

Abstract
Background
Congenital diaphragmatic hernia (CDH) is a common birth defect affecting 1 in 3,000 births. It is characterized by herniation of abdominal viscera through an incompletely formed diaphragm. Although chromosomal anomalies and mutations in several genes have been implicated, the cause for most patients is unknown.
Methods
We used whole exome sequencing in two families with CDH and congenital heart disease, and identified mutations in GATA6 in both.
Results
In the first family, we identified a de novo missense mutation (c.1366C>T, p.R456C) in a sporadic CDH patient with tetralogy of Fallot. In the second, a nonsense mutation (c.712G>T, p.G238*) was identified in two siblings with CDH and a large ventricular septal defect. The G238* mutation was inherited from their mother, who was clinically affected with congenital absence of the pericardium, patent ductus arteriosus, and intestinal malrotation. Deep sequencing of blood and saliva derived DNA from the mother suggested somatic mosaicism as an explanation for her milder phenotype, with only approximately 15% mutant alleles. To determine the frequency of GATA6 mutations in CDH, we sequenced the gene in 378 patients with CDH. We identified one additional de novo mutation (c.1071delG, p.V358Cfs34*).
Conclusions
Mutations in GATA6 have been previously associated with pancreatic agenesis and congenital heart disease. We conclude that, in addition to the heart and the pancreas, GATA6 is involved in development of two additional organs, the diaphragm and the pericardium. In addition we have shown that de novo mutations can contribute to the development of CDH, a common birth defect.
Keywords: Congenital diaphragmatic hernia, de novo mutation, GATA6, somatic mutation, whole exome sequencing
INTRODUCTION
Congenital diaphragmatic hernia (CDH [OMIM 142340]) is a birth defect characterized by the herniation of abdominal viscera into the chest cavity through the incomplete formation of the diaphragm. The prevalence of CDH is approximately 1 in 3,000 births.[1] Newborns with CDH often have severe respiratory distress resulting from pulmonary hypoplasia and pulmonary hypertension, which contribute significantly to its morbidity and mortality. CDH can occur as an isolated defect but occurs with other anomalies approximately 40% of the time.[1]
Chromosomal anomalies, including whole chromosome and segmental aneuplodies, are currently the most commonly recognized genetic cause of CDH.[2] Mutations in single genes have also been identified in patients with syndromic CDH.[3] However, the etiology remains unknown in most CDH patients. This is likely due to the historically high mortality in CDH, resulting in very few multiplex families amenable to traditional linkage analysis. De novo rare variants have been identified as a cause of sporadic diseases with decreased reproductive fitness,[4] and a pathogenic de novo variant in GATA4 was recently identified as a cause of CDH.[5] Lango Allen et al identified a de novo c.1516+4A>G mutation in GATA6 in a patient with pancreatic agenesis and CDH. [6] Whole exome sequencing (WES) has enabled the rapid and systematic identification of rare, causative variants in Mendelian disorders. We used WES in two families with CDH and congenital heart defects, and identified mutations in GATA binding protein 6 (GATA6) in both. Sequencing of a cohort of 378 patients with CDH identified one additional patient with a GATA6 mutation. This is, to our knowledge, the largest cohort of CDH to undergo targeted sequencing to date. Since less than 1% of patients with CDH have mutations in GATA6, our data suggest that CDH is genetically heterogeneous, and additional loci remain to be discovered.
MATERIALS AND METHODS
Participants
We studied the subset of patients without pathogenic chromosomal anomalies who were recruited as part of DHREAMS (Diaphragmatic Hernia Research & Exploration; Advancing Molecular Science) study, which was approved by the Institutional Review Boards at Columbia University, Washington University Medical Center/St. Louis Children’s Hospital, University of Pittsburgh, Cincinnati Children’s Hospital and Medical Center/University of Cincinnati, Omaha Children’s Hospital/University of Nebraska, University of Michigan/CS Mott Children’s Hospital, and Vanderbilt University Medical Center.[2] Medical records were reviewed and family history of diaphragm defects and major malformations was extracted from the medical record by project coordinators. Informed consent was obtained from all parents or guardians. Family 2 was recruited at Seattle Children’s Hospital after informed consent with approval by the Institutional Review Board at Seattle Children’s Hospital.
Clinical Information
We sought to identify mutations by exome sequencing of two families with CDH, one sporadic and one familial. In family 1 the proband (family 1:II-1) had a left sided CDH, tetralogy of Fallot (TOF) and a single umbilical artery. At the age of 3 his development has been normal, and he has had no hyperglycemia. His parents are unaffected. In family 2 the proband (family 2: III-1) presented perinatally with CDH and a large ventricular septal defect (VSD). No dysmorphic features or other anomalies were noted. He survived surgical repair of his CDH, but died shortly thereafter due to intractable pulmonary hypertension. His mother’s medical history was notable for the presence of patent ductus arteriosus (PDA) as a full-term infant, and congenital absence of the pericardium, which was asymptomatic but noted intraoperatively during her PDA repair. She was also found to have intestinal malrotation, diagnosed and repaired at 25 years of age. Her second pregnancy was terminated after prenatal diagnosis of left sided CDH and large VSD. Autopsy of that fetus also showed bilateral cervical ribs, absent right 12th rib and left ureteral duplication.
Exome Sequencing Analysis
Three members of family 1 and six members of family 2 were analyzed by exome sequencing (Figure 1). Genomic DNA extracted from whole blood was fragmented and exomes were captured using the Agilent SureSelect™ Human All Exon kit (family 1) and the Nimblegen SeqCap EZ Human Exome Library v2.0 (family 2). The captured DNA was sequenced with 100 bp paired-end reads (family 1) or 50 bp reads (family 2) on an Illumina HiSeq 2000 according to the manufacturer’s protocol. Sequencing reads were aligned to the human genome reference sequence human assembly hg19 using BWA (Version 0.5.9),[7] and GATK (Unified Genotyper; version 1.0) was used to refine local alignment of reads, recalibrate base quality score, and call variants (single nucleotide variants (SNVs)/indels) within targeted regions. In addition to the default filters in GATK, variants were further filtered for genotype minimum quality of 30, minimum quality over depth of 5, minimum strand bias −0.10, and maximum fraction of reads with mapping quality of zero at 10%. Annotated variants were subsequently filtered to exclude the variants greater or equal to 1% of minor allele frequency based on dbSNP135 and the 1000 genomes project (Family 1) and the NHLBI Exome Variant Server (Families 1 and 2). Variants predicted to be pathogenic by either SIFT or PolyPhen2 were further analyzed. De novo SNVs were identified when heterozygous in proband and homozygous for the common allele in the parents.
Figure 1. Pedigrees of CDH families analyzed by WES.

A. Filled, dark blue indicates subjects affected with CDH. Filled light blue indicates congenital absence of the pericardium. +/+ indicates reference genotype, −/+ indicates heterozygosity for the mutation. B. Sequence chromatograms of affected individuals of family 1 (c.1366C>T, p. R456C) and family 2 (c.712G>T, p.G238*) are shown. Note that the peak height of the mutant allele in II-2 is less than 50%. CDH (congenital diaphragmatic hernia), TOF (tetralogy of Fallot), SUA (single umbilical artery), PDA (patent ductus arteriosus), CAP (congenital absence of the pericardium), malro (intestinal malrotation), VSD (ventricular septal defect), ribs (bilateral cervical ribs and absent right 12th rib), dup ureter (duplicate left ureter).
After filtering, additional prediction tools were applied to obtain more detailed pathogenicity scores for each variant. These included SeattleSeq Annotation 134, ANNOVAR, Pmut, and MutationAssessor.
Mutation screening and variant validation
Primers were designed to confirm potentially pathogenic variants and to sequence all the coding exons and splice junctions of GATA6 in 104 CDH patients with congenital heart disease from DHREAMS by Sanger sequencing (Applied Biosystems, Foster City, CA). We screened an overlapping set of 357 CDH patients from the DHREAMS cohort, as well as 184 controls from Coriell plates NDPT020 and NDPT090, which together represent a group of unique and unrelated Caucasian males (n=59) and females (n=125) who were assessed as being neurologically normal at the time of collection (age 19–46 years). None have any first degree relative with a known primary neurological disorder. For more detailed information see (http://ccr.coriell.org). To avoid the large amount of time and money that would be required for Sanger sequencing all 541 samples we used Molecular Inversion Probes (MIPs) to capture all exons and exon/intron boundaries (5bp flanking) of GATA6.[8] This method requires minimal DNA input and due to the extensive multiplexing of samples prior to sequencing,[9] is cost effective. Briefly, pooled MIPs were used to capture 50 nanograms of each proband’s DNA. PCR was performed using universal primers with the introduction of unique eight-base barcodes on the tagged reverse primer. Pooled libraries were subject to massively parallel sequencing using 101 paired end protocol on an Illumina MiSeq. Candidate variants identified by MIP capture were confirmed by Sanger sequencing. Mutations are reported using standard HGVS nomenclature (GATA6 RefSeq NM005257.4).
Somatic mosaic analysis of II-1
An independent pair of MIPs was designed to capture the region around chr18:g.19751817G>T, where the pathogenic variant in GATA6 in family 2 was found. 100 nanograms of DNA derived from saliva or blood was used for capture, and experiments were performed in triplicate. The remainder of the procedure, including PCR and sequencing, was as described above under mutation screening.
RESULTS
We detected a total of 31,005 variants in the proband from family 1, including 25,014 single nucleotide variants (SNVs) and 5,991 indels in the proband (Figure 1, supplementary table 1). After filtering, 5 de novo SNVs and 15 indels remained. Three of the five germline missense de novo SNVs present in the blood (GATA6, c.1366C>T, p.R456C; SLC5A9, c.172C>T, p.R58C; EME1, c.634G>A, p.G212R) were confirmed by Sanger sequencing. Paternity and maternity were confirmed by segregation of multiple variants. None of 15 indels were confirmed by Sanger sequencing. Of the three de novo SNVs identified, GATA6 was identified as the likely pathogenic cause because the same rare variant has been previously reported in another two patients with pancreatic agenesis. In addition functional studies showed that this variant inhibited the ability of GATA6 to bind to and activate expression of target genes [6]. The heterozygous SNV c.634G>A (p.G212R) in EME1 is rs146272309 and has been identified in 2 of 2203 African Americans and none of the 4300 European Americans in EVS. The majority of the prediction algorithms suggest that the p.G212R is of low functional impact. Furthermore, the residue G212 is not highly conserved across species. The heterozygous SNV c.172C>T (p.R58C) in SLC5A9 has been identified in 1 of 2203 African Americans but not in 4300 European Americans in EVS. The residue R58 in SLC5A9 was predicted to be pathogenic by the majority of algorithms. This gene is not highly expressed in heart, lung, or muscle,[10] suggesting that this variant was less likely to be the pathogenic cause of CDH.
Family 2 was analyzed independently. A total of 98,955 variants were detected in these 6 individuals (Figure 1). Given the close embryonic relationship between the pericardium and the diaphragm, as well as the presence of a congenital heart defect in both the mother and her children, we hypothesized that a de novo mutation in II-2 that was subsequently inherited by III-1 and III-2 was likely. We filtered for de novo variants in II-2 that were not found in the NHLBI Exome Variant Server and were non-synonymous or potentially affected splicing, identifying a total of 6 candidate variants (see supplementary table 2), one of which was stop-gained (GATA6, c.712G>T; p.G238*). Sanger sequencing confirmed that this mutation was de novo in II-2 and inherited by both affected offspring (Figure 1B). Of the remaining 5 variants, 3 did not segregate to both affected children, one was present in 1000 genomes with a minor allele frequency of 0.16 (in a region not covered in the Exome Variant Sever), and the other was a conservative change present in other vertebrates and predicted to benign.
We additionally examined genes containing rare/novel and functional variants from the proband of family 1 with genes containing rare/novel and functional variants found in affected individuals from family 2. In addition to GATA6 there were 7 other overlapping genes, all of which were missense variants either not evolutionarily conserved or were predicted to be benign (see supplementary table 3). All of these variants were inherited from unaffected individuals and are unlikely to be contributing to our patient’s phenotypes.
We hypothesized that somatic mosaicism might explain the significantly milder phenotype seen in II-2 compared with her offspring. This was suggested by the decreased mutant peak height on Sanger sequencing (Figure 1B). To quantitatively measure the percent mosaicism in II-2, we performed targeted deep sequencing (average coverage of 450×) of the mutated region of GATA6 from both a single blood and saliva derived DNA sample. Only 16% of alleles from saliva and 15% of alleles from blood were mutant (Table 1), suggesting that the c.712G>T mutation is post-zygotic.
Table 1.
Evidence for somatic mosaicism in II-1 (family 2)*
| Source | Ref (G) reads | Alt (T) reads | % Alt reads | Total |
|---|---|---|---|---|
| blood | 399 | 26 | 6% | 425 |
| blood | 501 | 132 | 21% | 633 |
| blood | 66 | 9 | 12% | 75 |
| saliva | 451 | 109 | 19% | 560 |
| saliva | 497 | 54 | 10% | 551 |
| saliva | 358 | 95 | 21% | 453 |
| control | 519 | 0 | 0% | 519 |
The region of GATA6 containing the c.712G>T mutation (chr18:g.19751817G>T) was captured from blood- or saliva-derived DNA and sequenced at high depth. The number of reference and mutant alleles are shown. The weighted average percent of mutant alleles is 16% from saliva and 15% from blood. Results from each independent experiment (performed in triplicate) are shown.
We sequenced GATA6 in an additional 104 patients with CDH as well as congenital heart defects by Sanger sequencing, and did not identify any additional mutations. Mutations in GATA4, a paralogue of GATA6, have recently been identified in only 1 of a cohort of 96 patients with CDH,[5] so we reasoned that a larger cohort might be needed to find additional patients with GATA6 mutations. We took advantage of a recently described high throughput targeted sequencing approach,[8] using molecular inversion probes (MIPs) to screen a cohort of 357 patients with CDH (clinical information in supplementary Table 4), 83 of whom overlapped with those Sanger sequenced. In screening for all types of rare variants, we identified one additional de novo GATA6 mutation (c.1071delG, p.V358Cfs34*) in a patient with CDH and an atrial septal defect, which was not found in 1000 genomes or EVS. Sanger sequencing of the proband and both parents confirmed that the mutation was present and de novo. We used the same technique to screen a cohort of control individuals, and did not find any of the mutations reported here among 184 controls screened.
DISCUSSION
Here, we have used both WES and targeted capture to identify three unrelated families with CDH and GATA6 mutations. Although GATA6 mutations have previously been reported in patients with congenital heart disease and pancreatic agenesis,[6, 9] we have expanded the phenotypic spectrum to include CDH. We identified a somatic mutation in GATA6 in a patient with a PDA, intestinal malrotation, and congenital absence of the pericardium, who passed the mutation to two offspring who were affected with CDH and congenital heart disease. Lastly, we screened a total of 378 patients with CDH and found only one additional case with a GATA6 mutation, suggesting that mutations in GATA6 are likely to account for <1% of all patients with CDH. Given the embryonic lethality of GATA6 null mice,[11] it is possible that GATA6 mutations may result in spontaneous fetal demise which might account for the low frequency in livebirths.
Several lines of evidence support the pathogenicity of these mutations. The de novo missense variant (family 1: c.1366C>T p.R456C) is in a highly conserved zinc finger region of GATA6 (Figure 2). This mutation has been previously reported in two patients with pancreatic agenesis.[6] One of these patients also had truncus arteriosus, perimembranous vertricular septal defect, developmental delay and seizures. The other patient had TOF, developmental delay and an umbilical hernia. Our patient had no evidence of pancreatic agenesis or developmental delay, but did have CDH, TOF and a single umbilical artery.
Figure 2.
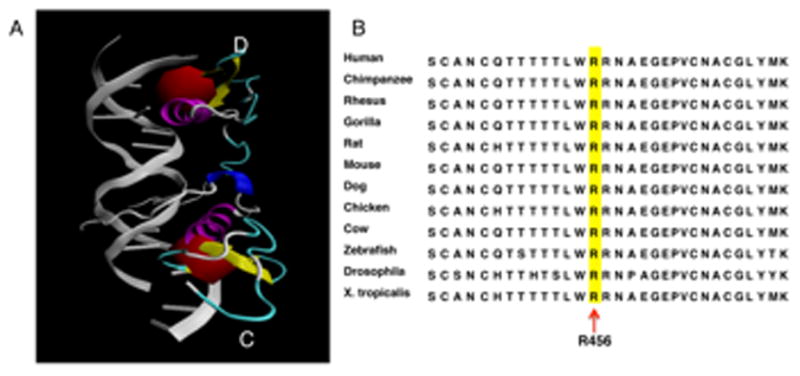
A. Protein structure of GATA6 C-terminal zinc finger bound to DNA. A. Protein Data Bank entry 3dfv, visualized using VMD1.9.1. [22, 23] Two C-terminal zinc finger domains of GATA6 (chains C and D) are shown bound to DNA (in grey). Residue Arg456, highlighted in red, participates directly in DNA binding. B. Amino acid alignment of a portion (Ser443 to Lys473) of C-terminal zinc finger in GATA6 from different species. Arginine 456 is highlighted.
The de novo frameshift mutation (c.1071delG, p.V358Cfs34*), identified in a patient with CDH and atrial septal defect, has not been previously reported, but two other frameshift mutations have been reported nearby (c.1036_1042del, p.T346Pfs*44 and c.1108_1121dup, p.G375Sfs*22).[6, 9] The T346Pfs*44 mutation was reported in a patient with pancreatic agenesis, congenital heart disease, and neurocognitive deficits.[9] Interestingly, this mutation was inherited from a father who was diagnosed with diabetes at 46 years of age, leading the authors to suggest that GATA6 mutations could contribute to later onset, milder phenotypes. The G375Sfs*22 mutation was de novo and the patient had pancreatic agenesis and TOF.[6] The most likely outcome of these frameshift mutations is mRNA instability via nonsense mediated decay (NMD), resulting in haploinsufficiency. The recent report of a 4.7 Mb interstitial deletion that contains GATA6 (and 24 other RefSeq genes) in a patient with complex congenital heart disease supports a haploinsufficient pathogenesis.[12]
The nonsense mutation (family 2: c.712G>T; p.G238*), identified in two siblings with CDH and a large VSD, has not been previously reported. The mother of these patients carried this mutation as well, but with a mutant allele frequency from saliva and blood of only ~15%. This suggests that her relatively milder phenotype, which included PDA, intestinal malrotation, and congenital absence of the pericardium, may be the result of somatic mosaicism. There are less than 400 cases of congenital absence of the pericardium reported in the literature, but it is often asymptomatic and discovered incidentally.[13] Congenital absence of the pericardium, in association with CDH, has been reported several times.[14, 15] This is the first report, to our knowledge, of a genetic cause for this rare condition. Additionally, since both the pericardium and the diaphragm are embryonically derived from the septum transversum, our results support a role for GATA6 in specification or maintenance of this embryonic structure.
Our results underscore the importance of sensitive phenotyping in gene discovery using exome analysis. Congenital absence of the pericardium may be underreported as it is most often asymptomatic and found incidentally,[13] as was the case with our patient (II-2) when it was discovered during surgery for her PDA. If we had not recognized II-2 as being phenotypically affected, we might not have included her parents in the exome analysis, and the family 2: c.712C>T variant we identified would have appeared to be inherited from an unaffected parent. This mechanism, in which post zygotic mutations contribute to mild or even subclinical phenotypes in one generation, but are then passed on as constitutional mutations causing more severe congenital phenotypes in the next, may be a more common inheritance mechanism for apparent “sporadic” birth defects than is currently appreciated.
There are 34 GATA6 mutations reported in the literature (Figure 3). Allen et al. identified 14 mutations (6 missense, 5 indel and 3 splice) in GATA6 in their pancreatic agenesis patients.[6] All of those patients had additional phenotypes, including congenital cardiac defects, developmental gastrointestinal disorders, neurocognitive abnormalities and additional endocrine abnormalities. Among them, one patient with a de novo mutation at c.1516+4A>G had a left CDH without congenital heart disease.[6] The other 20 mutations (3 nonsense, 7 missense and 7 indels and 3 splice) were identified in patients with congenital cardiovascular malformations, pancreatic insufficiency or neonatal diabetes.[9] In addition to the cardiac and pancreatic phenotypes associated with GATA6 mutations, we add CDH to the phenotypic spectrum. There was no evidence of hyperglycemia or pancreatic insufficiency in any of our patients. One of them (III-2) had a full autopsy which confirmed the presence of pancreatic tissue, though it was noted to be associated with the mesentery of the small bowel and not in its usual location. At present we do not have an explanation for the phenotypic variability among patients with GATA6 mutations.
Figure 3. GATA6 mutations.
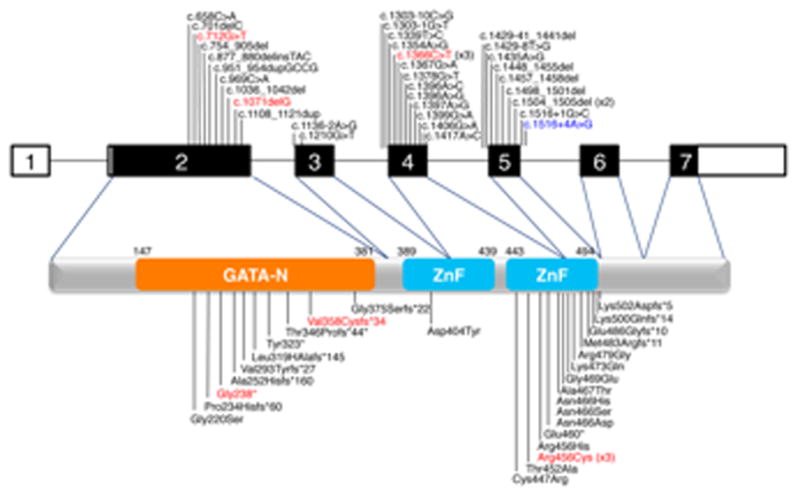
Filled squares are coding exons of GATA6. All identified disease causing mutations are indicated. Mutations identified in this study indicated in red. Mutations that have been identified in greater than one family are also indicated. The c. 1516+4A>G mutation, indicated in blue, was identified in a patient with pancreatic agenesis and left diaphragmatic hernia.[6] Mutations are according to RefSeq NM005257.4. See main text for references for other mutations.
GATA6 is a zinc finger containing transcription factor that plays a crucial role in differentiation and organogenesis. It is highly expressed in heart, lung, gut and gonads.[16] Similar to GATA4, GATA6 has two zinc fingers as a well as a GATA-N transactivation domain. The Arg456 residue is located at the surface of the C-terminal zinc finger (Figure 2). The R456C mutation, which is predicted to disrupt DNA binding,[6] has been reported in three unrelated patients (including this report), making it the most common mutation seen in GATA6 to date.
Genetic studies in mice have shown that GATA6 is essential for lung development[17] through regulation of Wnt signaling.[18] Chimeric and explant-based experiments in mice support a direct role for GATA6 in branching morphogenesis and surfactant expression.[17] These results suggest that, in some cases, the pulmonary hypertension and lung hypoplasia associated with CDH are due to an abnormal developmental program and not simply due to mechanical compression of herniated viscera against developing lung.
In mice, GATA6 is expressed in the septum transversum mesenchyme as well as the primordial diaphragmatic mesenchyme[19, 20]. Interestingly, GATA6 expression is restricted to the lateral pleuroperitoneal folds (PPFs) [19], suggesting a potential explanation for the observation that the vast majority of CDH is located in this posterolateral region. All of our CDH patients had left sided posterolateral diaphragmatic hernias. No pericardial abnormalities were identified in individuals II-1 (family 1) or III-1 and III-2 (family 2), and all three had either autopsy or surgeries that would have identified an absence of the pericardium. Although the mechanisms of embryonic development of the human diaphragm remain controversial[21], our demonstration that mutations in GATA6 can cause posterolateral CDH support the importance of the lateral pleuroperitoneal folds in human diaphragm development.
In summary, we used WES to identify de novo mutations in GATA6 in patients with CDH and congenital heart disease. Since all three GATA6 mutations were identified in CDH patients with other cardiac malformations, patients with CDH and congenital heart disease should be prioritized in screening for GATA6 mutations. Multiple phenotypes have been described with the same mutation in GATA6 suggesting that additional modifying genes or environmental factors determine which organ systems will be affected. Our data suggests that WES will provide a powerful tool to identify genes and mutations associated with CDH.
Supplementary Material
Acknowledgments
We greatly appreciate the families who participated in this study and all the clinical care teams who assisted with study coordination. We are grateful for the technical assistance provided by Patricia Lanzano, Jiancheng Guo and Liyong Deng from Columbia University. We also thank Jeannie Kreutzman and Robert Drongowski from University of Michigan; Trish Burns from Cincinnati Children’s Hospital Medical Center; Sheila Horak from University of Nebraska; Mary Dabrowiak from Monroe Carell Jr Children’s Hospital at Vanderbilt; and Laurie Luther from University of Pittsburgh. Study data were collected and managed using Research Electronic Data Capture (REDCap) electronic data capture tools hosted at Columbia University. REDCap is a secure, web-based application designed to support data capture for research studies. This work was supported by NIH grant R01 HD057036, Cherubs, Global CDH, and was supported in part by Columbia University’s CTSA grant UL1 RR024156 from NCATS-NCRR/NIH, a Burroughs Wellcome Fund Career Award for Medical Scientists (HCM), and Medical Genetics training grant T32GM007454 (JTB).
Abbreviations
- CDH
congenital diaphragmatic hernia
- WES
whole exome sequencing
- TOF
tetralogy of Fallot
- VSD
ventricular septal defect
- ASD
atrial septal defect
Footnotes
Contributors: LY, JTB, HM, WKC conceived the study. JTB, JW, GBM, KSA, TMC, DHC, DP, BWW, BB, CS, GA, MSA, HM, WKC contributed patients. UWCMG performed whole exome sequencing (Family 2). JTB and GLC designed and performed targeted resequencing. LY, JTB, GLC, YHC, YS, HM, WKC analysed the data. LY, JTB, HM, WKC wrote the paper. All authors read, revised and approved the final version of the manuscript.
Patient consent: Obtained.
Data sharing statement: The data presented in the manuscript are available on request
Competing interests: None.
Ethics approval: Institutional Review Boards at Columbia University, Washington University Medical Center/St. Louis Children’s Hospital, University of Pittsburgh, Cincinnati Children’s Hospital and Medical Center/University of Cincinnati, Omaha Children’s Hospital/University of Nebraska, University of Michigan/CS Mott Children’s Hospital, Vanderbilt University Medical Center, and Seattle Children’s Hospital.
Provenance and peer review: Not commissioned; externally peer reviewed
Web Resources: DHREAMS (www.cdhgenetics.com/)
dbSNP135 (http://www.genome.ucsc.edu/cgi-bin/hgGateway)
1000 genome project (www.1000genomes.org/)
NHLBI Exome Variant Server (EVS) (http://evs.gs.washington.edu/EVS/).
SIFT (http://sift.jcvi.org/)
PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)
SeattleSeq Annotation 134 (http://snp.gs.washington.edu/SeattleSeqAnnotation134/)
ANNOVAR (http://www.openbioinformatics.org/annovar/)
Pmut (http://mmb.pcb.ub.es)
MutationAssessor (http://mutationassessor.org/)
References
- 1.Pober BR. Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. Am J Med Genet C Semin Med Genet. 2007;145C(2):158–71. doi: 10.1002/ajmg.c.30126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yu L, Wynn J, Ma L, Guha S, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, LeDuc CA, Costa K, Stolar C, Aspelund G, Arkovitz MS, Chung WK. De novo copy number variants are associated with congenital diaphragmatic hernia. J Med Genet. 2012;49(10):650–9. doi: 10.1136/jmedgenet-2012-101135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Slavotinek AM. Single gene disorders associated with congenital diaphragmatic hernia. Am J Med Genet C Semin Med Genet. 2007;145C(2):172–83. doi: 10.1002/ajmg.c.30125. [DOI] [PubMed] [Google Scholar]
- 4.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13(8):565–75. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 5.Yu L, Wynn J, Cheung YH, Shen Y, Mychaliska GB, Crombleholme TM, Azarow KS, Lim FY, Chung DH, Potoka D, Warner BW, Bucher B, Stolar C, Aspelund G, Arkovitz MS, Chung WK. Variants in GATA4 are a rare cause of familial and sporadic congenital diaphragmatic hernia. Hum Genet. 2013;132(3):285–92. doi: 10.1007/s00439-012-1249-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lango Allen H, Flanagan SE, Shaw-Smith C, De Franco E, Akerman I, Caswell R, Ferrer J, Hattersley AT, Ellard S. GATA6 haploinsufficiency causes pancreatic agenesis in humans. Nat Genet. 2012;44(1):20–2. doi: 10.1038/ng.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.O’Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, Munson J, Hiatt JB, Turner EH, Levy R, O’Day DR, Krumm N, Coe BP, Martin BK, Borenstein E, Nickerson DA, Mefford HC, Doherty D, Akey JM, Bernier R, Eichler EE, Shendure J. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science. 2012;338(6114):1619–22. doi: 10.1126/science.1227764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Franco E, Shaw-Smith C, Flanagan SE, Shepherd MH, Hattersley AT, Ellard S. GATA6 mutations cause a broad phenotypic spectrum of diabetes from pancreatic agenesis to adult-onset diabetes without exocrine insufficiency. Diabetes. 2013;62(3):993–7. doi: 10.2337/db12-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tazawa S, Yamato T, Fujikura H, Hiratochi M, Itoh F, Tomae M, Takemura Y, Maruyama H, Sugiyama T, Wakamatsu A, Isogai T, Isaji M. SLC5A9/SGLT4, a new Na+-dependent glucose transporter, is an essential transporter for mannose, 1,5-anhydro-D-glucitol, and fructose. Life Sci. 2005;76(9):1039–50. doi: 10.1016/j.lfs.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 11.Morrisey EE, Tang Z, Sigrist K, Lu MM, Jiang F, Ip HS, Parmacek MS. GATA6 regulates HNF4 and is required for differentiation of visceral endoderm in the mouse embryo. Genes Dev. 1998;12(22):3579–90. doi: 10.1101/gad.12.22.3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bui PH, Dorrani N, Wong D, Perens G, Dipple KM, Quintero-Rivera F. First report of a de novo 18q11. 2 microdeletion including GATA6 associated with complex congenital heart disease and renal abnormalities. Am J Med Genet A. 2013;161:1773–8. doi: 10.1002/ajmg.a.35974. [DOI] [PubMed] [Google Scholar]
- 13.Verde F, Johnson PT, Jha S, Fishman EK, Zimmerman SL. Congenital absence of the pericardium and its mimics. J Cardiovasc Comput Tomogr. 2013;7(1):11–7. doi: 10.1016/j.jcct.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Verloes A, Perrin L, Delbecque K, Gonzales M, Demarche M, Dekoster G. Congenital absence of the left pericardium and diaphragmatic defect in sibs. Eur J Med Genet. 2010;53(3):133–5. doi: 10.1016/j.ejmg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 15.Patel Y, McNally J, Ramani P. Left congenital diaphragmatic hernia, absent pericardium, and liver heterotopia: a case report and review. J Pediatr Surg. 2007;42(5):E29–31. doi: 10.1016/j.jpedsurg.2007.03.033. [DOI] [PubMed] [Google Scholar]
- 16.Morrisey EE, Ip HS, Lu MM, Parmacek MS. GATA-6: a zinc finger transcription factor that is expressed in multiple cell lineages derived from lateral mesoderm. Dev Biol. 1996;177(1):309–22. doi: 10.1006/dbio.1996.0165. [DOI] [PubMed] [Google Scholar]
- 17.Keijzer R, van Tuyl M, Meijers C, Post M, Tibboel D, Grosveld F, Koutsourakis M. The transcription factor GATA6 is essential for branching morphogenesis and epithelial cell differentiation during fetal pulmonary development. Development. 2001;128(4):503–11. doi: 10.1242/dev.128.4.503. [DOI] [PubMed] [Google Scholar]
- 18.Zhang Y, Goss AM, Cohen ED, Kadzik R, Lepore JJ, Muthukumaraswamy K, Yang J, DeMayo FJ, Whitsett JA, Parmacek MS, Morrisey EE. A Gata6-Wnt pathway required for epithelial stem cell development and airway regeneration. Nat Genet. 2008;40(7):862–70. doi: 10.1038/ng.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell MK, Longoni M, Wells J, Maalouf FI, Tracy AA, Loscertales M, Ackerman KG, Pober BR, Lage K, Bult CJ, Donahoe PK. Congenital diaphragmatic hernia candidate genes derived from embryonic transcriptomes. Proc Natl Acad Sci U S A. 2012;109(8):2978–83. doi: 10.1073/pnas.1121621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao R, Watt AJ, Li J, Luebke-Wheeler J, Morrisey EE, Duncan SA. GATA6 is essential for embryonic development of the liver but dispensable for early heart formation. Mol Cell Biol. 2005;25(7):2622–31. doi: 10.1128/MCB.25.7.2622-2631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pober BR. Genetic aspects of human congenital diaphragmatic hernia. Clin Genet. 2008;74(1):1–15. doi: 10.1111/j.1399-0004.2008.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bates DL, Chen Y, Kim G, Guo L, Chen L. Crystal structures of multiple GATA zinc fingers bound to DNA reveal new insights into DNA recognition and self-association by GATA. J Mol Biol. 2008;381(5):1292–306. doi: 10.1016/j.jmb.2008.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–8. 27–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.