

Abstract
Ebola viruses remain a substantial threat to both civilian and military populations as bioweapons, during sporadic outbreaks, and from the possibility of accidental importation from endemic regions by infected individuals. Currently, no approved therapeutics exist to treat or prevent infection by Ebola viruses. Therefore, we performed an in vitro screen of Food and Drug Administration (FDA)– and ex–US-approved drugs and selected molecular probes to identify drugs with antiviral activity against the type species Zaire ebolavirus (EBOV). From this screen, we identified a set of selective estrogen receptor modulators (SERMs), including clomiphene and toremifene, which act as potent inhibitors of EBOV infection. Anti-EBOV activity was confirmed for both of these SERMs in an in vivo mouse infection model. This anti-EBOV activity occurred even in the absence of detectable estrogen receptor expression, and both SERMs inhibited virus entry after internalization, suggesting that clomiphene and toremifene are not working through classical pathways associated with the estrogen receptor. Instead, the response appeared to be an off-target effect where the compounds interfere with a step late in viral entry and likely affect the triggering of fusion. These data support the screening of readily available approved drugs to identify therapeutics for the Ebola viruses and other infectious diseases. The SERM compounds described in this report are an immediately actionable class of approved drugs that can be repurposed for treatment of filovirus infections.
INTRODUCTION
Filoviruses (Ebola virus and Marburg virus) are responsible for some of the most lethal viral hemorrhagic fevers. The genus Ebolavirus includes five species of Ebola virus with case fatality rates up to 90%, whereas the single Marburg virus has different isolates with differing mortality rates (20 to 90%). Natural outbreaks of filoviruses in humans have been reported in the Democratic Republic of the Congo, Republic of the Congo, Sudan, Uganda, Angola, and Gabon. Filovirus illness is characterized by fever, myalgia, headache, and gastrointestinal symptoms, and patients may also develop a maculopapular rash (1). Fatal outcomes correlate with increased viremia, convulsions, and disseminated intravascular coagulation (1). The filoviruses are grave viral threats that continue to infect humans as well as nonhuman primates (NHPs) (2). There is a great concern about the potential for accidental importation from endemic regions by infected humans before the onset or diagnosis of the disease, and that filoviruses may be used as a biological weapon (3).
Although effective drugs have been found to treat several other viral diseases, there are currently no approved therapeutics (small molecule or biologic) to prevent or treat filovirus infections. Therefore, it is important to develop therapeutics that can be used for prophylaxis and as antiviral agents against filovirus infection.
A high-throughput assay for Zaire ebolavirus (EBOV) has been developed using the recombinant EBOV engineered to express the enhanced green fluorescent protein (eGFP) established by Towner et al. (4). The insertion of the eGFP gene into the EBOV genome allows for the detection of infected cells by flow cytometry, fluorimetry, fluorescence microscopy, and high-content imaging. The eGFP-expressing EBOV retains the infection and replication characteristics of the parent virus in vitro (4). The eGFP-EBOV offers great utility for screening because this virus targets the complete virus life cycle and offers a higher throughput of drug screening than traditional plaque assays and yield reduction assays. Such a cell-based assay can be used to identify inhibitors that target both viral and host pathways relevant to viral replication, and the activity of “hit” compounds can be confirmed using native isotypes. The identification of active compounds from this type of screen also may be helpful in identifying the critical pathways or targets that are essential for viral replication.
We conducted a cell-based screen of Food and Drug Administration (FDA)– and ex–US-approved drugs and molecular probes to identify inhibitors of Ebola viruses using the eGFP-EBOV assay. This screen identified many approved drugs and probes with previously undocumented anti-EBOV activity, including the selective estrogen receptor modulators (SERMs) clomiphene and toremifene. SERM activity involves binding of the ligand SERM to the estrogen receptor (ER), a member of the nuclear receptor superfamily, causing conformational changes that facilitate interactions with coactivator or corepressor proteins and subsequently initiate or suppress transcription of target genes. SERM activity is intrinsic to each ER ligand, which accomplishes its profile by specific interactions in the target cell, leading to tissue-selective actions [reviewed in (5) and (6)]. Clomiphene (brand names Clomid and Serophene) is used to treat female infertility due to anovulation. Toremifene (brand name Fareston) is approved for the treatment of advanced metastatic breast cancer.
We used clomiphene and toremifene to further characterize the mechanism by which these drugs affected Ebola virus infection. We confirmed the anti-EBOV activity of clomiphene and toremifene in mouse infection models. We demonstrated that expression of the ER was not required for clomiphene inhibition of EBOV infection, suggesting that these drugs are not acting through their known targets. Follow-up work with EBOV virus-like particle (VLP) entry assays indicated that these drugs inhibit EBOV VLP entry after binding and internalization. Together, these findings suggest that clomiphene and toremifene inhibit EBOV infection through pathways unrelated to the classical estrogen pathway.
RESULTS
Clomiphene and toremifene inhibit EBOV infection in vitro
We identified a set of compounds known to modulate ER-α (fig. S1) from a screen of FDA- and ex–US-approved drugs and molecular probes (7). Some of the strongest antiviral activity was observed with the ER antagonists, including clomiphene, toremifene, tamoxifen, and raloxifene (fig. S1). The ER antagonist diethylstilbestrol also inhibited EBOV infection but to a lesser extent. Additionally, we observed antiviral activity with ER agonists: hydroxyprogesterone caproate, equillin, and quinestrol (fig. S1). Clomiphene and toremifene were selected for confirmatory studies using an eight-point dose response in Vero E6 cells, where the active drug concentrations could be refined and an accurate inhibitory concentration of 50% (IC50) determined (Fig. 1). We evaluated the antiviral activity in human HepG2 cells to rule out mechanisms that may be distinctive to the monkey-derived Vero E6 cell line (Fig. 1). The antiviral activities of both clomiphene and toremifene were confirmed in both the Vero E6 and HepG2 cell lines. Although toremifene showed no impact on cell proliferation in both Vero E6 cells and HepG2 cells, we did observe that clomiphene inhibited HepG2 cell proliferation at the higher concentrations evaluated.
Fig. 1. In vitro eight-point dose-response curves for clomiphene and toremifene.
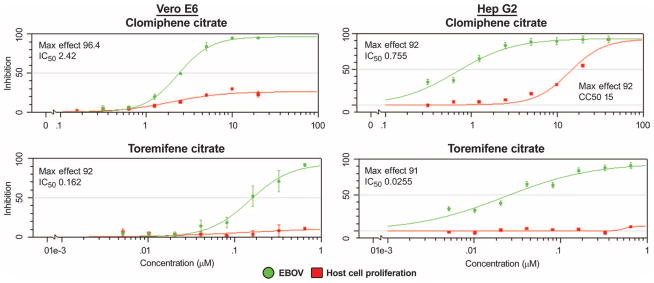
Compounds were evaluated in both the Vero E6 and HepG2 cell lines. The percent inhibition of the compound in the EBOV assay is shown in green, and the cytotoxic effect of the compounds on the host cell is shown in red. The maximum percent inhibition observed (Max Effect) and IC50 are indicated. Error bars indicate SEM. Results are from two replicates.
To exclude the possibility that antiviral activity observed with clomiphene and toremifene was specific to the engineered eGFP-EBOV strain, which is somewhat attenuated in vivo (8), we further tested the antiviral activity of clomiphene and toremifene against different species of Ebolavirus, namely, Zaire ebolavirus [EBOV/Kik (also known as EBOV-95) and EBOV/May (also known as EBOV-76)] and Sudan ebolavirus (SUDV), as well as two members of the species Marburg marburgvirus, Marburg virus (MARV) and Ravn virus (RAVV). The activity of the compounds against the native strains was evaluated by both cell-based enzyme-linked immunosorbent assay (ELISA) and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR). For the ELISA, viral isolates were used to infect Vero E6 cells in the presence of vehicle, clomiphene, or toremifene (concentration ranges of 0.08 to 20 μM); inhibition of viral infection was assessed at 48 hours after infection by fixing cells and staining with antibodies specific to the viral glycoproteins. The ELISAs demonstrated that the compounds inhibited viral infection of all the native strains tested (Figs. 2 and 3). Table 1 shows the IC50 and maximum inhibition observed with the compounds across the strains evaluated. The qRT-PCR results confirmed that clomiphene and toremifene were active against the native Ebola virus species as well as MARV (fig. S2). RAVV was not evaluated in this assay. These results indicate that clomiphene and toremifene broadly inhibit filovirus infection in vitro.
Fig. 2. In vitro eight-point dose-response curves for clomiphene evaluated against native filovirus strains, EBOV/Kik (EBOV-95), EBOV/May (EBOV-76), SUDV, MARV, and RAVV.

Results are from ELISA as described in Materials and Methods. Shown is the Max Effect (in % inhibition) along with the IC50 values (in μM). Vero E6 proliferation shows toxicity of clomiphene on uninfected cells. Results indicate that clomiphene is effective across all native virus strains evaluated. Results are from two or more replicates.
Fig. 3. In vitro eight-point dose-response curves for toremifene evaluated against native filovirus strains, EBOV/Kik, EBOV/May, SUDV, MARV, and RAVV.

Results are from ELISA as described in Materials and Methods. Shown is the Max Effect (in % inhibition) along with the IC50 values (in μM). Vero E6 shows the toxicity of toremifene on uninfected cells. Results indicate that toremifene is effective across all native virus strains evaluated. Results are from two or more replicates.
Table 1. Activity of clomiphene and toremifene across native filovirus strains.
IC50, the concentration at which 50% inhibition of viral infection was observed; Max effect, the maximum percent inhibition observed.
| Compound | EBOV/Kik
|
EBOV/May
|
SUDV
|
MARV
|
RAVV
|
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | Max effect | IC50 (μM) | Max effect | IC50 (μM) | Max effect | IC50 (μM) | Max effect | IC50 (μM) | Max effect | |
| Clomiphene | 11.1 | 89.4 | 3.83 | 97.8 | 4.96 | 97.6 | 5.82 | 97.0 | 7.18 | 96.0 |
| Toremifene | 1.73 | 96.6 | 0.973 | 3.83 | 3.96 | 97.8 | 5.73 | 95.7 | 6.17 | 95.6 |
Clomiphene and toremifene inhibit infection in vivo
To confirm that the antiviral activity observed with clomiphene and toremifene translated to in vivo EBOV infection, these compounds were evaluated in a murine EBOV infection model using a mouse-adapted viral strain (9, 10). The intraperitoneal inoculation of mouse-adapted virus results in the acute onset of severe illness 3 to 4 days after infection, with high-level viral replication observed in the liver and spleen, multifocal hepatic necrosis, and a rapid increase in viremia to titers >108 plaque-forming units (PFU)/ml (9, 10).
For these studies, female C57BL/6 mice (5 to 8 weeks old) were challenged via intraperitoneal injection with the target virus exposure dose of 1000 PFU of mouse-adapted Ebola virus (ma-EBOV). One hour after infection, the animals were treated with clomiphene, toremifene, or vehicle over a period of 10 days, and survival was monitored out to 28 days. Both compounds were given at a dose of 60 mg/kg by intra-peritoneal injection with dosing on days 0 and 1 and a regimen of every-other-day (QOD) dosing thereafter (a total of six treatments). The dose and regimen were selected on the basis of both the acute toxicity and the reported half-life of the compounds being greater than 24 hours (11–13). We attempted to select a dose that would avoid acute toxicity but could achieve high enough plasma exposure in the mice to observe a positive effect on survival.
For both clomiphene and toremifene, treatment of infected mice resulted in statistically significant survival benefit as shown in the survival plots in Fig. 4. For clomiphene, 90% of the treated animals survived (P < 0.0001). For toremifene, 50% (P = 0.0441) of the treated animals survived the EBOV exposure.
Fig. 4. Survival plots for clomiphene and toremifene in a mouse model of EBOV infection.

(A) A survival plot for clomiphene shows 90% of treated animals survived EBOV infection. All animals in the vehicle control group succumbed to disease by day 9. n = 10 for both the vehicle and clomiphene treatment groups. (B) A survival plot for toremifene indicating that 50% of the treated animals survived infection by EBOV. Animals in the vehicle control group succumbed to disease by day 7. n = 7 in the vehicle control treatment group and n = 10 in the toremifene treatment group. The P value for each study was determined by Fisher’s exact test. **P < 0.0001; *P < 0.0441.
An additional study was performed with clomiphene using the same dose and regimen to evaluate the treatment response of clomiphene in male versus female mice (fig. S3). Again, clomiphene treatment resulted in a statistically significant survival benefit compared to control animals for both male and female mice. Analysis of time to death indicated that there were no gender differences in response to the clomiphene treatment. In summary, the antiviral activity observed for clomiphene and toremifene translated to a significant survival benefit for mice in a murine EBOV infection model.
Clomiphene inhibition of EBOV infection is independent of ER expression
Considering the large number of SERM compounds that scored as active in our screen, we hypothesized that ER signaling may play a role in EBOV infection. However, ER signaling has not been reported to play a role in EBOV infection, and preliminary observations indicated no differences in response to clomiphene in infected male and female mice (fig. S3). Therefore, we investigated the involvement of the classical estrogen signaling pathway in EBOV infection.
We first determined whether ER-α was expressed in Vero E6 or HepG2 cells before or after clomiphene treatment. Cells were either untreated or treated with clomiphene or vehicle for 1 hour, and then probed for ER expression by Western blotting. We observed no expression of ER-α in untreated or clomiphene-treated Vero E6 or HepG2 cells. Additionally, we looked for expression of the ER-β. Expression was observed in both cell lines, although detected at a much lower level in Vero E6 cells (Fig. 5A). Treatment with clomiphene did not affect the levels of ER expression.
Fig. 5. ER expression in EBOV permissive cell lines.

(A) Western blot analysis of Vero E6 and HepG2 cells treated with clomiphene, dimethyl sulfoxide (DMSO) (vehicle control), or no treatment (NT) and probed for either ER-α or ER-β expression. MCF-7 and SK-BR-3 cell lysates were used as controls for ER-α and ER-β, respectively. (B) eGFP-EBOV was used to infect cell lines both positive and negative for ER-α and/or ER-β expression (as shown in fig. S4 and summarized in Table 2) that were treated with clomiphene. (C) Clomiphene and toremifene treatment of eGFP-EBOV–infected HUVECs that do not express ER-α or ER-β (fig. S5). Results indicate that both compounds inhibit virus infection similarly to cells that express ER receptors.
To further explore any possible role of ER signaling in EBOV infection, we used a panel of cell lines with varying combinations of ER expression (Table 2 and fig. S4). Cell lines were infected with eGFP-EBOV, and GFP expression was measured at 48 hours. Two cell lines, the breast cancer cell line ZR-75-1 and the non–small cell lung cancer (NSCLC) cell line H322, were not readily infected with EBOV. Further, two other breast cancer cell lines in the panel, MDA-MB-231 and SK-BR-3, only produced low levels of infection at the multiplicity of infection used for this study. No pattern was observed relating ER expression to infectibility of the cell lines.
Table 2.
Estrogen receptor expression and infection status in cell lines.
| Cell line | ER-α | ER-β | Infection |
|---|---|---|---|
| Vero E6 | − | + | Y |
| HepG2 | − | + | Y |
| ZR-75-1 | + | + | N |
| MDA-MB-231 | − | + | Low |
| MCF-7 | + | + | Y |
| SK-BR-3 | − | + | Low |
| A549 | − | + | Y |
| H460 | + | − | Y |
| H322 | − | + | N |
| H1650 | − | + | Y |
−, no expression; +, positive expression based on analysis by Western blotting; Y, cells that could be infected with eGFP-EBOV; N, cells resistant to infection with eGFP-EBOV; Low, a low level of eGFP-EBOV infection noted.
After clomiphene treatment of EBOV-susceptible cell lines, we observed a dose-dependent inhibition of eGFP-EBOV infection similar to that seen in Vero E6 and HepG2 cells (Fig. 5B). Greater than 50% inhibition was observed in all cell lines at the highest dose (3.2 μM), and a dose response was present in all cell lines regardless of the ER status.
Unstimulated human umbilical cord endothelial cells (HUVECs) have been reported to express no ER-α (14–16) and either no (14) or only low levels (16) of ER-β. In our hands, HUVECs did not express either isoform of ER (fig. S5). We found that HUVECs were also readily infected with eGFP-EBOV (fig. S5), and both clomiphene and toremifene inhibited infection with maximum inhibition of around 80% at 10 μM (IC50 values of 3.5 and 2.5, respectively) (Fig. 5C). Thus, the lack of ER expression had no impact on the ability of either clomiphene or toremifene to inhibit EBOV infection. Together, these data suggest that clomiphene and toremifene are acting through a pathway independent of the classical estrogen signaling pathway.
Clomiphene and toremifene inhibit EBOV entry at a step after internalization
After the observation that the antiviral effects of clomiphene and toremifene were independent of the classical estrogen signaling pathway, we sought to determine the mechanism of the antiviral activity of these compounds. Much progress has been made on characterizing the EBOV life cycle and interactions with the host cell (17, 18). As a first step to evaluate how clomiphene and toremifene affect the EBOV life cycle, we evaluated whether the compounds could be inhibiting viral entry, a process mediated exclusively by the EBOV glycoprotein (GP1,2) (18, 19).
After GP1,2-mediated binding to the host cell surface, EBOV is internalized into endosomes primarily through macropinocytosis (20). The particles then traffic to late endosomal compartments where low pH–dependent cellular proteases, cathepsins B and L, prime GP1,2 for subsequent fusion by generating an 18- to 19-kD form of GP1 (21–23). Next, an additional factor (or factors) triggers GP1,2 to mediate fusion with the endosomal membrane in a process that requires low pH (23–25). Host factors involved in the architecture and trafficking of endosomal/lysosomal compartments such as the subunits of the homotypic fusion and vacuole protein sorting (HOPS) complex and, in particular, Niemann-Pick C1 (NPC1), a membrane protein involved in cholesterol egress, have emerged as EBOV entry factors (26). Studies have shown that the processed GP1 specifically interacts with NPC1 protein, and expression of NPC1 is required for productive infection (26–28). Moreover, expression of NPC1 confers susceptibility to filovirus infection when it is expressed in nonpermissive reptilian cells (28). The GP1/NPC1 interaction appears to be a key step in the entry process, but additional events may also be required to generate a fusion-ready form of the glycoprotein. Once a fusion-ready form of the glycoprotein has been generated, the GP2 subunit mediates viral/cellular membrane fusion, and the EBOV nucleocapsids are released into the cell cytoplasm where synthesis of new viral components ensues.
EBOV entry was examined using VLPs expressing the EBOV glycoprotein (GP1,2) and containing a β-lactamase reporter (BLaM) (23, 29). For these experiments, we used human SNB19 cells, which are negative for ER-α expression and have a low level of ER-β expression according to the National Cancer Institute (NCI) database (http://dtp.cancer.gov/mtweb/search.jsp). Cells were pretreated with clomiphene, toremifene, or vehicle for 30 min and subsequently cooled on ice. VLPs were then added to the cells in the presence of the inhibitors. After allowing for binding at 4°C, the cells were placed at 37°C to allow for VLP entry and then loaded with a BLaM fluorescent substrate. With this system, compounds that inhibit viral entry will result in low or no BLaM signal. The results of the VLP entry assay in SNB19 cells show that both clomiphene and toremifene inhibit entry of EBOV VLPs (VLP-GP) (Fig. 6, A and B). Both compounds were evaluated with four-point, twofold dose titrations. Representative flow cytometry plots from these experiments are provided in fig. S6. For both clomiphene and toremifene, we observed a dose response where higher concentrations resulted in higher levels of entry inhibition. Evaluation of the effects of clomiphene and toremifene on the entry of VLPs containing the Sudan GP showed similar inhibition (fig. S7).
Fig. 6. Effect of clomiphene and toremifene on VLP entry.

(A and B) Clomiphene (A) and toremifene (B) were tested for their ability to inhibit VLPs with the EBOV GP1,2 glycoprotein (VLP-GP), the VSV-G glycoprotein (VLP-G), and LCMV GP (VLP-LCMV). Results indicate that both clomiphene and toremifene exhibit greater specificity to VLPs bearing the EBOV GP1,2 glycoprotein compared to VLPs bearing VSV-G or the LCMV GP. For each compound, assays with the different VLPs were performed in parallel. Error bars represent the SE for two or more replicates.
To evaluate whether the entry inhibition observed was specific to VLPs with GP1,2, we evaluated VLPs containing the vesicular stomatitis virus (VSV) glycoprotein G (VLP-G) and VLPs containing the lymphocytic choriomeningitis virus glycoprotein (VLP-LCMV) in parallel (Fig. 6, A and B). VSV viral entry is mediated largely in early endosomes soon after internalization, whereas LCMV entry is mediated in late endosomes (30). Both clomiphene and toremifene inhibited the entry of VLP-GPs with greater potency compared to the VLP-Gs and VLP-LCMVs. Treatment with clomiphene resulted in 93% inhibition of VLP-GP at 5 μM, compared to 25% inhibition of VLP-G entry and 73% inhibition of VLP-LCMV. Similar results were observed with toremifene. Treatment with 5 μM toremifene resulted in 95% VLP-GP entry inhibition, compared to 10% inhibition of VLP-G and 70% inhibition of VLP-LCMV. Lower concentrations of the compounds, 1 μM for clomiphene and 0.5 μM for toremifene, still strongly inhibited the entry of VLPs containing the EBOV GP1,2. In contrast, these concentrations did not affect the entry of the VLPs containing the VSV or the LCMV glycoproteins.
We next asked whether the compounds inhibit EBOV entry because they inhibit particle internalization. To examine this, we used VLPs that contained a fluorescently-tagged VP40 marker. After incubations for VLP binding (at 4°C) and internalization (at 37°C), SNB19 cells were treated with protease to remove VLPs remaining on the cell surface. The amount of fluorescence was then measured. Cells that retained fluorescently-tagged VP40 were therefore scored as positive for VLP internalization. For these experiments, SNB19 cells were treated with clomiphene (5 μM), toremifene (0.8 μM), vehicle, or 50 μM 5-N-ethyl-N-isopropyl-amiloride (EIPA), which is a known inhibitor of macropinocytosis and thus inhibits EBOV VLP internalization (20, 31). As shown in Fig. 7A, neither clomiphene nor toremifene inhibited VLP internalization. In contrast, treatment of cells with EIPA resulted in a greater than threefold reduction in VLP internalization. The fact that neither clomiphene nor toremifene inhibits VLP internalization implies that neither inhibits VLP binding to or internalization from the cell surface.
Fig. 7. Evaluation of clomiphene and toremifene on Ebola virus VLP-GP1,2 internalization and cathepsin processing.

(A) Clomiphene and toremifene were evaluated at 5 and 0.8 μM, respectively. EIPA is a known inhibitor of EBOV internalization. Results indicate that neither clomiphene nor toremifene inhibits EBOV internalization. (B) Clomiphene (5 μM) and toremifene (0.8 μM) were evaluated, as described in Materials and Methods, for their effects on cathepsin B (CatB) and cathepsin L (CatL) activity (singly and combined) in SNB19 cells. EST is a cysteine protease inhibitor that was included as a positive control for the assay. Data in the main plot are from the 1.5-hour time point. Data for CatL at 18 hours are shown in fig. S8.
Next, we tested whether either clomiphene or toremifene affected the activity of cathepsin B or cathepsin L, the endosomal proteases needed to prime GP1,2 for cellular entry (22, 23). Clomiphene and toremifene were added to cells for 3 hours at 5 and 0.8 μM, respectively. Epoxysuccinylleucylamido-3-methylbutane ethyl ester (EST; a cysteine protease inhibitor) was used as a positive control for cathepsin inhibition (23). The cells were lysed and assayed for enzyme activity (26, 32). We found that neither clomiphene nor toremifene inhibited the activity of either cathepsin B or cathepsin L (Fig. 7B and fig. S8). Thus, these compounds are not inhibiting the cathepsin enzymes that prime EBOV GP1.
We also evaluated whether clomiphene or toremifene affected endosome acidification, which is needed for EBOV entry for cathepsin priming of GP1 and perhaps for GP2-mediated viral fusion (24, 33). Endosome acidification was assessed in SNB19 cells with LysoTracker Red, which is a probe for low-pH organelles. Cells were treated with vehicle, clomiphene (5 μM), toremifene (0.8 μM), or NH4Cl (10 mM), the latter as a positive control for inhibition of acidification. Although we observed strong inhibition of acidification with NH4Cl, we observed no inhibition with clomiphene or toremifene (Fig. 8).
Fig. 8. Endosomal pH upon exposure to clomiphene and toremifene.

(A to D) SNB19 cells were preincubated in the presence of (A) DMSO vehicle, (B) 10 mM NH4Cl as a positive control for inhibition of acidification, (C) 5 μM clomiphene, or (D) 0.8 μM toremifene for 1 hour at 37°C. At this time, Lyso-Tracker Red (± inhibitor, as indicated) was added, and the cells were incubated for an additional 30 min at 37°C. At this time, the cells were fixed, viewed, and photographed with a confocal microscope. Images are representative of 10 fields observed per condition.
Our results show that clomiphene and toremifene inhibit EBOV VLP entry with some specificity to GP1,2. The inhibition observed with clomiphene and toremifene occurred after binding and internalization and was not due to effects on either endosomal cathepsins or endosomal acidification. This suggests that these compounds could be inhibiting either VLP trafficking to the late endosome where fusion occurs or a step in the triggering of fusion. To investigate these possibilities, we looked at the impact of the compounds on VLP trafficking to late endosomes/lysosomes.
SNB19 cells were allowed to internalize VLP-GP1,2 in the presence of clomiphene, toremifene, or vehicle. The colocalization of the VLPs with the late endosome/lysosome maker lysosomal membrane protein 1 (LAMP1) was then assessed (Fig. 9). The results indicate that neither clomiphene nor toremifene had a meaningful impact on VLP-GP1,2 trafficking to LAMP1+ late endosomes. The VLPs were observed to colocalize with LAMP1 in the late endosome.
Fig. 9. VLP-GP trafficking to late endosomes/lysosomes after clomiphene or toremifene treatment.

SNB19 cells were pretreated with either DMSO, 2.5 μM clomiphene, or 1 μM toremifene for 1 hour, followed by the addition of VLP-GP1,2 as described in Materials and Methods. The impact of clomiphene or toremifene treatment on trafficking was assessed with confocal microscopy to determine the localization of VLP-GP in relation to LAMP1. (A) Ten random image fields per sample were analyzed with the JACoP plugin on ImageJ. Auto-thresholds were subtracted for each component 8-bit channel. Colocalization of VLPs (red, marked by mCheery-VP40) with LAMP1 (green) is reported as the average Mander’s overlap coefficient. Error bars represent SD. (B) Representative images (enlarged regions from an image field) from each treatment are shown with the green channel thresholds raised to 255 to better reveal areas of colocalization. This adjustment was made uniformly in all three images. White arrows indicate examples of colocalization.
DISCUSSION
Multiple SERM compounds were identified as new and specific inhibitors of EBOV infection from a screen of approved drugs and mechanistic probes. The aim was to characterize the anti-filovirus activity of two ER antagonist compounds: clomiphene and toremifene (5). Our results indicate that both clomiphene and toremifene broadly inhibit filovirus infection, with in vitro activity observed against native isolates of EBOV/Kik, EBOV/May, SUDV, MARV, and RAVV. Moreover, a statistically significant survival benefit was observed for both clomiphene and toremifene in a murine EBOV infection model. Because of the long half-life of these compounds, they were administered in a modified QOD dosing regimen, giving rise to the potential for the development of a once-daily or QOD regimen to treat Ebola virus disease in humans.
Although we initially identified the ER antagonist compounds on the basis of their collective known mechanism of action, our results indicate that these compounds are mediating their antiviral effects through cell-based mechanisms unrelated to the classical estrogen signaling pathway. First, various cell lines were susceptible to EBOV infection regardless of their expression (or lack of expression) of ER-α or ER-β, or both isoforms. Second, a study in the murine infection model indicated no gender differences in the response to clomiphene treatment.
Our data with filamentous VLPs further demonstrate that these compounds inhibit viral infection through an ER-independent mechanism. The compounds inhibit viral entry of VLPs bearing Ebola GP1,2 in a dose-dependent manner in SNB19 cells. The observed entry inhibition occurs after viral binding and internalization and does not involve inhibition of cathepsin enzyme activity, endosome acidification, or trafficking to late endosomes/lysosomes. Thus, these compounds are likely affecting GP1,2 viral fusion.
The ER antagonist drugs identified in our screen—clomiphene, toremifene, tamoxifen, and raloxifene—are structurally similar (5) and can be classified as class II cationic amphiphiles (CADs). CADs contain a hydrophobic tertiary amine with clearly segregated hydrophobic and hydrophilic segments. Our results here along with other studies indicate that CADs inhibit EBOV entry (26, 32).
In further support of clomiphene and toremifene interfering with filoviral fusion, we have observed that higher concentrations of clomiphene and the other CADs are required for inhibition in EBOV pseudovirus infection studies in cells overexpressing the EBOV entry factor NPC1, compared to parental cells (32). These results support a mechanism where clomiphene and other CADs mediate EBOV inhibition at a later step in the entry process.
It is not known whether clomiphene and toremifene (or other CADs) interact directly with NPC1 or mediate their effects indirectly through NPC1. Coimmunoprecipitation assays indicate that clomiphene and CAD compounds do not disrupt the interaction between primed GP1 and NPC1, which occurs before viral fusion (32). The CAD compound U18666A has been shown to interact with purified NPC1 (34), but not block GP1 binding to NPC1 (27, 32). Clomiphene and toremifene could mediate the entry block indirectly through NPC1 by targeting other endosomal/lysosomal proteins involved in the cholesterol uptake pathway whose function may be regulated by NPC1 (32).
The modulation of these endosomal enzymes and the impact on endosomal membrane composition may account for the entry inhibition observed with VLP-LCMV after treatment with higher concentrations of clomiphene or toremifene. Like EBOV, LCMV entry relies on trafficking through the endosomal system with entry mediated through the late endosome. The endosomal membrane composition may be modified such that it affects LCMV fusion, perhaps in a manner similar to dengue virus, which is known to rely on negatively charged phospholipids, specifically lysobisphosphatidic acid, for fusion triggering (35). It will be interesting to evaluate the activity of other CADs in VLP-LCMV entry assays.
Although this characterization of anti-filovirus activity of clomiphene and toremifene shows the potential for their use to treat Ebola virus disease, additional evaluation of these drugs in the NHP infection model is required to fully validate their use in a clinical setting. Both drugs showed activity in the murine infection model; however, many drugs that are active in this model have not proved efficacious in the NHP model (36).
Still, the ER antagonists clomiphene and toremifene are FDA-approved drugs with oral availability, good safety, and tolerability profiles and a long history of use. These drugs have good plasma exposure and bioavailability, making them excellent candidates for repurposing efforts for use for the treatment of Ebola virus infection (11, 12). The study presented here highlights the benefits of screening approved drugs to identify candidates for drug repurposing for filoviruses. A successful drug repurposing effort for either clomiphene or toremifene for filovirus disease would support the use of this approach to identify therapeutics for other viral threats as well as for rare and neglected diseases. The approved drug status of clomiphene and toremifene may allow them to be rapidly developed for use with filovirus disease. The oral availability of these drugs offers great utility in the resource-constrained geographical regions where outbreaks of filoviral infection frequently occur. Current efforts are focused on the development of these drugs as a medical countermeasure alone or through synergistic combinations with other antiviral drugs identified in the same drug screen.
MATERIALS AND METHODS
Study design
Drug screening and the evaluation of clomiphene and toremifene for in vitro entry assays occurred in a blinded fashion, with the compound identities unknown to the experimenters. For all studies with live virus, a minimum of two replicates were performed on separate days. For entry studies, two to three replicates were performed on the same day. For in vitro studies with microscopy, 10 random image fields were analyzed. Outlier data points were defined as a value > median + 3σ and were excluded from calculations.
Raw phenotype measurements T from each treated well were converted to normalized fractional inhibition I = 1 – T/V relative to the median V of vehicle-treated wells arranged around the plate. After normalization, average activity values were calculated between replicate measurements at the same treatment doses along with σ1, the accompanying standard error estimates. Drug-response curves were represented by a logistic sigmoidal function with a maximal effect level (Amax), the concentration at half-maximal activity of the compound (EC50), and a Hill coefficient representing the sigmoidal transition. All curve fits were through the 0 concentration point; however, the display range may not show this in all cases. We used the fitted curve parameters to calculate the concentration (IC50) at which the drug response reached an absolute inhibition of 50%, limited to the maximum tested concentration for inactive compounds.
For in vivo studies, sample sizes were selected to minimize the number of animals needed while obtaining a statistically significant result. Animals were randomly assigned to study arms. Animal studies were not blinded to the study investigators but were blinded to personnel performing treatment injections. Cage weights were used to determine an average mouse weight for treatment purposes.
Ethics statement
Animal research was conducted under an Institutional Animal Care and Use Committee–approved protocol at the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) [U.S. Department of Agriculture (USDA) Registration Number 51-F-0021/728 and Office of Laboratory Animal Welfare (OLAW) Assurance Number A3473-01] in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals. The facility where this research was conducted is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care, International and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 2011.
Live virus experiments
Experiments using live filoviruses were performed in biosafety level 4 (BSL-4) facilities at USAMRIID. Personnel wear positive-pressure protective suits (ILC Dover) fitted with HEPA filters and umbilical-fed air. USAMRIID is registered with the Centers for Disease Control (CDC) Select Agent Program for the possession and use of biological select agents and toxins and has implemented a biological surety program in accordance with U.S. Army regulation AR 50-1 “Biological Surety.”
Reagents
Clomiphene citrate (CAS #50-41-9), tamoxifen citrate (CAS #54965-29-1), raloxifene hydrochloride (CAS #82640-04-8), diethylstilbestrol (CAS #56-53-1), and Quinestrol (CAS #152-43-2) were purchased from Sigma-Aldrich. Toremifene citrate (CAS #89778-27-8) was purchased from Sequoia Research Chemicals. Equilin (CAS #474-86-2) was purchased from ICN Biomedicals Inc. Hydroxyprogesterone caproate (CAS #630-56-8) was purchased from Professional Compounding Centers of America. DMSO was used as solvent for the high-throughput screening assay described below. DMSO (5%)/PBS (phosphate-buffered saline) was used as a solvent for the mouse infection studies described below. ER-α antibody was purchased from Millipore (cat. #04-820). Antibodies against ER-β and β-actin were purchased from Cell Signaling Technology (cat. #5513 and #3700, respectively). The 9G4 antibody was developed by Hevey et al. (USAMRIID, Fort Detrick) (37, 38). The 13C6 antibody was developed by Wilson et al. (USAMRIID, Fort Detrick) (39). The 3C10 antibody was provided by J. Dye (USAMRIID, Fort Detrick). The 9E12 antibody was developed and provided by D. Negley (USAMRIID, Fort Detrick).
Cells and viruses
Vero E6 cells [American Type Culture Collection (ATCC): CRL-1586] and HepG2 cells (ATCC: HB-8065) were maintained in Eagle’s minimum essential medium (Gibco Invitrogen) supplemented with 10% fetal bovine serum (FBS; Gibco Invitrogen). The breast cancer cell lines ZR-75-1, MDA-MB-231, MCF-7, and SK-BR-3 were purchased from the ATCC and maintained as above. The NSCLC cell lines A549, H460, H322, and H1650 were gifts from F. M. Johnson (M. D. Anderson Cancer Center, Houston, TX) and were maintained as above. SNB19 cells (ATCC: CRL-2219) were maintained in Opti-MEM medium (Gibco), and human embryonic kidney (HEK) 293T cells (ATCC: CRL-11268) were maintained in Dulbecco’s modified Eagle’s medium (DMEM, Gibco Invitrogen) supplemented with 10% FBS (Gibco Invitrogen) and 1% penicillin/streptomycin (Gibco Invitrogen). HUVECs were purchased from Lonza (cat. #CC-2517) and were maintained in endothelial basal medium (Lonza, cat. #CC-3121) supplemented with the EGM-MV bullet kit (Lonza, cat. #CC-3125).
Ebola virus isolates Kikwit (EBOV-95 or EBOV/Kik), Mayinga (EBOV-76 or EBOV/May), eGFP-EBOV, SUDV, MARV, and RAVV were replicated in Vero E6 cells at 90 to 100% confluency. Cells were inoculated with an approximate multiplicity of infection of 0.1 from historical stocks, and the medium was replaced 72 hours after inoculation. Cells were monitored for cytopathic effects, and the supernatant was collected once 95 to 100% of the cells had detached from the surface. The cell supernatant was clarified by centrifugation at 1200 rpm for 10 min at 4°C, and aliquots were placed at −80°C storage until further use.
Screening assay
The screening assay for EBOV used a genetically engineered Zaire strain of Ebola virus expressing eGFP, eGFP-EBOV, described in (4) and was provided by J. Towner of the CDC. For all screening experiments, Vero E6 or HepG2 cells were plated in 96-well plates at a density of 40,000 cells per well in a total volume of 100 μl per well and incubated overnight at 37°C, 5% CO2. Next, 50 μl of prediluted compounds was added at a 4× concentration to each well to achieve the desired final concentration. Finally, 50 μl of the indicated virus (corresponding to an approximate multiplicity of infection of 0.01) was added to cells. These assay plates were centrifuged at 2000 rpm for 5 min and were incubated for 48 hours at 37°C, 5% CO2. After this incubation, the amount of eGFP in each well of the infected plates was determined with a spectrofluorometer from Molecular Devices (excitation: 485 nm, emission: 515 nm, cutoff: 495 nm). Antiviral activity was calculated by the inhibition of eGFP compared to mock-treated control cells.
The compound responses for SERM compounds were tested at three concentrations in the preliminary screen for eGFP-EBOV. The in vitro anti-EBOV activity was confirmed by testing clomiphene and toremifene compounds at seven serially diluted doses in both Vero E6 and HepG2 cells.
To confirm that a decrease in fluorescence correlated with the inhibition of viral replication and not an increase in cell death, a counter screen was run in tandem using uninfected Vero E6 or HepG2 cells. Cells were seeded in 96-well plates as described above and incubated overnight at 37°C, 5% CO2. The following day, cells were treated with compound and mock-infected with medium. After 48 hours of incubation, cell viability was assessed with the Promega CellTiter-Glo Luminescent Cell Viability Assay Kit. This assay provides a quantitative measure of the levels of adenosine triphosphate (ATP) in the cell cultures in each well, with higher levels of ATP correlating with greater cellular viability. Thus, a compound with antiviral activity is expected to inhibit the levels of fluorescence measured with minimal effect on the ATP levels measured by the CellTiter-Glo assay.
Yield reduction assay
To exclude the possibility that antiviral activity observed with clomiphene and toremifene was specific to the engineered eGFP-EBOV strain, which is somewhat attenuated in vivo (8), we tested the antiviral activity of clomiphene and toremifene against two different species of Ebolavirus, namely, EBOV/Kik and SUDV, as well as in the single Marburgvirus species, Marburg marburgvirus (MARV). These viruses were used to infect Vero E6 cells in the presence of vehicle, clomiphene, or toremifene (concentration ranges of 0.2 to 50 μM); 48 hours after viral infection, cell culture supernatants were collected. To determine whether the cell culture supernatants treated with clomiphene or toremifene had an impact on viral proliferation in comparison to controls, we used qRT-PCR to evaluate virus titer.
Quantitative RT-PCR
Quantitative RT-PCR to detect the nucleoprotein of EBOV/Kik, SUDV, and MARV was performed as previously published (40). Viral supernatants were inactivated with Trizol-LS (Invitrogen; 10296-028) before removal from BSL-4, and viral RNA was purified using the Qiagen Viral RNA Mini Kit per manufacturer’s directions. Purified RNA was suspended in deoxyribonuclease- and ribonuclease-free water and stored at −80°C until further use. The qRT-PCR assay used was previously published (40). The primers and probes used in this assay are commercially available and were purchased from Life Technologies (Applied Biosciences): MARV primer (Life Technologies/ Applied Biosciences, reference F4573T and R4638G); MARV probe (Life Technologies/Applied Biosciences, reference MBG P461CT); EBOV primer (Life Technologies/Applied Biosciences, reference F1542 and R1621); EBOV probe (Life Technologies/Applied Biosciences, reference P597S); SUDV probe (Life Technologies/Applied Biosciences, reference P1079S); SUDV primer (Life Technologies/Applied Biosystems, reference R1625 and F1545). PFU equivalents were determined using a known virus concentration (determined by plaque assay) whose RNA was extracted and was 10-fold serially diluted. The sensitivity of the assay was determined to be 0.04 genome copies/ml.
Clomiphene and toremifene were evaluated across the viral species using a threefold dilution scheme of compound to generate six serial dilutions starting at 50 μM. Compound dilutions were added to the cell plate for 1 hour before viral infection; after infection, plates were incubated for 48 hours at 37°C, and then cell supernatants were collected for qRT-PCR. At least two or more experimental replicates with multiple internal replicates were performed for each of the compound treatments.
Native strain ELISA
Vero E6 cells were plated in 96-well plates at a density of 40,000 cells per well in a total volume of 100 μl per well and incubated overnight at 37°C, 5% CO2. Next, 50 μl of prediluted compounds was added at a 4× concentration to each well to achieve the desired final concentration. Finally, 50 μl of EBOV/Kik, EBOV/May, SUDV, MARV, or RAVV was added to cells. Cells were infected with a multiplicity of infection of 0.1 for SUDV, 0.5 for RAVV, 0.1 for MARV, and 0.4 for EBOV. After 48 hours of incubation, cells were fixed with 10% buffered formalin for 24 hours. Plates were then washed three times with PBS and blocked with 3% bovine serum albumin or 3% milk for 1 hour at room temperature. Virus was detected with an appropriate virus-specific monoclonal antibody (mAb) to probe for virus glycoprotein. For EBOV isolates, 13C6 antibody was used at 1:2000 dilution; for SUDV, the C310 mAb was used at 1:20,000 dilution; for MARV, the 9G4 mAb was used at 1:8000 dilution; and for RAVV, the 9E12 antibody was used at 1:2000 dilution. The assays were incubated with primary antibody for 2 hours at room temperature, followed by three PBS washes. A horseradish peroxidase (HRP)–conjugated goat anti-mouse secondary antibody (KPL, cat. #074-1802) was added at 1:4000 dilution for SUDV and EBOV, at 1:2000 dilution for RAVV, and at 1:8000 dilution for MARV. Assay plates were incubated for an additional hour at room temperature and then were washed three times with PBS. Cells expressing viral glycoprotein were detected with the Pierce SuperSignal ELISA Pico Chemiluminescent Substrate Kit (Thermo Scientific, cat. #37069), and assay plates were read on a luminescent plate reader (SpectraMax M5, Molecular Devices). Control plates with mock-infected Vero E6 cells were run in parallel as described above.
Western blotting
Vero E6 and HepG2 cells were treated with clomiphene (2.5 and 0.75 μM, respectively), vehicle (DMSO), or left untreated for 1 hour at room temperature. Adherent cells were washed with ice-cold PBS and collected in radioimmunoprecipitation assay buffer (Sigma-Aldrich) with HALT protease (10 μg/ml) and phosphatase inhibitor cocktail (Thermo Fisher). Lysates were held on ice for 10 min and then clarified at maximum speed in a tabletop centrifuge for 10 min. Lysates of untreated NSCLC cells, breast cancer cells, and HUVECs were collected in the same manner from untreated cells. With the exception of HUVEC lysates, protein concentration was determined with the Quick Start Bradford protein assay (Bio-Rad), and equal protein aliquots were resolved using Mini-PROTEAN TGX gels (Bio-Rad) and then transferred onto nitrocellulose using the iBlot system (Invitrogen). After transfer, the membranes were immunoblotted with primary antibody and detected with HRP-conjugated secondary antibody (Cell Signaling Technology) and enhanced chemiluminescence reagent (Thermo Fisher). Immunoblots were imaged using the ChemiDoc XRS+ system (Bio-Rad).
Murine Ebola infection model
The murine Ebola infection model has been described previously (9, 10). Ma-EBOV was obtained from M. Bray, Virology Division, USAMRIID. C57BL/6 mice (5 to 8 weeks old) were obtained from NCI (Frederick, MD) and housed under specific pathogen–free conditions. C57BL/6 mice were exposed with a target dose of 1000 PFU of the ma-EBOV by intraperitoneal injection in a BSL-4 laboratory. One hour after challenge, the animals were treated with either clomiphene or toremifene. The compounds were dissolved in DMSO at a 20× concentration and further diluted in PBS to a final formulation of 5% DMSO/PBS. Both compounds were administered to animals by intraperitoneal injection with a total injection volume of 200 μl. Animals in the vehicle control groups were administered a solution of 5% DMSO/PBS with no drug. Animals were treated on days 0, 1, 3, 5, 7, and 9 (a total of six separate treatments). Animals were monitored for survival for a total of 28 days after infection. The P value was calculated using Fisher’s exact test (with step-down Bonferroni adjustment where necessary) to compare the compound-treated groups and vehicle-treated control groups. A P value of <0.05 indicates a significant difference between experimental groups.
Statistical methods
For animal studies evaluating the efficacy of just clomiphene or toremifene in a single gender (female mice), the mean time to death was analyzed by analysis of variance comparing only those mice that succumbed before the study end. Due to the sample sized used for these studies, differences in overall survival between treatment and vehicle control groups were evaluated using Fisher’s exact test (with step-down Bonferroni adjustment where necessary). A P value of <0.5 indicates a significant difference between experimental groups.
For the animal study evaluating both genders, Kaplan-Meier curves were used to determine whether there were statistically significant differences in survival for the two treatment groups. The Kaplan-Meier curves were evaluated using the log-rank test for overall homogeneity. Pairwise comparisons were also made using the log-rank test with a post hoc Tukey-Kramer adjustment for multiple testing.
Time to death was analyzed by analysis of variance comparing only those mice that succumbed before the study end. A model fitting time to death using gender treatment and gender × treatment interaction was evaluated to determine whether gender × treatment interaction had an effect on time to death. In the absence of an interaction effect, the model was rerun without interaction.
As indicated above, the differences in overall survival between treatment and control groups were evaluated using Fisher’s exact test. All statistics conform to the journal’s policy.
Virus-like particle preparation
For VLP entry, internalization, and trafficking assays, Ebola virus VLPs were prepared by transfecting HEK 293T cells using polyethyle-nimine (41) with four plasmids: one encoding VP40 with β-lactamase fused to its N terminus, one encoding VP40 with mCherry fused to its N terminus (32), one encoding untagged VP40, and a fourth encoding codon-optimized full-length EBOV (Mayinga strain) GP1,2 (pDisplay/GPco). The plasmids were transfected at a ratio of 9:9:4:6, respectively. SUDV VLPs were generated in an identical fashion, but with the substitution of a plasmid encoding full-length SUDV GP (Boniface strain). VLPs with other viral glycoproteins were made by inclusion of plasmids encoding either VSV-G or LCMV GP (in place of a plasmid encoding a filoviral glycoprotein). For EBOV GP VLPs, transfected cells were treated with 10 mM sodium butyrate, at 4 hours posttransfection (hpt), to boost GP expression. Culture supernatant at 24 hpt was collected and replaced with fresh media (with sodium butyrate for EBOV GP VLPs). A second harvest was conducted at 48 hpt. Both harvests were pooled and cleared of cell debris by two sequential centrifugations at 1070g for 10 min at 4°C. VLPs in the cleared supernatant were then pelleted through 20% sucrose [in 20 mM Hepes, 130 mM NaCl, pH 7.4 (HNB)] at 112,398g (25,000 rpm) in an SW28 rotor for 2 hours at 4°C. Pelleted VLPs were then resuspended overnight in 10% sucrose (in HNB; 1/100 of original volume of culture medium collected), aliquoted, and stored at −80°C. All VLPs were assessed on Western blots for the presence of glycoproteins and VP40 proteins.
EBOV entry and internalization assays
The β-lactamase (BlaM) entry assay (42) was adapted for assessing effects of candidate inhibitory compounds on EBOV VLP entry as follows. About 24 hours before each experiment, 100,000 SNB19 cells were plated in each well of a 48-well dish. Cells were then pretreated for 30 min with the indicated concentration of inhibitor or with DMSO vehicle in Opti-MEM I (OMEM) medium (Gibco BRL). These drug concentrations were maintained in all subsequent steps of the experiment until CCF2/AM substrate loading. After the preincubation period, the cells were cooled on ice for 15 min and 10 μl of VLPs (in OMEM ± inhibitor) was added. VLPs were concentrated at the cell surface by spinfection at 4°C at 250g for 60 min. Cells were then washed twice with OMEM ± inhibitor and placed in a 37°C incubator for 3 hours to allow entry to occur. Cells were then washed with loading medium (phenol red–free DMEM containing 25 mM Hepes, 10% FBS, 2.5 mM probenecid, 200 nM bafilomycin, and 2 mM L-glutamine) and loaded with 1 μM CCF2/AM dye (Gene Blazer loading kit, cat. #K1095, Invitrogen) in loading medium for 1 hour in the dark at room temperature. Next, the plates were washed with loading medium and incubated overnight in the dark at room temperature in loading medium supplemented with 10% FBS. Cells were fixed and then analyzed (on the same day as fixation) by flow cytometry with a FACSCalibur flow cytometer (BD Biosciences). When CCF2/AM is cleaved by β-lactamase, its fluorescence shifts from green to blue (43). The percent of cells showing VLP entry was calculated as the ratio of the number of blue cells to the total number of cells gated (those containing CCF2/AM dye) subtracting off backgrounds in samples without VLPs.
VLP internalization was assessed using the same stock of VLPs, but relying on their content of mCherry-VP40 instead of β-lactamase–VP40 (32). In brief, after spinfection and a 1-hour warm-up period, cells were treated with protease to remove VLPs remaining on the cell surface. Samples were then analyzed by flow cytometry to determine the percentage of cells with fluorescent mCherry-VP40 signal, representing internalized VLPs. Cells maintained at 4°C (to prevent internalization) were used as controls to confirm efficacy of protease stripping of VLPs remaining on the cell surface. Cells were analyzed with a LSR Fortessa flow cytometer (BD Biosciences). All flow cytometric data for both the VLP internalization and entry assays were analyzed with the FlowJo software package.
Cathepsin activity and endosome acidification assays
Cathepsin B activity in cell lysates was assayed with the cathepsin B substrate Z-Arg-Arg-7-AMC (Calbiochem), as described in (44). Cathepsin L was assayed in the same manner as cathepsin B, but with the cathepsin L substrate Z-Phe-Arg-7-AMC (Calbiochem) in the presence of 1 μM CA074, a cathepsin B inhibitor (Calbiochem). Combined activity of cathepsin B plus cathepsin L was assayed as above using the cathepsin L substrate in the absence of any inhibitor. Endosomal acidification was assessed with LysoTracker Red (Molecular Probes) as a probe for low-pH organelles. Cells were pretreated with or without inhibitor (as indicated) for 1 hour at 37°C and then incubated with 50 nM LysoTracker Red for an additional 30 min (± inhibitor). Cells were fixed with 4% paraformaldehyde and analyzed by fluorescence microscopy.
In vitro EBOV trafficking assays
SNB19 cells were pretreated with either DMSO, 2.5 μM clomiphene, or 1 μM toremifene for 1 hour. VLP-GP was then spinfected onto cells for 1 hour, after which the cells were warmed for 2 hours (in the presence of DMSO or compound) and then fixed. The cells were then permeabilized and stained for LAMP1 with mAb H4A3 (Developmental Studies Hybridoma Bank) followed by Alexa Fluor 488 anti-mouse mAb (Invitrogen, Molecular Probes). The cells were mounted and then imaged by confocal microscopy. Ten random image fields per sample were analyzed using the JACoP plugin on ImageJ. Auto-thresholds were subtracted for each component 8-bit channel. Colocalization of VLPs (marked by mCherry-VP40) and LAMP (Alexa Fluor 488) was reported as the average Mander’s overlap coefficient.
Supplementary Material
Acknowledgments
We thank J. Towner for the eGFP-expressing Ebola virus Zaire; J. Dye for providing the mAb used in our Ebola virus Sudan screen; E. Ollman-Saphire for the EBOV-GP plasmid; Y. Kawaoka for the VP40 plasmid; and L. Rong for the β-lactamase–VP40 plasmid. We also thank D. Julias for assistance with the initial animal infection studies; K. Spurgers for help with establishing the filovirus cell-based ELISA; J. Pettitt, J. Grenier, K. DeRoche, M. B. Kasda, E. Nelson, and S. Stronsky for technical assistance.
Funding: This work was funded by DTRA project 4.10007_08_RD_B awarded to G.G.O. and subcontract (W81XWH-08-0051) awarded to L.M.J. from USAMRIID, as well as by NIH grant U54 AI057168 awarded to J.M.W. This research was performed while J.M.B. and L.E.D. held National Research Council Research Associateship Awards at USAMRIID. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. Army.
Footnotes
Author contributions: L.M.J., B.G.H., J.M.B., J.L., L.E.H., J.M.W., and G.G.O. designed the research; J.M.B., S.E.D., C.J.S., A.S., C.L., K.L.S., L.E.D., and C.S. performed the research; L.M.J., J.M.B., S.E.D., C.J.S., B.G.H., K.L.S., J.L., J.M.W., and G.G.O. analyzed the data; and L.M.J., J.M.B., J.M.W., and G.G.O. wrote the paper.
Competing interests: B.G.H. and J.L. were employed at Zalicus Inc. during the time the research was performed. L.M.J. is currently employed at Zalicus Inc. L.M.J. and G.G.O. hold a patent entitled “Composition and methods for the treatment of filovirus-mediated diseases,” Application No. 12710203. The other authors declare no competing interests. The authors confirm that this manuscript complies with the Science Translational Medicine materials and data sharing policy.
REFERENCES AND NOTES
- 1.Geisbert TW, Hensley LE. Ebola virus: New insights into disease aetiopathology and possible therapeutic interventions. Expert Rev Mol Med. 2004;6:1–24. doi: 10.1017/S1462399404008300. [DOI] [PubMed] [Google Scholar]
- 2.Mahanty S, Bray M. Pathogenesis of filoviral haemorrhagic fevers. Lancet Infect Dis. 2004;4:487–498. doi: 10.1016/S1473-3099(04)01103-X. [DOI] [PubMed] [Google Scholar]
- 3.Alibek K. The Soviet Union’s anti-agricultural biological weapons. Ann N Y Acad Sci. 1999;894:18–19. doi: 10.1111/j.1749-6632.1999.tb08038.x. [DOI] [PubMed] [Google Scholar]
- 4.Towner JS, Paragas J, Dover JE, Gupta M, Goldsmith CS, Huggins JW, Nichol ST. Generation of eGFP expressing recombinant Zaire Ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology. 2005;332:20–27. doi: 10.1016/j.virol.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 5.Shelly W, Draper MW, Krishnan V, Wong M, Jaffe RB. Selective estrogen receptor modulators: An update on recent clinical findings. Obstet Gynecol Surv. 2008;63:163–181. doi: 10.1097/OGX.0b013e31816400d7. [DOI] [PubMed] [Google Scholar]
- 6.Palacios S. The future of the new selective estrogen receptor modulators. Menopause Int. 2007;13:27–34. doi: 10.1258/175404507780456791. [DOI] [PubMed] [Google Scholar]
- 7.Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, III, Staunton JE, Jin X, Lee MS, Zimmermann GR, Borisy AA. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27:659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ebihara H, Theriault S, Neumann G, Alimonti JB, Geisbert JB, Hensley LE, Groseth A, Jones SM, Geisbert TW, Kawaoka Y, Feldmann H. In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J Infect Dis. 2007;196(Suppl 2):S313–S322. doi: 10.1086/520590. [DOI] [PubMed] [Google Scholar]
- 9.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 1998;178:651–661. doi: 10.1086/515386. [DOI] [PubMed] [Google Scholar]
- 10.Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis. 1999;179(Suppl 1):S248–S258. doi: 10.1086/514292. [DOI] [PubMed] [Google Scholar]
- 11.Mikkelson TJ, Kroboth PD, Cameron WJ, Dittert LW, Chungi V, Manberg PJ. Single-dose pharmacokinetics of clomiphene citrate in normal volunteers. Fertil Steril. 1986;46:392–396. doi: 10.1016/s0015-0282(16)49574-9. [DOI] [PubMed] [Google Scholar]
- 12.Morello KC, Wurz GT, DeGregorio MW. Pharmacokinetics of selective estrogen receptor modulators. Clin Pharmacokinet. 2003;42:361–372. doi: 10.2165/00003088-200342040-00004. [DOI] [PubMed] [Google Scholar]
- 13.Taras TL, Wurz GT, Linares GR, DeGregorio MW. Clinical pharmacokinetics of toremifene. Clin Pharmacokinet. 2000;39:327–334. doi: 10.2165/00003088-200039050-00002. [DOI] [PubMed] [Google Scholar]
- 14.Evans MJ, Harris HA, Miller CP, Karathanasis SK, Adelman SJ. Estrogen receptors α and β have similar activities in multiple endothelial cell pathways. Endocrinology. 2002;143:3785–3795. doi: 10.1210/en.2002-220356. [DOI] [PubMed] [Google Scholar]
- 15.Kim-Schulze S, McGowan KA, Hubchak SC, Cid MC, Martin MB, Kleinman HK, Greene GL, Schnaper HW. Expression of an estrogen receptor by human coronary artery and umbilical vein endothelial cells. Circulation. 1996;94:1402–1407. doi: 10.1161/01.cir.94.6.1402. [DOI] [PubMed] [Google Scholar]
- 16.Toth B, Saadat G, Geller A, Scholz C, Schulze S, Friese K, Jeschke U. Human umbilical vascular endothelial cells express estrogen receptor beta (ERβ) and progesterone receptor A (PR-A), but not ERα and PR-B. Histochem Cell Biol. 2008;130:399–405. doi: 10.1007/s00418-008-0426-7. [DOI] [PubMed] [Google Scholar]
- 17.Dolnik O, Kolesnikova L, Becker S. Filoviruses: Interactions with the host cell. Cell Mol Life Sci. 2008;65:756–776. doi: 10.1007/s00018-007-7406-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hunt CL, Lennemann NJ, Maury W. Filovirus entry: A novelty in the viral fusion world. Viruses. 2012;4:258–275. doi: 10.3390/v4020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White JM, Schornberg KL. A new player in the puzzle of filovirus entry. Nat Rev Microbiol. 2012;10:317–322. doi: 10.1038/nrmicro2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nanbo A, Imai M, Watanabe S, Noda T, Takahashi K, Neumann G, Halfmann P, Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010;6:e1001121. doi: 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brindley MA, Hughes L, Ruiz A, McCray PB, Jr, Sanchez A, Sanders DA, Maury W. Ebola virus glycoprotein 1: Identification of residues important for binding and postbinding events. J Virol. 2007;81:7702–7709. doi: 10.1128/JVI.02433-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308:1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J Virol. 2006;80:4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brecher M, Schornberg KL, Delos SE, Fusco ML, Saphire EO, White JM. Cathepsin cleavage potentiates the Ebola virus glycoprotein to undergo a subsequent fusion-relevant conformational change. J Virol. 2012;86:364–372. doi: 10.1128/JVI.05708-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dube D, Brecher MB, Delos SE, Rose SC, Park EW, Schornberg KL, Kuhn JH, White JM. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): Sequence and residues critical for host cell binding. J Virol. 2009;83:2883–2891. doi: 10.1128/JVI.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. Ebola virus entry requires the cholesterol transporter Niemann–Pick C1. Nature. 2011;477:340–343. doi: 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Côté M, Misasi J, Ren T, Bruchez A, Lee K, Filone CM, Hensley L, Li Q, Ory D, Chandran K, Cunningham J. Small molecule inhibitors reveal Niemann–Pick C1 is essential for Ebola virus infection. Nature. 2011;477:344–348. doi: 10.1038/nature10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, Ruthel G, Pfeffer SR, Dye JM, Whelan SP, Brummelkamp TR, Chandran K. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31:1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dube D, Schornberg KL, Shoemaker CJ, Delos SE, Stantchev TS, Clouse KA, Broder CC, White JM. Cell adhesion-dependent membrane trafficking of a binding partner for the ebolavirus glycoprotein is a determinant of viral entry. Proc Natl Acad Sci USA. 2010;107:16637–16642. doi: 10.1073/pnas.1008509107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kunz S. Receptor binding and cell entry of Old World arenaviruses reveal novel aspects of virus-host interaction. Virology. 2009;387:245–249. doi: 10.1016/j.virol.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 31.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010;6:e1001110. doi: 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, White JM. Multiple cationic amphiphiles induce a Niemann-Pick C phenotype and inhibit ebola virus entry and infection. PLoS One. 2013;8:e56265. doi: 10.1371/journal.pone.0056265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bale S, Liu T, Li S, Wang Y, Abelson D, Fusco M, Woods VL, Jr, Saphire EO. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl Trop Dis. 2011;5:e1395. doi: 10.1371/journal.pntd.0001395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu R, Lu P, Chu JW, Sharom FJ. Characterization of fluorescent sterol binding to purified human NPC1. J Biol Chem. 2009;284:1840–1852. doi: 10.1074/jbc.M803741200. [DOI] [PubMed] [Google Scholar]
- 35.Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010;6:e1001131. doi: 10.1371/journal.ppat.1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wahl-Jensen V, Bollinger L, Safronetz D, de Kok-Mercado F, Scott DP, Ebihara H. Use of the Syrian hamster as a new model of Ebola virus disease and other viral hemorrhagic fevers. Viruses. 2012;4:3754–3784. doi: 10.3390/v4123754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hevey M, Negley D, Geisbert J, Jahrling P, Schmaljohn A. Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology. 1997;239:206–216. doi: 10.1006/viro.1997.8883. [DOI] [PubMed] [Google Scholar]
- 38.Hevey M, Negley D, Schmaljohn A. Characterization of monoclonal antibodies to Marburg virus (strain Musoke) glycoprotein and identification of two protective epitopes. Virology. 2003;314:350–357. doi: 10.1016/s0042-6822(03)00416-1. [DOI] [PubMed] [Google Scholar]
- 39.Wilson JA, Hevey M, Bakken R, Guest S, Bray M, Schmaljohn AL, Hart MK. Epitopes involved in antibody-mediated protection from Ebola virus. Science. 2000;287:1664–1666. doi: 10.1126/science.287.5458.1664. [DOI] [PubMed] [Google Scholar]
- 40.Weidmann M, Mühlberger E, Hufert FT. Rapid detection protocol for filoviruses. J Clin Virol. 2004;30:94–99. doi: 10.1016/j.jcv.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 41.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cavrois M, De Noronha C, Greene WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat Biotechnol. 2002;20:1151–1154. doi: 10.1038/nbt745. [DOI] [PubMed] [Google Scholar]
- 43.Yonezawa A, Cavrois M, Greene WC. Studies of ebola virus glycoprotein-mediated entry and fusion by using pseudotyped human immunodeficiency virus type 1 virions: Involvement of cytoskeletal proteins and enhancement by tumor necrosis factor α. J Virol. 2005;79:918–926. doi: 10.1128/JVI.79.2.918-926.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ebert DH, Deussing J, Peters C, Dermody TS. Cathepsin L and cathepsin B mediate reovirus disassembly in murine fibroblast cells. J Biol Chem. 2002;277:24609–24617. doi: 10.1074/jbc.M201107200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.