

Abstract
Rhodopsins (Rh) are G-protein-coupled receptors that function as light sensors in photoreceptors. In humans, mutations in Rhodopsins cause retinitis pigmentosa, a degenerative disease that ultimately results in blindness. Studies in Drosophila have provided many insights into basic Rhodopsin biology and identified pathways that lead to retinal degeneration. It has been shown that because Rhodopsin is very abundant in photoreceptors, its accumulation in numerous organelles induces severe stress and results in degeneration of these cells. Moreover, genetic lesions that affect proper activation of membrane-bound Rh lead to disruption in Ca2+ homeostasis which also causes photoreceptor degeneration. Here, we review the molecular signals involved in Rhodopsin homeostasis and the mechanisms underlying retinal degeneration in flies, and discuss possible links to human diseases.
Introduction
Retinitis pigmentosa (RP), which affects ~1/4,000 individuals, is the most common form of retinal degeneration [1]. The symptoms start with night vision loss, followed by tunnel vision and eventually result in complete loss of vision. RP is typically hereditary and often autosomal dominant (ADRP). Over 120 mutations in Rhodopsin (Rh) have been associated with RP, accounting for ~25% of all cases [2]. Moreover, mutations in some other RP genes, such as PRPF31, TULP1 and Myosin VIIA, also affect Rh dynamics [3–5]. Hence, dissecting Rh function and homeostasis is central to understanding the pathology of RP. The Drosophila eye has been used to study Rh biology and photoreceptor degeneration [6, 7]. The compound eye consists of ~800 ommatidia, each of which contains 8 photoreceptors R1-R8 (Figure 1A & B). Each photoreceptor contains a microvillar structure of stacked membranes, the rhabdomere, where phototransduction proteins reside (Figure 1B). The rhadbdomeres are light-sensing organelles and functional equivalents of outer segments of mammalian cones and rods. In flies, retinal degeneration can be assessed by examining rhabdomere morphology (Figure 1C) or photoresponses (Figure 1D). Numerous mutations that cause retinal degeneration have been identified in screens using these assays.
Figure 1. Drosophila eye morphology and degeneration.

A. A scanning electron microscope picture of the Drosophila compound eye. Each eye consists of ~800 ommatidia, each of which contains 8 photoreceptors (R1-R8).
B. A transmission electron microscope picture of a cross section of an ommtidium. Since R7 reside on top of R8, in each section only seven photoreceptors are visible. Showing here is a section of R1-R7. Rhabdomeres (shown as round black structures in the picture) are the photosensing organelle that consists of densely stacked membranes supported by actin filaments.
C. An example of degenerated photoreceptors. Note that the rhabdomere morphology is severely disrupted.
D. Electroretinogram (ERG) can be used to assess retinal degeneration. Shown on the left is a normal response of fly eye when exposed to a brief light stimulation (typically 1 second); shown on the right is the response of a degenerated eye when exposed to the same light stimulation. Note that the amplitude of the ERG is largely reduced. Please refer to [94] for further details regarding the ERG recordings.
Rhs are evolutionarily conserved light sensors from Cnidaria to human [8]. In Drosophila, Rh1 is the major light sensor that is abundant in R1-R6 (Figure 2). Similar to vertebrate Rh, Drosophila Rh1 consists of a seven transmembrane protein, opsin [9, 10] and a chromophore, 11-cis-3-hydroxyretinal [11]. Upon photon absorption, 11-cis-retinal is isomerized to all-trans-retinal, inducing a conformational change of the opsin to produce metarhodopsin (Mrh) [12]. Mrh activates a G-protein-alpha subunit which activates phospholipase C (PLC) [13]. PLC hydrolyzes phosphatidylinositol 4,5-bisphosphate (PIP2) to produce diacylglycerol (DAG). DAG or its metabolites may trigger the opening of TRP channels and depolarization of photoreceptors [14–16] although recent evidence suggests that a decrease in PIP2 and the subsequent acidification also contribute to TRP activation [17, 18]. In the vertebrate eye, a similar phototransduction cascade has been identified in intrinsically photosensitive retinal ganglion cells, which function in circadian rhythm and iris reflex [19, 20]. Indeed, in these cells, melanopsin, a homolog of Drosophila Rh1, functions as a light sensor [21]. Moreover, a PLC and two TRP channels are required for the phototransduction cascade [22]. Although the light sensors in cones and rods are Rhs, the downstream events are different. In the dark, cones and rods are depolarized due to the opening of cGMP-gated cation channels. Light activation of Rh leads to activation of a phosphodiesterase, that hydrolyzes cGMP and causes closure of the cation channels, causing repolarization of these cells [23].
Figure 2. Phototransduction in Drosophila.
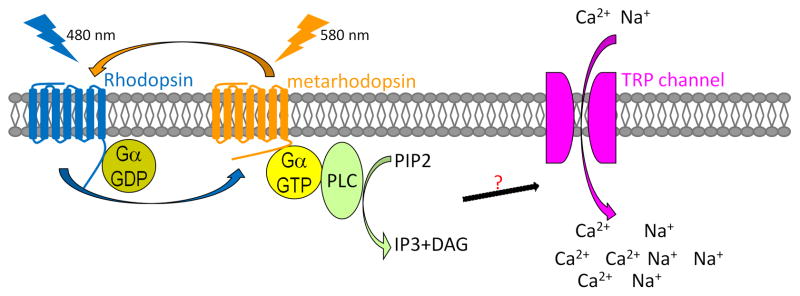
Upon absorption of a blue photon (480 nm), Rhodopsin undergoes a conformational change to the active form, metarhodopsin. Metarhodopsin activates G protein alpha subunit, which in turn activates phospholipase C (PLC). PLC hydrolyzes PIP2 to IP3 and DAG. PLC activation then leads to the opening of TRP channels and the influx of Ca2+ and Na+ into photoreceptor cells. Upon absorption of a second photon (580 nm), metarhodopsin is converted back to Rhodopsin and the response is terminated.
A cycle of Rh1 production and turnover has been unraveled in flies [6] (Figure 3). Rh1 is synthesized and folded in the ER and transported to rhabdomeres via vesicular trafficking through the Golgi. During its maturation, Rh1 undergoes glycosylation and deglycosylation. Upon binding to the chromophore, Rh1 is further folded and matures into its final conformation. In rhabdomeres, Rh1 functions as a light sensor and cycles between its inactive (Rh) and active form (Mrh). Moreover, a small portion of Rh1 is internalized and degraded upon light exposure. Hence, Rh1 needs to be continously produced and transported to the rhabdomeres to maintain homeostasis upon light stimulation. Here, we will discuss how disruption of maturation, transport, activation and degradation of Rh1 leads to retinal degeneration based on Drosophila research. We also explore the correlation between findings in Drosophila and mechanisms underlying retinitis pigmentosa in human.
Figure 3. Production and degradation of Rhodopsin.

Left panel. During development of photoreceptor cells, Rh1 is synthesized and folded in the ER. Two chaperones, NinaA and Calnexin, are required for Rh1 folding and its secretion from the ER. Rh1 is then transported to the Golgi through a Rab1 mediated process. In the Golgi, Rh1 is further processed and matured. Rh1 transport between different Golgi compartments is dependent on Rab6. Mature Rh1 is secreted from the Golgi and transported to the rhabdomeres via Rab11 mediated vesicular trafficking.
Right panel. In adult flies, Rh1 functions as light sensor and recycles in the rhabdomeres between an inactive (Rh) and active form (Mrh). Upon light exposure, a small portion of Rh1 is internalized and degraded through the endolysosomal pathway or autophagy pathway. Hence, upon light exposure, Rh1 needs to be constantly produced and transported to the rhabdomeres via a Crag/Rab11 dependent process to maintain its level.
Rhodopsin synthesis and maturation
Rh synthesis and maturation are essential for photoreceptors. Besides its light-sensing function, Rh1 is required for maintaining the structure of rhabdomeres. Rh1 is an essential component of the rhabdomere terminal web (RTW), an F-actin meshwork that provides a mechanical force to support the rhabdomeric membranes and mediates vesicular trafficking towards rhabdomeres [24, 25]. Hence, complete loss of Rh1 leads to collapse of rhabdomeres and partial loss of Rh1 leads to formation of smaller rhabdomeres [26–29]. Maturation of Rh encompasses a series of steps of post-translational modification, whose disruption can lead to accumulation of Rh in corresponding organelles. Since Rh is abundant, its aberrant accumulation in organelles usually leads to retinal degeneration. However, the consequences of Rh accumulation in different compartments dramatically affect the nature and speed of degeneration, a topic that we discuss here.
Transcription of Drosophila Rh1 is controlled by Eyeless/Pax-6 [30]. Upon translation, folding of Rh1 requires chaperones, including NinaA, Calnexin and XPORT. NinaA is a homolog of a vertebrate cyclophilin, which functions as a peptidylprolyl cis-trans-isomerase [31, 32]. NinaA is predominantly localized to the ER and secretory vesicles and forms a complex with Rh1 to promote its maturation and transport [33–35]. Mutations in ninaA cause a severe reduction of Rh1 levels, accumulation of immature Rh1 in the ER and ER expansion. Interestingly, photoreceptor degeneration is not obvious in ninaA mutants as determined by rhabdomere morphology [36]. We therefore propose that the photoreceptors are able to degrade immature Rh1 through an unfolded protein response (UPR) and ER-associated degradation (ERAD) to prevent degeneration.
Unlike ninaA, mutations in Calnexin, another chaperone located in the ER that promotes folding of Rh1 in flies, cause a light-enhanced retinal degeneration. Loss of Calnexin leads to an accumulation of immature Rh1 in the ER and reduced Rh1 levels in rhabdomeres similar to loss of NinaA. In the dark, only a subtle retinal degeneration is observed in Calnexin mutants, but the degeneration is much enhanced by light [36]. Besides its chaperone function, Calnexin also functions to buffer Ca2+ [36]. In a double mutant of PLC and Calnexin, in which the Ca2+ influx is blocked, the light-induced degeneration rate is much slower than in the single Calnexin mutants. These results indicate that defects in Ca2+ buffering contribute to photoreceptor degeneration in calnexin mutants when exposed to light.
XPORT functions as a chaperone for both Rh1 and TRP, and loss of xport leads to a light-enhanced retinal degeneration [37]. In the xport mutant, both Rh1 and TRP levels are reduced, and the remaining proteins are mostly accumulated in the cytosol. In the dark, xport mutant photoreceptors exhibit ER accumulations and Golgi dilation, however rhabdomere morphology is preserved. Upon light exposure, xport mutants undergo retinal degeneration. Since loss of trp causes a light-induced retinal degeneration, whereas reduction of Rh1 levels does not cause degeneration, the degeneration of xport photoreceptors is likely due to loss of TRP in rhabdomeres and a disruption of Ca2+ influx, which will be discussed later.
In humans, mutations in Rh that affect folding usually cause ADRP. Expression of the most common human Rh1 mutant, P23H (P37H in Drosophila) in wild type photoreceptors leads to an accumulation of Rh1 in ER and age-dependent retinal degeneration [38]. Fly geneticists also performed screens to isolate dominant mutations in Rh1 that cause photoreceptor degeneration [39, 40]. Interestingly, some amino acid changes in dominant Rh1 mutants are identical to those found in ADRP patients, including G119E, P184L, E194K and G195S [39]. Surprisingly, none of the dominant Rh1 mutations cause degeneration in homozygous mutants, although Rh1 is dramatically reduced [39, 40]. Thus, these data indicate that degeneration of photoreceptors is dependent on the existence of both wild-type and mutant Rh1. This implies that mutant and wild-type Rh1s form a complex that is resistant to degradation and is toxic to the photoreceptors. Indeed, boosting ERAD by overexpression of Hrd1, an ERAD-associated E3 ubiquitin-protein ligase, or EDEM2 (ER degradation enhancer mannosidase alpha-like 2) reduces mutant Rh1 levels in dominant Rh1 mutants and delays retinal degeneration [41], providing a potential strategy for treating RP.
Glycosylation promotes proper folding of various proteins. Rh1 is glycosylated at Asn20 in the ER at NXS/T site, which is an essential determinant for N-linked protein glycosylation [42, 43]. Mutations of Asn20 lead to a reduction of the Rh1 levels, accumulation of Rh1 in the ER, and eventually degeneration of photoreceptors [42, 44, 45]. Since this glycosylation is required for the binding of Rh1 to Calnexin but not to NinaA [36, 46], it’s possible that the binding of unglycosylated Rh1 to NinaA prevents an efficient UPR and ERAD in the ER and causes an accumulation of immature Rh1 and degeneration. In vertebrates, N-glycosylation on Asn15 has also been shown to be required for stability of Rhodopsin [47]. A missense mutation which disrupts the NXS/T consensus sequence also causes ADRP in patients [48]. These findings suggest that N-glycosylation is a conserved process that is required for the maturation of Rhodopsin.
Mature Rh1 is not N-glycosylated, indicating that sugars are removed in the Golgi. Indeed, a metallophosphoesterase, dMPPE, is invovled in the deglycosylation of Rh1 [49]. dMPPE functions as a Mn2+/Zn2+-dependent phosphoesterase and mediates the dephosphorylation and activation of a Golgi glycoprotein mannosidase, alpha-Man-II, which is required for the removal of the Rh1 oligosaccharide chain. Interestingly, defects in deglycosylation do not affect Rh1 localization or signaling. However, loss of dMPPE leads to enhanced degradation of endocytosed Rh1 and light-dependent retinal degeneration. Therefore, deglycosylation of Rh1 is required for its stability.
An important step of Rh1 maturation is linking opsin to the chromophore. Defects in chromophore biosynthesis affect Rh1 deglycosylation and transport, and lead to a reduction of Rh1 levels [50]. The biosynthetic pathway of the chromophore is summarized in [6]. A recent study shows that Neurexin I, a cell adhesion molecule involved in synapse formation, forms a complex with retinoid- and fatty-acid-binding protein apolipoprotein I, which is required for the transport of 11-cis-retinal from the pigment cells to photoreceptors [51]. Mutations that affect chromophore biosynthesis lead to a severe reduction of Rh1 as well as smaller rhabdomeres [48]. However, the photoreceptors do not degenerate. Hence, opsins that lack the chromophore can be efficiently degraded to prevent ER stress.
In summary, accumulation of unfolded Rh1 usually leads to degeneration of the photoreceptors. Degradation of immature Rh1 seems to serve as a protective mechanism and prevent degeneration. ERAD may be one of the most effective ways to degrade unfolded Rh1 without causing photoreceptor degeneration. Although Rh1 is essential for photoreceptors, a reduction of misfolded Rh1 levels is beneficial for photoreceptor integrity.
Transport of Rh1 between different compartments
Rh1 is transported to the rhabdomeres through the secretory pathway, and many factors are involved in this process (Figure 3). One of these, Arf72A, a small GTPase, functions as a quality control checkpoint for Rh1-bearing vesicles when they bud from the ER [52]. Loss of Arf72A in Rh1 dominant mutants reduces Rh1 accumulation in the ER and delays retinal degeneration. Rab proteins are small GTPases that control membrane trafficking processes and several Rabs also regulate Rh1 trafficking. Indeed, Rab1 is required for transport of Rh1 from the ER to the Golgi [53] and expression of a dominant negative Rab1 (DN-Rab1) leads to a swollen rough ER, accumulation of glycoslated Rh1, and degeneration of photoreceptor [53]. These findings indicate that the accumulation of unfolded Rh1 is the primary cause of retinal degeneration in these mutants.
Sorting of Rh1 between the Golgi compartments is dependent on Rab6 [54]. Expression of a dominant negative Rab6 in photoreceptors leads to an accumulation of glycosylated Rh1, a reduction of mature Rh1 levels, formation of smaller rhabdomeres and eventually degeneration of photoreceptors. Hence, accumulation of immature Rh1 in the Golgi also induces retinal degeneration. A recent study shows that glycosylphosphatidylinositol (GPI) and some GPI-anchored proteins are required for Rh1 sorting in the trans-Golgi [55]. Loss of a GPI-transamidase subunit leads to mis-sorting of Rh1 into the endolysosomal pathway upon its secretion from the trans-Golgi, which leads to a breakdown of rhabdomeres. Hence, proper sorting of Rh1 in the Golgi is critical for photoreceptor development and maintenance as well.
The transport of Rh1-bearing-post-Golgi vesicles to rhabdomeres is mediated by Rab11 [56] (Figure 3). During development, inhibition of Rab11 activity leads to defects in rhabdomere formation and accumulation of Rh1-bearing vesicles in the cytosol. MyosinV and dRip11 form a complex with Rab11 and loss of MyosinV or dRip11 leads to similar phenotypes as loss of Rab11 [57]. It was proposed that this complex delivers Rh1-bearing vesicles to rhabdomeres along actin filaments. The delivery of Rh1-bearing vesicles is also dependent on the exocyst complex, which may be a Rab11 effector, as loss of Sec6 leads to accumulation of Rh1 in the cytosol [58]. Therefore, Rab11 regulates both the transport and fusion of Rh1-bearing vesicles to the rhabdomere membrane. Loss of Rab11 causes accumulation of Rh1 in the cytosol and loss of Rh1 in the rhabdomeres, which are probably at the root of the demise of the photoreceptors [56].
In humans, mutations in crumbs cause RP [59, 60]. Crumbs is one of the major components of Crumbs complex, that regulates cell junction formation and cell polarity in flies [61]. It was also shown that crumbs is involved in stabilizing the adherens junctions between photoreceptors and Müller cells in mammalian retina [62]. Recent studies in Drosophila show that Crumbs interacts and stabilizes myosin V thereby promoting Rh1 trafficking to the rhabdomeres [63]. Loss of crumbs causes a severe reduction in MyosinV levels and an age-dependent cytoplamic accumulation of Rh1 and retinal degeneration [63, 64]. Whether Crumbs is also required for Rh1 transport in vertebrates has not yet been established.
In summary, mutations that affect the transport of Rh1 generally lead to retinal degeneration. However, when Rh1 maturation is affected and Rh1 levels are reduced, retinal degeneration is mild or absent. We therefore propose that photoreceptor can be preserved when misfolded Rh1 are degraded through UPR and ERAD. However, accumulation of properly folded Rh1 in the secretory pathway, which cannot be efficiently removed, leads to a severe retinal degeneration.
Rhodopsin cycles upon light stimulation
Light stimulation induces isomerization of the chromophore thereby inducing a conformational change of Rh1 to Mrh (Figure 4) In vertebrate cones and rods, light-activated chromophore is released from the opsins and recycled via an enzymatic pathway [65]. However, Drosophila Mrh is thermally stable and can be converted to Rh1 upon absorption of another photon [12]. This recycling process is critical for both function and maintenance of photoreceptors.
Figure 4. Rhodopsin cycle upon light exposure.

Rhodopsin (blue) is converted into metarhodopsin (orange) upon absorption of a photon (480nm). Metarhodopsin is subsequently phosphorylated by GPRK1and deactivated upon Arr2 binding. Upon absorption of a second photon, metarhodopsin is converted back into Rhodopsin. Arr2 is then phosphorylated by CaMKII and released from Rhodopsin. Finally, Rhodopsin is dephosphorylated by RdgC.
The recycling of Rh1 can be roughly divided into two parts, inactivation of Mrh and regeneration of Rh1. Following activation, Mrh is rapidly phosphorylated by a G-protein-coupled receptor kinase GPRK1 in a cluster of serine-threonine residues in its C-terminal [66–68]. This phosphorylation is arguably an inhibitory feedback as lower GPRK1 activity leads to larger amplitudes of light responses. Arrestin2 (Arr2) then binds Mrh to terminate the photoresponse by preventing Mrh from activating more Gqα[69]. Loss of Arr2 leads to a prolonged deactivation time. Interestingly, Arr2 is mostly localized in the cytoplasm in dark raised animals, and is translocated to rhabdomeres upon light exposure [70, 71]. In dim light, low Arr2 levels in rhabdomeres enhance the photoreceptor sensitivity, whereas in strong light, high Arr2 levels in rhabdomeres prevent hyperactivity of photoreceptors. Loss of Arr2 leads to a light-dependent retinal degeneration possibly due to an overload of Ca2+, which is toxic to cells and may induce apoptosis [72]. Regeneration of Rh1 is initiated by absorption of another photon (580 nm), which induces a conformational change of Mrh to Rh1. Calmodulin-dependent kinase II (CaMKII) phosphorylates Arr2 and promotes the dissociation of Arr2 from Rh1 [73, 74]. Once Arr2 is released, Retinal degeneration C (RdgC) dephosphorylates Rh1 [75–77]. It was proposed that loss of rdgC leads to hyperphosphorylation of Rh1 and prolonged deactivation time leading to degeneration of photoreceptors. However, based on intracellular recordings, the deactivation time of rdgC photoreceptor is indistinguishable from wild-type [78]. It is therefore possible that hyperphosphorylated Rh1 forms a stable complex with Arr2 that causes degeneration, a topic discussed below. Upon dephosphorylation, Rh1 is regenerated and ready to sense the next photon. In summary, this recycling process is required for the rapid termination of photoresponse and maintenance of the proper Rh1 pool in the rhabdomeres.
The light induced Ca2+ influx plays pivotal roles in Rh1 recycling by modulating the activity of at least three players [79]. Ca2+ influx promotes Arr2 translocation from the cytoplasm to the rhabdomeres, which is critical for the termination of Mrh activity [71]. Calmodulin (CaM) functions as a Ca2+ sensor and regulates the activity of various factors. Ca2+/CaM activates CaMKII to promote the release of Arr2 from Rh1 [74]. Ca2+/CaM also activates RdgC to promote the dephosphorylation of Rh1 [80]. Therefore, CaM is required for the regeneration of Rhodopsin. Furthermore, Ca2+/CaM also activates the transcription factor dCAMTA (calmodulin binding transcription activator) to promote production of dFBX14, a potential F-box containing E3 ubiquitin ligases that is proposed to reduce Rh1 activity through ubiquitination [81]. Loss of dCAMTA leads to a slow termination of the light response, which can be rescued by overexpression of dFBX14 [81]. In summary, light induced Ca2+ influx and CaM activation facilitates the termination of the light response and regeneration of Rh1.
Mutations in genes that are involved in the Rh1 cycle, especially those involved in the termination of Mrh activity, including arr2, rdgC and dCAMTA, typically cause light-dependent retinal degeneration [69, 75, 81]. In the dark, the photoreceptors are not affected since recycling of Rh1 is not required. A possible mechanism underlying the degeneration is the overload of Ca2+ influx, which is toxic to cells and may induce apoptosis [72]. In some cases, where Mrh cannot be converted back to Rhodopsin, excessive endocytosis of Mrh may also contribute to the degeneration, which is discussed in the next section.
Rhodopsin internalization, degradation and replenishment
Under normal physiological conditions, a fraction of Mrh is endocytosed upon light activation [82] (Figure 3). It is proposed that this process scavenges damaged or constitutively active Mrh to prevent photoreceptor hyperactivity. Endocytosed Mrh is then degraded and newly synthesized Rh is transported to the rhabdomeres to compensate for the loss. Therefore, endocytosis and exocytosis of Rh are both required for Rh1 homeostasis upon light exposure.
The mechanism of Mrh endocytosis is not fully understood yet. However it is known that C-terminal phosphorylation of Mrh triggers its endocytosis and that Arr1 and Arr2 are involved. Arr1 binds to phosphorylated Mrh and initiates endocytosis [82]. Hence, mutations in Arr1 lead to a reduction of endocytosis of Mrh. Arr2 forms a stable complex with phosphorylated Mrh and also promotes its endocytosis [83]. Arr2 binds to AP-2, an adaptor protein involved in clathrin-mediated endocytosis, which is required for Mrh endocytosis [84]. Since the Mrh C-terminal phosphorylation is a transient process and persists only when Mrh cannot be converted back to Rh1, it was proposed that Arr1/Arr2 scavenges damaged or constitutively active Mrh. Indeed, disrupting the binding between Arr2 and AP-2 reduces Mrh endocytosis and induces a light-dependent retinal degeneration [84]. However, loss of arr1 leads to a light-independent retinal degeneration [82], suggesting that Arr1 mediates other functions than Mrh endocytosis. Similarly, inhibition of Mrh endocytosis using a temperature-sensitive Dynamin (Shibirets) or a dominant negative Rab5 induces retinal degeneration [24]. However, in some contexts, inhibiton of endocytosis delays degeneration, an issue that will be discussed later. In summary, these findings support an important role for endocyosis of Mrh in photoreceptor maintenance. Impairing the process leads to accumulation of defective Mrh in the rhabdomeres and enhanced signaling, and eventually photoreceptor degeneration.
Upon internalization, Rh1 is degraded to prevent its accumulation in the cytoplasm (Figure 3). The majority of Rh1 is degraded through the endolysosomal pathway, and impairing this process leads to photoreceptor degeneration. Indeed, mutations in carnation, carmine, light, synaptobrevin or fatp, which are all involved in endolysosomal trafficking, cause endosomal accumulation of Rh1 and light-dependent retinal degeneration [85–88]. Moreover, mutation in Sunglasses, a tetraspanin protein localized in late endosomes and lysosomes that directly interacts with Rh1 and promotes its trafficking, also causes a light-dependent degeneration [89]. Collectively, the data indicate that the endolysosomal pathway is required for the degradation of Rh1 and defects in this pathway usually lead to a severe light-dependent retinal degeneration.
In addition to lysosomal degradation, the autophagy pathway has also been implicated in the clearance of endocytosed Mrh. Inducing autophagy by inhibiting TOR signaling suppresses the retinal degeneration associated with PLC mutants in which Mrh is excessively endocytosed whereas suppressing autophagy by promoting TOR activity leads to retinal degeneration in wild type photoreceptors [90]. Similarly, loss of essential autophagy components, such as atg7 and atg8, causes accumulation of Rh1 in the endosomes and retinal degeneration [91]. These results support a protective role of autophagy in photoreceptors by promoting the degradation of Rhodpsin in flies. In vertebrates, an autophagy pathway is required for the degradation of the outer segments in the retinal pigment epithelium. This process is required for the maintenance of the proper chromophore levels in photorepetrors [92].
Although a basal level of endocytosis is necessary, excessive endocytosis of Mrh is highly toxic to photoreceptors. An extreme example is the rapid retinal degeneration induced by blue light stimulation. Blue light converts Rh1 to Mrh, and Mrh requires orange light to be converted back into Rh1. In the absence of orange light, blue light coverts Rh1 to Mrh and triggers massive endocytosis of Mrh [93, 94]. Loss of Rh1 from the rhabdomeres causes breakdown of the membranes, whereas endocytosed Mrh stresses the endolysomal pathway. Hence, prolonged exposure to blue light leads to retinal degeneration. In norpA or trp mutants, where Rh1 can still be converted to Mrh upon light exposure but the Ca2+ influx is not induced, the lack of the Ca2+ influx blocks the dephosphorylation of Mrh and induces formation of stable Mrh-Arr2 complex [70, 83, 95]. Hence, in these mutants, accumulation of Mrh triggers massive endocytosis and retinal degeneration. Inhibition of Mrh endocytosis using shibirets or by removing Arr2 suppresses the degeneration in these mutants [83], indicating that excessive endocytosis of Mrh is the major cause of degeneration. Moreover, reducing Rh1 levels by raising flies on food that has severely reduced levels of β-carotene, which is the major precursor of 11-cis-retinal and critical for Rh1 synthesis [96], also suppresses the degeneration associated with the loss of norpA or trp, suggesting that the endolysosomal pathway is unable to cope with high levels of endocytosed Mrh in these mutants. However, aberrant trafficking of Rh1 may not be the only cause of photoreceptor degeneration in these mutants [97]. Interestingly, in a mouse model of ADRP, RhK296E forms a stable complex with Arrestin and accumulates in the inner segment of the photoreceptors [98], suggesting that endocytosis of abnormally formed Rhodopsin/Arrestin complex maybe a conserved mechanism in retinal degeneration.
In vertebrate photoreceptors, the chromophore is released from the opsins and recycled through an enzymatic pathway after each cycle of Rhodopsin activation [65]. In Drosophila, although the majority of all-trans-retinal is converted back to all-cis-retinal in the rhabdomeres, recent studies show that all-trans-retinal can be released from opsin upon degradation of internalized Mrh and all-cis-retinal can be regenerated. At least two enzymes, Pigment cell dehydrogenase (PDH) and Retinal dehydrogenase (RDHB), are invovled in this process [99, 100]. When flies are raised in food supplemented with β-carotene, loss of either pdh or rdhb does not affect de novo synthesis of the chromophore during development. However, their loss leads to reduction of chromophore and Rh1 levels in the adult stage upon light stimulation. Moreover, when wild-type flies are kept on food deprived of β-carotene starting in adulthood, they can maintain their Rh1 levels for several weeks in a light-dark cycle. These data support the idea that recycling instead of de novo synthesis of the chromophore is required for its homeostasis in adults. When the chromophore levels are reduced during development, small rhabdomeres are formed, but the photoreceptors do not degenerate. However, loss of either pdh or rdhb leads to a light-dependent retinal degeneration in adults [99, 100]. We therefore propose that when photoreceptors develop normally and the rhabdomeres expand to their normal size, the Rh1 levels must be maintained to support rhabdomere structure in adult flies. In pdh or rdhb mutant, the gradual reduction of Rh1 levels leads to a breakdown of the rhabdomeres and excessive internalization of rhabdomere membranes and actin filaments. These in turn stress the endolysosomal pathway and cause cell death. These data also indicate that the levels of the chromophore are regulated via different mechanisms during developmental and adult stages.
Internalization of Mrh leads to a gradual decrease of Rh1 levels in the rhabdomere upon light stimulation. Hence, the rhabdomeric Rh1 pool needs to be replenished. Consistent with this model, transcription of Rh1 is maintained at a basal level during the light dark cycles [101], indicating that Rh1 is continuously synthesized to offset its loss by endocytosis and degradation. A recently study shows that Crag, a DENN domain containing protein that functions as a GEF for Rab11, is required for the transport of Rh1-bearing-post-Golgi vesicles to the rhabdomeres upon light stimulation [94]. Loss of Crag leads to a light-induced accumulation of newly synthesized Rh1 in the cytoplasm and eventually leads to photoreceptor degeneration. Crag was shown to bind to CaM in a Ca2+ dependent manner and CaM binding enhances the GEF activity of Crag in an in vitro assay [94, 102]. It is therefore possible that upon photoactivation, the Ca2+ influx activates CaM which in turn activates Crag and promotes the transport of Rh1 to the rhabdomeres to maintain the Rh1 levels.
Concluding remarks
In Drosophila photoreceptors, mutations that disrupt the homeostasis of Rh1 often lead to retinal degeneration. One of the main reasons may relate to the fact that Rh1 is one of the most abundant proteins in photoreceptors, suggesting that their demise may occur because of mistrafficking and accumulation of Rh1 in some subcellular compartments. Accumulation of Rh1 in the secretory pathway or endolysomal pathway usually leads to a degeneration of photoreceptors, most likely because the levels of Rh1 exceed the capacity of these trafficking pathways. Interestingly, in many cases, defects in the maturation of Rh1 in the ER do not cause an obvious degenerative phenotype, indicating that the UPR and ERAD response are able to cope relatively well with the unfolded Rh1 to relieve ER stress.
Another key player is intracellular Ca2+ homeostasis. Elevated Ca2+ levels are highly toxic. However, low Ca2+ levels are also detrimental as they induce massive endocytosis of Rh1 because of the dysregulation of the recycling of Mrh into Rh, resulting in an accumulation of Mrh in the endolysomal pathway. Since the Ca2+ influx is induced by light exposure, mutants that affect Ca2+ homeostasis usually cause a light dependent retinal degeneration. One or more of these defects may be responsible for the retinal degenerations observed in different mutants.
There are both similarities and differences between the Rh-associated photoreceptor degenerations observed in flies and humans. Maturation defects of Rh cause photoreceptor degeneration in both systems. Moreover, mutations in several key amino acids, which are conserved and essential for proper folding of Rh, lead to photoreceptor degeneration in both flies and humans. However, specific chaperones that are required for Rh folding may not be conserved, for instance Calnexin is not required for rod opsin biogenesis [103]. Unfortunately, the pathways and players that regulate the deposition of vertebrate Rh in rods and cones are poorly characterize, although some factors that are involved in Rh trafficking in flies have been implicated in the same process in vertebrates, including Rab11 [104]. Although the process of recycling between Rh and Mrh is not conserved in vertebrate rods and cones,, some of the factors involved in this process in flies, such as Arr2, have also been implicated in the regulation of photoactivation in vertebrates. Finally, some components of the autophagy pathway are found to be required for the maintenance of visual function in both flies and vertebrates, although the cellular process may be different. In summary, studies in flies have provided a molecular framework that allows a better understanding of the mechanisms underlying some human retinal degeneration diseases as many of the mechanisms underlying retinal degeneration seem to be conserved between flies and human.
Outstanding Questions.
Past studies in flies with viable mutants have provided a basic understanding of the molecular framework underlying the visual transduction pathway. However, it is likely that numerous essential genes are also involved. To systematically identify potentially important regulators of Rhodopsin function systematic ERG screens based on clonal analysis of essential genes may be the most effective methods to isolate these players.
It is known that a portion of Rhodopsin is internalized and degraded in the lysosome. Is there a recycling pathway to deliver endocytosed Rhodopsin back to the rhabdomere membrane?
How is transcription and translation of Rhodopsin regulated? Since the turnover rate of Rhodopsin is much high in the light than in the dark, it will be interesting to examine how light exposure promotes the production of Rhodopsin.
Although Rhodopsin trafficking has been studied in flies, how Rhodopsin is transported through different cellular compartment in vertebrates is mostly unknown. Can the observations in flies be translated in vertebrate rod and cone cells?
A common theme of Rhodopsin-induced retinal degeneration relates to aberrant accumulation of Rhodopsin in subcellular organelles. How can we develop specific treatments to promote the clearance of these proteins? Promoting the ERAD or autophagy pathway may lead to promising strategies.
Highlights.
Rhodopsin are universal light sensors and very abundant in photoreceptors
Disruption of Rhodopsin homeostasis cause retinal degeneration in flies and humans
Accumulation of Rhodopsin due to trafficking defects leads to severe cellular stress
Misregulation of intracellular Calcium homeostasis disrupts Rhodopsin cycle
Acknowledgments
We thank Dr. Kartik Venkatachalam, Dr. Manish Jaiswal and Shiuan Wang for discussion and suggestions. B.X. was supported by the Houston Laboratory and Population Science Training Program in Gene-Environment Interaction from Burroughs Wellcome Fund (BWF Grant No.1008200). This work was supported in part by the NIH (1RC4GM096355-01) to H.J.B. H.J.B. is an investigator of the HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hartong DT, et al. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- 2.Briscoe AD, et al. The spectrum of human rhodopsin disease mutations through the lens of interspecific variation. Gene. 2004;332:107–118. doi: 10.1016/j.gene.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 3.Liu X, et al. Myosin VIIa, the product of the Usher 1B syndrome gene, is concentrated in the connecting cilia of photoreceptor cells. Cell Motil Cytoskeleton. 1997;37:240–252. doi: 10.1002/(SICI)1097-0169(1997)37:3<240::AID-CM6>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 4.Yuan L, et al. Mutations in PRPF31 inhibit pre-mRNA splicing of rhodopsin gene and cause apoptosis of retinal cells. J Neurosci. 2005;25:748–757. doi: 10.1523/JNEUROSCI.2399-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grossman GH, et al. Immunocytochemical evidence of Tulp1-dependent outer segment protein transport pathways in photoreceptor cells. Exp Eye Res. 2011;93:658–668. doi: 10.1016/j.exer.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang T, Montell C. Phototransduction and retinal degeneration in Drosophila. Pflugers Arch. 2007;454:821–847. doi: 10.1007/s00424-007-0251-1. [DOI] [PubMed] [Google Scholar]
- 7.Montell C. Drosophila visual transduction. Trends Neurosci. 2012;35:356–363. doi: 10.1016/j.tins.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alvarez CE. On the origins of arrestin and rhodopsin. BMC Evol Biol. 2008;8:222. doi: 10.1186/1471-2148-8-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuker CS, et al. Isolation and structure of a rhodopsin gene from D. melanogaster. Cell. 1985;40:851–858. doi: 10.1016/0092-8674(85)90344-7. [DOI] [PubMed] [Google Scholar]
- 10.O’Tousa JE, et al. The Drosophila ninaE gene encodes an opsin. Cell. 1985;40:839–850. doi: 10.1016/0092-8674(85)90343-5. [DOI] [PubMed] [Google Scholar]
- 11.Goldsmith TH, et al. Separation and identification of geometric isomers of 3-hydroxyretinoids and occurrence in the eyes of insects. Vision Res. 1986;26:1763–1769. doi: 10.1016/0042-6989(86)90126-4. [DOI] [PubMed] [Google Scholar]
- 12.Kiselev A, Subramaniam S. Activation and regeneration of rhodopsin in the insect visual cycle. Science. 1994;266:1369–1373. doi: 10.1126/science.7973725. [DOI] [PubMed] [Google Scholar]
- 13.Scott K, et al. Gq alpha protein function in vivo: genetic dissection of its role in photoreceptor cell physiology. Neuron. 1995;15:919–927. doi: 10.1016/0896-6273(95)90182-5. [DOI] [PubMed] [Google Scholar]
- 14.Leung HT, et al. DAG lipase activity is necessary for TRP channel regulation in Drosophila photoreceptors. Neuron. 2008;58:884–896. doi: 10.1016/j.neuron.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hardie RC, Minke B. The trp gene is essential for a light-activated Ca2+ channel in Drosophila photoreceptors. Neuron. 1992;8:643–651. doi: 10.1016/0896-6273(92)90086-s. [DOI] [PubMed] [Google Scholar]
- 16.Montell C, Rubin GM. Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron. 1989;2:1313–1323. doi: 10.1016/0896-6273(89)90069-x. [DOI] [PubMed] [Google Scholar]
- 17.Hardie RC, Franze K. Photomechanical responses in Drosophila photoreceptors. Science. 2012;338:260–263. doi: 10.1126/science.1222376. [DOI] [PubMed] [Google Scholar]
- 18.Huang J, et al. Activation of TRP channels by protons and phosphoinositide depletion in Drosophila photoreceptors. Curr Biol. 2010;20:189–197. doi: 10.1016/j.cub.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 19.Pickard GE, Sollars PJ. Intrinsically photosensitive retinal ganglion cells. Rev Physiol Biochem Pharmacol. 2012;162:59–90. doi: 10.1007/112_2011_4. [DOI] [PubMed] [Google Scholar]
- 20.Schmidt TM, et al. Intrinsically photosensitive retinal ganglion cells: many subtypes, diverse functions. Trends Neurosci. 2011;34:572–580. doi: 10.1016/j.tins.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melyan Z, et al. Addition of human melanopsin renders mammalian cells photoresponsive. Nature. 2005;433:741–745. doi: 10.1038/nature03344. [DOI] [PubMed] [Google Scholar]
- 22.Xue T, et al. Melanopsin signalling in mammalian iris and retina. Nature. 2011;479:67–73. doi: 10.1038/nature10567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kefalov VJ. Rod and cone visual pigments and phototransduction through pharmacological, genetic, and physiological approaches. J Biol Chem. 2012;287:1635–1641. doi: 10.1074/jbc.R111.303008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinal N, Pichaud F. Dynamin- and Rab5-dependent endocytosis is required to prevent Drosophila photoreceptor degeneration. J Cell Sci. 2011;124:1564–1570. doi: 10.1242/jcs.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chang HY, Ready DF. Rescue of photoreceptor degeneration in rhodopsin-null Drosophila mutants by activated Rac1. Science. 2000;290:1978–1980. doi: 10.1126/science.290.5498.1978. [DOI] [PubMed] [Google Scholar]
- 26.Kumar JP, et al. Rhodopsin replacement rescues photoreceptor structure during a critical developmental window. Dev Biol. 1997;188:43–47. doi: 10.1006/dbio.1997.8636. [DOI] [PubMed] [Google Scholar]
- 27.Kumar JP, Ready DF. Rhodopsin plays an essential structural role in Drosophila photoreceptor development. Development. 1995;121:4359–4370. doi: 10.1242/dev.121.12.4359. [DOI] [PubMed] [Google Scholar]
- 28.O’Tousa JE, et al. Morphological defects in oraJK84 photoreceptors caused by mutation in R1-6 opsin gene of Drosophila. J Neurogenet. 1989;6:41–52. doi: 10.3109/01677068909107099. [DOI] [PubMed] [Google Scholar]
- 29.Leonard DS, et al. Degeneration of photoreceptors in rhodopsin mutants of Drosophila. J Neurobiol. 1992;23:605–626. doi: 10.1002/neu.480230602. [DOI] [PubMed] [Google Scholar]
- 30.Sheng G, et al. Direct regulation of rhodopsin 1 by Pax-6/eyeless in Drosophila: evidence for a conserved function in photoreceptors. Genes Dev. 1997;11:1122–1131. doi: 10.1101/gad.11.9.1122. [DOI] [PubMed] [Google Scholar]
- 31.Schneuwly S, et al. Drosophila ninaA gene encodes an eye-specific cyclophilin (cyclosporine A binding protein) Proc Natl Acad Sci U S A. 1989;86:5390–5394. doi: 10.1073/pnas.86.14.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shieh BH, et al. The ninaA gene required for visual transduction in Drosophila encodes a homologue of cyclosporin A-binding protein. Nature. 1989;338:67–70. doi: 10.1038/338067a0. [DOI] [PubMed] [Google Scholar]
- 33.Baker EK, et al. The cyclophilin homolog NinaA functions as a chaperone, forming a stable complex in vivo with its protein target rhodopsin. EMBO J. 1994;13:4886–4895. doi: 10.1002/j.1460-2075.1994.tb06816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colley NJ, et al. The cyclophilin homolog ninaA is required in the secretory pathway. Cell. 1991;67:255–263. doi: 10.1016/0092-8674(91)90177-z. [DOI] [PubMed] [Google Scholar]
- 35.Ondek B, et al. Genetic dissection of cyclophilin function. Saturation mutagenesis of the Drosophila cyclophilin homolog ninaA. J Biol Chem. 1992;267:16460–16466. [PubMed] [Google Scholar]
- 36.Rosenbaum EE, et al. Calnexin is essential for rhodopsin maturation, Ca2+ regulation, and photoreceptor cell survival. Neuron. 2006;49:229–241. doi: 10.1016/j.neuron.2005.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosenbaum EE, et al. XPORT-dependent transport of TRP and rhodopsin. Neuron. 2011;72:602–615. doi: 10.1016/j.neuron.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galy A, et al. Rhodopsin maturation defects induce photoreceptor death by apoptosis: a fly model for RhodopsinPro23His human retinitis pigmentosa. Hum Mol Genet. 2005;14:2547–2557. doi: 10.1093/hmg/ddi258. [DOI] [PubMed] [Google Scholar]
- 39.Colley NJ, et al. Defective intracellular transport is the molecular basis of rhodopsin-dependent dominant retinal degeneration. Proc Natl Acad Sci U S A. 1995;92:3070–3074. doi: 10.1073/pnas.92.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurada P, O’Tousa JE. Retinal degeneration caused by dominant rhodopsin mutations in Drosophila. Neuron. 1995;14:571–579. doi: 10.1016/0896-6273(95)90313-5. [DOI] [PubMed] [Google Scholar]
- 41.Kang MJ, Ryoo HD. Suppression of retinal degeneration in Drosophila by stimulation of ER-associated degradation. Proc Natl Acad Sci U S A. 2009;106:17043–17048. doi: 10.1073/pnas.0905566106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huber A, et al. Opsin of Calliphora peripheral photoreceptors R1-6. Homology with Drosophila Rh1 and posttranslational processing. J Biol Chem. 1990;265:17906–17910. [PubMed] [Google Scholar]
- 43.O’Tousa JE. Requirement of N-linked glycosylation site in Drosophila rhodopsin. Vis Neurosci. 1992;8:385–390. doi: 10.1017/s0952523800004910. [DOI] [PubMed] [Google Scholar]
- 44.Brown G, et al. Receptor demise from alteration of glycosylation site in Drosophila opsin: electrophysiology, microspectrophotometry, and electron microscopy. Vis Neurosci. 1994;11:619–628. doi: 10.1017/s0952523800002509. [DOI] [PubMed] [Google Scholar]
- 45.Katanosaka K, et al. N-linked glycosylation of Drosophila rhodopsin occurs exclusively in the amino-terminal domain and functions in rhodopsin maturation. FEBS Lett. 1998;424:149–154. doi: 10.1016/s0014-5793(98)00160-4. [DOI] [PubMed] [Google Scholar]
- 46.Webel R, et al. Role of asparagine-linked oligosaccharides in rhodopsin maturation and association with its molecular chaperone, NinaA. J Biol Chem. 2000;275:24752–24759. doi: 10.1074/jbc.M002668200. [DOI] [PubMed] [Google Scholar]
- 47.Tam BM, Moritz OL. The role of rhodopsin glycosylation in protein folding, trafficking, and light-sensitive retinal degeneration. J Neurosci. 2009;29:15145–15154. doi: 10.1523/JNEUROSCI.4259-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li ZY, et al. Autosomal dominant retinitis pigmentosa caused by the threonine-17-methionine rhodopsin mutation: retinal histopathology and immunocytochemistry. Exp Eye Res. 1994;58:397–408. doi: 10.1006/exer.1994.1032. [DOI] [PubMed] [Google Scholar]
- 49.Cao J, et al. A Drosophila metallophosphoesterase mediates deglycosylation of rhodopsin. EMBO J. 2011;30:3701–3713. doi: 10.1038/emboj.2011.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ozaki K, et al. Maturation of major Drosophila rhodopsin, ninaE, requires chromophore 3-hydroxyretinal. Neuron. 1993;10:1113–1119. doi: 10.1016/0896-6273(93)90059-z. [DOI] [PubMed] [Google Scholar]
- 51.Tian Y, et al. Neurexin Regulates Visual Function via Mediating Retinoid Transport to Promote Rhodopsin Maturation. Neuron. 2013;77:311–322. doi: 10.1016/j.neuron.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 52.Lee J, Ju BG. Drosophila arf72A acts as an essential regulator of endoplasmic reticulum quality control and suppresses autosomal-dominant retinopathy. Int J Biochem Cell Biol. 2011;43:1392–1401. doi: 10.1016/j.biocel.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 53.Satoh A, et al. In situ inhibition of vesicle transport and protein processing in the dominant negative Rab1 mutant of Drosophila. J Cell Sci. 1997;110 ( Pt 23):2943–2953. doi: 10.1242/jcs.110.23.2943. [DOI] [PubMed] [Google Scholar]
- 54.Shetty KM, et al. Rab6 regulation of rhodopsin transport in Drosophila. J Biol Chem. 1998;273:20425–20430. doi: 10.1074/jbc.273.32.20425. [DOI] [PubMed] [Google Scholar]
- 55.Satoh T, et al. GPI biosynthesis is essential for rhodopsin sorting at the trans-Golgi network in Drosophila photoreceptors. Development. 2013;140:385–394. doi: 10.1242/dev.083683. [DOI] [PubMed] [Google Scholar]
- 56.Satoh AK, et al. Rab11 mediates post-Golgi trafficking of rhodopsin to the photosensitive apical membrane of Drosophila photoreceptors. Development. 2005;132:1487–1497. doi: 10.1242/dev.01704. [DOI] [PubMed] [Google Scholar]
- 57.Li BX, et al. Myosin V, Rab11, and dRip11 direct apical secretion and cellular morphogenesis in developing Drosophila photoreceptors. J Cell Biol. 2007;177:659–669. doi: 10.1083/jcb.200610157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beronja S, et al. Essential function of Drosophila Sec6 in apical exocytosis of epithelial photoreceptor cells. J Cell Biol. 2005;169:635–646. doi: 10.1083/jcb.200410081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.den Hollander AI, et al. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12) Nat Genet. 1999;23:217–221. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 60.den Hollander AI, et al. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the crumbs homologue 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Izaddoost S, et al. Drosophila Crumbs is a positional cue in photoreceptor adherens junctions and rhabdomeres. Nature. 2002;416:178–183. doi: 10.1038/nature720. [DOI] [PubMed] [Google Scholar]
- 62.Gosens I, et al. Composition and function of the Crumbs protein complex in the mammalian retina. Exp Eye Res. 2008;86:713–726. doi: 10.1016/j.exer.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 63.Pocha SM, et al. Crumbs regulates rhodopsin transport by interacting with and stabilizing myosin V. J Cell Biol. 2011;195:827–838. doi: 10.1083/jcb.201105144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnson K, et al. Drosophila crumbs is required to inhibit light-induced photoreceptor degeneration. Curr Biol. 2002;12:1675–1680. doi: 10.1016/s0960-9822(02)01180-6. [DOI] [PubMed] [Google Scholar]
- 65.Saari JC. Vitamin A metabolism in rod and cone visual cycles. Annu Rev Nutr. 2012;32:125–145. doi: 10.1146/annurev-nutr-071811-150748. [DOI] [PubMed] [Google Scholar]
- 66.Lee SJ, et al. Rhodopsin kinase activity modulates the amplitude of the visual response in Drosophila. Proc Natl Acad Sci U S A. 2004;101:11874–11879. doi: 10.1073/pnas.0402205101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Doza YN, et al. Characterization of fly rhodopsin kinase. Eur J Biochem. 1992;209:1035–1040. doi: 10.1111/j.1432-1033.1992.tb17379.x. [DOI] [PubMed] [Google Scholar]
- 68.Cassill JA, et al. Isolation of Drosophila genes encoding G protein-coupled receptor kinases. Proc Natl Acad Sci U S A. 1991;88:11067–11070. doi: 10.1073/pnas.88.24.11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dolph PJ, et al. Arrestin function in inactivation of G protein-coupled receptor rhodopsin in vivo. Science. 1993;260:1910–1916. doi: 10.1126/science.8316831. [DOI] [PubMed] [Google Scholar]
- 70.Kiselev A, et al. A molecular pathway for light-dependent photoreceptor apoptosis in Drosophila. Neuron. 2000;28:139–152. doi: 10.1016/s0896-6273(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 71.Satoh AK, et al. Arrestin translocation is stoichiometric to rhodopsin isomerization and accelerated by phototransduction in Drosophila photoreceptors. Neuron. 2010;67:997–1008. doi: 10.1016/j.neuron.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Orrenius S, et al. Cell death mechanisms and their implications in toxicology. Toxicol Sci. 2011;119:3–19. doi: 10.1093/toxsci/kfq268. [DOI] [PubMed] [Google Scholar]
- 73.Alloway PG, Dolph PJ. A role for the light-dependent phosphorylation of visual arrestin. Proc Natl Acad Sci U S A. 1999;96:6072–6077. doi: 10.1073/pnas.96.11.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kahn ES, Matsumoto H. Calcium/calmodulin-dependent kinase II phosphorylates Drosophila visual arrestin. J Neurochem. 1997;68:169–175. doi: 10.1046/j.1471-4159.1997.68010169.x. [DOI] [PubMed] [Google Scholar]
- 75.Steele F, O’Tousa JE. Rhodopsin activation causes retinal degeneration in Drosophila rdgC mutant. Neuron. 1990;4:883–890. doi: 10.1016/0896-6273(90)90141-2. [DOI] [PubMed] [Google Scholar]
- 76.Vinos J, et al. A G protein-coupled receptor phosphatase required for rhodopsin function. Science. 1997;277:687–690. doi: 10.1126/science.277.5326.687. [DOI] [PubMed] [Google Scholar]
- 77.Steele FR, et al. Drosophila retinal degeneration C (rdgC) encodes a novel serine/threonine protein phosphatase. Cell. 1992;69:669–676. doi: 10.1016/0092-8674(92)90230-a. [DOI] [PubMed] [Google Scholar]
- 78.Liu CH, et al. Ca2+-dependent metarhodopsin inactivation mediated by calmodulin and NINAC myosin III. Neuron. 2008;59:778–789. doi: 10.1016/j.neuron.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.O’Tousa JE. Ca2+ regulation of Drosophila phototransduction. Adv Exp Med Biol. 2002;514:493–505. [PubMed] [Google Scholar]
- 80.Lee SJ, Montell C. Regulation of the rhodopsin protein phosphatase, RDGC, through interaction with calmodulin. Neuron. 2001;32:1097–1106. doi: 10.1016/s0896-6273(01)00538-4. [DOI] [PubMed] [Google Scholar]
- 81.Han J, et al. The fly CAMTA transcription factor potentiates deactivation of rhodopsin, a G protein-coupled light receptor. Cell. 2006;127:847–858. doi: 10.1016/j.cell.2006.09.030. [DOI] [PubMed] [Google Scholar]
- 82.Satoh AK, Ready DF. Arrestin1 mediates light-dependent rhodopsin endocytosis and cell survival. Curr Biol. 2005;15:1722–1733. doi: 10.1016/j.cub.2005.08.064. [DOI] [PubMed] [Google Scholar]
- 83.Alloway PG, et al. The formation of stable rhodopsin-arrestin complexes induces apoptosis and photoreceptor cell degeneration. Neuron. 2000;28:129–138. doi: 10.1016/s0896-6273(00)00091-x. [DOI] [PubMed] [Google Scholar]
- 84.Orem NR, et al. An essential role for endocytosis of rhodopsin through interaction of visual arrestin with the AP-2 adaptor. J Cell Sci. 2006;119:3141–3148. doi: 10.1242/jcs.03052. [DOI] [PubMed] [Google Scholar]
- 85.Chinchore Y, et al. Accumulation of rhodopsin in late endosomes triggers photoreceptor cell degeneration. PLoS Genet. 2009;5:e1000377. doi: 10.1371/journal.pgen.1000377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lloyd V, et al. Not just pretty eyes: Drosophila eye-colour mutations and lysosomal delivery. Trends Cell Biol. 1998;8:257–259. doi: 10.1016/s0962-8924(98)01270-7. [DOI] [PubMed] [Google Scholar]
- 87.Haberman A, et al. The synaptic vesicle SNARE neuronal Synaptobrevin promotes endolysosomal degradation and prevents neurodegeneration. J Cell Biol. 2012;196:261–276. doi: 10.1083/jcb.201108088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dourlen P, et al. Drosophila fatty acid transport protein regulates rhodopsin-1 metabolism and is required for photoreceptor neuron survival. PLoS Genet. 2012;8:e1002833. doi: 10.1371/journal.pgen.1002833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Xu H, et al. A lysosomal tetraspanin associated with retinal degeneration identified via a genome-wide screen. EMBO J. 2004;23:811–822. doi: 10.1038/sj.emboj.7600112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang T, et al. TOR-mediated autophagy regulates cell death in Drosophila neurodegenerative disease. J Cell Biol. 2009;186:703–711. doi: 10.1083/jcb.200904090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Midorikawa R, et al. Autophagy-dependent rhodopsin degradation prevents retinal degeneration in Drosophila. J Neurosci. 2010;30:10703–10719. doi: 10.1523/JNEUROSCI.2061-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kim JY, et al. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stark WS, Carlson SD. Blue and ultraviolet light induced damage to the Drosophila retina: ultrastructure. Current eye research. 1984;3:1441–1454. doi: 10.3109/02713688409000840. [DOI] [PubMed] [Google Scholar]
- 94.Xiong B, et al. Crag Is a GEF for Rab11 Required for Rhodopsin Trafficking and Maintenance of Adult Photoreceptor Cells. PLoS Biol. 2012;10:e1001438. doi: 10.1371/journal.pbio.1001438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Orem NR, Dolph PJ. Loss of the phospholipase C gene product induces massive endocytosis of rhodopsin and arrestin in Drosophila photoreceptors. Vision Res. 2002;42:497–505. doi: 10.1016/s0042-6989(01)00229-2. [DOI] [PubMed] [Google Scholar]
- 96.Isono K, et al. Dependency on light and vitamin A derivatives of the biogenesis of 3-hydroxyretinal and visual pigment in the compound eyes of Drosophila melanogaster. J Gen Physiol. 1988;92:587–600. doi: 10.1085/jgp.92.5.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sengupta S, et al. Depletion of PtdIns(4,5)P(2) underlies retinal degeneration in Drosophila trp mutants. J Cell Sci. 2013;126:1247–1259. doi: 10.1242/jcs.120592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Chen J, et al. Stable rhodopsin/arrestin complex leads to retinal degeneration in a transgenic mouse model of autosomal dominant retinitis pigmentosa. J Neurosci. 2006;26:11929–11937. doi: 10.1523/JNEUROSCI.3212-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wang X, et al. The Drosophila visual cycle and de novo chromophore synthesis depends on rdhB. J Neurosci. 2012;32:3485–3491. doi: 10.1523/JNEUROSCI.5350-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, et al. Requirement for an enzymatic visual cycle in Drosophila. Curr Biol. 2010;20:93–102. doi: 10.1016/j.cub.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hartman SJ, et al. Expression of rhodopsin and arrestin during the light-dark cycle in Drosophila. Mol Vis. 2001;7:95–100. [PubMed] [Google Scholar]
- 102.Xu XZ, et al. Retinal targets for calmodulin include proteins implicated in synaptic transmission. J Biol Chem. 1998;273:31297–31307. doi: 10.1074/jbc.273.47.31297. [DOI] [PubMed] [Google Scholar]
- 103.Kosmaoglou M, Cheetham ME. Calnexin is not essential for mammalian rod opsin biogenesis. Mol Vis. 2008;14:2466–2474. [PMC free article] [PubMed] [Google Scholar]
- 104.Mazelova J, et al. Ciliary targeting motif VxPx directs assembly of a trafficking module through Arf4. EMBO J. 2009;28:183–192. doi: 10.1038/emboj.2008.267. [DOI] [PMC free article] [PubMed] [Google Scholar]