activity prevents GFP-Bad localization to the PV.
PKA RI localizes to the PV in a T4SS-dependent fashion
Due to Bad presence at the PV membrane, we predicted PKA must localize accordingly to access Bad for phosphorylation. PKA localization is critical for proper function and A-kinase anchoring proteins (AKAPs) serve as scaffolding molecules that tether PKA subunits to specific subcellular locations (Wong et al., 2004). Three PKA catalytic subunit isoforms (α, β, and γ) and four regulatory subunit isoforms (RIα, RIβ, RIIα, and RIIβ) have been described (Shabb, 2001). All isoforms are ubiquitously expressed except catalytic subunit isoform γ, which is testes-specific (Beebe et al., 1990). Because Bad localizes to the PV membrane in a PKA-dependent fashion, we investigated whether PKA also traffics to the PV using RI as a readout of protein localization. THP-1 cells were infected with wild type NMII C. burnetii or T4SS-defective organisms (IcmD mutant) (Beare et al., 2011) and processed for fluorescence microscopy at the indicated times post-infection for quantification of RI-positive PV. As shown in Fig. 7, RI was present on ~ 50% of wild type C. burnetii PV at 48 hpi and this trafficking required an active T4SS as only ~ 7% of IcmD mutant-containing phagosomes decorated with RI throughout infection. R1 still localized to the PV membrane in H-89-treated cells (Fig. 7), further indicating that C. burnetii secretion is not prevented in the presence of H-89. Interestingly, RI localization correlates with PKA activation during infection, and ~ 85% of PV decorated with RI by 96 hpi (Fig. 7). These results suggest C. burnetii uses T4SS effectors to coordinate PKA-directed signaling at the PV membrane.
Fig. 7.

PKA RI localizes to the PV membrane in a T4SS-dependent manner. THP-1 cells were infected with wild type NMII or IcmD mutant organisms and processed for fluorescence microscopy at 72 hpi. DNA (blue) was labeled with DAPI, and PKA RI (green) and C. burnetii (red) were detected with antibodies. The percentage of RI-positive PV was quantified from 500 cells and experiments were performed in triplicate. n.s. = not significant, * indicates p<0.05, and ** indicates p<0.01 as analyzed by the Student’s t test, and error bars represent standard deviation from the mean. Bar, 10 μM. Indicated cells were treated with H-89 24 h prior to processing. PKA RI localizes to the PV in NMII wild type-infected cells and H-89-treated cells, but not phagosomes harboring T4SS-defective bacteria.
Discussion
Here, we show that eukaryotic PKA activity is required for C. burnetii-mediated survival of mammalian cells, and the pathogen recruits pro-apoptotic Bad to the PV where the protein is phosphorylated and inactivated. PKA inhibition significantly increases death of infected THP-1 cells and primary hAMs, implicating the in vivo importance of this signaling cascade. Together with our previous studies (MacDonald et al., 2012, Hussain et al., 2010), we propose that PKA serves a prominent dual role in C. burnetii infection, controlling PV expansion and host cell survival. No previous reports have directly shown a role for PKA in intracellular pathogen anti-apoptotic activity. However, Bartonella relies on increased cAMP levels to efficiently inhibit host cell death (Schmid et al., 2006). Furthermore, Bartonella T4SS-secreted BepA inhibits endothelial cell apoptosis by promoting increased cAMP production (Schmid et al., 2006) and C. burnetii anti-apoptotic activity also requires a functional T4SS (Beare et al., 2011). Our current results suggest Bartonella may also benefit from PKA activity triggered by increased concentrations of cAMP. Thus, PKA represents a versatile new target of intracellular bacterial pathogens to regulate mitochondrial anti-apoptotic events, effectively preventing cytochrome c release and downstream caspase activity.
During C. burnetii infection, Bad is phosphorylated at serine residues 136 and 155. Interestingly, Ser136 phosphorylation increases at 12 hpi, coinciding with Akt activation needed for efficient inhibition of THP-1 cell death by C. burnetii (Voth et al., 2009a). Ser155 phosphorylation increases between 24 and 48 hpi, coinciding with PKA activation, and PKA inhibition prevents Ser155 phosphorylation. Bad activity is also controlled by phosphorylation at Ser112 in some systems. We did not detect Ser112 phosphorylation during infection (data not shown), and kinases reported to phosphorylate Bad at Ser112, including p38 MAPK, are not activated throughout infection (Voth et al., 2009a). However, RSK kinase phosphorylation increases slightly during infection (MacDonald et al., 2012, Voth et al., 2009a), suggesting Ser112 phosphorylation may occur and RSK may play a synergistic role with PKA and Akt in apoptosis prevention by C. burnetii. Furthermore, Raf reportedly phosphorylates all three Bad serine residues in vitro (Rapp et al., 2007); however, Raf activation has not been reported during C. burnetii infection.
Bad and its adaptor protein 14-3-3β traffic to the PV membrane during infection and Bad is retained at the PV following induction of apoptosis. These results suggest C. burnetii permanently retains Bad at the PV membrane, preventing interaction of the protein with anti-apoptotic mitochondrial proteins that promote cytochrome c release. By tethering 14-3-3β to the PV membrane, the pathogen uses the host’s natural Bad-recruiting protein for this process. Taken together, our results suggest a situation in which C. burnetii first activates Akt [25], which in turn phosphorylates Bad at Ser136, promoting loose association with 14-3-3 [38]. Bad then undergoes a structural change allowing T4SS effector-recruited PKA to phosphorylate Ser155 [38], promoting tight interaction between Bad and 14-3-3 present at the PV membrane. Thus, we predict PKA plays a two-fold role in preventing apoptosis during infection. First, PKA phosphorylates Bad at Ser155, blocking interaction with pro-survival mitochondrial proteins. Second, phosphorylation promotes mis-localization via interaction with 14-3-3β, trafficking Bad to the PV membrane and sequestering the protein from mitochondrial proteins. Because the balance of pro- and anti-apoptotic mitochondrial proteins is critical in deciding cell fate, blocking Bad interaction with Bcl-2 proteins by phosphorylation and altered trafficking heavily favors survival of C. burnetii-infected cells. By targeting both PKA and Akt, C. burnetii can efficiently and permanently prevent Bad pro-apoptotic activity.
Chlamydia trachomatis, the intracellular agent of trachoma, also targets Bad activity during infection. Inhibition of phosphatidylinositol-3 kinase (PI3K) upstream of Akt increases death of HeLa cells infected with C. trachomatis, suggesting Akt is required for apoptosis prevention during infection. Activation of Akt from 16–32 hpi is followed by increased Bad phosphorylation at Ser136 from 24–48 hpi with C. trachomatis. Bad phosphorylation is maintained in the presence of staurosporine and decreases upon addition of an Akt inhibitor, similar to our PKA studies with C. burnetii and Bad Ser155 phosphorylation. Additionally, Bad and 14-3-3β localize to the chlamydial inclusion, in an Akt-dependent process requiring C. trachomatis secreted IncG-mediated recruitment of 14-3-3β (Verbeke et al., 2006, Scidmore et al., 2001). Akt is also required for C. burnetii inhibition of apoptosis, but PKA has not previously been implicated in apoptosis prevention strategies by intracellular bacterial pathogens. Although C. burnetii lacks Inc proteins for recruiting 14-3-3β, the pathogen uses a T4SS to deliver proteins to the host cytosol where they interact with host proteins to promote infection. Recent studies identified three T4SS effectors that decorate the PV (Voth et al., 2011, Voth et al., 2009b) and these and other effectors are candidates for recruiting 14-3-3β and Bad.
In addition to recruiting Bad and 14-3-3β, C. burnetii coordinates PKA trafficking to allow efficient access to Bad. PKA localization is controlled by AKAPs that regulate PKA regulatory subunit trafficking, allowing catalytic subunits to act on local substrates. PKA RI localizes to the PV membrane in a T4SS-dependent manner, implicating a C. burnetii protein effector(s) as a critical component of PKA-associated cell survival. Likewise, we are currently investigating whether a C. burnetii effector is required for 14- 3-3β and Bad tethering to the PV membrane. In addition, 14-3-3 proteins preferentially localize to cholesterol-rich membranes (Rapp et al., 2007, Hekman et al., 2006), and the PV membrane is sterol-rich, decorating with the sterol probe filipin (Howe et al., 2006). Thus, we predict that inhibiting cholesterol biosynthesis would also prevent 14-3-3β localization and disrupt Bad recruitment to the PV membrane.
PKA RI localization to the PV membrane suggests a scaffolding complex composed of PKA catalytic subunits and AKAPs is present on the PV membrane. Interestingly, AKAP-Lbc binds to PKA and 14-3-3 proteins in other systems (Diviani et al., 2004), establishing precedent for a scaffolding complex mediating PKA-directed cell death events during infection. We previously reported that PKA is involved in PV formation and may act through Rho signaling (MacDonald et al., 2012). PKA phosphorylation of AKAP-Lbc regulates 14-3-3 recruitment and inhibits Rho-GEF activity on Rho GTPases (Diviani et al., 2004), proteins that also regulate PV formation (Aguilera et al., 2009). Therefore, C. burnetii recruitment of PKA and 14-3-3β may allow PKA to function in both apoptosis and PV formation by phosphorylating Bad and altering Rho-GEF signaling, respectively.
It is currently unknown if C. burnetii also recruits Akt to the PV membrane. However, canonical phagosome maturation relies on Akt’s upstream regulator, PI3K to generate a phagolysosome (Vieira et al., 2001, Fratti et al., 2001, Franke et al., 1995, Burgering et al., 1995). The C. burnetii PV transits through the typical phagolysosome maturation pathway and ultimately displays characteristics of secondary lysosomes. Thus, Akt may be recruited to the PV membrane to influence vacuole biogenesis and/or Bad activity, and this scenario awaits further testing. Regardless, Akt clearly contributes to the pathogen’s anti-apoptotic activity (Voth et al., 2009a) and phosphorylated levels of the Akt-specific Bad residue Ser136 increase throughout infection, further implicating Akt in the survival response of C. burnetii-infected macrophages.
Importantly, virulent C. burnetii activates PKA during growth in primary hAMs (MacDonald et al., 2012) and prevents staurosporine-induced apoptosis of these cells. This anti-apoptotic activity apparently involves Bad inactivation, as the protein is phosphorylated throughout infection. Apoptosis is a critical process in the lung environment and macrophages tightly regulate survival to appropriately respond to invading respiratory pathogens. Macrophages are typically long-lived cells that process pathogen components and present antigens to other cells of the immune system (Marriott et al., 2007). However, macrophages infected with certain pathogens trigger apoptosis to allow removal of the pathogen and infected macrophage (Marriott et al., 2007, Ashida et al., 2011). Respiratory pathogens, such as M. tuberculosis and L. pneumophila, confront this response by manipulating macrophage survival pathways to allow replication. In contrast to these two pathogens, C. burnetii replicates for many days in the same cell, doubling at a slow rate of ~ 12 h (Coleman et al., 2004). This lifestyle requires sustained viability of the host cell and C. burnetii actively promotes survival in a T4SS-dependent manner (Beare et al., 2011). Therefore, C. burnetii has evolved methods for preventing the innate alveolar macrophage response to infection, allowing outgrowth of the bacterial population prior to hematogenous spread to other sites.
In conclusion, we demonstrate the novel anti-apoptotic use of PKA by an intracellular bacterial pathogen and present a temporal mechanism for survival kinase activity in C. burnetii antagonism of macrophage apoptosis. To our knowledge, this is also the first study to demonstrate PKA localization to a bacterial replication vacuole. Future studies will further investigate the dual role of PKA in C. burnetii infection and identify T4SS effectors that regulate PKA and Bad trafficking. Furthermore, the pro-survival role of PKA should be explored in other intracellular bacterial pathogen systems as a mechanism for subverting host cell apoptosis.
Experimental procedures
C. burnetii and mammalian cell culture
C. burnetii phase II (RSA439) organisms (NMII) were cultured in Vero cells (American Type Culture Collection [ATCC]), and purified as previously described (Coleman et al., 2004). C. burnetii Nine Mile phase I (RSA493) organisms (NMI) were cultured in acidified citrate cysteine medium (ACCM) and purified as previously described (Omsland et al., 2009, MacDonald et al., 2012). For R1 localization assays, NMII wild type and icmD::Tn mutant organisms were cultured in ACCM medium as described previously [7], with mutant cultures containing kanamycin (350 μg/ml). Human monocytic THP-1 cells, HeLa (human epithelial) cells, and Vero (green monkey kidney) cells (ATCC) were cultured in RPMI1640 medium (Invitrogen) containing 10% fetal bovine serum (FBS; Invitrogen) at 37°C in 5% CO2. THP-1 cells were differentiated into adherent, macrophage-like cells by treatment for 18 h with phorbol 12-myristate 13- acetate (PMA; 200 nM; EMD Biosciences) (Loeuillet et al., 2006) and cultured in 6- or 24-well plates for infections. Prior to infection, medium containing PMA was replaced with PMA-free medium. THP-1 and HeLa cells were infected with C. burnetii strains at a multiplicity of infection (MOI) ~ 10 by addition of organisms to medium (time = 0).
Primary human alveolar macrophages (hAMs) were collected from lung tissue obtained post-mortem (National Disease Research Interchange) and harvested by bronchioalveolar lavage as previously described (Graham et al., 2012). Cells were cultured in Dulbecco’s Modified Eagle medium/F-12 medium containing 10% FBS, penicillin (50 U/mL), streptomycin sulfate (50 μg/mL), gentamicin sulfate (50 μg/ml), and amphotericin B (0.25 μg/mL) (Invitrogen) and incubated for 2 h at 37 °C in 5% CO2. Cells were washed vigorously 3 times with fresh medium containing antibiotics to remove non-adherent cells. After removal of antibiotics, hAMs were infected with NMI at an MOI ~ 10 by addition of organisms to medium (time = 0). All virulent isolate handling and infections were performed in the Centers for Disease Control and Prevention-approved biosafety level-3 facility at the University of Arkansas for Medical Sciences.
Staurosporine (EMD Biosciences) was added to cell cultures at a final concentration of 750 nM to induce intrinsic apoptosis. PKA activity was inhibited by addition of H-89 (10 μM; Sigma-Aldrich) or Rp-cAMPS (250 μM; Enzo Life Sciences) at the indicated times post-infection.
Fluorescence microscopy
For apoptosis assays, PKA was inhibited by H-89 or Rp-cAMPS treatment of infected cells, apoptosis induced with staurosporine, and cells processed for microscopy. Cells were fixed and permeabilized with 100% ice-cold methanol for 3 min, then blocked for 1 h in phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) at room temperature. Cells were incubated with rabbit anti-cleaved PARP (Cell Signaling Technology) and guinea pig anti-C. burnetii primary antibodies for 1 h at room temperature. Cells were then washed with PBS and incubated in PBS with 0.5% BSA and Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor-594 anti-guinea pig secondary antibodies (Invitrogen) for 1 h at room temperature. Bacterial and eukaryotic DNA was stained with 4′, 6′-diamidino-2-phenylindole dilactate (DAPI; Invitrogen) for 5 minutes, then samples were mounted in MOWIOL. Samples were observed using a Ti-U microscope with a 60X oil immersion objective (Nikon). Images were obtained with a D5-QilMc digital camera and analyzed with NIS-Elements software (Nikon). Approximately 950 cells were assessed to determine the percentage of fragmented nuclei or cleaved PARP-positive cells within each cell culture.
For RI localization assays, THP-1 cells were infected with NMII wild type or icmD::Tn organisms and processed for microscopy at 96 hpi. Cells were fixed with 4% paraformaldehyde and blocked in PBS containing 0.5% BSA and 0.03% Triton-X 100 at room temperature 1 h. Cells were then incubated with mouse monoclonal anti-PKA RI (Clone 18/PKA[R1]; BD Biosciences) primary antibody and rabbit anti-C. burnetii antibody for 1 h at room temperature. Cells were washed with PBS and incubated in PBS with 0.5% BSA and Alexa Fluor 488-conjugated anti-mouse and Alexa Fluor 594-conjugated anti-rabbit secondary antibodies (Invitrogen) for 1 h at room temperature. Bacterial and eukaryotic DNA was stained with DAPI for 5 minutes. Samples were analyzed as described above using a 40X objective, and approximately 500 cells were scored for RI localization to the PV membrane.
Immunoblot analysis
Cells were washed twice with PBS, harvested in buffer containing 1% sodium dodecyl sulfate (SDS; 50 mM Tris-HCl [pH 7.4], 5 mM EDTA, 1% SDS, protease and phosphatase inhibitor cocktail [Sigma]), passaged 10 times through a 26-gauge needle, and boiled for 5 minutes in SDS loading buffer. Equal amounts of total protein as determined by the DC Protein Assay (Bio-Rad) were separated by 15% SDS-polyacrylamide gel electrophoresis, transferred to a 0.2-μM pore size polyvinylidene fluoride membrane (Bio-Rad), and blocked in Tris-buffered saline (150 mM NaCl, 100 mM Tris-HCl [pH 7.6]) containing 0.1% Tween 20 and 5% nonfat milk for 1 h at room temperature. Equal protein loading was assessed using a mouse monoclonal anti-β-tubulin primary antibody (Clone SAP.4G5; Sigma) overnight at 4°C. Lysates were probed for PARP processing using rabbit primary antibodies directed against total PARP (Cell Signaling Technology). Lysates were also probed for Bad phosphorylation using rabbit primary antibodies directed against total or phosphorylated Bad (Ser155 or Ser136) (Cell Signaling Technology). Anti-rabbit or anti-mouse secondary antibodies conjugated to horseradish peroxidase (Cell Signaling Technology) were used to detect primary antibody using Femto enhanced chemiluminescence reagent (Pierce) and exposure to film. Immunoblots were imaged using an Alpha Innotech FluorChem F2 and analyzed using AlphaView band analysis.
Effector translocation assay
Differentiated THP-1 cells were infected with C. burnetii expressing CyaA-CpeE as previously described (Voth et al., 2011). At 48 hpi, indicated cells were treated with H- 89, then harvested, cAMP extracted, and samples processed using the cAMP Enzymeimmunoassay (GE Healthcare) at 72 hpi. As in previous studies, secretion was scored as ≥ 2.5-fold more cAMP than cells infected with C. burnetii expressing CyaA alone.
Subcellular localization of Bad
Human bad was amplified from a HeLa cDNA library (Invitrogen) using the forward primer CACCATGGGCTTCCAGATCCCAGAGT and reverse primer TTACTGGGAGGGGGCGGAGCTTCCC (Integrated DNA Technologies). Correctly sized products were inserted into the pENTR/D-TOPO vector and transformed into TOP10 E. coli (Invitrogen). Inserts from correct pENTR/D-TOPO clones were then sub-cloned into the pcDNA6.2-N-EmGFP vector using Gateway LR clonase II (Invitrogen) to generate a construct encoding GFP fused in frame to the N-terminus of Bad and transformed into TOP10 E. coli. Plasmids were isolated from E. coli and sequences confirmed by DNA sequencing. Transfection of HeLa cells with correctly sequenced plasmids was performed using Effectene (Qiagen) and 100 ng of plasmid DNA. At 18 h post-transfection, cells were fixed with 4% paraformaldehyde and blocked with PBS containing 0.5% BSA and 0.03% Triton-X 100 at room temperature 1 h. Cells were then incubated with mouse monoclonal anti-LAMP-1 primary antibody (Clone H4A3-S; Developmental Studies Hybridoma Bank) for 1 h at room temperature. Cells were washed with PBS and incubated in PBS with 0.5% BSA and Alexa Fluor 647-conjugated anti-mouse secondary antibody (Invitrogen) and rabbit anti-14-3-3β TRITC-conjugated primary antibody (Santa Cruz Biotechnology) for 1 h at room temperature. Bacterial and eukaryotic DNA was stained with DAPI for 5 minutes. At least 100 cells expressing GFP or GFP-Bad were analyzed for PV localization as described above.
Acknowledgments
This research was supported by funding to D. E. V. from the NIH/NIAID (R01AI087669 and R21AI107148), the American Heart Association (BGIA3080001), and the Arkansas Biosciences Institute, and to R. C. K. from the UAMS Translational Research Institute (1UL1RR029884). We thank Caylin Winchell for critical review of the manuscript.
References
- Aguilera M, Salinas R, Rosales E, Carminati S, Colombo MI, Beron W. Actin dynamics and Rho GTPases regulate the size and formation of parasitophorous vacuoles containing Coxiella burnetii. Infect Immun. 2009;77:4609–4620. doi: 10.1128/IAI.00301-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174:595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- Ashida H, Mimuro H, Ogawa M, Kobayashi T, Sanada T, Kim M, Sasakawa C. Cell death and infection: a double-edged sword for host and pathogen survival. J Cell Biol. 2011;195:931–942. doi: 10.1083/jcb.201108081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banga S, Gao P, Shen X, Fiscus V, Zong WX, Chen L, Luo ZQ. Legionella pneumophila inhibits macrophage apoptosis by targeting pro-death members of the Bcl2 protein family. Proc Natl Acad Sci U S A. 2007;104:5121–5126. doi: 10.1073/pnas.0611030104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, et al. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. Mbio. 2011;2:e00175–00111. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beebe SJ, Oyen O, Sandberg M, Froysa A, Hansson V, Jahnsen T. Molecular cloning of a tissue-specific protein kinase (C gamma) from human testis--representing a third isoform for the catalytic subunit of cAMP-dependent protein kinase. Mol Endocrinol. 1990;4:465–475. doi: 10.1210/mend-4-3-465. [DOI] [PubMed] [Google Scholar]
- Behar SM, Divangahi M, Remold HG. Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat Rev Microbiol. 2010;8:668–674. doi: 10.1038/nrmicro2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell D, McDermott BJ. Use of the cyclic AMP antagonist, Rp-cAMPS, to distinguish between cyclic AMP-dependent and cyclic AMP-independent contractile responses in rat ventricular cardiomyocytes. J Mol Cell Cardiol. 1994;26:1439–1448. doi: 10.1006/jmcc.1994.1163. [DOI] [PubMed] [Google Scholar]
- Beron W, Gutierrez MG, Rabinovitch M, Colombo MI. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect Immun. 2002;70:5816–5821. doi: 10.1128/IAI.70.10.5816-5821.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- Cain K, Bratton SB, Cohen GM. The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie. 2002;84:203–214. doi: 10.1016/s0300-9084(02)01376-7. [DOI] [PubMed] [Google Scholar]
- Carey KL, Newton HJ, Luhrmann A, Roy CR. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 2011;7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, et al. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc Natl Acad Sci U S A. 2010;107:21755–21760. doi: 10.1073/pnas.1010485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, et al. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol. 2004;186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Control CfD. Q fever--California, Georgia, Pennsylvania, and Tennessee, 2000–2001. MMWR Morb Mortal Wkly Rep. 2002;51:924–927. [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14–3–3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Diviani D, Abuin L, Cotecchia S, Pansier L. Anchoring of both PKA and 14–3–3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004;23:2811–2820. doi: 10.1038/sj.emboj.7600287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enserink M. Infectious diseases. Questions abound in Q-fever explosion in the Netherlands. Science. 2010;327:266–267. doi: 10.1126/science.327.5963.266-a. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, et al. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol. 2001;154:631–644. doi: 10.1083/jcb.200106049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallucci S, Lolkema M, Matzinger P. Natural adjuvants: endogenous activators of dendritic cells. Nat Med. 1999;5:1249–1255. doi: 10.1038/15200. [DOI] [PubMed] [Google Scholar]
- Graham JG, Macdonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol. 2012 doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackstadt T, Williams JC. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci USA. 1981;78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect Immun. 1996;64:796–809. doi: 10.1128/iai.64.3.796-809.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hekman M, Albert S, Galmiche A, Rennefahrt UE, Fueller J, Fischer A, et al. Reversible membrane interaction of Bad requires two C-terminal lipid binding domains in conjunction with 14–3–3 protein binding. J Biol Chem. 2006;281:17321–17336. doi: 10.1074/jbc.M600292200. [DOI] [PubMed] [Google Scholar]
- Howe D, Heinzen RA. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell Microbiol. 2006;8:496–507. doi: 10.1111/j.1462-5822.2005.00641.x. [DOI] [PubMed] [Google Scholar]
- Howe D, Mallavia LP. Coxiella burnetii exhibits morphological change and delays phagolysosomal fusion after internalization by J774A.1 cells. Infect Immun. 2000;68:3815–3821. doi: 10.1128/iai.68.7.3815-3821.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SK, Broederdorf LJ, Sharma UM, Voth DE. Host kinase activity is required for Coxiella burnetii parasitophorous vacuole formation. Front Microbiol. 2010;1:137. doi: 10.3389/fmicb.2010.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, El-Deiry WS. Overview of cell death signaling pathways. Cancer Biol Ther. 2005;4:139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Klingenbeck L, Eckart RA, Berens C, Luhrmann A. The Coxiella burnetii type IV secretion system substrate CaeB inhibits intrinsic apoptosis at the mitochondrial level. Cell Microbiol. 2012 doi: 10.1111/cmi.12066. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Laguna RK, Creasey EA, Li Z, Valtz N, Isberg RR. A Legionella pneumophila-translocated substrate that is required for growth within macrophages and protection from host cell death. Proc Natl Acad Sci U S A. 2006;103:18745–18750. doi: 10.1073/pnas.0609012103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Manipulation of host cell death pathways during microbial infections. Cell Host Microbe. 2010;8:44–54. doi: 10.1016/j.chom.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Loeuillet C, Martinon F, Perez C, Munoz M, Thome M, Meylan PR. Mycobacterium tuberculosis subverts innate immunity to evade specific effectors. J Immunol. 2006;177:6245–6255. doi: 10.4049/jimmunol.177.9.6245. [DOI] [PubMed] [Google Scholar]
- Losick VP, Haenssler E, Moy MY, Isberg RR. LnaB: a Legionella pneumophila activator of NF-kappaB. Cell Microbiol. 2010;12:1083–1097. doi: 10.1111/j.1462-5822.2010.01452.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrmann A, Nogueira CV, Carey KL, Roy CR. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc Natl Acad Sci USA. 2010;107:18997–19001. doi: 10.1073/pnas.1004380107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrmann A, Roy CR. Coxiella burnetii inhibits activation of host cell apoptosis through a mechanism that involves preventing cytochrome c release from mitochondria. Infect Immun. 2007;75:5282–5289. doi: 10.1128/IAI.00863-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald LJ, Kurten RC, Voth DE. Coxiella burnetii alters cyclic AMP-dependent protein kinase signaling during growth in macrophages. Infect Immun. 2012;80:1980–1986. doi: 10.1128/IAI.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marriott HM, Dockrell DH. The role of the macrophage in lung disease mediated by bacteria. Exp Lung Res. 2007;33:493–505. doi: 10.1080/01902140701756562. [DOI] [PubMed] [Google Scholar]
- Monack DM, Mecsas J, Ghori N, Falkow S. Yersinia signals macrophages to undergo apoptosis and YopJ is necessary for this cell death. Proc Natl Acad Sci USA. 1997;94:10385–10390. doi: 10.1073/pnas.94.19.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, et al. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc Natl Acad Sci U S A. 2009;106:4430–4434. doi: 10.1073/pnas.0812074106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth K, Palmer LE, Bao ZQ, Stewart S, Rudolph AE, Bliska JB, Dixon JE. Inhibition of the mitogen-activated protein kinase kinase superfamily by a Yersinia effector. Science. 1999;285:1920–1923. doi: 10.1126/science.285.5435.1920. [DOI] [PubMed] [Google Scholar]
- Raoult D, Marrie T, Mege J. Natural history and pathophysiology of Q fever. Lancet Infect Dis. 2005;5:219–226. doi: 10.1016/S1473-3099(05)70052-9. [DOI] [PubMed] [Google Scholar]
- Rapp UR, Fischer A, Rennefahrt UE, Hekman M, Albert S. BAD association with membranes is regulated by Raf kinases and association with 14- 3–3 proteins. Adv Enz Reg. 2007;47:281–285. doi: 10.1016/j.advenzreg.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Schmid MC, Scheidegger F, Dehio M, Balmelle-Devaux N, Schulein R, Guye P, et al. A translocated bacterial protein protects vascular endothelial cells from apoptosis. PLoS Pathog. 2006;2:e115. doi: 10.1371/journal.ppat.0020115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T. Mammalian 14–3–3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–1650. doi: 10.1046/j.1365-2958.2001.02355.x. [DOI] [PubMed] [Google Scholar]
- Shabb JB. Physiological substrates of cAMP-dependent protein kinase. Chem Rev. 2001;101:2381–2411. doi: 10.1021/cr000236l. [DOI] [PubMed] [Google Scholar]
- Shimizu S, Tsujimoto Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc Natl Acad Sci USA. 2000;97:577–582. doi: 10.1073/pnas.97.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Youle RJ, Tjandra N. Structure of Bax: coregulation of dimer formation and intracellular localization. Cell. 2000;103:645–654. doi: 10.1016/s0092-8674(00)00167-7. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440–442. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]
- Vazquez CL, Colombo MI. Coxiella burnetii modulates Beclin 1 and Bcl-2, preventing host cell apoptosis to generate a persistent bacterial infection. Cell Death Differ. 2009;17:421–438. doi: 10.1038/cdd.2009.129. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying S, Hansen J, Hacker G, Darville T, Ojcius DM. Recruitment of Bad by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006;2:e45. doi: 10.1371/journal.ppat.0020045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol. 2001;155:19–25. doi: 10.1083/jcb.200107069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DC, et al. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J Bacteriol. 2011;193:1493–1503. doi: 10.1128/JB.01359-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Heinzen RA. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell Microbiol. 2007a;9:829–840. doi: 10.1111/j.1462-5822.2007.00901.x. [DOI] [PubMed] [Google Scholar]
- Voth DE, Heinzen RA. Sustained activation of Akt and Erk1/2 is required for Coxiella burnetii antiapoptotic activity. Infect Immun. 2009a;77:205–213. doi: 10.1128/IAI.01124-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Howe D, Beare PA, Vogel JP, Unsworth N, Samuel JE, Heinzen RA. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm- mediated secretion. J Bacteriol. 2009b;191:4232–4242. doi: 10.1128/JB.01656-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE, Howe D, Heinzen RA. Coxiella burnetii inhibits apoptosis in human THP-1 cells and monkey primary alveolar macrophages. Infect Immun. 2007b;75:4263–4271. doi: 10.1128/IAI.00594-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Wong W, Scott JD. AKAP signalling complexes: focal points in space and time. Nat Rev Mol Cell Biol. 2004;5:959–970. doi: 10.1038/nrm1527. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Osipov K, Jockel J, Waksman G, Korsmeyer SJ. BH3 domain of Bad is required for heterodimerization with BCL-XL and pro-apoptotic activity. J Biol Chem. 1997;272:24101–24104. doi: 10.1074/jbc.272.39.24101. [DOI] [PubMed] [Google Scholar]
- Zhu H, Fearnhead HO, Cohen GM. An ICE-like protease is a common mediator of apoptosis induced by diverse stimuli in human monocytic THP.1 cells. FEBS Lett. 1995;374:303–308. doi: 10.1016/0014-5793(95)01116-v. [DOI] [PubMed] [Google Scholar]