

Abstract
Alpha-herpesviruses can produce more severe infections in non-natural host species than in their natural host. Isolates of the baboon alpha-herpesvirus Papiine herpesvirus 2 (HVP2) are either very neurovirulent in mice (subtype nv) or non-virulent (subtype ap), but no such difference is evident in the natural baboon host. Comparative genome sequencing was used to identify subtype-specific sequence differences (SSDs) between HVP2nv and HVP2ap isolates. Some genes were identified that despite exhibiting sequence variation among isolates did not have any SSDs, while other genes had comparatively high levels of SSDs. Construction of genomic recombinants between HVP2nv and HVP2ap isolates mapped the mouse neurovirulence determinant to within three genes. Construction of gene-specific recombinants demonstrated that the UL39 ORF is responsible for determining the lethal neurovirulence phenotype of HVP2 in mice. These results demonstrate that differences in a single viral gene can determine the severity of herpesvirus infection in a non-natural host species.
Keywords: Herpesvirus, zoonoses, neurovirulence, HVP2
Introduction
Zoonotic infections are a major public health concern and constitute the majority of "emerging infectious diseases". Despite the extremely serious nature of many zoonotic infections, it is likely that the vast majority of human exposures to nonhuman viruses result in very mild or abortive infections in which the virus is never able to establish itself in the human host. Understanding why some viruses are able to cross the species barrier and produce serious disease in non-natural hosts while others cannot is central to being able to treat and even prevent such infections.
Macacine herpesvirus 1 (monkey B virus; BV) is an α-herpesvirus that is indigenous in macaque monkeys (Macaca spp.). The BV genome is orthologous to that of herpes simplex virus type 2 (HSV2) and the chimpanzee virus (ChHV), having long and short unique regions (UL and US, resp.) that are each flanked by repeat sequences (RL and RS, resp.) (Dolan et al., 1998; Perelygina et al., 2003; and Severini et al., 2013). BV also exhibits substantial antigenic cross-reactivity with HSV (Eberle and Hilliard, 1995 and Eberle and Black, 1999). In its natural macaque host BV behaves very much as HSV does in humans: primary infections are usually asymptomatic, latent infection is established in sensory neurons, and in response to stress the virus may reactivate from latency with shedding of infectious virus. In stark contrast to the self-limiting nature of infections in its natural macaque host, a very different disease picture results when BV is transmitted to humans, usually via a monkey bite or scratch (Davidson and Hummeler, 1960; Elmore and Eberle, 2008; Huff and Barry, 2003; Weigler et al., 1992). BV invades peripheral sensory neurons, but instead of establishing a latent infection in sensory ganglia BV rapidly invades and spreads within the CNS, ascending the spinal cord to the brainstem and destroying CNS tissue. Untreated, about 80% of human BV patients die, and survivors frequently experience progressive neurological sequelae. Why BV behaves so very differently in macaques vs. humans remains unknown.
Phylogenetically and antigenically, the α-herpesvirus most closely related to BV is Papiine herpesvirus 2 (HVP2) (Eberle et al., 1995; Eberle and Hilliard, 1995; and Tyler and Severini, 2006). The HVP2 genome is orthologous with that of BV, having ~85% sequence identity to BV, a similarly high G+C content (76.5% vs 74.5% for BV) and a very high GC-codon bias (Tyler and Severini, 2006). HVP2 is indigenous in baboons (Papio spp.) in which it behaves the same as BV in macaques or HSV in humans (Eberle et al., 2008). Despite the very close relatedness of BV and HVP2, there are no known cases of human HVP2 infections. However, HVP2 does have the potential to produce fatal neurological infections as evidenced by lethal infection of a colobus monkey (Colobus guereza) that bore all the hallmarks of a human BV infection (Troan et al., 2007). Why BV and HVP2 should be so very divergent in regard to their apparent neurovirulence in humans is unknown.
Based on its very close similarity to BV, HVP2 has found use as a model for BV. While HVP2 isolates are equally pathogenic in their natural baboon host, testing of >20 HVP2 isolates in mice revealed that there are only two very distinct phenotypes with regard to their neurovirulence: either extremely neuroinvasive and lethal or nonvirulent (Rogers et al., 2003, 2005 and 2006). Neurovirulent HVP2 strains (HVP2nv) rapidly invade the CNS with destruction of CNS tissue that reflects that of BV in humans, while nonvirulent HVP2 strains (HVP2ap) invade the CNS much less efficiently, do not spread efficiently within the CNS, and cause only limited destruction of CNS tissue (Rogers et al., 2006). Since mouse-HVP2 neurovirulence is a simple 2-character state model, it represents an ideal model system in which viral determinants that facilitate severe cross-species infections can be investigated. Here we report the results of studies aimed at identifying specific gene(s) of HVP2 that determine the lethal neurovirulence of the virus in a non-natural host species.
Material and Methods
Viruses and cells
The origins of HVP2 isolates have been reported elsewhere, as has their neurovirulence in mice (Eberle et al., 1995, 1998, and Rogers et al., 2003). HVP2nv strains OU1-76 & X313 and HVP2ap strains OU2-5 & A951 were used for most experiments. Virus stocks were grown and titrated in Vero cells. Some experiments utilized primary mouse dermal fibroblast (PMDF) cell cultures prepared from mouse skin as previously described (Rogers et al. 2007).
Genomic DNA sequencing
Viral genome sequences were determined as described (Severini et al., 2013, Tyler and Severini, 2006; and Tyler et al., 2011). Briefly, initial sequencing was performed on a Genome Sequencer 20 (Roche) using the manufacturer's shotgun sequencing protocol. Sequencing was performed to an approximate depth of coverage of 40x and the data assembled with the GS De Novo Assembler (Roche). To fill sequence gaps, a conventional plasmid based shotgun library was prepared using the TOPO Shotgun Subcloning Kit (Invitrogen). Plasmid DNA was end sequenced, and clones of interest fully sequenced. Gaps in regions not covered by spanning clones were closed using conventional PCR. The UL39 ORFs of a number of HVP2 isolates were determined by direct sequencing of PCR products. All sequence data have been submitted to the Genbank database (UL39 sequences: accession nos. KF790618-KF790630; HVP2 genome sequences accession nos. KF908239-KF908244).
To identify regions of the HVP2 genome potentially associated with neurovirulence, open reading frames (ORFs) were aligned and nucleotide differences were classified as either subtype-specific differences (SSDs (the same in all HVP2ap isolates but different from HVP2nv isolates) or as isolate-specific differences (present in one or more isolates but not correlating with the neurovirulence phenotype). SSDs were further subdivided into those that resulted in a no change in amino acid (AA) sequence, a conservative AA change, or a non-conservative AA change. To assess the distribution of types of sequence differences across the HVP2 genome, ORF variation scores were calculated by assigning SSDs that resulted in no AA change a value of 0, a conservative AA change a value of 1, a non-conservative change a value of 2, and those resulting in an insertion/deletion a value of 2. To account for the varying size of ORFs, the total score for each ORF was divided by the ORF size (number of AAs) and multiplied by 100.
Construction of recombinant viruses
Viral DNA was purified from infected cells on NaI gradients as described (d'Offay et al., 1993 and Walboomers and Schegget, 1976). Viral DNA from an HVP2nv and an HVP2ap strain (2.5 μg each) was digested for 1 hr with Xba1. All HVP2 strains have a single Xba1 site located in the UL33 ORF. After digestion, viral DNAs were mixed at a 1:1 ratio, precipitated, resuspended in 10 mM Tris/0.1 mM EDTA, and 5 μg transfected into subconfluent Vero cells using the Geneporter reagent (Genlantis, San Diego, CA). When CPE was evident (24–36 hrs post transfection) cell cultures were subjected to 2 freeze/thaw cycles, clarified by centrifugation at 14,000 x g for 15 sec, the supernatant diluted and plated onto Vero cell monolayers, incubated at 37°C for 1 hr, and cultures overlaid with medium containing 1% methylcellulose. Once plaques developed (48 hrs), individual plaques were picked and inoculated onto Vero cells. When CPE was complete, infected cultures were subjected to 2 freeze/thaw cycles, clarified by centrifugation, the supernatant saved as stock, and DNA extracted from the pellet using DNAzol per the manufacturer's instructions (Molecular Research Center, Inc., Cincinnati, OH). Using primers located on either side of the unique Xba1 site in the UL33 ORF, sequence spanning the Xba1 site was amplified by PCR and sequenced, allowing HVP2nv/ap recombinants to be identified based in single nucleotide sequence differences that distinguished the two parental viruses. Recombinants were further characterized by PCR/sequencing to determine which parental virus the RL2 (ICP0), US4, UL5 and UL51 genes were derived from. Additional mapping of the UL33-UL56 region of some recombinants was performed using PCR/sequencing with primers located at approximately 5 kbp intervals.
Gene-specific recombinants were constructed using a GFP expression cassette insertion/replacement approach as previously described (Rogers et al., 2009). Briefly, sequences flanking the UL39 ORF 5’ (1.7 kbp) and 3’ (2.0 kbp) as well as the UL39 ORF (2.85 kbp) were amplified by high fidelity PCR incorporating unique restriction sites at their termini. The 5’ and 3’ flanking sequences were cloned into pLitmus28i and a fluorescent protein (GFP or RFP) expression cassette (Black et al., 2002) was inserted between the two flanking sequences. To delete the UL39 ORF the construct was transfected into Vero cells, the cultures infected with virus 24 hrs later, and fluorescent viral plaques picked and plaque purified. Gene-specific UL39 recombinants were subsequently produced by inserting a UL39 ORF between the two flanking sequences, the construct transfected into cells, cells infected with a fluorescent UL39-deletion virus, non-fluorescent plaques picked, and the virus then plaque purified. All mutant viruses were sequenced across the entire region (5’ flanking, ORF, and 3’ flanking) to confirm the sequence integrity of the region.
Mouse inoculations
All animal experiments were reviewed and approved by the OSU IACUC. Mice were inoculated to assess neurovirulence of viruses as described (Rogers et al., 2003). Briefly, the left flank of 10–12 gm Balb/c mice was shaved, the skin lightly scarified, and 105 PFU of virus applied. Mice were observed for clinical symptoms of neurological infection for 2 weeks, and any mice showing signs of serious CNS involvement were humanely euthanized.
Statistical analyses
Statistical analyses were performed using the software program SigmaPlot (version 11.0, Systat Software Inc., Chicago, IL). The effects of viral strain and of recombination on viral titer or IFN-β production were each tested by a two-way analysis of variance (ANOVA). Viral titers were log10 transformed to meet normality assumptions. Further comparisons of recombinants versus the wild-type virus were performed using the Holm-Sidak multiple comparison procedure with the overall significant level set to 0.05.
Results
Comparative genome sequencing and analysis
Comparison of sequences of closely related viruses to identify genetic differences that underlie variant phenotypes is a simplistic approach that may not be effective for a number of reasons. For example, different mutations, even different mutations within different genes, may result in a similar phenotype in different viral isolates. Also, multiple genetic differences may define a phenotype. It is even possible that mutations resulting in different codon usage, changes in non-translated regions of mRNA, or even varied expression of a gene could alter a phenotype. Assessing >20 strains of HVP2 for neurovirulence in mice revealed only two very distinct and consistent phenotypes – neurovirulent and rapidly lethal or non-lethal. Furthermore, the neurovirulence phenotype of HVP2 strains has correlated 100% with the ability of an isolate to replicate efficiently and suppress the IFN-β response in PMDF cells (Rogers et al., 2003; 2007; 2009). The lack of variability in this phenotype among so many HVP2 strains suggested that there may be only a single, consistent difference between HVP2nv and HVP2ap strains that underlies the mouse neurovirulence phenotype. We therefore took the approach of comparative genome sequencing to determine if this approach had value in identifying phenotype-defining genetic differences among herpesvirus isolates.
The complete genome sequence of six HVP2 isolates was determined which together with the previously published sequence for strain X313 (Tyler and Severini, 2006) resulted in genomic sequences for three HVP2nv strains and four HVP2ap strains. For each coding sequence, nucleotide differences that resulted in changes in the AA sequence of the encoded protein were divided into two groups: sequence differences that were the same in all HVP2ap isolates but were different from HVP2nv isolates were classified as SSDs, while those that were present in one or more isolates but did not correlate with the neurovirulence subtype were classified as isolate-specific differences. To assess the distribution of types of sequence differences across the HVP2 genome, ORF variation scores were calculated and plotted along the viral genome map (Fig 1).
Figure 1. ORF variation scores based on SSDs identified from comparison of 7 HVP2 genomic sequences.
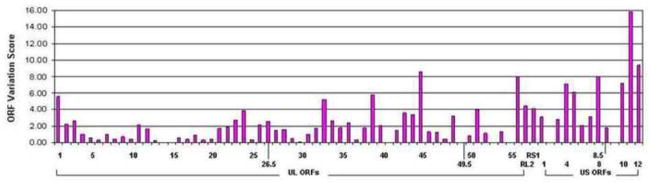
Genome sequences were aligned, SSDs identified and those located with coding sequences assigned a weighted value as described in Materials & Methods to arrive at an ORF variation score for each gene.
The distribution of SSDs was not uniform along the HVP2 genome; there were fewer SSDs located in genes located in the left part of the UL region (genes UL4-20) than in the right part of the UL region or the US region, and overall the US region had a higher density of SSDs than the UL region. Of the 73 ORFs assessed, 8 had scores of 0 as they contained no SSDs (UL14, UL15, UL41, UL49.5, UL53, UL55, US2 & US9). A number of other genes had very low scores (i.e., only a few subtype-specific conservative AA substitutions). The ORFs having the highest scores were UL1, UL33, UL39, UL45, UL56, US4, US5, US8, and US10-12. Gene function was not associated with a high or low ORF score; groups of genes involved in similar functions (DNA replication, protein modification, regulatory, capsid structure, DNA packaging, glycoproteins) exhibited a spectrum of scores ranging from none to very high. We could not identify any characteristic that related to the level of SSDs in different genes.
Although only 3–4 genomes of each HVP2 subtype were sequenced, the above analyses completely eliminated 8 viral genes (with scores of 0) from possibly influencing the mouse neurovirulence phenotype of HVP2. There were 11 genes that had only one SSD resulting in a non-conservative AA change and an additional 11 that had only two. Assuming that such conserved genes are less likely to underlie the pathogenic phenotype of the virus than are genes with more SSDs, a total of 30 genes out of the 72 known genes had a low probability of being a potential determinant of cross-species neurovirulence.
To determine if additional genome sequences could eliminate additional genes through greater discrimination between SSDs and non-SSDs, we focused on a single gene (UL39) that had a comparatively high ORF variation score of 6 (50 total SSDs + 4 gaps/2985 bp). The UL39 gene sequence was determined for a total of 12 HVP2 isolates (7 from complete genome sequencing and 5 by PCR/sequencing, 6 of each neurovirulence subtype). DNA sequences of the UL39 ORF, the 5’ intergenic flanking region (413 nucleotides), and the 3’ flanking intergenic region (69 nucleotides) were aligned sequentially, adding one pair of sequences (one of each subtype) at a time. At each step the total number of nucleotide sequence differences and the number of these that were subtype-specific were tabulated as were gaps (insertions/deletions) in the aligned sequences. Starting with comparison of two sequences (one of each subtype), all sequence differences appear as subtype-specific (Table 1). As additional sequence pairs are added to the alignment, the total number of sequence differences increases, but the ability to discriminate between isolate-specific and subtype-specific differences also increases. Despite the sequence variation in the UL39 ORF, alignment of only five sequence pairs was required before no further reduction in SSDs occurred. Sequence variation in the UL39 ORF thus appears associated with the neurovirulence phenotype and is not due to a high degree of sequence variation among individual HVP2 isolates. For the two flanking intergenic regions, even fewer sequence pairs were required before stabilization in the number of SSDs occurred. Assembling alignments by sequential addition of sequence pairs using a different order of sequence pairs did have a small effect on the number of pairs required to reach a stable endpoint in the number of SSDs; initiating the process using sequences from isolates most different from each other (strains isolated years apart at different institutions) resulted in the most rapid identification of final SSDs.
Table 1.
Effect of the Number of Gene Sequence Pairs on Differentiating Isolate-Specific from Subtype-Specific Sequence Differences
| No. Seq Pairs | 5’ Intergenic Region (413 nuc) | UL39 ORF(2973 nuc) | ||||||
|---|---|---|---|---|---|---|---|---|
| Substitutions | Gaps | Substitutions | Gaps | |||||
| Total | SSD | Total | SSD | Total | SSD | Total | SSD | |
| 1 | 27 | 27 | 13 | 13 | 75 | 75 | 18 | 18 |
| 2 | 32 | 24 | 24 | 10 | 81 | 63 | 36 | 15 |
| 3 | 38 | 20 | 19 | 8 | 93 | 52 | 39 | 6 |
| 4 | 39 | 20 | 19 | 8 | 101 | 50 | 39 | 3 |
| 5 | 39 | 20 | 19 | 8 | 104 | 49 | 39 | 3 |
| 6 | 39 | 20 | 19 | 8 | 105 | 49 | 39 | 3 |
DNA sequences were aligned sequentially, adding one pair of sequenced at a time (one ap & one nv). At each step the total number of nucleotide sequence differences (total) and the number of these that were subtype-specific differences (SSD) were tabulated. Gaps (insertions/deletions) in the aligned sequences were similarly tabulated. The point at which the number of SSDs stabilized is indicated by numbers in bold.
Mapping the neurovirulence phenotype with inter-typic recombinants
While comparative sequencing does appear to have the potential to reduce the number of potential candidate genes conferring the cross-species neurovirulence phenotype on HVP2, three genome pairs was not sufficient to accomplish this goal. As a second approach to identify a gene(s) responsible for the mouse neurovirulence phenotype of HVP2, recombinant viruses were constructed. Taking advantage of a unique Xba1 site located in the UL33 ORF of all HVP2 isolates, recombinant viruses were generated between two separate pairs of HVP2nv and HVP2ap strains. An initial set of recombinants (series XX) was made using strains OU1-76 (HVP2nv) and OU2-5 (HVP2ap) as parental viruses. Viral DNAs were digested with Xba1, the two DNAs combined, transfected into Vero cells, and infectious virus recovered. Recombinants were identified by PCR/sequencing of 570 bp spanning the unique Xba1 site, with sequence differences between the two parental strains allowing the virus strain origin of sequences on each side of the Xba1 site to be determined. The genomes of recombinants were further characterized by PCR/sequencing of 300–500 bp in the UL5, UL51, RL2 and US4 genes to map in a very broad sense what areas of the genome were derived from which parental virus. Genome maps of several representative XX recombinants are shown in Fig 2.
Figure 2. Genomic maps of inter-typic HVP2 recombinants.
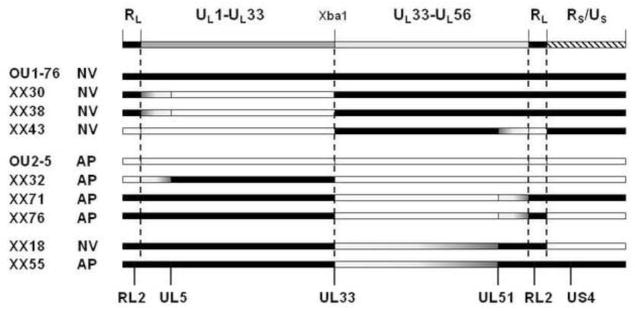
Recombinant HVP2 strains were constructed and the parental origin of UL5, UL51, RL2, US4 and the UL33 ORF on both sides of the unique Xba1 site in UL33 were determined by sequencing of PCR products. Regions of the genomes derived from the neurovirulent parent are shown in black and regions from the apathogenic parent virus are in white. Representative recombinants are shown, and are grouped with the parental virus (OU1-76 or OU2-5) having the same neurovirulence phenotype as indicated at left. Recombinants XX18 and XX55 are representative recombinants having a second recombination point located between the UL33 and UL51 genes (indicated by gradient coloring).
A total of 24 XX recombinants were generated. While some of the recombinants had complete halves of the UL region (UL5-UL33 or UL33-UL51) derived from each parental virus (eg. XX30, XX38, XX43, XX32, XX71 & XX76 in Fig. 2), other recombinants had a second recombination point located in one or both halves of the UL region (eg. XX18 & XX55 in Fig. 2). All recombinants had the long repeat (RL) regions of only one parental virus. The neurovirulence of 22 XX recombinants was assessed by inoculating mice with 105 PFU by skin scarification. Using this virus dose the two parental strains exhibit very distinct neurovirulence phenotypes in mice (Rogers et al., 2003). All recombinants tested displayed one of the two parental neurovirulence phenotypes (Fig 3 shows results for the representative recombinants diagramed in Fig 2). Mice inoculated with some recombinants rapidly succumbed to lethal CNS infection, developing hind limb paralysis and death within 5–7 days, while others exhibited minimal lethality (that followed a more extended development timeframe) or very mild/no clinical signs of CNS infection. All mice surviving to 14 days PI developed an anti-HVP2 IgG response, confirming that they had been infected.
Figure 3. Neurovirulence of inter-typic HVP2 recombinants.
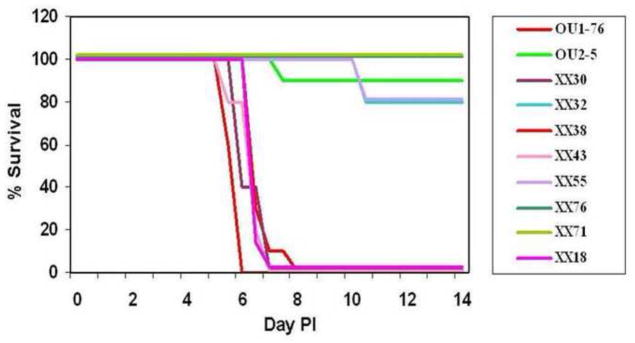
Mice were infected with 105 PFU of recombinants by skin scarification on the hind flank and observed for 14 days. Results for the representative viruses in Figure 2 are shown here. The RL, US/RS, and UL1-UL33 regions did not correlate with neurovirulence. In contrast, all recombinants having the UL33-UL51 region of the genome derived from the neurovirulent parent virus developed severe neurological infections requiring euthanasia by 8 days PI while viruses having the UL33-UL51 region derived from the apathogenic parent virus survived to 14 day PI.
To identify HVP2 gene(s) potentially associated with lethal neurovirulence, genome maps of recombinant viruses (Fig 2) were compared with their in vivo neurovirulence phenotype (Fig 3). Based on recombinants XX18, XX55 & XX71 it was evident that the parental virus origin of the US region did not correlate with the neurovirulence phenotype. Similarly, recombinants XX43, XX55, XX71 & XX76 indicated that the parental origin of the RL region did not correlate with neurovirulence. Correlations are more complicated for the much larger UL region due to secondary recombination points. However the parental origin of the left half of the UL region (UL5-UL33) did not correlate with neurovirulence of recombinants. Recombinants XX32, XX30, XX38, XX43, XX55, XX71 & XX76 had the entire region between UL5 and UL33 derived from one parental virus, and the parental virus origin of this region did not correlate with the neurovirulence of the virus. In contrast recombinants XX30, XX32, XX38, XX43, XX71 & XX76 all had the right half of the UL region (UL33-UL51) derived from a single parental virus and the neurovirulence of these recombinants directly correlated with the parental virus origin of this region. These results suggest that the gene(s) determining the murine lethal neurovirulence phenotype lies within the UL33-Ul51 region of the HVP2 genome.
To confirm these results, a second set of recombinants (series AX) was generated using two different virus strains (HVP2nv strain X313 and HVP2ap strain A951). Recombinants were generated, genomes mapped, and neurovirulence assessed as described above. A total of 20 AX recombinants were tested to assess their neurovirulence in mice. Once again, neurovirulence did not correlate with the parental origin of the RL region, the US region, or the UL1-33 region while the parental virus origin of the UL33-UL56 region showed 100% correlation with the neurovirulence phenotype (not shown). From these results we conclude that the gene(s) underlying the neurovirulence phenotype of HVP2 lies within the UL33-UL51 region of the genome.
While many recombinants had the majority of each half of the UL region derived from different parent viruses, 18 recombinants (11 from XX series, 7 from AX series) had a second recombination point within the UL33-UL51 region (eg. recombinants XX18 & XX55 in Fig. 2). To further map the neurovirulence determinant within this region, PCR/sequence analysis using primer pairs sited at ~5 kbp intervals between UL33 and UL51 were used to spot-check for the parental virus sequence and so localize the second recombination point (Fig 4). Based on the neurovirulence phenotype of these recombinants and the location of recombination points within the UL33-UL51 region of the genome, the neurovirulence phenotype was mapped to a 9.31 kbp region extending from the 5’ 200 bp of the UL36 ORF to the midpoint of the UL39-UL40 intergenic region.
Figure 4. Localization of the HVP2 murine neurovirulence determinant.

The general layout of the HVP2 genome and genes in the right half of the UL region are shown at top. The parental origin of sequences in all nine recombinants that had a second recombination site between UL33 and UL51 was determined by sequencing of PCR products generated at approximately 5 kb intervals (indicated by triangles). Sequences from the neurovirulent parent virus are indicated by black and those from the apathogenic parent virus by white; gray areas represent areas between PCR points in which the second recombination point lies. The neurovirulence phenotype of each virus is indicated at right. For all nine recombinants, the parental origin of the region of the HVP2 genome between the ends of the UL36 and UL40 genes correlates with the neurovirulence phenotype.
Identification of UL39 as the primary determinant of neurovirulence
While nucleotide sequence differences that alter transcription of a gene could affect neurovirulence, we initially focused on differences resulting in a change in the AA sequence of encoded polypeptides. Focusing on the UL36-UL39/40 intergenic region, comparative sequence analysis showed that the 5’ 200 bp of the UL36 ORF contained no SSDs, eliminating UL36 as a potential neurovirulence determinant. All three of the remaining coding sequences (UL37, UL38 & UL39) had SSDs that resulted in changes of their AA sequences. Alignment of 10 UL37 sequences identified a total of 3 subtype-specific AA differences (2 conservative and 1 non-conservative) in the 1207 AA alignment (0.2% subtype-specific sequence divergence). Similarly, alignment of 10 UL38 sequences identified only 5 subtype-specific AA substitutions (2 conservative and 3 nonconservative) in the 457 AA alignment (1.1% subtype-specific sequence divergence). In contrast, comparison of 13 UL39 sequences revealed a total of 38 subtype-specific AA substitutions (24 conservative and 14 non-conservative) along the 989 AA sequence alignment (3.8% subtype-specific sequence divergence; see supplemental material). Based on this, the UL39 gene seemed to be the most probable candidate gene for determination of the cross-species neurovirulence phenotype.
To test this prediction, gene-specific UL39 recombinants were constructed. Nomenclature used for these recombinants consisted of an A, N or F [designating the phenotype origin of the UL39 ORF (apathogenic, neurovirulent, or deletion via replacement with GFP or RFP, resp.] plus flanking ‘a’ or ‘n’ to designate the parental phenotype of the flanking genomic sequences. The UL39 ORF of HVP2ap strain OU2-5 was deleted and replaced with a GFP expression cassette (mutant aFa), with the parental HVP2ap UL39 ORF (revertant aAa) or with the HVP2nv UL39 ORF (recombinants aNa). A similar set of UL39 mutants was constructed on the HVP2nv OU1-76 background (designated nFn, nNn & nAn, resp.). The neurovirulence of the UL39 mutants was then tested in mice (Fig 5). Mice infected with the neurovirulent parental OU1-76 virus or revertant nNn developed initial neurological symptoms at 4-5 DPI that rapidly progressed to death by 6-7 DPI while mice infected with the apathogenic parental OU2-5 virus or revertant aAa survived, developing only mild neurological symptoms that improved over time. Infection with mutants lacking a UL39 ORF (nFn or aFa) failed to produce any neurological symptoms in infected mice, indicating that UL39 is important for neurovirulence. Mice infected with two different recombinant apathogenic viruses carrying the UL39nv ORF (aNa) developed clinical signs of neurological involvement and died at 7-8 DPI while two neurovirulent recombinant viruses carrying the UL39ap ORF (nAn) survived infection with only mild neurological symptoms that improved with time like the parental HVP2ap virus. All survivors had anti-HVP2 serum IgG, confirming their infection. These results demonstrate that the UL39 ORF determines the ability of HVP2 to produce lethal neurological infections in mice. However, the slightly delayed death of mice infected with the aNa recombinants relative to WT HVP2nv (~1 day) suggests that some factor other than UL39 may affect the rate at which the infection progresses.
Figure 5. The UL39 ORF determines neurovirulence of HVP2 in mice.
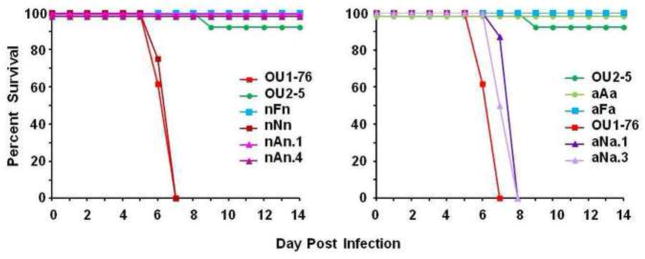
UL39 mutants constructed on the neurovirulent OU1-76 background (designated nXn) are shown at left, and those on the apathogenic OU2-5 background (designated aXa) are at right. Mice were inoculated by skin scarification with 105 PFU of virus and observed for 14 days. All viruses with the UL39nv ORF were neurovirulent while those with the UL39ap ORF or with the UL39 ORF deleted were not pathogenic.
Previous studies have shown that the mouse neurovirulence phenotype of HVP2 strains correlated with their ability to replicate productively and to suppress an IFN-β response in primary mouse skin fibroblast cultures (Rogers et al., 2009). Since UL39 determines lethal neurovirulence in mice, UL39 recombinants were tested for their ability to replicate in PMDF cultures and to suppress an IFN-β response.
To assess replication, PMDF cultures were infected at a low MOI (0.01 PFU/cell), observed daily for plaque formation, and at 96 hrs PI infectious virus was quantitated. As previously reported (Rogers et al., 2007) OU2-5 and revertant aAa formed small foci of infection by 24 hrs PI but CPE did not spread with time. Conversely, OU1-76 and revertant nNn both formed plaques by 24 hrs PI and CPE rapidly progressed to involve the entire cell monolayer within 48 hrs PI. OU2-5 mutants with UL39nv (aNa) also formed plaques that rapidly spread to destroy the cell cultures by 48 hrs PI. In contrast, OU1-76 mutants with UL39 (nAn) initially formed small plaques by 24 hr PI, but these failed to enlarge past 24 hrs PI. Consistent with these observations, the two deletion mutants that lacked a UL39 ORF (aFa & nFn) produced only small initial foci of infection that failed to enlarge past 24 hr PI. Quantitative assessment of infectious virus at 96 hrs PI reflected these observations (Fig 6, left). Viruses that could form large plaques produced significantly greater amounts of infectious virus than did viruses that did not. These results show that UL39 ORF imparts upon the virus the ability to replicate in primary murine cells, which correlates with their neurovirulence in mice.
Figure 6. UL39 determines the ability of HVP2 to replicate efficiently in primary murine cells but not suppression of IFN-β response.
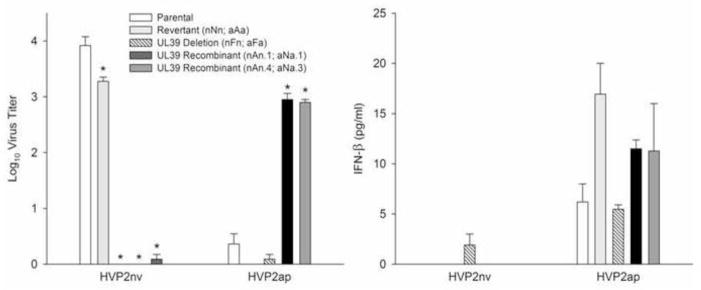
The ability of UL39 recombinants to replicate in primary murine cells was assessed by infecting confluent PMDF cultures at an MOI = 0.01 PFU/cell and quantitation of infectious virus at 96 hrs PI by plaque assay on Vero cells (left panel). Neurovirulent virus carrying the UL39ap ORF (nAn) or with the UL39 ORF deleted (nFn) produced significantly less infectious virus than the parental wild-type or revertant viruses, while apathogenic virus carrying the UL39nv ORF (aNa) produced significantly higher levels of infectious virus than the apathogenic wild-type, revertant (aAa) or UL39 deletion virus (aFa). To assess the role of UL39 in suppressing the IFN-β response, PMDF cultures were infected at an MOI = 0.4 PFU/cell and IFN-β present in the extracellular medium assayed by ELISA at 24 hrs PI (right panel). The background of the recombinant viruses but not the UL39 ORF correlated with the ability to effectively suppress the IFN-β response. In each graph, the background of each set of viruses (WT, revertant, UL39 deletion and two UL39 recombinants) are indicated below the graph. Data were analyzed using a two-way ANOVA and compared to the parental virus with significance set to 0.05. Asterisks denote significant differences between recombinants and their respective parental wild type virus.
To assess the ability of UL39 mutants to suppress an IFN-β response by the infected cell, confluent PMDF cultures were infected and IFN-β excreted into the extracellular medium assayed by ELISA at 24 hrs PI (Fig 6, right). Consistent with previous reports (Rogers et al., 2009), the parental apathogenic strain OU2-5 and revertant aAa both induced IFN-β production while the parental neurovirulent OU1-76 strain and revertant nNn did not (P<0.001). Both of the neurovirulent mutants carrying the UL39ap ORF (nAn) also failed to induce an IFN-β response, while apathogenic parental recombinants carrying the UL39nv ORF (aNa) did. This suggests that the ability to suppress an IFN-β response is not a function of the UL39 gene, but rather lies elsewhere within the parental background of the virus. However, the HVP2nv mutant lacking a UL39 ORF (nFn) consistently produced low levels of IFN-β, suggesting that UL39 does to some extent suppress the IFN-β response. The consistently lower level of IFN-3 produced by cells infected with the nFn mutant relative to all HVP2ap mutants implies that some other factor present in HVP2nv but not in HVP2ap also has a suppressive effect which, together with that of UL39, results in complete suppression of the IFN-β response.
Discussion
Advances in genome sequencing technology have made sequencing of large genomes feasible. This in turn makes possible comparative analysis of multiple genomic sequences as a means of identifying sequence differences or genes associated with specific phenotypes. This approach has been used to identify nucleotides changes associated with attenuation of varicella zoster virus (Peters et al., 2006; Tyler et al., 2007) and to differentiate vaccine and field isolates of BHV1 and to explore the viral origins of vaccination-associated disease outbreaks (Fulton et al., 2013). Here we used this approach in an attempt to identify genes of HVP2 that are associated with extreme neurovirulence in a non-natural host species.
The very high G+C content of the HVP2 genome (>75%) resulted in less efficient sequencing than for other microbial agents, so only 6 HVP2 isolates were sequenced. This comparatively small number of genome sequences (3 pairs) together with the greater inter-strain sequence plasticity of simplexvirus genomes as compared to varicellovirus genomes did not allow identification of specific genes that were associated with the mouse neurovirulence phenotype. However, a significant number of genes could be eliminated as being associated with the phenotype by virtue of their lack of any SSDs. Analysis of gene sequences for one highly variable gene (UL39) indicated that as few as 6-7 genome pairs would likely provide the power necessary to distinguish all SSDs from isolate-specific sequence differences, thereby further narrowing the number of genes potentially associated with mouse neurovirulence.
Construction of recombinant viruses has long been used to identify regions of herpesvirus genomes and to confirm the role of specific genes associated with specific traits. Using this approach the mouse neurovirulence phenotype was mapped to the UL36-UL40 region of the HVP2 genome. Applying a comparative sequence approach to this region of the genome completely eliminated one ORF and an intergenic region from consideration and allowed prioritization of the remaining genes as to the likelihood of their affecting neurovirulence, UL39 being the most likely candidate. Subsequent construction and testing of UL39-specific recombinants confirmed these predictions and validated the use of predictive comparative sequence analysis.
The UL39 gene encodes the large subunit of the ribonucleotide reductase (RR) enzyme. RR converts ribonucleotides (CDP, ADP & GDP) into deoxyribonucleotides for use in DNA synthesis. The poor replication of UL39-deficient HSV mutants in confluent, non-dividing cells where intracellular dNTP pools are low (Goldstein and Weller, 1988, and Smith et al., 1998) suggests that the viral RR functions in vivo by enabling the virus to replicate in non-dividing cells like neurons. Consistent with this assumption, HSV UL39 mutants do not productively replicate in peripheral neural tissue but do establish latent infections (Cameron et al., 1988, and Jacobson et al., 1989). The UL39 gene of cytomegalovirus has also been shown to be a pathogenic determinant despite lacking any RR enzymatic activity (Lembo et al., 2004). Thus, the UL39 gene being a determinant of cross-species neuropathogenicity of HVP2 is consistent with previous studies on HSV and CMV.
Unlike the large subunit of cellular dimeric RRs, the large RR subunit polypeptides of herpesviruses have a large N-terminal domain unrelated to other known proteins (Conner et al., 1994b, and Lembo and Brune, 2009). The RR active site, RR consensus motif, and other RR associated functions are located in the conserved C-terminal domain of the UL39 protein suggesting that the N-terminal region does not function in RR enzymatic activity. Like HSV UL39 mutants, HVP2ap strains replicate slightly less well in densely packed, non-dividing Vero cells and exhibit abortive replication in primary murine cells (Rogers et al., 2007). Previous studies have also demonstrated that while HVP2ap strains can replicate in the skin of infected mice, they are less efficient than HVP2nv strains at invading the CNS and spreading within it (Rogers et al., 2009). The results of experiments presented here indicate that the UL39 gene is responsible for this difference in lethal neurovirulence of HVP2 strains in mice. Furthermore, the strongly conserved nature of the C-terminal region of the HVP2 UL39 ORF among all strains suggests that the N-terminal region likely determines the ability to replicate in primary murine cells and to efficiently invade and replicate within the CNS of mice. Further experiments are required to test this.
Previous studies on the ability of HVP2 strains to replicate in primary murine cells found that efficient replication correlated with the ability of the virus to suppress an IFN-β response; an IFN-β response was induced by HVP2ap infection while HVP2nv strains effectively suppressed any such response (Rogers et al., 2009). This suggested that the innate immune response, in particular the IFN-β response, was important in determining the ability of HVP2 to cause serious CNS disease. However, the results of experiments presented here with UL39 mutants are not entirely inconsistent with this prediction. The UL39 ORF (ap vs. nv) is not solely responsible for suppressing the IFN-β response, although it does apparently play some role as evidenced by the low level but consistent IFN-β response induced following infection with the HVP2nv nFn mutant. Since deletion of the UL39 ORF from either HVP2 subtype results in a failure to sustain replication in primary murine cells, the UL39 ORF/protein is clearly required for this. While the nFn deletion mutant did allow a low IFN-β response, HVP2ap mutants carrying the UL39nv ORF did not, suggesting that some function of UL39 unrelated to complete suppression of the IFN-β response determines the ability of the virus to replicate in primary murine cells and invade/spread within the CNS.
The HVP2 UL39 protein is closely related to the homologous HSV protein, and homologues of all domains and specific AA residues associated with various functions or motifs in HSV are readily evident in the HVP2 protein. If RR enzymatic activity were to impart on the virus the ability to replicate in neurons, the lack of RR activity in HVP2ap isolates would be a ready explanation for their lack of neurovirulence. However, the vast majority of AA SSDs in the HVP2 UL39 are located in the N-terminal region of the protein, only two conserved substitutions being present within the C-terminal domain where RR enzymatic activity has been localized. Also, both HVP2ap and HVP2nv isolates are equally pathogenic in the natural baboon host (Rogers et al., 2005). This suggests that the N-terminal domain of the HVP2 UL39 protein in some way affects neurovirulence of HVP2 in mice, likely independently of RR enzymatic activity. Furthermore, the lack of any detectable pathogenic difference of HVP2 isolates in their natural baboon host suggests that whatever mechanism is involved in determining neurovirulence of HVP2 in mice is a host species-specific function, and it is difficult to see how RR enzymatic activity would be species-specific. Additional studies are clearly needed to dissect the mechanism of HVP2 UL39-determined neurovirulence.
While much effort has gone into associating a critical neurovirulence function with the large N-terminal region of the HSV UL39 protein, none has yet been identified. Initially, UL39 was thought to have protein kinase (PK) activity (Smith and Aurelian, 1997), but further research established that the UL39 protein does not itself have PK activity but rather is an excellent PK substrate (Conner, 1999). The HSV UL39 protein is cleaved in infected cells (Conner et al., 1994a, 1994b, and Smith and Aurelian, 1997) but the significance of this is unknown. Sequence analysis indicates that the N-terminal region of the UL39 protein has what could be a membrane signal sequence and a transmembrane domain, and the UL39 protein has been detected associated with cellular membranes as well as the nucleus (Smith et al., 1994), but again the significance of this remains unknown. There is a RIP homotypic interaction motif (RHIM) present near the N-terminus that allows the UL39 protein of CMV to interact with receptor-interacting protein 1 (RIP1), thereby acting as a RIP1 inhibitor and impeding RIP1-dependent processes such as NF-κB activation after TNFα or Toll-like receptor 3 stimulation and activation of p38 mitogen-activated protein kinase (MAPK) and caspase-independent programmed cell death (Mack et al., 2008, Upton et al., 2008). Although recent studies have demonstrated that the HSV UL39 protein has chaperone-like activity and blocks TNFα-induced apoptosis (Chabaud et al., 2003 and 2007), the overall function of the large N-terminal region of UL39 in neurovirulence remains unresolved. The HVP2-mouse system provides an ideal model with which both the function of the N-terminal region of the UL39 protein and the mechanism of inter-species neurovirulence of herpesviruses can be investigated.
Supplementary Material
The genetic basis of HVP2 lethality for mice was examined
Construction of inter-typic recombinants mapped virulence to 3 genes
Gene-specific recombinants revealed that UL39 determines lethal neurovirulence
UL39 does not play a major role in suppression of the interferon-beta response
Acknowledgments
Funding: This work was partially supported by grants 2P40 OD010988 and 1P40 OD01031 from the National Institutes of Health (DB & RE) and Grant-in Aid for Scientific Research B (19300148) from the Ministry of Education, Culture, Sport, Science and Technology of Japan (KO).
We thank Geoff Peters and Christine Bonner for technical assistance with the sequencing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Black DH, Saliki JT, Eberle R. Development of a green fluorescent protein reporter cell line to reduce biohazards associated with detection of infectious Cercopithecine herpesvirus 1 (monkey B virus) in clinical specimens. Compar Med. 2002;52:534–542. [PubMed] [Google Scholar]
- Cameron JM, McDougall I, Marsden HS, Preston VG, Ryan DM, Subak-Sharpe JH. Ribonucleotide reductase encoded by herpes simplex virus is a determinant of the pathogenicity of the virus in mice and a valid antiviral target. J Gen Virol. 1988;69 ( Pt 10):2607–2612. doi: 10.1099/0022-1317-69-10-2607. [DOI] [PubMed] [Google Scholar]
- Chabaud S, Lambert H, Sasseville AM, Lavoie H, Guilbault C, Massie B, Landry J, Langelier Y. The R1 subunit of herpes simplex virus ribonucleotide reductase has chaperone-like activity similar to Hsp27. FEBS Letters. 2003;545:213–218. doi: 10.1016/s0014-5793(03)00547-7. [DOI] [PubMed] [Google Scholar]
- Chabaud S, Sasseville AM, Elahi SM, Caron A, Dufour F, Massie B, Langelier Y. The ribonucleotide reductase domain of the R1 subunit of herpes simplex virus type 2 ribonucleotide reductase is essential for R1 antiapoptotic function. J Gen Virol. 2007;88:384–394. doi: 10.1099/vir.0.82383-0. [DOI] [PubMed] [Google Scholar]
- Conner J. The unique N terminus of herpes simplex virus type 1 ribonucleotide reductase large subunit is phosphorylated by casein kinase 2, which may have a homologue in Escherichia coli. J Gen Virol. 1999;80 ( Pt 6):1471–1476. doi: 10.1099/0022-1317-80-6-1471. [DOI] [PubMed] [Google Scholar]
- Conner J, Cross A, Murray J, Marsden H. Identification of structural domains within the large subunit of herpes simplex virus ribonucleotide reductase. J Gen Virol. 1994a;75 ( Pt 12):3327–3335. doi: 10.1099/0022-1317-75-12-3327. [DOI] [PubMed] [Google Scholar]
- Conner J, Marsden H, Clements JB. Ribonucleotide reductase of herpesviruses. Rev Med Virol. 1994b;4:25–34. [Google Scholar]
- d'Offay JM, Mock RE, Fulton RW. Isolation and characterization of encephalitic bovine herpesvirus type 1 isolates from cattle in North America. Am J Vet Res. 1993;54:534–539. [PubMed] [Google Scholar]
- Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. The genome sequence of herpes simplex virus type 2. J Virol. 1998;72:2010–2021. doi: 10.1128/jvi.72.3.2010-2021.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberle R, Black DH, Lehenbauer TW, White GL. Shedding and transmission of baboon Herpesvirus papio 2 (HVP2) in a breeding colony. Lab Anim Sci. 1998;48:23–28. [PubMed] [Google Scholar]
- Eberle R, Black DH, Lipper S, Hilliard JK. Herpesvirus papio 2, an SA8-like alpha-herpesvirus of baboons. Arch Virol. 1995;140:529–545. doi: 10.1007/BF01718429. [DOI] [PubMed] [Google Scholar]
- Eberle R, Blas-Machado U, Wolf R, White G. Microbiology of Captive Baboons. In: VandeBerg J, Williams-Blangero S, Tardif S, editors. The Baboon in Biomedical Research. Springer; New York, NY: 2008. pp. 111–138. [Google Scholar]
- Eberle R, Hilliard JK. The simian herpesviruses: a review. Infect Agents Dis. 1995;4:55–70. [PubMed] [Google Scholar]
- Elmore D, Eberle R. Monkey B virus (Cercopithecine herpesvirus 1) Compar Med. 2008;58:11–21. [PMC free article] [PubMed] [Google Scholar]
- Fulton RW, d'Offay JM, Eberle R. Bovine herpesvirus-1: comparison and differentiation of vaccine and field strains based on genomic sequence variation. Vaccine. 2013;31:1471–1479. doi: 10.1016/j.vaccine.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Weller SK. An ICP6 lacZ insertional mutagen is used to demonstrate that the UL52 gene of herpes simplex virus type 1 is required for virus growth and DNA synthesis. J Virol. 1988;62:2970–2977. doi: 10.1128/jvi.62.8.2970-2977.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JL, Barry PA. B-virus (Cercopithecine herpesvirus 1) infection in humans and macaques: potential for zoonotic disease. Emerg Infect Dis. 2003;9:246–250. doi: 10.3201/eid0902.020272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson JG, Leib DA, Goldstein DJ, Bogard CL, Schaffer PA, Weller SK, Coen DM. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virol. 1989;173:276–283. doi: 10.1016/0042-6822(89)90244-4. [DOI] [PubMed] [Google Scholar]
- Lembo D, Donalisio M, Hofer A, Cornaglia M, Brune W, Koszinowski U, Thelander L, Landolfo S. The ribonucleotide reductase R1 homolog of murine cytomegalovirus is not a functional enzyme subunit but is required for pathogenesis. J Virol. 2004;78:4278–4288. doi: 10.1128/JVI.78.8.4278-4288.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lembo D, Brune W. Tinkering with a viral ribonucleotide reductase. Trends in Biochem Sci. 2009;34:25–32. doi: 10.1016/j.tibs.2008.09.008. [DOI] [PubMed] [Google Scholar]
- Mack C, Sickmann A, Lembo D, Brune W. Inhibition of proinflammatory and innate immune signaling pathways by a cytomegalovirus RIP1-interacting protein. PNAS (USA) 2008;105:3094–3099. doi: 10.1073/pnas.0800168105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters GA, Tyler SD, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. A full-genome phylogenetic analysis of varicella-zoster virus reveals a novel origin of replication-based genotyping scheme and evidence of recombination between major circulating clades. J Virol. 2006;80:9850–9860. doi: 10.1128/JVI.00715-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KM, Black DH, Eberle R. Primary mouse dermal fibroblast cell cultures as an in vitro model system for the differential pathogenicity of cross-species herpesvirus papio 2 infections. Arch Virol. 2007;152:543–552. doi: 10.1007/s00705-006-0865-1. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Deatheridge M, Breshears MA, Chapman S, Black D, Ritchey JW, Payton M, Eberle R. Type I IFN response to Papiine herpesvirus 2 (Herpesvirus papio 2; HVP2) determines neuropathogenicity in mice. Virol. 2009;386:280–289. doi: 10.1016/j.virol.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KM, Ealey KA, Ritchey JW, Black DH, Eberle R. Pathogenicity of different baboon Herpesvirus papio 2 isolates is characterized by either extreme neurovirulence or complete apathogenicity. J Virol. 2003;77:10731–10739. doi: 10.1128/JVI.77.20.10731-10739.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers KM, Ritchey JW, Payton M, Black DH, Eberle R. Neuropathogenesis of Herpesvirus papio 2 in Mice Parallels Cercopithecine herpesvirus 1 (B Virus) Infections in Humans. J Gen Virol. 2006;87:267–276. doi: 10.1099/vir.0.81476-0. [DOI] [PubMed] [Google Scholar]
- Rogers KM, Wolf RF, White GL, Eberle R. Experimental infection of baboons (Papio cynocephalus anubis) with apathogenic and neurovirulent subtypes of Herpesvirus papio 2. Compar Med. 2005;55:425–430. [PubMed] [Google Scholar]
- Severini A, Tyler SD, Peters GA, Black D, Eberle R. Genome sequence of a chimpanzee herpesvirus and its relation to other primate alphaherpesviruses. Arch Virol. 2013 doi: 10.1007/s00705-013-1666-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Aurelian L. The large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is associated with the virion tegument and has PK activity. Virol. 1997;234:235–242. doi: 10.1006/viro.1997.8645. [DOI] [PubMed] [Google Scholar]
- Smith CC, Luo JH, Hunter JC, Ordonez JV, Aurelian L. The transmembrane domain of the large subunit of HSV-2 ribonucleotide reductase (ICP10) is required for protein kinase activity and transformation-related signaling pathways that result in ras activation. Virol. 1994;200:598–612. doi: 10.1006/viro.1994.1223. [DOI] [PubMed] [Google Scholar]
- Smith CC, Peng T, Kulka M, Aurelian L. The PK domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) is required for immediate-early gene expression and virus growth. J Virol. 1998;72:9131–9141. doi: 10.1128/jvi.72.11.9131-9141.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troan BV, Perelygina L, Patrusheva I, van Wettere A, Hilliard J, Loomis M, De Voe R. Naturally Transmitted Herpesvirus papio 2 Infection in a Black and White Colobus Monkey. JAVMA. 2007;231:1878–1883. doi: 10.2460/javma.231.12.1878. [DOI] [PubMed] [Google Scholar]
- Tyler S, Severini A, Black D, Walker M, Eberle R. Structure and sequence of the saimiriine herpesvirus 1 genome. Virol. 2011;410:181–191. doi: 10.1016/j.virol.2010.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyler SD, Peters GA, Grose C, Severini A, Gray MJ, Upton C, Tipples GA. Genomic cartography of varicella-zoster virus: a complete genome-based analysis of strain variability with implications for attenuation and phenotypic differences. Virol. 2007;359:447–458. doi: 10.1016/j.virol.2006.09.037. [DOI] [PubMed] [Google Scholar]
- Tyler SD, Severini A. The complete genome sequence of Herpesvirus papio 2 (Cercopithecine herpesvirus 16) shows evidence of recombination events among various progenitor herpesviruses. J Virol. 2006;80:1214–1221. doi: 10.1128/JVI.80.3.1214-1221.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upton JW, Kaiser WJ, Mocarski ES. Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J Biol Chem. 2008;283:16966–16970. doi: 10.1074/jbc.C800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walboomers JM, Schegget JT. A new method for the isolation of herpes simplex virus type 2 DNA. Virol. 1976;74:256–258. doi: 10.1016/0042-6822(76)90151-3. [DOI] [PubMed] [Google Scholar]
- Weigler BJ, Hird DW, Hilliard JK, Lerche NW, Roberts JA, Scott LM. Epidemiology of Cercopithecine herpesvirus 1 (B virus) infection and shedding in a large breeding cohort of rhesus macaques. J Infect Dis. 1993;167:257–263. doi: 10.1093/infdis/167.2.257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.