

Abstract
Increasing evidence suggests that extracellular Vpr could contribute to HIV pathogenesis through its effect on bystander cells. Soluble forms of Vpr have been detected in the sera and cerebrospinal fluids of HIV-1-infected patients, and in vitro studies have implicated extracellular Vpr as an effector of cellular responses, including G2 arrest, apoptosis and induction of cytokines and chemokines production, presumably through its ability to transduce into multiple cell types. However, the mechanism underlying Vpr release from HIV-1-producing cells remains undefined and the biological modifications that the extracellular protein may undergo are largely unknown. We provide evidence indicating that soluble forms of Vpr are present in the extracellular medium of HIV-1-producing cells. Release of Vpr in the extracellular medium did not originate from decaying or disrupted HIV-1 virions that package Vpr but rather appeared associated with HIV-1-mediated cytopathicity. Interestingly, Vpr was found to undergo proteolytic processing at a very well conserved proprotein convertase (PC) cleavage site, R85QRR88↓, located within the functionally important C-terminal arginine-rich domain of the protein. Vpr processing occurred extracellularly upon close contact to cells and most likely involved a cell surface-associated PC. Consistently, PC inhibitors suppressed Vpr processing, while expression of extracellular matrix-associated PC5 and PACE4 enhanced Vpr cleavage. PC-mediated processing of extracellular Vpr led to the production of a truncated Vpr product that was defective for the induction of cell cycle arrest and apoptosis when expressed in human cells. Collectively, these results suggest that cell surface processing of extracellular Vpr by PCs might regulate the levels of active soluble Vpr.
Keywords: HIV-1, Vpr, Extracellular processing, Proprotein convertase, Protease, Extracellular matrix
Introduction
HIV-1 encodes four accessory gene products – Vif, Vpr, Vpu and Nef – which are thought to collectively manipulate host cell biology in order to promote viral replication, persistence and immune escape (reviewed in Emerman and Malim, 1998). One of these accessory proteins, Vpr, is a 96 amino acid polypeptide that is highly conserved both among the primate lentiviruses HIV-1, HIV-2 and the simian immunodeficiency virus (Tristem et al., 1998), supporting the notion that it plays an important role during viral infection in vivo. Indeed, deletion of vpr and the related vpx genes in simian immunodeficiency virus (SIV) severely compromises virus burden and disease progression in experimentally infected monkeys (Gibbs et al., 1995; Hirsch et al., 1998). Despite its small size, Vpr induces multiple effects in host cells in culture that may contribute to the phenotypic effects observed in vivo (reviewed in Andersen and Planelles, 2005; Sherman et al., 2002a). First, Vpr has been shown to act early in viral infection as a facilitator of HIV-1 preintegration complex (PIC) entry through the limiting nuclear pore (Heinzinger et al., 1994; Popov et al., 1998). This activity is thought to be responsible for Vpr’s ability to enhance HIV-1 replication in nondividing cells, most notably in terminally differentiated macrophages (Nitahara-Kasahara et al., 2007; Vodicka et al., 1998). Consistent with this function, Vpr is packaged in relatively large amounts into viral particles through an interaction with the carboxy-terminal of the p6 late domain of the Pr55gag polyprotein precursor (Lu et al., 1995), contains nuclear targeting sequences (Jenkins et al., 2001; Nitahara-Kasahara et al., 2007; Sherman et al., 2001) and is present in PICs (Popov et al., 1998). Second, expression of Vpr was reported to induce cell cycle arrest in G2 by recruiting the DDB1-CUL4 VPRBPE3 ubiquitin ligase complex (Belzile et al., 2007; Hrecka et al., 2007; Le Rouzic et al., 2007; Schrofelbauer et al., 2007; Wen et al., 2007) and by activating the ATR (for Ataxia-Telangiectasia mutated and Rad3-related) checkpoint signaling pathway, a signaling event that is normally part of the cell response system to DNA damage and DNA replication stress (Roshal et al., 2003). Such arrested cells were shown to ultimately die as a result of apoptosis (Andersen et al., 2006; Zhu et al., 2001). Finally, Vpr was also reported to act as a transcriptional activator of the HIV-1 LTR as well as host cell genes (Cohen et al., 1990; Kino et al., 2002; Muthumani et al., 2006; Sherman et al., 2000).
Interestingly, beside being found in virions and in cells, Vpr and Vpr cleavage products have been shown to exist as free molecules in the plasma and the cerebrospinal fluids of HIV-1-infected patients (Hoshino et al., 2007; Levy et al., 1994), indicating that Vpr may be released extracellularly and may exert its biological function beyond infected cells. In that regard, extracellular recombinant Vpr was shown to transduce cells in vitro, apparently via an energy- and receptor-independent process (Henklein et al., 2000). Following cellular uptake, recombinant Vpr was shown to retain the ability to localize to the nucleus and to induce G2 arrest and apoptosis (Henklein et al., 2000; Majumder et al., 2007; Sherman et al., 2002b), thus raising the possibility that circulating forms of Vpr observed in HIV-1-infected patients may exert biological effects on a broad range of host cells. Indeed, a large number of studies have reported that treatment of cells with recombinant Vpr or C-terminal Vpr fragments resulted in apoptosis (Ayyavoo et al., 1997; Jacotot et al., 2000; Patel et al., 2000) and cytotoxic effects (Huang et al., 2000; Piller et al., 1999) in a variety of cell types. Furthermore, recombinant Vpr was also shown to activate AP-1, JNK, and NF-κB in primary macrophages (Varin et al., 2005), to impair cytokine production in DC (Majumder et al., 2007) and to enhance replication in chronically infected cells and in acutely infected primary macrophages (Levy et al., 1995; Sherman et al., 2002b; Varin et al., 2005). However, it has not yet been clearly established how Vpr is released from HIV-1-infected cells. Furthermore, it is unclear whether Vpr proteins released in the extracellular milieu remain in a fully functional form.
In the present work, we provide evidence indicating that Vpr is released in the extracellular milieu of HIV-1-producing cells via a process that is most likely associated to HIV-1-mediated cytopathicity. Interestingly, extracellular Vpr was cleaved by cell surface-associated PCs at a site, R85QRR88↓, located within the functionally important C-terminal arginine-rich motif. The resulting truncated Vpr product was found to be defective for the induction of cell cycle arrest and apoptosis, suggesting that cell surface processing of soluble Vpr by PC might represent a mechanism to control the level of functionally active extracellular Vpr during HIV-1 infection.
Results
HIV-1 Vpr and C-terminally cleaved products are detected in the extracellular medium of HIV-1-producing cells
To determine whether Vpr can be detected in the extracellular medium of HIV-1-producing cells, we initially transfected human embryonic kidney (HEK) 293T cells with a proviral construct (HxBruBH10.R+) expressing Vpr in cis or with a Vpr-defective provirus (HxBruBH10.R+) complemented in trans with an expressor plasmid encoding Vpr. Forty hours post-transfection, cells were isolated by low-speed centrifugation while cell-free culture supernatants were fractionated by ultracentrifugation to separate viral particles from virus-free extracellular medium. The presence of native Vpr was analyzed in HIV-1-producing cells, pelleted virions and virus-free extracellular medium as described in Materials and methods. As shown in Fig. 1A, Vpr was detected as a single band of approximately 16 kDa in lysates from Vpr+ HIV-1-producing cells (Fig. 1A upper panel, lanes 3–4) as well as in lysates from Vpr+ HIV-1 virions (Fig. 1A upper panel, lanes 7–8). Interestingly, upon more sensitive immunoprecipitation/western blot (IP/WB) analysis of cell lysates, a discrete band of lower molecular weight reacting specifically with murine anti-Vpr monoclonal antibodies (mAb) 9F2 was detected in addition to native Vpr regardless of whether Vpr was expressed in cis or in trans (Fig. 1A lower panel, lanes 3–4). Importantly, Vpr as well as a fast migrating Vpr-related band was detected in the virus-free extracellular medium of Vpr+ HIV-1-producing cells by IP/WB analysis (Fig. 1A lower panel, lanes 7–8). In that regard, it is interesting to note that the levels of fast and slow migrating Vpr species were inverted in the extracellular medium as compared to cell lysates (Fig. 1A lower panel, compare lanes 3–4 to lanes 7–8).
Fig. 1.

HIV-1 Vpr and cleaved products are found in the extracellular medium of HIV-1-producing cells. (A) Native Vpr and cleaved products are detected in the extracellular medium of HIV-1-producing 293T cells. 293T cells were mock-transfected (lanes 1 and 5) or transfected with HxBruBH10.R- (lanes 2 and 6) or HxBruBH10.R- with SV CMV Vpr (lanes 3 and 7), or the HxBruBH10.R+ provirus (lanes 4 and 8). Forty hours post-transfection, cells, pelleted virus particles and virus-free extracellular medium were isolated, lysed and analyzed for the presence of native Vpr directly by western blot (upper panel) or by IP/WB (lower panel). Samples corresponding to 5% of the original crude cell (lanes 1–4) and viral (lanes 5–8) lysates were analyzed for the presence of Vpr and CA by western blot using murine anti-Vpr mAb 9F2 and CA mAb, respectively (upper panels). In parallel, cell lysates (50% of total cell lysates) (lanes 1–4) and virus-free extracellular medium (50% of total medium recovered) (lanes 5–8) were immunoprecipitated (IP) with rabbit anti-Vpr pAb and immunocomplexes analyzed by western blot using anti-Vpr mAb 9F2 (lower panel). (B) Extracellular release of Vpr and C-terminally cleaved products requires expression of viral proteins and is independent of Vpr virion incorporation. 293T cells were mock-transfected (lanes 1 and 6) or transfected with HxBruBH10.R- (lanes 2 and 7) or HxBruBH10.3HAR+ (lanes 3 and 8) or HxBruBH10.3HAR+/p6 (1–17) (lanes 4 and 9), or SV CMV 3HA-Vpr (lanes 5 and 10). Forty hours post-transfection, cells, pelleted virus particles and virus-free extracellular medium were isolated as described. Levels of HA-tagged-Vpr (3HA-Vpr) and CA were determined in equivalent proportion of cell and viral lysates by direct western blot using anti-CA or anti-HA mAb (upper panel). In addition, the presence of 3HA-Vpr in cell lysates and virus-free extracellular medium was analyzed by IP/WB using anti-HA mAb (lower panel). (C) Cleaved Vpr is associated to the external surface of HIV-1-producing cells. 293T cells transfected with the indicated proviral constructs or expression plasmids were extensively washed with PBS 40 h post-transfection prior to a 10-min treatment with 0.25% trypsin. Following addition of 5 ml DMEM supplemented with 10% FCS to stop trypsin digestion, cells were extensively washed with PBS and lysed in NP40 lysis buffer. The presence of 3HA-Vpr and cleaved products in cell lysates was analyzed by western blot using anti-HA mAb.
To increase the sensitivity of Vpr immunodetection and to analyze further Vpr release and possible processing, we constructed proviral (HxBruBH10.3HAR+) and expression (SV CMV 3HA-Vpr) plasmids encoding a Vpr protein containing hemagglutinin (HA) epitope tags fused to the protein N-terminus (3HA-Vpr). The HxBruBH10.3HAR+ provirus was still able to replicate and produce infectious virus in Jurkat T cell even though the C-terminus of Vif had to be truncated in the process of engineering the construct (data not shown). Experiments similar to the one described in Fig. 1A led to essentially the same observations with, however, a drastic increase in sensitivity of Vpr species immunodetection (Fig. 1B). Both slow and fast migrating forms of Vpr were detected in cell and viral lysates as well as in extracellular medium from HxBruBH10.3HAR+-producing cells (Fig. 1B upper and lower panels, lanes 3 and 8). The fact that the fast migrating Vpr band was specifically detected with anti-HA antibodies indeed suggested that full-length Vpr undergoes processing at a putative cleavage site located at the Vpr C-terminus since the 3HA tags were fused at the N-terminal-end of the protein. To examine how Vpr products were released extracellularly, we analyzed Vpr release in cell cultures transfected with either the p6-defective proviral construct, HxBruBH10.3HAR+/p6 (1–17), which is unable to efficiently package Vpr into virions (Checroune et al., 1995) or with an expression plasmid encoding 3HA-Vpr only (SV CMV 3HA-Vpr). Even though the p6-defective virus was unable to efficiently incorporate Vpr (Fig. 1B upper panel, compare lanes 8 and 9), we could still detect large amounts of forms of Vpr in the extracellular medium (Fig. 1B lower panel, compare lanes 8 and 9), thus indicating that extracellular Vpr does not primarily originate from disrupted or decaying viral particles. Interestingly, expression of Vpr alone did not lead to extracellular release of Vpr nor to efficient Vpr processing (Fig. 1B upper panel, lane 5 and lower panel lanes 5 and 10). In fact, co-expression of proviral constructs encoding Gag, Pol Vif, Tat and Rev was found to be necessary and sufficient to detect release of soluble Vpr in the extracellular medium (data not shown). As described for native Vpr in Fig. 1A, the ratio of full-length 3HA-Vpr/cleaved Vpr was inverted in cell lysates and extracellular medium from HIV-1-producing 293T cells (Fig. 1B lower panel, compare lanes 8–9 to lanes 3–4). To examine whether the C-terminally cleaved Vpr products detected in cell lysates were present intracellularly or were externally associated to cells, we treated HxBruBH10.3HAR+-transfected 293T cells with 0.25% trypsin prior to lysis and 3HA-Vpr detection by western blot. Fig. 1C clearly reveals that proteolytic treatment of cells with trypsin prior to lysis drastically decreased detection of the fast Vpr migrating band while keeping levels of full-length Vpr intact (Fig. 1C, compare lanes 4 and 3), thus indicating that truncated Vpr products associated with cells are located on the external surface. Moreover, these results suggest that Vpr undergoes processing at its C-terminus extracellularly.
Finally, to ensure that these observations were reproducible in the context of infected CD4+ T cells, we monitored the presence of extracellular forms of Vpr in infected Jurkat T cell cultures in which viral infection was let to spread over an 8-day period as well as in HIV-1-infected peripheral blood mono-nuclear cells (PBMCs). As shown in Fig. 2, results of these experiments clearly reveal that Vpr as well as Vpr C-terminal cleaved products are detected in the extracellular medium during HIV-1 infection of Jurkat T cells (Fig. 2A lower panel, lane 6) and primary PBMCs (Fig. 2B lower panel, lane 6). Furthermore, as shown in HIV-1-producing 293T cells (Fig. 1), some cleaved Vpr products were detected in association with infected T cells (Fig. 2 lower panel A and B, lanes 3), most probably representing extracellular forms of Vpr associated with the cell surface.
Fig. 2.

Vpr and C-terminal cleaved products are detected in the extracellular medium of HIV-1-infected Jurkat T cell and human PBMC cultures. (A) Jurkat T cells were mock-transfected (lanes 1 and 4) or transfected with HxBruBH10.R-(lanes 2 and 5) or HxBruBH10.3HAR+ proviral constructs (lanes 3 and 6). (B) PBMCs were mock-infected (lanes 1 and 4) or infected with HxBruBH10.R-(lanes 2 and 5) or HxBruBH10.3HAR+ virus (lanes 3 and 6). The presence of 3HA-Vpr and 3HA-Vpr cleaved products in infected cells, viral lysates or virus-free extracellular medium was determined 4 days post-infection for infected PBMCs and 8 days post-transfection the infected Jurkat T cell cultures.
Taken altogether, these results indicate that Vpr is released in the extracellular medium of HIV-1-producing cells by a process that requires expression of viral proteins but that is independent of Vpr virion incorporation. Furthermore, released Vpr appears to undergo processing at a putative cleavage site located within the C-terminal domain of the protein.
Vpr is cleaved at a proprotein convertase processing site located at the protein C-terminus
Having obtained evidence that Vpr undergoes processing at the protein C-terminus, we next performed a deletion analysis in order to map the putative cleavage site. Expression plasmids encoding 3HA-Vpr harboring C-terminal deletions from amino acid (aa) residue 79 to 96 (SV CMV 3HA-Vpr (1–78)) and from residue 89 to 96 (SV CMV 3HA-Vpr (1–88)) were generated and used to trans-complement a Vpr-defective HxBruBH10. R- provirus in transfection assays in 293T cells. As shown in Fig. 3A, both 3HA-Vpr deletion mutants were expressed at levels comparable to the wild type (Wt) 3HA-Vpr in transfected cells (lanes 3–4) and were efficiently released in the extracellular medium (lanes 9–10), thus indicating that the C-terminal domain of Vpr is dispensable for extracellular release. Importantly, none of the extracellular Vpr deletion mutants displayed any detectable processing (Fig. 3A, lanes 9–10). Indeed, processed 3HA-Vpr was found to migrate very closely to 3HA-Vpr (1–88) (Fig. 3A, compare lanes 11 and 10) but slower than 3HA-Vpr (1–78) (Fig. 3A, compare lanes 11 and 9) indicating that the putative Vpr processing site most probably lies between residues 78 and 88 and very close to residue 88. Consistently, a putative basic aa-specific PC cleavage site (R/K)-Xn-(R/K)↓ (n=0, 2, 4, 6) (Seidah and Chretien, 1999) was identified at Vpr R85QRR88↓ positions using the Prop v.10b Propeptide cleavage site prediction program (Duckert et al., 2004). Alignments of HIV-1 Vpr from different viral isolates and clades show that the putative P1 (R88) and P2 (R87) cleavage positions are very well conserved, while the P4 (R85) position reveals a degree of variation (Fig. 3B). To confirm whether this predicted PC processing site was indeed functional, we selectively substituted aa residues within the putative cleavage site using site-directed mutagenesis. Given that the P4 position is important for processing by furin while the P1 and P2 position are critical for PCs in general (Thomas, 2002), we focused our mutagenesis on these positions and generated 3HA-tagged Vpr mutants R85Q (this amino acid substitution at the P4 position is often found in viral isolates from clade B) and RR87/88AA (substitution mutations at the P1 and P2 positions) (Fig. 3C). Both mutants were efficiently expressed in HxBruBH10.R- provirus-cotransfected 293T cells and released in the extracellular medium (Fig. 3C, lanes 3–4 and 9–10). Interestingly, while extracellular 3HA-VprR85Q mutant exhibited a two-fold decrease in processing as compared to Wt 3HA-Vpr (Fig. 3C, compare lanes 11 to 9), processing of the double mutant RR87/88AA was reduced by at least six-fold (lane 10). These results strongly suggest that Vpr contains a PC processing site, R85QRR88↓(V/A/G)R, within the protein C-terminal arginine-rich region that is recognized by a PC that is most probably distinct from furin. Furthermore, the presence of hydrophobic aa residues such as valine, in some viral strains, just following the cleavage site (i.e., at P1′) would be indicative of either a PC5 or PACE4-generated cleavage (Seidah and Chretien, 1999).
Fig. 3.

Vpr is cleaved at a PC processing site located within the arginine-rich C-terminal domain. (A) Deletion mapping of Vpr cleavage site. 293T cells were mock-transfected (lanes 1 and 7) or transfected with HxBruBH10.R- (lanes 2 and 8), or cotransfected with HxBruBH10.R- and SV CMV 3HA-Vpr (1–78) (lanes 3 and 9), SV CMV 3HA-Vpr (1–88) (lanes 4 and 10) or SV CMV 3HA-Vpr (lanes 5 and 11). As an additional control, cells were transfected with HxBruBH10.3HAR+ (lanes 6 and 12). Forty hours post-transfection, the presence of 3HA-Vpr and cleavage products was analyzed in cells and virus-free extracellular medium by IP/WB as indicated. (B) Alignment of HIV-1 Vpr putative proprotein cleavage sites from different HIV-1 subtypes. The consensus Vpr aa sequences between amino acid residue 80 and 90 are derived from the HIV sequence database, Los Alamos National Laboratory (http://hiv-web.lanl.gov/content/hiv%1Edb/mainpage.html). Predicted PC processing site aa residues are underlined and their positions indicated above. (C) Mutagenic analysis of the putative Vpr PC cleavage site. 293T cells were mock-transfected (lanes 1 and 7), or transfected with HxBruBH10.R- (lanes 2 and 8) or HxBruBH10.3HAR+ (lanes 6 and 12), or cotransfected with HxBruBH10.R-provirus and SV CMV 3HA-Vpr R85Q (lanes 3 and 9), SV CMV 3HA-Vpr RR87/88AA (lanes 4 and 10) or SV CMV 3HA-Vpr (lanes 5 and 11). Forty hours post-transfection, 3HA-Vpr expression and extracellular release were analyzed in cells and virus-free extracellular medium by IP/WB as indicated. (D) Analysis of extracellular Vpr products by mass spectrometry. Soluble Vpr and cleavage products from virus-free extracellular medium of mock-transfected or HxBruBH10.R- or HxBruBH10.3HAR+ transfected 293Tcell were immunopurified. Eluted proteins with molecular mass ranging from 10 to 22 kDa were then analyzed by SELDI-TOF-MS using the Protein-Chip Reader (Ciphergen Biosystem Inc., Fremont, CA).
To further identify the exact Vpr cleavage site, we immunopurified extracellular forms of 3HA-Vpr from virus-free cell culture supernatant of HxBruBH10.3HAR+ provirus-transfected 293T cells by immunoprecipitation using anti-HA mAb and analyzed the molecular mass (MM) of proteins eluted from the immunocomplexes by SELDI-TOF (surface-enhanced laser desorption/ionization time-of-flight) mass spectrometry (Ciphergen Biosystem Inc., Fremont, CA). Data of Fig. 3D reveal the presence of two specific peaks of isolated proteins. The first peak was found to correspond to a protein having a molecular mass 16189.6 Da, which is indeed very close to the predicted molecular mass of full-length 3HA-Vpr (theoretical MM: 16166.0 Da). The second peak consisted of a mix of proteins having MM ranging from 15355.6 to 15034.6 Da, which are indeed very close to the predicted MM for of 3HA-Vpr (1–88) (theoretical MM 15365.6 Da) and 3HA-Vpr (1–86) (theoretical MM: 15054.4 Da), respectively.
Taken altogether, the results obtained by mutagenesis and mass spectrometry are consistent with a sequential proteolysis process of Vpr. First, the protein is likely cleaved at a PC processing site located at position 85–88, R85Q RR88↓, to lead Vpr cleavage product 1–88. Then, the two terminal arginine residues are most probably removed by a basic-aa specific carboxypeptidases, such as carboxypeptidase D (CPD) (Fricker and Leiter, 1999), thus leading to fully processed Vpr (1–86).
Proprotein convertases mediate extracellular HIV-1 Vpr processing
To determine if PCs have an exclusive role in extracellular Vpr processing, 293T cells were cotransfected with HxBru-BH10.3HAR+ and expression plasmids encoding the protein-based PC inhibitors α1-PDX or Spn4A (Fig. 4A). α1-PDX, which is an α1-antitrypsin variant serine protease inhibitor (serpin), contains the minimal consensus furin cleavage site (R-I-P-R) in its reactive site loop and functions as a suicide substrate inhibitor of PCs, including furin, PC5, PC3 and PACE4 (Jean et al., 1998; Thomas, 2002; Tsuji et al., 1999). Likewise, Spn4A is a secretory pathway serpin from Drosophila melanogaster that contains a consensus furin cleavage site, R-R-K-R, in its reactive site loop. Spn4A inhibits human furin and Drosophila PC2 by a slow-binding mechanism characteristic of serpin molecules and forms kinetically trapped SDS-stable complex with each enzyme (Richer et al., 2004). Fig. 4A reveals that extracellular Vpr processing was significantly reduced in cells expressing α1-PDX or Spn4A as compared to control cells expressing empty vectors (compare lanes 12 and 11, and lanes 14 and 13). Interestingly, Spn4A appeared to have a more potent inhibitory effect on extracellular Vpr processing than α1-PDX in several reproducible experiments. To ensure that these inhibitory effects were specific, we analyzed the processing of HIV-1 envelope gp160 precursor, a known substrate of furin (Thomas, 2002), in cell lysates from the same cell transfectants. As expected, gp160 processing was inhibited in cells expressing α1-PDX or Spn4A, with α1-PDX showing a more potent inhibition of gp160 processing than Spn4A (data not shown). In a second approach, HxBruBH10.3HAR+ provirus-transfected 293T cells were treated 40 h post-transfection for 7 h with 10 μM of the membrane soluble peptide PC inhibitor decanoyl-Arg-Val-Lys-Arg-chloromethyl ketone (dec-RVKR-cmk) (Jean et al., 1998). Processing of extracellular Vpr was specifically inhibited by at least 6-fold in presence of 10 μM dec-RVKR-cmk (Fig. 4B, right panel, compare lanes 8 and 7; left panel). The fact that both protein-based (α1-PDX and Spn4A) and peptide-based (dec-RVKR-cmk) PC inhibitors inhibited extracellular Vpr processing strongly suggests that basic aa-specific PCs mediate extracellular Vpr processing during HIV-1 infection.
Fig. 4.

Proprotein convertases mediate HIV-1 Vpr processing. (A) Extracellular Vpr processing is inhibited by PCs inhibitors α1-PDX and Spn4A. 293T cells were mock-transfected (lanes 1 and 8), or transfected with HxBruBH10.R- (lanes 2 and 9) or HxBruBH10.3HAR+ (lanes 3 and 10), or cotransfected with 10 μg of HxBruBH10.3HAR+ and 1 μg of pIR v5 empty vector (lanes 4 and 11) or pIR PDX+v5 (lanes 5 and 12), or SV CMVexPA control vector (lanes 6 and 13), or pShuttle CMV Spn4A (lanes 7 and 14). Forty hours post-transfection, the presence of 3HA-Vpr products was analyzed in cells and virus-free supernatants by IP/WB as indicated. (B) Extracellular Vpr processing is inhibited by the PC inhibitor dec-RVKR-cmk. 293T cells were mock-transfected (lanes 1 and 5), or transfected with either HxBruBH10.R- (lanes 2 and 6) or HxBruBH10.3HAR+ (lanes 3–4 and 7–8). Two days later, culture media were replaced with fresh medium and cells were cultured for 7 h in presence (lanes 4 and 8) or in absence (lanes 1–3 and 5–7) of 10 μM dec-RVKR-cmk. 3HA-Vpr and cleaved products levels were analyzed in cell lysates and virus-free extracellular medium by IP/WB as indicated (left panel). Vpr processing efficiency was analyzed by quantitating the density of each Vpr-related band using an AGFA Duoscan T1200 scanner. Densitometric analysis of WB results was performed with Image Quant 5.0 from Molecular Dynamics. Vpr processing efficiency is expressed as the percentage of cleaved-Vpr over total Vpr products in the extracellular medium. (C) PC5A and PACE4 expression increases Vpr processing in different cell lines. Hela-CCR5, COS-1 and 293T cells were mock-transfected (lanes 1 and 7) or transfected with HxBruBH10.R- (lanes 2 and 8) or cotransfected with HxBruBH10.3HAR+ and pIR v5 empty vector (lanes 3 and 9) or pIR mPC5A+v5 (lanes 4 and 10) or pIR hFurin FL (lanes 5 and 11) or pIR hPACE4+v5 (lanes 6 and 12). Forty hours post-transfection, levels of 3HA-Vpr products were analyzed in cells and virus-free supernatants as indicated.
We next investigated which widely expressed PCs associated with the constitutive secretory pathway could mediate processing of extracellular Vpr. Furin, PC5 and PACE4 were selected because of their potential role in proteolytic processing of Env gp160 during HIV-1 infection (Thomas, 2002), but more importantly because of their presence at the cell surface as well as in the extracellular medium as enzymatically active shed forms (Koo et al., 2006; Mayer et al., 2004; Tsuji et al., 2003). Plasmids expressing human furin, mouse PC5A or human PACE4 were cotransfected with HxBruBH10.3HAR+ in different cell lines including Hela-CCR5, COS-1 and 293T and 48 h post-transfection, levels of 3HA-Vpr and cleaved products were analyzed in cell lysates and virus-free extracellular medium by western blot or IP/WB (Fig. 4C). When PC5A or PACE4 were co-expressed with 3HA-Vpr-expressing virus, the level of fast migrating Vpr-processed forms detected in the extracellular medium of all transfected cell lines was significantly increased (Fig. 4C, compare lanes 12 and 10 with lane 9). Interestingly, this increased detection of Vpr processed forms in the extracellular medium was also accompanied by a similar increase in the detection of cleaved product externally associated with cells (Fig. 4C, compare lanes 6 and 4 with lane 3) as shown in Fig. 1C. In contrast, co-expression of furin in HxBruBH10.3HAR+-producing cell lines had a marginal effect on basal Vpr processing (Fig. 4C, compare lanes 11 and 9 as well as lanes 5 and 3). Overall, these results suggest that proprotein convertases PC5 and PACE4 can efficiently process extracellular Vpr.
Extracellular Vpr is processed by a PC that is cell surface-associated
The fact that full-length Vpr is detected mainly intracellularly while processed-Vpr is found primarily in the extracellular medium suggests that Vpr proteolytic processing occurs extracellularly—i.e. at the cell surface or in the extracellular medium. To examine these possibilities, we first performed a time course analysis of 3HA-Vpr release from HIV-1-producing 293T cells. Equal amounts of HxBruBH10.3HAR+ provirus-transfected 293T cells plated in 6 wells-plate (106 cells/2 ml media per well) were extensively washed 40 h post-transfection and incubated with fresh culture medium. Over a 3-h period and at different time intervals, the presence of 3HA-Vpr and cleavage products was analyzed in cell and virus-free extracellular medium by western blot and IP/WB as described in Materials and methods. Under these conditions, Vpr was first released out in the extracellular medium mostly as a full-length protein as illustrated by detection of full-length 3HA-Vpr as early as 15 min post-culture medium change (Fig. 5A, lane 3). By 30 min, 3HA-Vpr cleavage products started being clearly detected and by 3 h almost 50% of extracellular Vpr consisted of Vpr processed products (Fig. 5A, lanes 4–6), thus providing evidence that Vpr processing occurs extracellularly once full-length Vpr has been released out of infected cells.
Fig. 5.
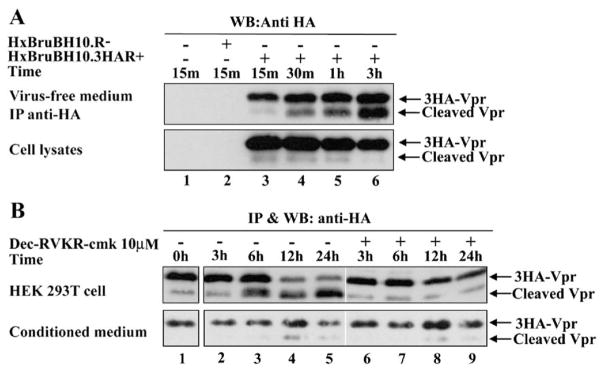
Vpr is processed extracellularly by a PC that is cell surface associated. (A) HIV-1 Vpr is processed in the extracellular medium. 293T cells were mock-transfected (lane 1) or transfected with HxBruBH10.R- (lane 2) or HxBruBH10.3HAR+ (lanes 3–6). Two days later, media were replaced with fresh culture medium and cells were further cultured for different intervals of time as indicated. Cells and virus-free culture media were then harvested at the indicated times and analyzed for the presence of 3HA-Vpr as indicated. (B) Extracellular Vpr is efficiently processed by a cell surface-associated PC. Equivalent amounts (approximately 1 μg) of in vitro translated Vpr were added to 293T cells (2×105/2 ml of culture media) or to cell-free conditioned media (2 ml) collected from 293T cell overnight cultures in presence or absence of 10 μM dec-RVKR-cmk. Cell-free supernatants (upper panel) and conditioned media (lower panel) were collected at the indicated time intervals and analyzed for the presence of 3HA-Vpr and cleavage products by IP/WB.
To characterize whether Vpr was processed by a PC at the cell surface or in the extracellular medium, equivalent amounts of exogenous in vitro translated 3HA-Vpr was added to 293T cell or to conditioned media for various time intervals. As shown in Fig. 5B (upper panel), addition of in vitro translated 3HA-Vpr to 293T cell culture led to processing of exogenous 3HA-Vpr in the cell supernatants starting at 6 h and reaching a peak by 12 h post-incubation (compare lanes 2–5 to lane 1). This processing was PC-specific since addition of 10 μM dec-RVKR-cmk to the culture media reduced drastically the accumulation of Vpr cleavage products (Fig. 5B, compare lanes 6–9 to lanes 2–5). In contrast, only trace amount of Vpr processing was detected upon addition of exogenous 3HA-Vpr to 293T conditioned medium (Fig. 5B lower panel, compare lanes 2–5 to lane 1). Importantly, this residual processing of Vpr in conditioned medium was not affected by addition of PC inhibitor. Similar results were obtained when baculovirus-expressed recombinant Vpr was used in similar experiments (data not shown). Taken together, these results suggest that Vpr undergoes proteolytic processing extracellularly upon close contact with cells, most likely by a PC that is predominantly cell-surface associated.
Vpr processing is detrimental for induction of G2 cell cycle arrest and apoptosis
It was previously reported that exogenous Vpr can transduce cells and cause G2 arrest and apoptosis in HeLa and CD4+ T cells (Henklein et al., 2000; Sherman et al., 2002b). Despite all of our attempts, we were unable to demonstrate that soluble Vpr released in the extracellular medium of HIV-1-producing cells could actually cause G2 arrest and apoptosis when transferred to uninfected Jurkat target cells (data not shown). This lack of detectable activity may be related to the diluted amounts of protein in the extracellular medium and/or the efficient proteolytic processing that is taking place extracellularly. To determine whether fully processed Vpr present in the extracellular medium retained its ability to induce G2 cell cycle arrest upon transduction of bystander cells, we analyzed the effect of Vpr (1–86) expression on the cell cycle. 293T cells were cotransfected with a green fluorescent protein (GFP)-expressing construct as well as with expression plasmids encoding Vpr (SV CMV Vpr Wt) or fully processed Vpr (1–86) (SV CMV Vpr (1–86) or the G2 arrest-defective mutant R80A (SV CMV Vpr R80A) and the cell cycle profile of the GFP-positive cell population was analyzed by flow cytometry 48 h post-transfection as described in Materials and methods. As shown in Fig. 6A, Vpr (1–86) was unable to induce a G2 cell cycle arrest as compared to full-length Vpr (Vpr (1–86) G2/M:G1 ratio=0.4 versus 3.5 for Wt Vpr) even though both proteins were expressed at similar levels (Fig. 6B). Indeed the degree of G2 arrest induced by Vpr (1–86) was similar to that of Vpr R80A, a well characterized G2 arrest-defective Vpr mutant (Belzile et al., 2007). Finally, to determine whether proteolytic processing of Vpr could modulate Vpr-mediated apoptosis, HeLa cells were transfected with plasmids co-expressing GFP and Wt or truncated Vpr, and apoptosis was monitored by PI and annexin V staining 48 h post-transfection. Results presented in Table 1 reveal that truncated Vpr (1–86), in contrast to Wt Vpr, was unable to induce apoptosis. In presence of Vpr (1–86) approximately 10% of cells were PI/annexin V positive while approximately 26% of cells were apoptotic in presence of Wt Vpr. Indeed, the level of apoptotic cells detected in Vpr (1–86)-expressing cells was similar to that obtained in control cells (Vpr−). Overall, these results indicate that processing of Vpr at aa residue 86 abolishes the ability of the protein to induce a G2 cell cycle arrest and mediate apoptosis.
Fig. 6.

Effect of processing on Vpr-mediated cell cycle arrest. (A) 293T cells were cotransfected with GFP-expressing plasmid (pQBI 25) and expression plasmids encoding Wt Vpr, Vpr (1–86)-truncated mutant and Vpr R80A. Forty-eight hours post-transfection, cell-associated DNA content of GFP-expressing cells was analyzed by PI staining and FACS analysis. The ability of Wt Vpr, Vpr (1–86) or Vpr R80A to induce G2 arrest was determined by calculating the G2/M:G1 ratio. Similar results were obtained in 3 independent experiments. (B) Vpr levels in each cell transfectant were analyzed by western blot using murine anti-Vpr mAb 9F2.
Table 1.
Effect of Vpr-processing on Vpr-mediated apoptosis
| PI(+)/annexin V(−) (%) | PI(+)/annexin V(+) (%) | |
|---|---|---|
| SV CMV Vpr−/GFP | 2.9±0.5 | 9.2±0.4 |
| SV CMV Vpr (1–86)/GFP | 3.2±0.3 | 9.6±1.3 |
| SV CMV Vpr Wt/GFP | 7.9±1.4 | 25.9±3.3 |
Discussion
In this study, we provide evidence indicating that Vpr as well as Vpr cleaved products are detected in the extracellular medium of several cell lines, including HEK 293T, HeLa, COS-1 and CD4+ Jurkat T cells, producing HIV-1 in vitro (Figs. 1, 2A and 4) as well as in HIV-1-infected primary PBMCs (Fig. 2B). Interestingly, Vpr expression alone, in the absence of any other viral products, did not lead to efficient Vpr extracellular release even though the protein has the ability to kill cell by apoptosis (Andersen et al., 2006; Stewart et al., 1997). Apparently, efficient Vpr release requires co-expression of viral proteins suggesting that other viral components or/and host cell responses may be necessary for Vpr extracellular release. Since Vpr is primarily located in the nucleus when expressed alone (Sherman et al., 2001), it is possible that expression of other nucleocytoplasmic shuttling viral proteins such as the Gag polyprotein precursor might be necessary to transport the protein in the cytosol near the plasma membrane. In that regard, it was previously reported that during viral infection, Vpr was redistributed from the nucleus to the cytosol and membrane compartments by a process that was independent from its interaction with the p6 domain of Gag (Jenkins et al., 2001). In addition to nuclear export and transport near the plasma membrane, HIV-1-mediated cytopathicity, including plasma membrane disruptions due to viral egress (Fauci, 1988) or the combined proapoptotic/toxic function of HIV-1 gene products (Gougeon, 2003), might be necessary for release of Vpr in the extracellular medium. Nevertheless, our data clearly reveal that Vpr release in the extracellular medium does not rely on the protein ability to be packaged into nascent viral particles since extracellular Vpr was detected in culture infected with p6-defective viral particles even though these do not package Vpr (Fig. 1B); as such, extracellular Vpr originates most likely from infected cells undergoing HIV-1-related cytopathicity rather than from disrupted or decaying virions.
Our data also provide evidence that Vpr undergoes processing extracellularly. Deletion mapping analysis is consistent with Vpr processing occurring within the C-terminal arginine-rich domain of the protein and specifically at a very well conserved putative PC motif located at position R85QRR88 (Fig. 3A). Indeed, site-directed mutagenesis of this putative PC processing site reveals that double mutations of the conserved basic arginine residues located at the P1 and P2 position for alanine (Vpr RR87/88AA) drastically reduced Vpr processing, while substitution of the less conserved arginine residue for a glutamine (R85Q) at the P4 position, attenuated Vpr processing (Fig. 3C). These results indicate that the basic arginine residues in the P1 and P2 position are critical for Vpr processing, while the arginine located in the P4 position is not essential but seems to modulate the extent of Vpr processing. In this regard, it is interesting to note that several Vpr alleles encoded by laboratory-adapted strains such as LAI or HIV-1 primary isolates from different clades contain a glutamine or a proline residue instead of an arginine at that position (Fig. 3B), thus raising the possibility that soluble Vpr encoded by these alleles might be less prone to efficient cleavage. Evidence that Vpr is undergoing processing at this site is also supported by our mass spectrometry analysis of extracellular Vpr species, which revealed the presence of primarily two Vpr cleavage products with predicted molecular mass corresponding to products undergoing cleavage at positions 88 (Vpr 1–88) and 86 (Vpr 1–86) in addition to full-length Vpr (Fig. 3D). As it has been previously reported that the last two basic aa residues of the released N-terminal peptide generated upon PC cleavage are generally removed by carboxypeptidases (Fricker and Leiter, 1999), our results are consistent with a Vpr processing occurring at the conserved double arginine residues (R87R88) in the P1 and P2 position, thus generating an 88-aa polypeptide that subsequently undergoes a rapid trimming of the two basic pair residues leading to an 86-aa fully processed Vpr product. The fact that fully processed Vpr was shown to migrate slightly faster than the Vpr (1–88) deletion mutant (Fig. 3A) supports such a sequential proteolytic process.
Furthermore, our results reveal that Vpr is initially released as a full-length protein and subsequently processed extracellularly by cell surface PCs (Fig. 5). PCs belong to a family of evolutionary conserved dibasic- and monobasic-specific Ca2+-dependent subtilisin-like serine proteinases related to the yeast kexin enzyme that constitute the major endoproteolytic processing enzymes of the secretory pathway in mammals (Seidah and Chretien, 1999; Steiner, 1998). Traditionally, PCs have been shown to cleave their substrates intracellularly. This is particularly true for furin, the best known member of the protease family (Thomas, 2002). Furin is known to exist at the cell surface while other PCs, such as PACE4 and PC5, are known to be secreted and anchored in the extracellular matrix (Nour et al., 2005; Tsuji et al., 2003) and are therefore presumed to have extracellular substrates. The inhibition of extracellular Vpr processing using PC inhibitors such as dec-RVKR-cmk or serpins α1-PDX and Spn4A strongly suggests that PCs are involved in Vpr processing (Figs. 4A–B). Furthermore, transient co-expression of PCs in several HIV-1-producing cell lines (293T, Hela and COS-1) reveals that PC5 and PACE4 efficiently processed extracellular Vpr, while furin expression had only a marginal effect (Fig. 5C). These results are consistent with the fact that mutation of the arginine residue at the P4 position of the processing site, a residue critical for processing by furin (Thomas, 2002), was not essential for Vpr cleavage.
Our data suggest that PC5 and PACE4 might be involved in extracellular Vpr processing. This is especially relevant since these enzymes can tolerate an aliphatic residue (e.g., Val or Leu) at P1′ just following the cleavage site, whereas furin does not (Seidah and Chretien, 1999). Interestingly, PC5 is expressed in freshly isolated human CD4 T-lymphocytes, the natural host cells of HIV-1 (Decroly et al., 1996; Hallenberger et al., 1997). Although PACE4 is not expressed in peripheral blood lymphocytes (PBL), it is highly expressed in lymphoid tissues such as thymus, lymph node and spleen (Hallenberger et al., 1997) and as such might be present at the cell surface of bystander cells located in close proximity of HIV-1-producing cells in vivo. However, we cannot rule out at this point that other PCs displaying similar substrate specificity and cellular location may also participate to Vpr processing in vivo.
We were unable to demonstrate that the levels of extracellular Vpr released from HIV-1-producing cells could actually cause G2 arrest and apoptosis when transferred to uninfected target T cells. The absence of detectable activity may be due to the diluted amounts of Vpr in the extracellular medium but might also result from the efficient processing that undergoes upon contact of the protein with target cells. Indeed, efficient processing of Vpr by PCs generates a truncated Vpr (1–86) that is defective for G2 cell cycle arrest and unable to induce apoptosis upon expression in cells (Fig. 6 and Table 1). These findings raise the possibility that processing of extracellular Vpr by cell-surface PCs, such as PC5 and PACE4, might indeed represent a host cell process to functionally inactivate the cytostatic and cytotoxic activities of extracellular Vpr. In addition to inactivating Vpr-mediated G2 arrest and proapoptotic activities, processing of extracellular Vpr C-terminal may also represent a mechanism to prevent or limit the uptake of the protein by target cells. The carboxy-terminus domain of Vpr contains six highly conserved arginine residues between residues 73 and 96. This domain shows similarity with those of arginine-rich protein transduction domains and may explain the transducing properties of Vpr, including its ability to cross the cell membrane lipid bilayer (Coeytaux et al., 2003; Henklein et al., 2000; Sherman et al., 2002b). In fact, transduction of exogenous protein has been shown to very often involve the attachment of arginine-rich domain of proteins to membrane HSPG (Melikov and Chernomordik, 2005; Richard et al., 2005). Likewise, soluble Vpr may interact with membrane HSPG via its C-terminal arginine-rich domain as a first step prior to internalization as was reported for other transducing proteins (Melikov and Chernomordik, 2005). Given that PCs such as PC5 and PACE4 are located in the extracellular matrix, it is possible that processing of extracellular Vpr may occur upon attachment of the protein to HSPG. Processing of extracellular Vpr by PCs will delete four of the six conserved arginine residues and as such is likely to interfere with the transduction properties of the protein (Sherman et al., 2002b). More studies will be required to analyze soluble Vpr interactions with cell surface HSPG and its implications in terms of protein cellular uptake. Importantly, additional studies using recombinant Vpr proteins will be needed to directly demonstrate that the level of extracellular Vpr biological activity correlates with the degree of protein processing.
It is interesting that Vpr is not the only HIV-transducing protein undergoing processing by PCs. Extracellular Tat was reported to be cleaved by furin (Tikhonov et al., 2004). Furin processing did not affect the rates of N-terminal cleavage product uptake and nuclear localization but greatly reduced the protein transactivation activity. It is thought that furin processing is a mechanism to inactivate extracellular Tat protein. Although our results point towards a role of Vpr processing by PCs as a mean to inactivate Vpr, we cannot entirely rule-out that soluble truncated Vpr (1–86) may still have other biological activities and function important for HIV infection in vivo.
In conclusion, this study provides evidence that HIV-1 Vpr is released in the extracellular milieu of HIV-producing cells where it undergoes processing by cell surface PCs such as PC5 or PACE4. PC processing of extracellular Vpr occurs at a very well conserved processing site located in the C-terminal arginine-rich domain of the protein and leads to the production of a truncated Vpr product that is unable to induce G2 cell cycle arrest and apoptosis. We propose that PC processing of extracellular Vpr might represent a mechanism to inactivate soluble Vpr.
Materials and methods
Plasmids and proviral DNA constructs
The expression plasmid SV CMV 3HA-Vpr was constructed by inserting three consecutive hemagglutinin tags (3HA) to the N-terminus of Vpr in SV CMV Vpr WT (Yao et al., 1998) using a two-steps PCR approach. The introduced 3HA tag contains 49 amino acids (aa): MASVSYPYDVPDYASLGG-PSSVSYPYDVPDYASLGGPSSVYPYDVPDYA (HA epitope sequences are underlined). The single mutant 3HA-Vpr R85Q and the double mutant 3HA-RR87/88AAwere produced by PCR-mutagenesis using SV CMV 3HA-Vpr as template. SV CMV 3HA-Vpr (1–88), (1–86) and (1–78) as well as SV CMV Vpr (1–86) were generated by introducing premature stop codons at aa position 88, 86 or 78 of Vpr using PCR-mutagenesis. The SV CMV Vpr− and R80A constructs as well as the bi-cistronic expressors SV CMV Vpr/GFP, Vpr−/GFP were described previously (Hrimech et al., 1999). The SV CMV Vpr (1–86)/GFP plasmid was constructed by inserting a BglII/DraIII fragment from pQB25I (Quantum Biotechnologies, Montreal, Que, Canada) encoding the GFP coding sequence preceded by the cytomegalovirus (CMV) early promoter into the BamHI sites of SV CMV Vpr (1–86). The pET21c 3HA-Vpr plasmid was generated by subcloning a DNA fragment encoding 3HA-Vpr from SV CMV 3HA-Vpr into pET21c (Novagen, Madison, WI, USA). SV CMVexPAvector was previously described (Yao et al., 1998). HxBruBH10.R+ (gag+-pol+-vif+-vpr+-tat+-rev+-vpu+-env+-nef−) and Vpr-defective HxBruBH10.R- proviral constructs were generated by introducing a SalI/KpnI fragment encoding Vpu from HxBH10 (Levesque et al., 2003) into HxBru or HxBru R- (Yao et al., 1995). The HxBruBH10.3HAR+ proviral construct was derived from the HxBruBH10.R+ provirus. In the process of introducing the DNA fragment encoding 3HA-Vpr, an NheI site was created at nucleotide position 5140 (+1=start of Bru transcription initiation site) before the Vpr ATG by PCR-mutagenesis. The creation of the NheI site led to the introduction of a frameshift mutation at aa position 174 in the Vif open reading frame that resulted in a truncated Vif protein. The proviral construct HxBruBH10.3HAR+/p6 (1–17) encodes a truncated p6gag domain (premature stop codon at aa 17). It was generated by replacing an ApaI (nucleotide position 2011)–BstZ17I (nucleotide position 2967) DNA fragment from HxBruBH10.3HAR+ by the corresponding fragment from HxBru.R+.s17stop (Checroune et al., 1995). The PC-expressing plasmids pIR hfurin, pIR mPC5A+v5 and pIR hPACE4+v5 encoding human furin, murine PC5A and human PACE4, respectively, were described previously (Bergeron et al., 2005). Plasmid pIR-PDX-v5 encoding the PC inhibitor α1-PDX fused to a v5 tag was described elsewhere (Bergeron et al., 2003), while pShuttle CMV Spn4A encoding the PC inhibitor Spn4A fused to a Flag tag was kindly provided by Dr. François Jean (Department of Microbiology and Immunology, University of British Columbia, Vancouver, Canada). All plasmid constructions were analyzed and confirmed by automated DNA sequencing.
Cells and reagents
HEK 293T, Hela-CCR5 and COS-1 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS). Jurkat T-lymphoid cells were maintained in RPMI-1640 medium supplemented with 10% FCS. PBMCs were isolated from volunteers by Ficoll–Paque centrifugation as recommended by the manufacturer (Amersham, Baie d’Urfé, Quebec, Canada). Cells were then cultured in the presence of phytohemagglutinin-L (5 μg/ml) (Sigma-Aldrich Canada Ltd, Oakville, Ontario, Canada) and interleukin 2 (IL-2) (20 U/ml) (Sigma-Aldrich) for 48 h to activate lymphocytes. Following activation, cells were washed with complete media to remove lectins and maintained in RPMI plus 20% FCS supplemented with 20 U/ml of IL-2. All cells were cultured at 37 °C under a 5% CO2 atmosphere. Anti-HA mAb were produced from hybridoma 12CA5, while anti-V5 and anti-Flag mAb (M2) were purchased from Invitrogen (San Diego, CA, USA) and Sigma-Aldrich, respectively. The anti-Vpr mAb 9F12 directed against an epitope comprising aa 4–16 was kindly provided by Dr. Jeffrey B. Kopp (Kidney Disease Branch, National Institutes of Health, Bethesda, USA). Rabbit anti-Vpr polyclonal antibodies (pAb) were previously described (Yao et al., 1995). Anti-HIV-1 capsid (CA or p24) protein mAb were produced from hybridoma HB9725 (ATCC; American Type Culture Collection, Manassas, VA, USA) while anti-gp120 mAb recognizing both gp120 and precursor gp160 were obtained from the NIH AIDS Research and Reference Reagents Program. The PC inhibitor dec-RVKR-cmk was purchased from Bachem Bioscience Inc. (King of Prussia, PA, USA). A 10-mM stock solution was prepared in DMSO and was further diluted in tissue culture medium to give the required final concentration.
Transfection
293T and COS-1 cells were transfected by the standard calcium phosphate DNA precipitation method. For detection of extra-cellular 3HA-Vpr, 106cells were either transfected with 10 μg of 3HA-Vpr proviral DNA plasmid or cotransfected with 10 μg of Vpr-defective proviral DNA plasmid and 5 μg of 3HA-Vpr expression vector. For native Vpr proteolytic processing detection, 293T (4×106cells) were either transfected with 40 μg of proviral DNA plasmid or cotransfected with 40 μg of Vpr-defective proviral DNA plasmid and 20 μg of Vpr expression plasmid vector. Hela-CCR5 cells (4×105 in 6 wells plate) were transfected with 4 μg of 3HA-Vpr proviral DNA construct and 0.5 μg of PC expression plasmids using Lipofectamine 2000 (Invitrogen, San Diego, CA, USA) according to the manufacturer instructions.
Analysis of Vpr processing in HIV-1-producing cells, extracellular medium and viral particles
Transfected or infected cells were separated from virion-containing supernatants by low-speed centrifugation and lysed in 1% NP40 lysis buffer (140 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 1% NP40, 0.5% SDS, pH7.2) supplemented with a protease inhibitor cocktail (Roche Diagnostics Canada, Laval, Que, Canada). Vpr was immunoprecipitated from cell lysates using anti-HA or anti-Vpr antibodies as described (Yao et al., 1995). For detection of extracellular Vpr, 16 ml of culture supernatants was first centrifuged at 2095×g for 10 min and then passed through a 0.45-μm filter to eliminate cell debris. Viral particles were separated from the extracellular medium by ultra centrifugation onto a 20% sucrose cushion at 126087×g for 1.5 h. Eight milliliters of virus-free extracellular medium treated with 5-fold concentrated NP-40 lysis buffer was immunoprecipitated with anti-HA or anti-Vpr antibodies as described (Yao et al., 1995). Pelleted virus was lysed directly in 1% NP40 lysis buffer. Cell and viral lysates as well as immunocomplexes resulting from immunoprecipitations were separated by 14% SDS–PAGE, transferred to a nitrocellulose membrane and analyzed by western blotting using antibodies against HIV-1 capsid p24, HA or Vpr as described previously (Levesque et al., 2003). Bound antibodies were revealed using the 3,3′-diaminobenzidine detection system, as recommended by the manufacturer (ICN Biomedicals, Irvine, CA, USA). For detection of extracellular native Vpr, supersensitive Immobilon™ Western Chemiluminescent HRP Substrate (Millipore, Billerica, MA, USA) was used according to the manufacturer instructions.
Analysis of extracellular Vpr processing in HIV-1-infected Jurkat T cells and human PBMCs
HIV-1-infected Jurkat T cells cultures were generated by transfecting Jurkat T cells (4×106) with 20 μg of Vpr− or 3HA-Vpr HIV-1 provirus using the DEAE-dextran method (Levesque et al., 2003). Analysis of Vpr processing was conducted at the peak of virus production (8 days post-transfection) as described above. HIV-1 virus stocks were generated by transfection of proviral constructs in 293T cells and titered by MAGI assay as described previously (Levesque et al., 2003). PBMCs (5×106) were infected at a multiplicity of infection (MOI) of 0.25 four days post-activation as described before (Yao et al., 1998). Analysis of Vpr processing was conducted 4 days post-infection as described above.
Surface-enhanced laser desorption/ionization time-of-flight mass spectrometry (SELDI-TOF-MS) analysis
3HA-Vpr was isolated from virus-free culture supernatant of HxBruBH10.3HAR+-transfected 293T cells by immunoprecipitation using anti-HA antibodies. 3HA-Vpr was eluted from anti-HA antibodies bound to protein A sepharose beads using trifluoroacetic acid (TFA). The 3HA-Vpr-containing eluate was combined with a similar volume of binding buffer (100 mM PBS and 0.5 M NaCl pH 7.0), applied onto a NP20 Protein-Chip spot (Ciphergen Biosystems Inc., Fremont, CA, USA) and incubated at room temperature for 20 min to dry. Unbound samples on the chip spot were washed-off following three washes of 5 min with binding buffer. The NP20 chip was then rinsed with 20 μl of distilled H2O and air dried. A matrix consisting of a saturated solution of 3,5-dimethoxy-4-hydro-xycinnamic acid (SPA) in 50% acetonitrile and 0.5% TFA was added to the chip surface before SELDI-TOF-MS analysis. The SELDI-TOF-MS analysis was performed using a Ciphergen Protein-Chip Reader (Ciphergen Biosystem Inc., Fremont, CA, USA). Cytochrome c (12 kDa) was used as the molecular mass calibrator.
In vitro translation
3HA-Vpr was in vitro translated from pET21c 3HA-Vpr using the Active Pro™ In vitro translation kit (Ambion, Austin, TX, USA) according to manufacturer instructions.
Cell cycle profiling and analysis of apoptosis
Cell cycle analysis was performed as described (Nishizawa et al., 2000). Briefly, 293Tcells were cotransfected with 10 μg of expression plasmids encoding Wt Vpr, truncated Vpr (1–86) or Vpr R80A and 0.5 μg of GFP-expressing plasmids. Cells were harvested 40 h later, fixed in 1% paraformaldehyde during 10 min and incubated in 70% ethanol for an additional 10 min. Fixed cells were then treated with phosphate-buffered saline (PBS) containing propidium iodine (PI) (50 μg/ml), RNase A (50 μg/ml) and FCS (1%, vol/vol) for 60 min at room temperature. The DNA content of GFP-positive cells was analyzed on a FACScan flow cytometer using the Cell QUEST software (Becton Dickinson, Franklin Lakes, NJ, USA). Relative numbers of cells in the G2/M and G1 phases of the cell cycle (G2/M: G1 ratios) were calculated using the ModFit LT software (Verity Software House, Topsham, ME, USA). Apoptotic cells were detected using the Annexin V-Alexa Fluor 647 assay (Invitrogen, San Diego, CA, USA). Briefly, 40 h post-transfection 0.25×106 Hela cells were washed once with PBS and then incubated for 20 min in annexin V binding buffer (2.5 μl of Annexin V-Alexa Fluro 647 per ml, 10 mM HEPES–NaOH [pH 7.4], 150 mM NaCl, 5 mM KCl, 1 mM MgCl2, 1.8 mM CaCl2 and 1 μg/ml of PI). Apoptotic cells, which stained positive for PI and annexin V, were detected using a FACScalibur (Becton Dickinson, Franklin Lakes, NJ, USA).
Acknowledgments
We thank Dr. Ghislaine Duisit and Jean-Philippe Belzile for helpful discussions and critical review the manuscript. HIV-1 IIIB gp120 Hybridoma 902 recognizing both gp120 and precursor gp160 was obtained from the NIH AIDS research and reference reagent program while the anti-Vpr mAb 9F12 was kindly provided by Dr. Jeffrey B. Kopp (NIH). EAC and NGS are recipients of the Canada Research Chairs in Human Retrovirology and Precursors Proteolysis, respectively.
This work was performed by YX in partial fulfillment of his doctoral thesis and was supported by grants from the Canadian Institute of Health Research to EAC (NRF-12381) and NGS (CIHR MGP-44363) and from the Fonds de la Recherche en Santé du Québec-AIDS network to EAC.
References
- Andersen JL, Planelles V. The role of Vpr in HIV-1 pathogenesis. Curr HIV Res. 2005;3 (1):43–51. doi: 10.2174/1570162052772988. [DOI] [PubMed] [Google Scholar]
- Andersen JL, Dehart JL, Zimmerman ES, Ardon O, Kim B, Jacquot G, Benichou S, Planelles V. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog. 2006;2 (12):e127. doi: 10.1371/journal.ppat.0020127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayyavoo V, Mahboubi A, Mahalingam S, Ramalingam R, Kudchodkar S, Williams WV, Green DR, Weiner DB. HIV-1 Vpr suppresses immune activation and apoptosis through regulation of nuclear factor kappa B. Nat Med. 1997;3 (10):1117–1123. doi: 10.1038/nm1097-1117. [DOI] [PubMed] [Google Scholar]
- Belzile JP, Duisit G, Rougeau N, Mercier J, Finzi A, Cohen EA. HIV-1 Vpr-mediated G2 arrest involves the DDB1-CUL4A(VPRBP) E3 ubiquitin ligase. PLoS Pathog. 2007;3 (7):e85. doi: 10.1371/journal.ppat.0030085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron E, Basak A, Decroly E, Seidah NG. Processing of alpha4 integrin by the proprotein convertases: histidine at position P6 regulates cleavage. Biochem J. 2003;373 (Pt 2):475–484. doi: 10.1042/BJ20021630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergeron E, Vincent MJ, Wickham L, Hamelin J, Basak A, Nichol ST, Chretien M, Seidah NG. Implication of proprotein convertases in the processing and spread of severe acute respiratory syndrome coronavirus. Biochem Biophys Res Commun. 2005;326 (3):554–563. doi: 10.1016/j.bbrc.2004.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checroune F, Yao XJ, Gottlinger HG, Bergeron D, Cohen EA. Incorporation of Vpr into human immunodeficiency virus type 1: role of conserved regions within the P6 domain of Pr55gag. J Acquir Immune Defic Syndr Human Retrovirol. 1995;10 (1):1–7. [PubMed] [Google Scholar]
- Coeytaux E, Coulaud D, Le Cam E, Danos O, Kichler A. The cationic amphipathic alpha-helix of HIV-1 viral protein R (Vpr) binds to nucleic acids, permeabilizes membranes, and efficiently transfects cells. J Biol Chem. 2003;278 (20):18110–18116. doi: 10.1074/jbc.M300248200. [DOI] [PubMed] [Google Scholar]
- Cohen EA, Terwilliger EF, Jalinoos Y, Proulx J, Sodroski JG, Haseltine WA. Identification of HIV-1 vpr product and function. J Acquir Immune Defic Syndr. 1990;3 (1):11–18. [PubMed] [Google Scholar]
- Decroly E, Wouters S, Di Bello C, Lazure C, Ruysschaert JM, Seidah NG. Identification of the paired basic convertases implicated in HIV gp160 processing based on in vitro assays and expression in CD4(+) cell lines. J Biol Chem. 1996;271 (48):30442–30450. doi: 10.1074/jbc.271.48.30442. [DOI] [PubMed] [Google Scholar]
- Duckert P, Brunak S, Blom N. Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel. 2004;17 (1):107–112. doi: 10.1093/protein/gzh013. [DOI] [PubMed] [Google Scholar]
- Emerman M, Malim MH. HIV-1 regulatory/accessory genes: keys to unraveling viral and host cell biology. Science. 1998;280 (5371):1880–1884. doi: 10.1126/science.280.5371.1880. [DOI] [PubMed] [Google Scholar]
- Fauci AS. The human immunodeficiency virus: infectivity and mechanisms of pathogenesis. Science. 1988;239 (4840):617–622. doi: 10.1126/science.3277274. [DOI] [PubMed] [Google Scholar]
- Fricker LD, Leiter EH. Peptides, enzymes and obesity: new insights from a ‘dead’ enzyme. Trends Biochem Sci. 1999;24 (10):390–393. doi: 10.1016/s0968-0004(99)01448-6. [DOI] [PubMed] [Google Scholar]
- Gibbs JS, Lackner AA, Lang SM, Simon MA, Sehgal PK, Daniel MD, Desrosiers RC. Progression to AIDS in the absence of a gene for vpr or vpx. J Virol. 1995;69 (4):2378–2383. doi: 10.1128/jvi.69.4.2378-2383.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gougeon ML. Apoptosis as an HIV strategy to escape immune attack. Nat Rev Immunol. 2003;3 (5):392–404. doi: 10.1038/nri1087. [DOI] [PubMed] [Google Scholar]
- Hallenberger S, Moulard M, Sordel M, Klenk HD, Garten W. The role of eukaryotic subtilisin-like endoproteases for the activation of human immunodeficiency virus glycoproteins in natural host cells. J Virol. 1997;71 (2):1036–1045. doi: 10.1128/jvi.71.2.1036-1045.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzinger NK, Bukinsky MI, Haggerty SA, Ragland AM, Kewalramani V, Lee MA, Gendelman HE, Ratner L, Stevenson M, Emerman M. The Vpr protein of human immunodeficiency virus type 1 influences nuclear localization of viral nucleic acids in nondividing host cells. Proc Natl Acad Sci U S A. 1994;91 (15):7311–7315. doi: 10.1073/pnas.91.15.7311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henklein P, Bruns K, Sherman MP, Tessmer U, Licha K, Kopp J, de Noronha CM, Greene WC, Wray V, Schubert U. Functional and structural characterization of synthetic HIV-1 Vpr that transduces cells, localizes to the nucleus, and induces G2 cell cycle arrest. J Biol Chem. 2000;275 (41):32016–32026. doi: 10.1074/jbc.M004044200. [DOI] [PubMed] [Google Scholar]
- Hirsch VM, Sharkey ME, Brown CR, Brichacek B, Goldstein S, Wakefield J, Byrum R, Elkins WR, Hahn BH, Lifson JD, Stevenson M. Vpx is required for dissemination and pathogenesis of SIV(SM) PBj: evidence of macrophage-dependent viral amplification. Nat Med. 1998;4 (12):1401–1408. doi: 10.1038/3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshino S, Sun B, Konishi M, Shimura M, Segawa T, Hagiwara Y, Koyanagi Y, Iwamoto A, Mimaya J, Terunuma H, Kano S, Ishizaka Y. Vpr in plasma of HIV type 1-positive patients is correlated with the HIV type 1 RNA titers. AIDS Res Hum Retroviruses. 2007;23 (3):391–397. doi: 10.1089/aid.2006.0124. [DOI] [PubMed] [Google Scholar]
- Hrecka K, Gierszewska M, Srivastava S, Kozaczkiewicz L, Swanson SK, Florens L, Washburn MP, Skowronski J. Lentiviral Vpr usurps Cul4-DDB1[VprBP] E3 ubiquitin ligase to modulate cell cycle. Proc Natl Acad Sci U S A. 2007;104 (28):11778–11783. doi: 10.1073/pnas.0702102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrimech M, Yao XJ, Bachand F, Rougeau N, Cohen EA. Human immunodeficiency virus type 1 (HIV-1) Vpr functions as an immediate-early protein during HIV-1 infection. J Virol. 1999;73 (5):4101–4109. doi: 10.1128/jvi.73.5.4101-4109.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MB, Weeks O, Zhao LJ, Saltarelli M, Bond VC. Effects of extracellular human immunodeficiency virus type 1 vpr protein in primary rat cortical cell cultures. J Neurovirol. 2000;6 (3):202–220. doi: 10.3109/13550280009015823. [DOI] [PubMed] [Google Scholar]
- Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HL, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, Kroemer G. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med. 2000;191 (1):33–46. doi: 10.1084/jem.191.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean F, Stella K, Thomas L, Liu G, Xiang Y, Reason AJ, Thomas G. alpha1-Antitrypsin Portland, a bioengineered serpin highly selective for furin: application as an antipathogenic agent. Proc Natl Acad Sci U S A. 1998;95 (13):7293–7298. doi: 10.1073/pnas.95.13.7293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins Y, Sanchez PV, Meyer BE, Malim MH. Nuclear export of human immunodeficiency virus type 1 Vpr is not required for virion packaging. J Virol. 2001;75 (17):8348–8352. doi: 10.1128/JVI.75.17.8348-8352.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kino T, Gragerov A, Slobodskaya O, Tsopanomichalou M, Chrousos GP, Pavlakis GN. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J Virol. 2002;76 (19):9724–9734. doi: 10.1128/JVI.76.19.9724-9734.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo BH, Longpre JM, Somerville RP, Alexander JP, Leduc R, Apte SS. Cell-surface processing of pro-ADAMTS9 by furin. J Biol Chem. 2006;281 (18):12485–12494. doi: 10.1074/jbc.M511083200. [DOI] [PubMed] [Google Scholar]
- Le Rouzic E, Belaidouni N, Estrabaud E, Morel M, Rain JC, Transy C, Margottin-Goguet F. HIV1 Vpr arrests the cell cycle by recruiting DCAF1/VprBP, a receptor of the Cul4-DDB1 ubiquitin ligase. Cell Cycle. 2007;6 (2):182–188. doi: 10.4161/cc.6.2.3732. [DOI] [PubMed] [Google Scholar]
- Levesque K, Zhao YS, Cohen EA. Vpu exerts a positive effect on HIV-1 infectivity by down-modulating CD4 receptor molecules at the surface of HIV-1-producing cells. J Biol Chem. 2003;278 (30):28346–28353. doi: 10.1074/jbc.M300327200. [DOI] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, MacGregor RR, Weiner DB. Serum Vpr regulates productive infection and latency of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1994;91 (23):10873–10877. doi: 10.1073/pnas.91.23.10873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DN, Refaeli Y, Weiner DB. Extracellular Vpr protein increases cellular permissiveness to human immunodeficiency virus replication and reactivates virus from latency. J Virol. 1995;69 (2):1243–1252. doi: 10.1128/jvi.69.2.1243-1252.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YL, Bennett RP, Wills JW, Gorelick R, Ratner L. A leucine triplet repeat sequence (LXX)4 in p6gag is important for Vpr incorporation into human immunodeficiency virus type 1 particles. J Virol. 1995;69 (11):6873–6879. doi: 10.1128/jvi.69.11.6873-6879.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumder B, Venkatachari NJ, Schafer EA, Janket ML, Ayyavoo V. Dendritic cells infected with Vpr-positive human immunodeficiency virus type 1 induce CD8+ T-cell apoptosis via upregulation of tumor necrosis factor alpha. J Virol. 2007;81 (14):7388–7399. doi: 10.1128/JVI.00893-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer G, Boileau G, Bendayan M. Sorting of furin in polarized epithelial and endothelial cells: expression beyond the Golgi apparatus. J Histochem Cytochem. 2004;52 (5):567–579. doi: 10.1177/002215540405200502. [DOI] [PubMed] [Google Scholar]
- Melikov K, Chernomordik LV. Arginine-rich cell penetrating peptides: from endosomal uptake to nuclear delivery. Cell Mol Life Sci. 2005;62 (23):2739–2749. doi: 10.1007/s00018-005-5293-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthumani K, Choo AY, Zong WX, Madesh M, Hwang DS, Premkumar A, Thieu KP, Emmanuel J, Kumar S, Thompson CB, Weiner DB. The HIV-1 Vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of PARP-1. Nat Cell Biol. 2006;8 (2):170–179. doi: 10.1038/ncb1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishizawa M, Kamata M, Katsumata R, Aida Y. A carboxy-terminally truncated form of the human immunodeficiency virus type 1 Vpr protein induces apoptosis via G(1) cell cycle arrest. J Virol. 2000;74 (13):6058–6067. doi: 10.1128/jvi.74.13.6058-6067.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitahara-Kasahara Y, Kamata M, Yamamoto T, Zhang X, Miyamoto Y, Muneta K, Iijima S, Yoneda Y, Tsunetsugu-Yokota Y, Aida Y. Novel nuclear import of Vpr promoted by importin alpha is crucial for human immunodeficiency virus type 1 replication in macrophages. J Virol. 2007;81 (10):5284–5293. doi: 10.1128/JVI.01928-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nour N, Mayer G, Mort JS, Salvas A, Mbikay M, Morrison CJ, Overall CM, Seidah NG. The cysteine-rich domain of the secreted proprotein convertases PC5A and PACE4 functions as a cell surface anchor and interacts with tissue inhibitors of metalloproteinases. Mol Biol Cell. 2005;16 (11):5215–5226. doi: 10.1091/mbc.E05-06-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel CA, Mukhtar M, Pomerantz RJ. Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J Virol. 2000;74 (20):9717–9726. doi: 10.1128/jvi.74.20.9717-9726.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piller SC, Ewart GD, Jans DA, Gage PW, Cox GB. The amino-terminal region of Vpr from human immunodeficiency virus type 1 forms ion channels and kills neurons. J Virol. 1999;73 (5):4230–4238. doi: 10.1128/jvi.73.5.4230-4238.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov S, Rexach M, Zybarth G, Reiling N, Lee MA, Ratner L, Lane CM, Moore MS, Blobel G, Bukrinsky M. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 1998;17 (4):909–917. doi: 10.1093/emboj/17.4.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard JP, Melikov K, Brooks H, Prevot P, Lebleu B, Chernomordik LV. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J Biol Chem. 2005;280 (15):15300–15306. doi: 10.1074/jbc.M401604200. [DOI] [PubMed] [Google Scholar]
- Richer MJ, Keays CA, Waterhouse J, Minhas J, Hashimoto C, Jean F. The Spn4 gene of Drosophila encodes a potent furin-directed secretory pathway serpin. Proc Natl Acad Sci U S A. 2004;101 (29):10560–10565. doi: 10.1073/pnas.0401406101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem. 2003;278 (28):25879–25886. doi: 10.1074/jbc.M303948200. [DOI] [PubMed] [Google Scholar]
- Schrofelbauer B, Hakata Y, Landau NR. HIV-1 Vpr function is mediated by interaction with the damage-specific DNA-binding protein DDB1. Proc Natl Acad Sci U S A. 2007;104 (10):4130–4135. doi: 10.1073/pnas.0610167104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidah NG, Chretien M. Proprotein and prohormone convertases: a family of subtilases generating diverse bioactive polypeptides. Brain Res. 1999;848 (1–2):45–62. doi: 10.1016/s0006-8993(99)01909-5. [DOI] [PubMed] [Google Scholar]
- Sherman MP, de Noronha CM, Pearce D, Greene WC. Human immunodeficiency virus type 1 Vpr contains two leucine-rich helices that mediate glucocorticoid receptor coactivation independently of its effects on G(2) cell cycle arrest. J Virol. 2000;74 (17):8159–8165. doi: 10.1128/jvi.74.17.8159-8165.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MP, de Noronha CM, Heusch MI, Greene S, Greene WC. Nucleocytoplasmic shuttling by human immunodeficiency virus type 1 Vpr. J Virol. 2001;75 (3):1522–1532. doi: 10.1128/JVI.75.3.1522-1532.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman MP, De Noronha CM, Williams SA, Greene WC. Insights into the biology of HIV-1 viral protein R. DNA Cell Biol. 2002a;21(9):679–688. doi: 10.1089/104454902760330228. [DOI] [PubMed] [Google Scholar]
- Sherman MP, Schubert U, Williams SA, de Noronha CM, Kreisberg JF, Henklein P, Greene WC. HIV-1 Vpr displays natural protein-transducing properties: implications for viral pathogenesis. Virology. 2002b;302 (1):95–105. doi: 10.1006/viro.2002.1576. [DOI] [PubMed] [Google Scholar]
- Steiner DF. The proprotein convertases. Curr Opin Chem Biol. 1998;2 (1):31–39. doi: 10.1016/s1367-5931(98)80033-1. [DOI] [PubMed] [Google Scholar]
- Stewart SA, Poon B, Jowett JB, Chen IS. Human immunodeficiency virus type 1 Vpr induces apoptosis following cell cycle arrest. J Virol. 1997;71 (7):5579–5592. doi: 10.1128/jvi.71.7.5579-5592.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3 (10):753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonov I, Ruckwardt TJ, Berg S, Hatfield GS, David Pauza C. Furin cleavage of the HIV-1 Tat protein. FEBS Lett. 2004;565 (1–3):89–92. doi: 10.1016/j.febslet.2004.03.079. [DOI] [PubMed] [Google Scholar]
- Tristem M, Purvis A, Quicke DL. Complex evolutionary history of primate lentiviral vpr genes. Virology. 1998;240 (2):232–237. doi: 10.1006/viro.1997.8929. [DOI] [PubMed] [Google Scholar]
- Tsuji A, Hashimoto E, Ikoma T, Taniguchi T, Mori K, Nagahama M, Matsuda Y. Inactivation of proprotein convertase, PACE4, by alpha1-antitrypsin Portland (alpha1-PDX), a blocker of proteolytic activation of bone morphogenetic protein during embryogenesis: evidence that PACE4 is able to form an SDS-stable acyl intermediate with alpha1-PDX. J Biochem (Tokyo) 1999;126 (3):591–603. doi: 10.1093/oxfordjournals.jbchem.a022491. [DOI] [PubMed] [Google Scholar]
- Tsuji A, Sakurai K, Kiyokage E, Yamazaki T, Koide S, Toida K, Ishimura K, Matsuda Y. Secretory proprotein convertases PACE4 and PC6A are heparin-binding proteins which are localized in the extracellular matrix. Potential role of PACE4 in the activation of proproteins in the extracellular matrix. Biochim Biophys Acta. 2003;1645 (1):95–104. doi: 10.1016/s1570-9639(02)00532-0. [DOI] [PubMed] [Google Scholar]
- Varin A, Decrion AZ, Sabbah E, Quivy V, Sire J, Van Lint C, Roques BP, Aggarwal BB, Herbein G. Synthetic Vpr protein activates activator protein-1, c-Jun N-terminal kinase, and NF-{kappa}B and stimulates HIV-1 transcription in promonocytic cells and primary macrophages. J Biol Chem. 2005;280 (52):42557–42567. doi: 10.1074/jbc.M502211200. [DOI] [PubMed] [Google Scholar]
- Vodicka MA, Koepp DM, Silver PA, Emerman M. HIV-1 Vpr interacts with the nuclear transport pathway to promote macrophage infection. Genes Dev. 1998;12 (2):175–185. doi: 10.1101/gad.12.2.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Duus KM, Friedrich TD, de Noronha CM. The HIV1 protein VPR acts to promote G2 cell cycle arrest by engaging a DDB1 and cullin4A containing ubiquitin ligase complex using VPRBP/DCAF1 as an adaptor. J Biol Chem. 2007 doi: 10.1074/jbc.M703955200. [DOI] [PubMed] [Google Scholar]
- Yao XJ, Subbramanian RA, Rougeau N, Boisvert F, Bergeron D, Cohen EA. Mutagenic analysis of human immunodeficiency virus type 1 Vpr: role of a predicted N-terminal alpha-helical structure in Vpr nuclear localization and virion incorporation. J Virol. 1995;69 (11):7032–7044. doi: 10.1128/jvi.69.11.7032-7044.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao XJ, Mouland AJ, Subbramanian RA, Forget J, Rougeau N, Bergeron D, Cohen EA. Vpr stimulates viral expression and induces cell killing in human immunodeficiency virus type 1-infected dividing Jurkat T cells. J Virol. 1998;72 (6):4686–4693. doi: 10.1128/jvi.72.6.4686-4693.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Gelbard HA, Roshal M, Pursell S, Jamieson BD, Planelles V. Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J Virol. 2001;75 (8):3791–3801. doi: 10.1128/JVI.75.8.3791-3801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]