

Abstract
Activation of the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and reactive oxygen species (ROS) promote neointimal hyperplasia after vascular injury. CaMKII can be directly activated by ROS through oxidation. In this study, we determined whether abolishing the oxidative activation site of CaMKII alters vascular smooth muscle cell (VCMC) proliferation, migration and apoptosis in vitro and neointimal formation in vivo. VSMC isolated from a knock-in mouse with oxidation-resistant CaMKIIδ (CaMKII M2V) displayed similar proliferation but decreased migration and apoptosis. Surprisingly, ROS production and expression of the NADPH oxidase subunits p47 and p22 were decreased in M2V VSMC, whereas superoxide dismutase 2 protein expression was upregulated. In vivo, after carotid artery ligation, no differences in neointimal size or remodeling were observed. In contrast to VSMC, CaMKII expression and autonomous activity were significantly higher in M2V compared to WT carotid arteries, suggesting an autoregulatory mechanism determines CaMKII activity in vivo. Our findings demonstrate that preventing oxidative activation of CaMKII decreases migration and apoptosis in vitro and suggest that CaMKII regulates ROS production. Our study presents novel evidence that CaMKII expression in vivo is regulated by a negative feedback loop following oxidative activation.
Keywords: CaMKII, reactive oxygen species, proliferation, apoptosis, migration
1. Introduction
Neointimal hyperplasia and remodeling play a key role in the development of in-stent restenosis, intimal proliferation after vein grafting and atherosclerosis. Vascular smooth muscle (VSMC) proliferation and migration are central components in the response to vascular injury. A large body of literature demonstrates that reactive oxygen species (ROS) are elevated in the vascular wall after injury [1,2]. Despite the clear connection between elevated ROS in the vascular wall and neointimal hyperplasia, the molecular pathways induced by ROS and therapeutic approaches addressing the role of ROS after vascular injury remain uncertain.
The Ca2+ and calmodulin-dependent protein kinase II (CaMKII) is a master regulatory molecule and an established signal for promoting diseases where elevated ROS contribute to disease initiation and progression [3-6]. Under resting conditions, CaMKII is maintained in an inactive state. Increased levels of calcified calmodulin (Ca2+/CaM) bind to and activate CaMKII [7]. Once activated, CaMKII retains activity via autophosphorylation at threonine 287, even in the absence of Ca2+/CaM binding. More recently, it was discovered that CaMKII is alternatively activated by methionine 281/282 oxidation (ox-CaMKII), which, like Thr287 autophosphorylation, maintains CaMKII in an active state [8,9]. In VSMC, active CaMKII catalyzes the phosphorylation of proteins that increase cell proliferation [10] and ion channel activity [11]. We recently demonstrated that CaMKII is oxidized in vivo after vascular injury and in vitro after stimulation with cytokines [12], suggesting that ox-CaMKII is sufficient to induce neointimal hyperplasia in response to vascular injury through regulation of VSMC proliferation and migration.
In this study, we investigated whether neointimal hyperplasia in vivo and relevant VSMC phenotypes in vitro are dependent on oxidative CaMKII activation. First we established whether CaMKII is activated in VSMC by oxidation or autophosphorylation under standard growth conditions. We identified ox-CaMKII-dependent phenotypes in VSMC isolated from a knock-in model in which the oxidative activation site of CaMKIIδ at methionines 281,282 are mutated to valines (M2V). In addition, we examined whether abolishing oxidative activation of CaMKII influences ROS production and expression of NADPH oxidase subunits. Lastly, we characterized the effect of the M2V mutation on neointimal hyperplasia and CaMKII expression and activity in vivo. Here we show that ox-CaMKII plays a critical role in VSMC functions and ROS generation.
2. Methods
2.1. Reagents
The following antibodies were used in this study: anti-α-smooth muscle actin, anti-BrdU (Invitrogen), anti-annexin V (Abcam), anti-p-Thr287 CaMKII (Cell Signaling Technology), anti-p47, anti-catalase, anti-SOD1, anti-p22, anti-GAPDH (Santa Cruz), anti-Nox1 (BD Biosciences), anti-SOD2 (Calbiochem) and anti-Nox4 (Epitomics). The generation of the anti-CaMKII [13] and anti-ox-Met 281/282 CaMKII [9] antibodies was described previously.
2.2. Mice
Male and female CaMKII M2V mice and wild-type littermates (WT) at 10- to 12-weeks of age were used [6]. This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Iowa (IACUC# 0905097 and 1111234).
2.3. Carotid injury model
Mice were anesthetized with ketamine and xylazine (2 mg and 0.3 mg respectively, intraperitoneal (IP)). The left common carotid artery was ligated through a midline neck incision [14]. Fourteen or 28 days after injury, all animals were given a lethal dose of pentobarbital and perfused at physiological pressure with PBS followed by 4% paraformaldehyde for 3 min. The carotid arteries were excised, paraffin-embedded and subjected to immunohistochemical analysis.
2.4. Morphological and histochemical analyses
Five micron cross sections were obtained at 0.5 mm from the carotid ligation site for Verhoeff–van Gieson elastin staining. Total vessel, luminal, intimal, and medial areas were measured using NIH Image J. Intimal and medial VSMC were counted after nuclear staining with DAPI. At 2-4 weeks following ligation, some animals received two subcutaneous injections of the thymidine analogue BrdU (2 mg per injection). These injections were given 12 h and 1 h before the mice were sacrificed. TUNEL staining was performed with the Roche Apoptosis kit as per the manufacturer's instructions.
2.5. Cell culture
Mouse aortic smooth muscle cells were isolated by enzymatic dispersion as previously described [15]. Cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 8 mM HEPES, and 2 mM L-glutamine at 37°C in a humidified 95% air and 5% CO2 incubator. The purity of the mouse VSMC preparation in culture was confirmed by immunocytochemistry for α-smooth muscle actin. Mouse VSMC at passages 5-12 were used for experiments, with passage number for WT and CaMKII M2V VSMCs matched with each experiment.
2.6. PCR genotyping
DNA lysates from growing cells and clipped mouse tails were prepared with DirectorAmp tissue kit (Denville Scientific) per manufacturer's recommendations. DNA was amplified with the primers mC2D-F18762-scr (5′-AAG GCC ATC AGG TGA TGC ATG TCC TTA GTC-3′) and mC2D-R19012-scr (5′-AAG TCT ATC TGT CTG TCT GTC TCT CCC CTC-3′), which amplify a 251bp fragment of DNA if WT, 327bp if M2V, and 251/327bp if heterozygous.
2.7. Immunoblotting
VSMC lysates were prepared in ice-cold RIPA lysis buffer and centrifuged at 4°C for 15 min at maximal speed and the supernatant was stored at -80°C for further analysis. Protein concentrations were determined by modified Lowry assay (Bio-Rad DC protein assay). Samples were separated by SDS-PAGE (4-20%) and transferred to a polyvinylidene difluoride membrane. After blocking with 5% BSA, the membranes were incubated with different primary antibodies for 1 h or overnight. The blots were then incubated with a horseradish peroxidase-conjugated secondary antibody and developed by achemiluminescence detection system (Santa Cruz Biotechnology).
2.8. Detection of intracellular ROS
Superoxide production was assessed with the CellRox FarRed reagent (Invitrogen) as recommended by the manufacturer. WT and M2V VSMC proliferating in DMEM with 10% FBS were treated with CellRox for 18 h. The slides were fixed in 5% paraformaldehyde for 5 min and rinsed in PBS. The nuclei were counterstained with DAPI. The number of CellRox-positive cells relative to the number of total cells (by DAPI) was determined in 6 fields each in 3 independent experiments.
2.9. Detection of mitochondrial ROS
VSMC were treated with a cytokine mixture of IFNγ (100 ng/mL, R&D), IL-1β (100 ng/mL, R&D), and TNFα (100 ng/mL, R&D) for 30 min. ROS was measured in live cells using mitoSOX Red (5 μM, Invitrogen). The mitochondrial localization of staining was confirmed by colocalizing with mitoTracker green (50 nM, Invitrogen). All images were taken at the same time and using the same imaging settings. Data are presented as mean fluorescent intensity per μm2 and normalized to control levels.
2.10. Adenoviral transduction
WT VSMC at about 30% confluence were infected with adenoviruses expressing, HA-CaMKIIN or control adenovirus at a MOI of 500 overnight [13]. The cells were in DMEM/10% FBS for 72h, growth arrested for 24 h, and treated as indicated for the specific experiment.
2.11. CaMKII activity assays
CaMKII activity assays were performed in lysates from CaMKII M2V and WT carotid arteries and from VSMC. CaMKII activity was measured by incorporation of 32P into the synthetic substrate syntide-2 as previously described [11]. Carotid and cell lysates were mixed with buffers containing Ca2+ and calmodulin (Ca2+-activated activity) or EGTA (Ca2+-independent activity). The samples were incubated at 30°C for 10min and spotted on P81 ion exchange paper. The paper was rinsed to remove unincorporated ATP, and radioactivity quantified using a liquid scintillator. Data were normalized to total protein.
2.12. Proliferation assays
WT and M2V VSMC were seeded in 12-well plates at 15,000 cells per well in a volume of 2 mL and cultured in DMEM containing 10% FBS. Cells were in counted in triplicate for 4 consecutive days using a Beckman Coulter automated cell counter.
For MTT assays, 12 mM MTT (3-(4,5-dimethylthazol-2-yl)-2,5-diphenyltetrazolium bromide) solution was added to 12-well plates prepared as described above and absorbance was measured at 560 nm.
2.13. Migration assays
VSMC migration assays were performed using the modified Boyden chamber method as previously described [12]. WT or CaMKII M2V VSMC (105 cells/well) were added to the upper chamber of the transwell and allowed 30 minutes to attach. The chambers contained DMEM with 0.1% FBS. Migration of VSMC was induced by the addition of DMEM with 10% FBS to the lower compartment. After 6 h, non-migrated cells were removed from the upper chamber. VSMC migrating to the lower surface of the membrane were fluorescently stained with DAPI, imaged and quantified using Cell Profiler software.
For scratch wound assays, the serum-starved VSMC monolayer was disrupted with a sterile cell scraper to create a cell-free zone. Cells were then washed with medium and treated in DMEM containing 10% FBS. Images were taken immediately following the scratch and 24 hours after injury using a phase-contrast microscope at 10× equipped with a digital camera. Pictures taken at comparable positions along the scratch line were evaluated. The migration distance was determined by subtracting the distance from the marker line at the same location at 24 hours from the distance at baseline.
2.14. Adhesion assays
WT and M2V VSMC were seeded at 100,000 cells/well in a 24-well plate with DMEM containing 10% FBS in a volume of 1 mL per well. After 30, 60, and 90 min, all wells were rinsed with PBS to remove non-adherent cells. Adherent cells were fixed with 4% paraformaldehyde for 10 min. Fixed cells were stained with 0.5% crystal violet in 2% ethanol for 10 min, then soluble dye was extracted by incubation with 1% SDS for 30 min. The absorbance was measured at 600 nm.
2.15. Quantitative RT-PCR (qRT-PCR)
Total RNA was isolated from VSMC per manufacturer's recommendation (Qiagen) followed by digestion with proteinase K and DNase I to eliminate genomic DNA contamination. cDNA was transcribed from 1 μg RNA using Superscript III enzyme (Invitrogen) and random nanomer primers. Message expression was quantified using an iQ Lightcycler (Bio-Rad) with SYBR Green with primers for CaMKIIγ (forward: 5′-ttg aac aag aag tcg gat gg-3′ reverse: 5′-gct ggg ctt acg aga ctg tt-3′) or CaMKIIδ (forward: 5′-gct aga cgg aaa ctg aag gg-3, reverse: 5′-cct caa tgg tgg tgt ttg ag-3′) and normalized to acidic ribosomal phosphoprotein (ARP) mRNA.
2.16. Apoptosis assays
WT and M2V VSMC were seeded in an 8-well Lab-Tek II Chamber slide system in DMEM containing 10% FBS and treated with 100, 250 or 500 μM H2O2 for 2 hours at 37°C. TUNEL-positive apoptotic cells were detected with the Cell Death Detection kit (Roche) as specified by the manufacturer.
2.17. Statistical analysis
Data are shown as mean± SE unless noted otherwise. The GraphPad statistical software package was used for the quantitative analyses of parameters such as EEL perimeter, staining intensity and number of migrated cells (ANOVA and Student's t test). Bonferroni post-hoc tests were performed for multiple comparisons. Student's t test was used for Figures 2, 4A-C, 6A-D. ANOVA was used for Figures 1B, 3A-F, 4D, 5 B-C, 6E-F. A probabilityvalue <0.05 was considered significant.
Figure 2. Characterization of CaMKIIδ M2V VSMC.
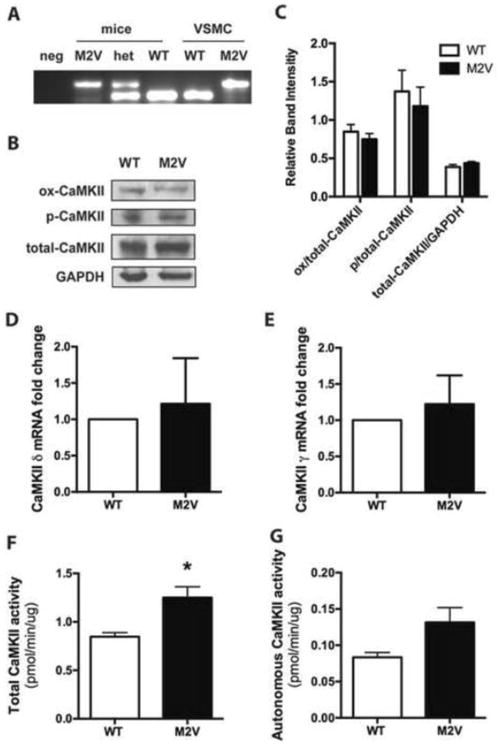
A. Genotyping of DNA from proliferating VSMC and mouse tail clips. B. Immunoblot of WT and M2V VSMC for oxidized CaMKII (ox-CaMKII), autophosphorylated CaMKII (p-CaMKII), and total CaMKII. C. Quantitation of data in (B) (n=4). D, E. qRT-PCR for CaMKIIδ (D) and CaMKIIγ (E) normalized to expression of acidic ribosomal protein P1 in WT and M2V VSMC (n=7) F. Ca2+/CaM-activated (total) and G. Ca2+/CaM-independent (autonomous) CaMKII activity assays as determined by incorporation of 32P into synthetic substrate syntide-2 (*p<0.05, n=3).
Figure 4. M2V VSMC have decreased ROS production.
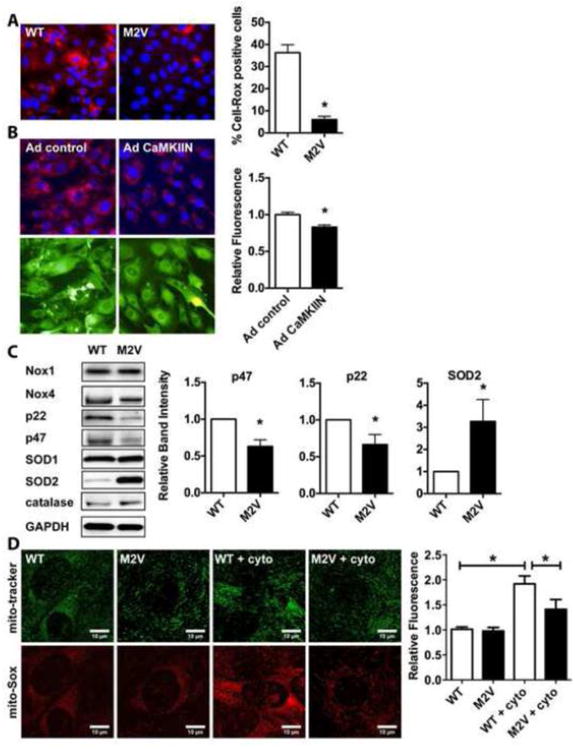
A, B. Assessment of ROS production using 5 μM CellRox in proliferating WT and M2V VSMC (A) or in (B) WT VSMC infected with Ad control or Ad CaMKIIN. Nuclei were counterstained with DAPI (20× magnification). Quantification was performed using 6 random fields per condition; data are representative of 3 independent experiments (*p<0.05 vs. WT) C. Representative immunoblots and quantification of different NADPH oxidase subunits, SOD and catalase. Densitometry was performed on eight samples per genotype and normalized to GAPDH protein levels. D. Mitochondrial ROS production by mito-Sox staining at baseline and following stimulation with cytokine mixture (cyto) for 24 h (n=3 independent experiments). * p<0.05.
Figure 6. CaMKII transcript levels and activity are upregulated in M2V mice in vivo.
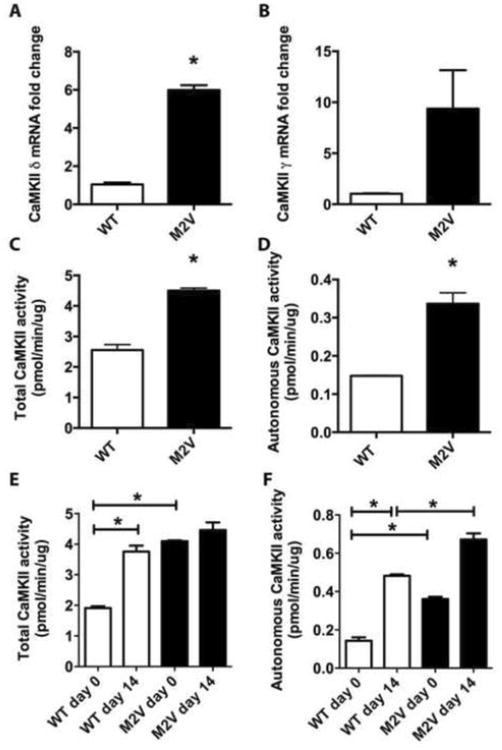
A, B. qRT-PCR for CaMKIIδ (A) and CaMKIIγ (B) normalized to expression of acidic ribosomal protein P1 in WT and M2V carotid artery lysates (n=3). C, D. Baseline Ca2+/CaM-activated (total, C) and Ca2+/CaM-independent (autonomous, D) CaMKII activity assays as determined by incorporation of 32P into synthetic substrate syntide-2. E, F. CaMKII total (E) and autonomous (F) activity assays 0 and 14 days after ligation. (*p<0.05, for C-F, 8 carotid arteries were pooled for each sample).
Figure 1. CaMKII activation in VSMC under standard growth conditions.
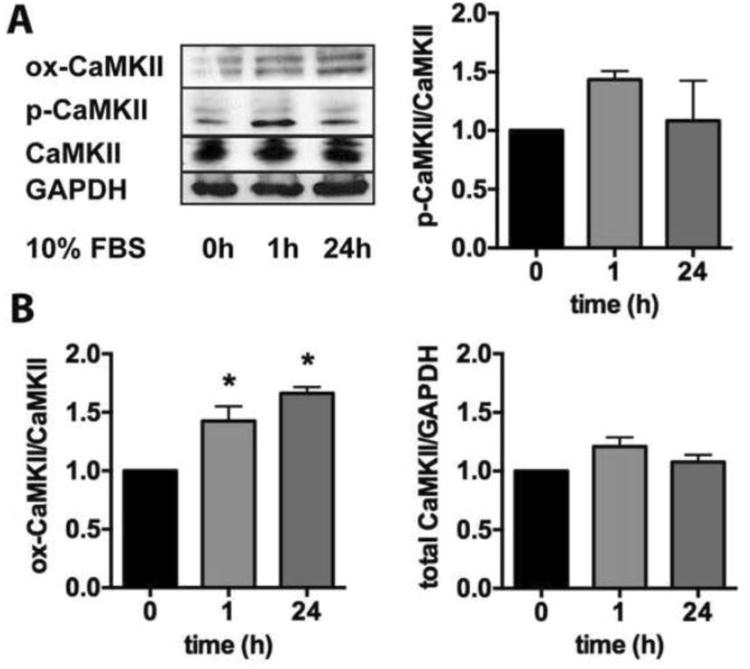
A. Immunoblots for p-, ox-, CaMKII and GAPDH in WT VSMC that were grown to 80% confluence, growth-arrested in serum-free media for 24 h and incubated with DMEM/10% FBS for in indicated times. (B) Quantification of 4 independent experiments (*p<0.05, n=4).
Figure 3. M2V VSMC display decreased migration and apoptosis and increased adhesion, but similar proliferation as compared to WT VSMC.
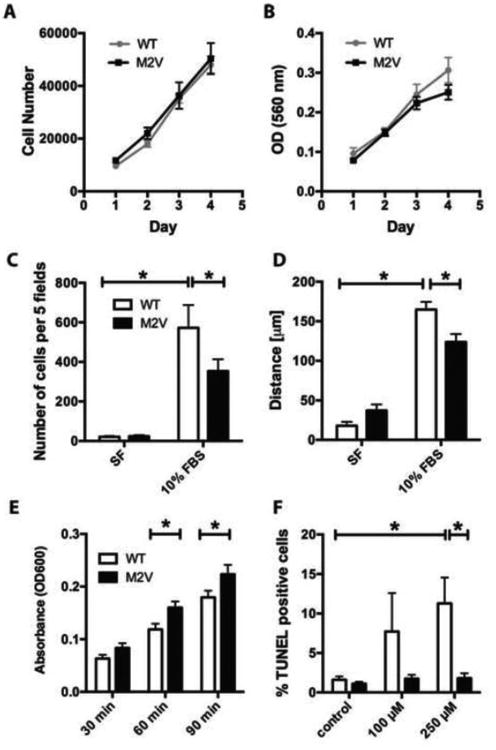
A. Cell counts (n=18) and B. MTT assay (n=9) of WT and M2V VSMC plated at low density and grown in 10% FBS-containing media. C. Modified Boyden chamber (n=5, *p<0.05) and D. Scratch wound assay of WT and M2V VSMCs grown in serum free (SF) media or with 10% FBS (n=3 *p< 0.05). Data are representative of 3 independent experiments. E. Adhesion of WT and M2V VSMC grown in 10% FBS media at 30, 60, and 90 minutes (n=4). F. TUNEL staining of WT and M2V VSMC treated with indicated concentrations of H2O2 for 2 h (n=4 per treatment, *p<0.05 vs. control).
Figure 5. Inhibition of oxidative CaMKII activation does not alter neointimal area, proliferation or apoptosis following vascular injury.
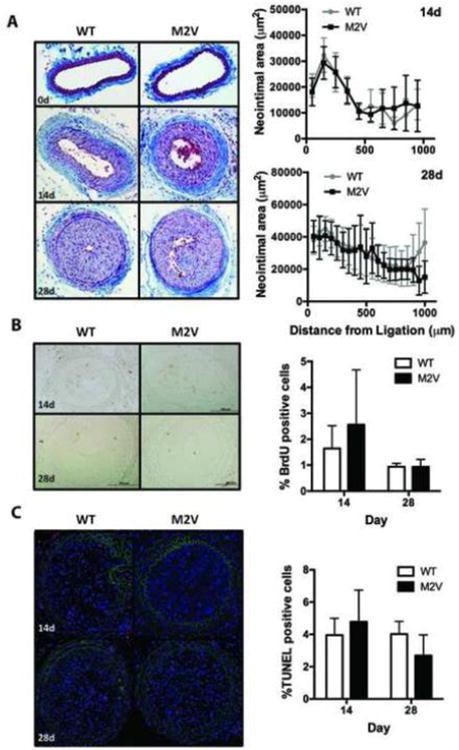
A. Masson's trichrome staining of representative sections of WT and M2V samples 0, 14, and 28 days after carotid ligation (20× magnification) with morphometric analysis of neointimal and media areas. Sections at 50 or 100μm intervals starting at 50μm from the ligation site were analyzed. N=9 per group. B. Quantification of BrdU-labeled cells in four sections per sample (n=6-10 samples) C. TUNEL-staining 14 and 28 days after ligation. TUNEL-labeled cells were counted in sections of 5-6 mice per group at 50-600 μm from the ligation.
3. Results
3.1. Ox-CaMKII and p-CaMKII are present in proliferating VSMC
Numerous agonists such as angiotensin-II, vasopressin, PDGF and TNF-α induce short-term CaMKII activation in VSMC through oxidation and auto-phosphorylation [16]. What remains unknown is whether these mechanisms are responsible for long-term CaMKII activity under standard culture conditions in vitro. We addressed this question by performing immunoblots for p-CaMKII and ox-CaMKII after incubation with DMEM/10% FBS at various time points (Figure 1A). At 1 h after the addition of DMEM/10% FBS, p- and ox-CaMKII are present, whereas after 24h, only ox-CaMKII levels were still significantly elevated (Figure 1B). CaMKIIδ regulates VSMC proliferation and migration in vitro and in vivo [16], phenotypes that require extended incubation times. Thus, these data provide a rationale for studying a mutant CaMKIIδ that cannot be activated by oxidation (CaMKII M2V) [6].
3.2. Characterization of CaMKII M2V VSMC
Given the evidence for oxidative activation of CaMKII in VSMC, we next extended studies to VSMC isolated from mice expressing the CaMKII M2V mutant [6]. Each genotype was verified in isolated aortic VSMC (Figure 2A), and total CaMKII protein expression was similar in WT and CaMKII M2V VSMC (Figure 2B, C). We did not detect a compensatory increase in phosphorylated CaMKII in M2V cells. A non-significant trend towards decreased ox-CaMKII in M2V compared to WT cells was observed (Figure 2B, C), which is likely due to oxidative activation of CaMKIIγ that is expressed along with the CaMKIIδ isoform in VSMC [17,18]. To assess whether the CaMKII M2V mutation affects CaMKII mRNA transcription, we performed qRT-PCR for the CaMKII isoforms γ and δ. Neither CaMKIIγ nor δ transcript levels were significantly increased (Figure 2D, E).
We also evaluated CaMKII activity in WT and M2V VSMC. The Ca2+/CaM-activated (total) activity, which is indicative of total levels of CaMKII in a sample, was increased by 1.5-fold in M2V cells (Figure 2F). The Ca2+/CaM-independent activity, which reflects the amount of active/autonomous CaMKII, was increased but this trend was not statistically significant (Figure 2G). The increase in in vitro activity that exceeds the difference in protein levels raises the question whether the M2V mutant has higher intrinsic activity, at least for the peptide substrate, as it may facilitate the activation of the adjacent autophosphorylation site. However, under our experimental conditions, abrogation of CaMKII oxidative activation does not substantially impact intrinsic CaMKII activity in VSMC (Figure 2G).
3.3. CaMKII M2V cells exhibit decreased migration and increased adhesion in vitro
Deletion or knockdown of CaMKIIδ significantly reduces VSMC proliferation [10]. Thus, we assessed whether CaMKII M2V is sufficient to decrease proliferation of VSMC. No difference in proliferation between M2V and WT VSMC was detected by cell counting or MTT assays regardless of passage number (Figures 3A, B). This suggests that while CaMKIIδ is a regulator of cell proliferation, oxidative activation of CaMKIIδ is not required to mediate proliferation or that increases in total CaMKII activity in M2V VSMC compensate for loss of oxidative activation of CaMKIIδ.
CaMKII has a well-established role in VSMC migration [12,19-21]. Thus, we performed migration assays to establish whether M2V VSMC are migration-deficient. In modified Boyden chamber experiments, we detected a decrease in migration in M2V VSMC compared to WT (Figure 3C). In scratch wound assays, the migration distance was significantly shorter in M2V compared to WT VSMC (Figure 3D). Thus, CaMKII oxidative activation contributes to induction of VSMC migration. These data are consistent with our previous findings that adenoviral overexpression of CaMKIIδ in CaMKIIδ-/- VSMC recovers migration in vitro, but overexpression of CaMKIIδ M2V does not [12].
Since CaMKII has been reported to regulate cell adhesion through integrin-mediated signaling [22,23], we examined whether the impaired migration in M2V cells is caused by differences in cell adhesion. We quantified the number of cells adherent to a collagen-coated surface and found significantly increased cell adhesion at 60, and 90 minutes in M2V as compared to WT VSMC (Figure 3E).
3.4. Impaired oxidative activation of CaMKIIδ in VSMC decreases apoptosis in vitro
Several studies have demonstrated that CaMKII regulates apoptosis of cardiac myocytes [9,24,25], and abolishing oxidative activation of CaMKII decreases apoptosis and mortality after myocardial infarction [6]. Increased rates of apoptosis may contribute to late stage remodeling of the neointima after vascular injury, but it is unknown whether CaMKII controls apoptosis in cells with potentially higher antioxidant capacity and less prominent Ca2+ fluctuations, such as VSMC. Thus, we tested whether CaMKII M2V VSMC are protected from apoptotic cell death in response to concentrations of H2O2 known to induce apoptosis. At 250 μM of H2O2, significantly fewer M2V cells were TUNEL-positive as compared to WT cells (Figure 3F). Similar though not significant trends were observed with 100 μM H2O2. M2V VSMC treated with a cytokine mixture of IFN-γ, TNF-α, and IL-1β also exhibited significantly reduced apoptosis rates (data not shown).
3.5. ROS production is decreased in CaMKII M2V VSMC
Apoptosis and migration are ROS-dependent VSMC phenotypes and our studies to this point were designed to detect the potential role of CaMKII in pathological responses to ROS in VSMCs. Next we next tested whether the M2V mutation affects ROS production. While CaMKII is regarded as a downstream effector of ROS generated by NADPH oxidase, two recent studies have reported decreased ROS production under CaMKII inhibition [26,27]. Accordingly, we have recently reported that CaMKIIδ-/- arteries display decreased superoxide production in flow-mediated remodeling and express less p47, a NADPH oxidase subunit that associates with Nox1 and 2 [5]. This suggests that ROS production may depend upon CaMKII. Using the fluorogenic probe CellRox, we detected greater superoxide production in proliferating WT compared to M2V VSMC (Figure 4A). After transient overexpression of the CaMKII peptide inhibitor CaMKIIN that inhibits all CaMKII isoforms regardless of activation mechanism, ROS levels were significantly lower than in control cells (Figure 4B).
Since the difference in cytoplasmic ROS levels with CaMKIIN was not as marked as under chronic inhibition of oxidative CaMKII activation, we hypothesized that the changes in M2V cells may be related to long-term effects. Therefore, we next assessed the expression of various NADPH oxidase subunits by immunoblotting. In M2V VSMC, protein expression of subunits p47 and p22 were significantly lower whereas levels of superoxide dismutase (SOD) 2 were increased (Figure 4C). In contrast, expression of Nox 1 and 4, catalase and SOD1 were not different. In qRT-PCR experiments, we did not detect any differences in mRNA levels of p22, p47 or SOD2 in CaMKII M2V compared to WT cells (data not shown). As SOD2 is mainly localized in mitochondria, we examined ROS production in mitochondria. Accordingly, we detected lower levels of ROS production in M2V VSMCs in response to cytokines as evidenced by mito-SOX staining (Figure 4D).
3.6. In vivo effect of M2V on neointimal hyperplasia
CaMKIIδ-/- mice are protected against neointimal hyperplasia following carotid ligation [10]. Since M2V and WT VSMC differed in cell migration and apoptosis rates in vitro, we sought to characterize the role of oxidative activation of CaMKII in vivo using a murine model of acute vascular injury. We performed complete ligations of the left common carotid artery in CaMKII M2V and WT mice and, 14 and 28 days later, determined neointimal area at various distances from the site of ligation. In contrast to our working hypothesis, no differences in morphology were seen. WT and M2V carotid arteries showed similar neointima and media areas and intima/media ratios (Figure 5A). Similarly, no statistically significant difference in external elastic lamina perimeters was detected (data not shown).
Next, we evaluated in vivo proliferation and apoptosis. We found that a similar percentage of WT and M2V cells were BrdU-positive at 14 and 28 days post-ligation (Figure 5B). We did not detect any BrdU-staining in the neointima of non-ligated, day 0 carotids. The percentage of apoptotic cells at 14 and 28 days following ligation was similar between genotypes (Figure 5C). Thus, in vivo, no difference in neointimal size or extent was detected when the oxidative activation of CaMKII was abrogated. In contrast to our in in vitro findings, apoptosis in the neointima was not increased after injury.
3.7. CaMKII mRNA expression and activity are significantly increased in vivo
Since we unexpectedly did not detect any differences in the neointima size in M2V compared to WT arteries, we evaluated whether CaMKII expression and/or activity were increased to compensate for decreased oxidative activation in vivo. Baseline mRNA levels CaMKIIδ were significantly 6-fold increased in M2V aortic artery samples compared to WT (Figure 6A), which is in contrast to WT and M2V VSMC where no differences in CaMKIIγ and δ mRNA transcript levels were detected (Figure 2D, E). CaMKIIγ transcripts were also increased by 9.4-fold (Figure 6B). Similarly, in murine carotid samples, baseline CaMKII activity was significantly increased in M2V samples (Figure 6C, D). CaMKIIδ mRNA and protein levels have been shown to be greatly increased in the neointima after vascular injury [10,28]. In accordance, the total CaMKII activity rose 1.9-fold in WT carotid arteries by day 14 after ligation compared to day 0 (Figure 6E). In M2V carotid arteries, on day 14, the total activity was not greater than at baseline. The autonomous activity reflects the in vivo activation state of CaMKII, and may therefore be more relevant for an understanding of the in situ conditions. Here, we detected a 3-fold increase in autonomous activity in WT carotid arteries on day 14, with a more pronounced 4.6-fold increase in M2V carotid arteries compared to WT control (Figure 6F). Thus, on day 14 after ligation, the autonomous CaMKII activity is significantly increased in vivo in M2V compared to WT carotid arteries. These differences point towards a compensatory upregulation of CaMKII activity in M2V carotid arteries, which may account for the surprising lack of an in vivo phenotype.
4. Discussion
ROS and CaMKII are upregulated in response to vascular injury [1,10]. Given the recent discovery of the oxidative activation pathway for CaMKII, we investigated its role in neointimal formation after vascular injury using a new knock in mouse model expressing oxidation-resistant CaMKIIδ. We hypothesized that CaMKIIδ is activated by oxidation of Met281/282 in vascular injury and that the oxidative activation of CaMKII is necessary for VSMC proliferation, migration, apoptosis, and thus, neointimal hyperplasia. We found that cellular processes that require elevated levels of ROS, such as migration and apoptosis, were significantly impaired in M2V cells. In addition, M2V VSMC had decreased expression of NADPH oxidase cytosolic subunits and ROS generation.
In contrast to our hypothesis, however, we did not find differences in neointimal and media thickness nor outward remodeling of murine carotids from WT versus M2V mice following carotid ligation. Consistent with these findings, we detected no differences in proliferation or apoptosis in vivo between the two groups. These findings may seem to suggest that the absence of oxidized CaMKII does not affect VSMC growth or survival. However, total CaMKII mRNA expression and activity were increased in M2V arteries, suggesting oxidative CaMKII activation provides a negative feedback mechanism for CaMKII gene expression. Thus, compensatory increases in CaMKII activity in M2V mice in vivo may overcome the protective benefits of the M2V mutation evidenced in vitro. Differential CaMKII isoform expression has been reported in intact arteries versus dedifferentiated VSMC in culture [18]. Thus, a stronger regulatory effect of the M2V mutation in vivo compared to in vitro may not be surprising.
CaMKIIδ expression is increased in the media and intima following vascular injury with carotid ligation or balloon angioplasty [10]. Inhibition of CaMKIIδ by siRNA prevents the increase of CaMKIIδ, resulting in decreased cell proliferation of both layers and markedly reduced neointimal formation [10,17]. CaMKIIδ-/- VSMC have decreased cell proliferation and migration compared with WT VSMC [10,12]. In contrast, M2V VSMC showed no difference in proliferation compared to WT, whereas they had decreased migration, implying that oxidative activation of CaMKII is necessary for migration rather than proliferation. Similarly, we previously reported that overexpression of a CaMKIIδ M2V mutant in CaMKIIδ-/- cells does not recover migration [12]. Our findings that the CaMKII M2V mutation decreased VSMC adhesion are in agreement with an earlier study that reported decreased adhesion of CHO cells to fibronectin in the presence of constitutively active CaMKII [22].
In contrast to proliferation that only requires low concentrations of ROS, VSMC migration and adhesion are heavily ROS-dependent [2,29,30]. Likewise, apoptosis, another ROS-dependent disease mechanism [31], was significantly decreased in M2V VSMC, consistent with data in CaMKIIδ M2V cardiac pacemaker cells [6].
Since we observed reduced migration and apoptosis, which are ROS-dependent, we also investigated the levels of ROS in M2V compared to WT VSMC. Classically, CaMKII is believed to be a downstream target of NADPH oxidase-dependent ROS production [3]. In a recent study, we observed decreased ROS production in vivo in CaMKIIδ-/- mice [5]. Our findings herein that ROS production is decreased in the cytoplasm and mitochondria lend further credence to the emerging concept that CaMKII also regulates ROS production [27]. In support of these data, we also observed decreased expression of NAPDH oxidase subunits p47, p22 and increased expression of SOD2, which dismutates superoxide into hydrogen peroxide and diatomic oxygen. Interestingly, Nishio and colleagues recently reported decreased expression of the subunits p47 and p67 in cardiac myocytes under CaMKII inhibition [26]. Our findings that SOD2 levels are increased also suggests a role of mitochondrial ROS production since changes in mitochondrial ROS (due to increases in SOD2) may lead to decreased cytoplasmic ROS levels via disruption of ROS-induced ROS production. Our data indicate that activation of CaMKII contributes to ROS production by altering expression of NADPH oxidases and SOD2, which then affects migration and apoptosis. CaMKII has been proposed to control migration through leading edge formation [32] and regulation of MMP9 [12]. Whether CaMKII-mediated ROS production serves as an upstream master regulator for these processes remains to be determined.
Our study also found that CaMKII M2V mice display similar neointimal increases, cell proliferation and apoptosis when compared to WT mice after injury. This is in contrast to CaMKIIδ-/- mice that display reduced proliferation and neointimal formation after vascular injury [10]. These findings suggest that inhibition of CaMKIIδ oxidative activation is not sufficient to abrogate the response to injury despite differences in apoptosis and migration rates in vitro. There are several possible reasons for the differences between the in vivo and in vitro phenotypes. First, our data suggest that there is a compensatory increase in CaMKIIδ transcript levels and autonomous CaMKII activity in M2V arteries in vivo, but not in VSMC in vitro. The enhanced CaMKII expression and activity in M2V carotid arteries may compensate for the lack of oxidized activation of CaMKII and may explain the normalization of neointimal size. Expression of other proteins important in vascular remodeling may also differ between VSMC in vitro and in vivo. Second, since proliferation is less ROS-dependent than migration or apoptosis [2], and we did not detect a difference in proliferation rates between M2V and WT VSMC, decreased VSMC migration may not be sufficient to prevent neointimal hyperplasia after carotid ligation injury in M2V mice. Third, distinct environmental signals that contribute to in vivo formation of neointimal hyperplasia may be absent in vitro in culture. VSMC co-exist with endothelial cells and extracellular matrix and are capable of major phenotypic changes depending on environmental cues such as growth factors/inhibitors, inflammatory mediators, mechanical changes, cell-to-cell and cell-to-matrix interactions [33]. Paracrine effects of bone marrow-derived stem cells [34] or endothelial cells [35] contribute to neointimal hyperplasia. As such, other cells and their interactions with VSMC may promote neointimal formation in M2V mice in vivo [36]. Lastly, the time points that were examined for in vitro assays differed from in vivo assays. For instance, apoptosis in vivo was only evaluated at 14 and 28 days post-ligation, and it is possible that apoptosis in vivo was decreased at other time points. Apoptosis occurs at early and late time points after vascular injury [37] and likely promotes neointimal regression [38]. Thus, based on our in vitro data, we speculated that in M2V carotid arteries, decreased VSMC apoptosis and migration could have opposing effects on neointimal size. However, in our analysis at 14 and 28 days after ligation, no differences in apoptosis in vivo were detected.
In our characterization of M2V carotid arteries, we surprisingly detected significant increases in CaMKIIδ mRNA transcripts. These results support the existence of a compensatory mechanism for increasing CaMKII activity in response to decreased oxidative activation in vivo through increased CaMKII gene expression. Transcriptional or post-trancriptional regulators of CaMKII isoforms remain unknown at this point and warrant further investigation.
We also observed a trend for increased mRNA transcripts of CaMKIIγ, another isoform of CaMKII that is abundantly expressed in the vasculature. It could be argued that CaMKIIγ upregulation compensates for decreased oxidative activation of CaMKIIδ and contributes to neointimal formation. However, recently generated CaMKIIγ-deficient mice exhibit enhanced neointimal size with increased proliferation and no change in apoptosis [39]. These data indicate opposing effects for the CaMKII isoforms. Thus, upregulation of CaMKIIγ isoform would, if anything, result in less neointimal hyperplasia.
In summary, this study provides evidence that inhibition of oxidative activation of CaMKIIδ alone is not sufficient to protect against neointimal formation after vascular injury. The CaMKIIδ M2V mutation causes decreased migration and apoptosis in vitro, but no differences in neointimal size, outward vascular remodeling, proliferation and apoptosis in vivo. Interestingly, blockade of ox-CaMKII increases overall CaMKII expression and activity in vivo, suggesting that oxidative CaMKII activation provides a negative feedback mechanism for CaMKII gene expression. As a result, any development of CaMKII inhibitors to prevent vascular occlusive disease will need to target the phosphorylation site, if not both activation sites.
Acknowledgments
We are indebted to Dr. Chantal Allamargot (Central Microscopy Research Facilities, University of Iowa) for expert technical support and to Dr. Kristina W. Thiel for assistance in the preparation of the manuscript.
Grants: This work was supported in part by funding from the following grants: VA Merit grant 01BX000163 by the Office of Research and Development, Department of Veterans Affairs (IMG), by NIH grants HL 079031 (IMG), and R01 HL 70250 (MEA); and the University of Iowa Cardiovascular Center Interdisciplinary Research Fellowship (PJK and AMP).
Footnotes
Disclosures: None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation. 2003;108:2034–2040. doi: 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- 2.Papaharalambus CA, Griendling KK. Basic mechanisms of oxidative stress and reactive oxygen species in cardiovascular injury. Trends Cardiovasc Med. 2007;17:48–54. doi: 10.1016/j.tcm.2006.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Erickson JR, He BJ, Grumbach IM, Anderson ME. CaMKII in the cardiovascular system: sensing redox states. Physiol Rev. 2011;91:889–915. doi: 10.1152/physrev.00018.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders PN, Koval OM, Jaffer OA, Prasad AM, Businga TR, Scott JA, et al. CaMKII Is Essential for the Proasthmatic Effects of Oxidation. Sci Transl Med. 2013;5:195ra97. doi: 10.1126/scitranslmed.3006135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scott JA, Klutho PJ, El Accaoui R, Nguyen E, Venema AN, Xie L, et al. The Multifunctional Ca(2+)/Calmodulin-Dependent Kinase IIdelta (CaMKIIdelta) Regulates Arteriogenesis in a Mouse Model of Flow-Mediated Remodeling. PLoS One. 2013;8:e71550. doi: 10.1371/journal.pone.0071550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–1274. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Payne ME, Fong YL, Ono T, Colbran RJ, Kemp BE, Soderling TR, et al. Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. J Biol Chem. 1988;263:7190–7195. [PubMed] [Google Scholar]
- 8.Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, et al. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 2010;8:e1000426. doi: 10.1371/journal.pbio.1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–474. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li W, Li H, Sanders PN, Mohler PJ, Backs J, Olson EN, et al. The Multifunctional Ca2+/Calmodulin-dependent Kinase II {delta} (CaMKII{delta}) Controls Neointima Formation after Carotid Ligation and Vascular Smooth Muscle Cell Proliferation through Cell Cycle Regulation by p21. J Biol Chem. 2011;286:7990–7999. doi: 10.1074/jbc.M110.163006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Prasad AM, Nuno DW, Koval OM, Ketsawatsomkron P, Li W, Li H, et al. Differential Control of Calcium Homeostasis and Vascular Reactivity by Ca2+/Calmodulin-Dependent Kinase II. Hypertension. 2013;62:434–441. doi: 10.1161/HYPERTENSIONAHA.113.01508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott JA, Xie L, Li H, Li W, He JB, Sanders PN, et al. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol Heart Circ Physiol. 2012;302:H1953–64. doi: 10.1152/ajpheart.00978.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Li W, Gupta AK, Mohler ME, Anderson IM, Grumbach IM. Calmodulin kinase II is required for angiotensin II-mediated vascular smooth muscle hypertrophy. Am J Physiol Heart Circ Physiol. 2010;298:H688–98. doi: 10.1152/ajpheart.01014.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–2244. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 15.Ray JL, Leach R, Herbert JM, Benson M. Isolation of vascular smooth muscle cells from a single murine aorta. Methods Cell Sci. 2001;23:185–188. doi: 10.1023/a:1016357510143. [DOI] [PubMed] [Google Scholar]
- 16.Scott JA, Xie L, Li H, Li W, He JB, Sanders PN, et al. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol Heart Circ Physiol. 2012;302:H1953–64. doi: 10.1152/ajpheart.00978.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.House SJ, Singer HA. CaMKII-delta isoform regulation of neointima formation after vascular injury. Arterioscler Thromb Vasc Biol. 2008;28:441–447. doi: 10.1161/ATVBAHA.107.156810. [DOI] [PubMed] [Google Scholar]
- 18.House SJ, Ginnan RG, Armstrong SE, Singer HA. Calcium/calmodulin-dependent protein kinase II-delta isoform regulation of vascular smooth muscle cell proliferation. Am J Physiol Cell Physiol. 2007;292:C2276–87. doi: 10.1152/ajpcell.00606.2006. [DOI] [PubMed] [Google Scholar]
- 19.Pfleiderer PJ, Lu KK, Crow MT, Keller RS, Singer HA. Modulation of vascular smooth muscle cell migration by calcium/calmodulin-dependent protein kinase II-delta 2. Am J Physiol Cell Physiol. 2004;286:C1238–45. doi: 10.1152/ajpcell.00536.2003. [DOI] [PubMed] [Google Scholar]
- 20.Pauly RR, Bilato C, Sollott SJ, Monticone R, Kelly PT, Lakatta EG, et al. Role of calcium/calmodulin-dependent protein kinase II in the regulation of vascular smooth muscle cell migration. Circulation. 1995;91:1107–1115. doi: 10.1161/01.cir.91.4.1107. [DOI] [PubMed] [Google Scholar]
- 21.Yang M, Kahn AM. Insulin-inhibited and stimulated cultured vascular smooth muscle cell migration are related to divergent effects on protein phosphatase-2A and autonomous calcium/calmodulin-dependent protein kinase II. Atherosclerosis. 2008;196:227–233. doi: 10.1016/j.atherosclerosis.2007.04.050. [DOI] [PubMed] [Google Scholar]
- 22.Bouvard D, Block MR. Calcium/calmodulin-dependent protein kinase II controls integrin alpha5beta1-mediated cell adhesion through the integrin cytoplasmic domain associated protein-1alpha. Biochem Biophys Res Commun. 1998;252:46–50. doi: 10.1006/bbrc.1998.9592. [DOI] [PubMed] [Google Scholar]
- 23.Takahashi K, Suzuki K. Regulation of protein phosphatase 2A-mediated recruitment of IQGAP1 to beta1 integrin by EGF through activation of Ca2+/calmodulin-dependent protein kinase II. J Cell Physiol. 2006;208:213–219. doi: 10.1002/jcp.20657. [DOI] [PubMed] [Google Scholar]
- 24.Velez Rueda JO, Palomeque J, Mattiazzi A. Early apoptosis in different models of cardiac hypertrophy induced by high renin-angiotensin system activity involves CaMKII. J Appl Physiol. 2012;112:2110–2120. doi: 10.1152/japplphysiol.01383.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salas MA, Valverde CA, Sanchez G, Said M, Rodriguez JS, Portiansky EL, et al. The signalling pathway of CaMKII-mediated apoptosis and necrosis in the ischemia/reperfusion injury. J Mol Cell Cardiol. 2010;48:1298–1306. doi: 10.1016/j.yjmcc.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishio S, Teshima Y, Takahashi NT, Thuc LC, Saito S, Fukui A, et al. Activation of CaMKII as a key regulator of reactive oxygen species production in diabetic rat heart. J Mol Cell Cardiol. 2012;52:1103–1111. doi: 10.1016/j.yjmcc.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 27.Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, et al. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol. 2009;46:989–997. doi: 10.1016/j.yjmcc.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, et al. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ Res. 2011;109:534–542. doi: 10.1161/CIRCRESAHA.111.246777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deshpande NN, Sorescu D, Seshiah P, Ushio-Fukai M, Akers M, Yin Q, et al. Mechanism of hydrogen peroxide-induced cell cycle arrest in vascular smooth muscle. Antioxid Redox Signal. 2002;4:845–854. doi: 10.1089/152308602760599007. [DOI] [PubMed] [Google Scholar]
- 30.Chiarugi P. Reactive oxygen species as mediators of cell adhesion. Ital J Biochem. 2003;52:28–32. [PubMed] [Google Scholar]
- 31.Simon HU, Haj-Yehia A, Levi-Schaffer F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis. 2000;5:415–418. doi: 10.1023/a:1009616228304. [DOI] [PubMed] [Google Scholar]
- 32.Mercure MZ, Ginnan R, Singer HA. CaM kinase II delta2-dependent regulation of vascular smooth muscle cell polarization and migration. Am J Physiol Cell Physiol. 2008;294:C1465–75. doi: 10.1152/ajpcell.90638.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 34.Daniel JM, Bielenberg W, Stieger P, Weinert S, Tillmanns H, Sedding DG. Time-course analysis on the differentiation of bone marrow-derived progenitor cells into smooth muscle cells during neointima formation. Arterioscler Thromb Vasc Biol. 2010;30:1890–1896. doi: 10.1161/ATVBAHA.110.209692. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Keiser JA. Vascular endothelial growth factor upregulates the expression of matrix metalloproteinases in vascular smooth muscle cells: role of flt-1. Circ Res. 1998;83:832–840. doi: 10.1161/01.res.83.8.832. [DOI] [PubMed] [Google Scholar]
- 36.Tang Z, Wang A, Yuan F, Yan Z, Liu B, Chu JS, et al. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat Commun. 2012;3:875. doi: 10.1038/ncomms1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000;130:947–962. doi: 10.1038/sj.bjp.0703407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pollman MJ, Hall JL, Mann MJ, Zhang L, Gibbons GH. Inhibition of neointimal cell bcl-x expression induces apoptosis and regression of vascular disease. Nat Med. 1998;4:222–227. doi: 10.1038/nm0298-222. [DOI] [PubMed] [Google Scholar]
- 39.Saddouk FZ, Jiang M, Sun LY, Schwarz JJ, Singer HA. Ca2+/calmodulin-dependent Protein Kinase II γ Gene Deletion Enhances Vascular Remodeling in Response to Injury. Arterioscler Thromb Vasc Biol. 2013;33:A94. [Google Scholar]