

Abstract
Programmed death-1 (PD-1) plays an important role in mediating immune tolerance through mechanisms that remain unclear. Herein, we investigated whether PD-1 prevents excessive host tissue damage during infection with the protozoan parasite, Toxoplasma gondii. Surprisingly, our results demonstrate that PD-1-deficient mice have increased susceptibility to T. gondii, with increased parasite cyst counts along with reduced type-1 cytokine responses (IL-12 and IFN-γ). PD-1−/− DCs showed no cell intrinsic defect in IL-12 production in vitro. Instead, PD-1 neutralization via genetic or pharmacological approaches resulted in a striking increase in IL-10 release, which impaired type-1-inflammation during infection. Our results indicate that the absence of PD-1 increases IL-10 production even in the absence of infection. Although the possibility that such increased IL-10 protects against autoimmune damage is speculative, our results show that IL-10 suppresses the development of protective Th1 immune response after T. gondii infection.
Keywords: IL-10, PD-1, Toxoplasma gondii
Introduction
Infection with the intracellular protozoan Toxoplasma gondii initiates a proinflammatory response that is matched by an anti-inflammatory process that ensures host survival. Parasite replication is controlled via a type-1 response characterized by IL-12/IFN-γ production 1–3. IL-12 is primarily derived from DCs, but neutrophils, macrophages, and inflammatory monocytes are also important sources 1,4–6. IL-12 drives expansion of the IFN-γ-producing NK cells, CD4+ and CD8+ T cells that protect against this parasite 7,8. In the absence of adequate immune counter regulation, as seen in IL-10−/− or 5-LO−/− mice, exacerbated type-1 responses in T. gondii-infected mice cause severe immunopathology and mortality 7,9,10. Therefore, it is important to understand the cellular interactions that promote clearance of pathogens with minimal damage to the host.
Regulation of proinflammatory cytokine production in response to T. gondii infection involves interactions among several molecules, including coreceptors expressed by both APCs and T cells. Among these, a CD28/B7 interaction was shown to promote type-1 inflammatory responses 11. On the other hand, negative regulators of T-cell activation, such as CTLA-4, limit type-1 responses to a number of protozoan parasitic, bacterial, and viral infections 12–14. In contrast, little is known about how programmed death-1 (PD-1), a B7-family member, regulates type-1 responses to intracellular infections.
Previously, the PD-1 pathway has been described to limit the inflammatory response in multiple disease models 15. PD-1 (CD279/Pdcd1) expression is inducible on a number of leukocytes, including T cells, B cells, NK cells, DCs, and monocytes 15–17. PD-1 inhibits antiviral T-cell responses during chronic infection with LCMV, HIV, and HCV via interaction with its two known ligands PD-L1 (B7-H1/CD274) and PD-L2 (B7-DC/CD273) 18,19. Multiple reports suggest that PD-1 attenuates inflammation by suppressing cytokine production from innate immune cells or limiting T-cell proliferation and effector functions 16. However, a broad characterization of the mechanisms by which PD-1 regulates pro- and anti-inflammatory cytokine responses has not been reported.
Here we postulate that PD-1 plays a regulatory role during T. gondii infection, similar to its role in other microbial infections 20–23. To test this hypothesis we examined the outcome of systemic T. gondii infection in PD-1-deficient mice. Unexpectedly, PD-1−/− animals were highly susceptible to T. gondii infection with increased parasite replication and lower type-1 cytokine production. Paradoxically, we found increased baseline IL-10 levels in both PD-1−/− mice and anti-PD-1 mAb-treated naïve WT mice. Such elevated IL-10 in naïve animals limited the ability of these mice to generate the potent type-1 cytokine response that is essential for control of parasite replication and survival upon infection. Indeed, neutralization of IL-10 receptor or reconstitution with recombinant IL-12 prior to infection restored protective immunity in PD-1−/− mice. Furthermore, we found that the lack of PD-1 resulted in increased IL-10 production from the CD4+ CD25− and CD8+ T-cell populations in naïve mice. Collectively, this study reveals an as-yet undefined host feedback response to the absence of PD-1 signaling via the production of IL-10 with direct consequences for immune therapies that block PD-1.
Results
PD-1 deficient mice are susceptible to T. gondii infection
Control of excessive inflammation is critical for host survival following T. gondii infection. Therefore, we asked whether PD-1 played a critical role in the suppression of proinflammatory responses to T. gondii infection. Given the counter-regulatory activity of PD-1, we hypothesized that PD-1−/− mice would control parasite replication better than their WT counterparts. To test this hypothesis, naïve WT and PD-1−/− mice were infected i.p. with the avirulent ME49 strain of T. gondii (50 cysts/mouse) and monitored for survival. While all WT mice survived at least 50 days after infection, PD-1−/− mice had significant early mortality with a median survival time of 13 days (Fig. 1) and infection with only 20 cysts was lethal for PD-1−/− mice (data not shown).
Figure 1.
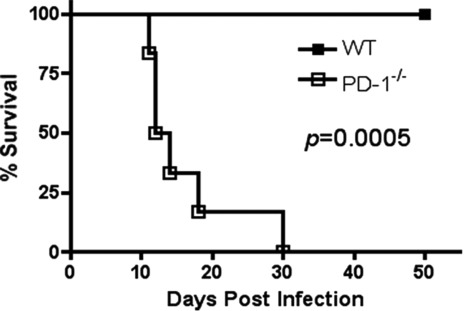
PD-1 deficient mice are susceptible to T. gondii infection. Survival of WT and PD-1−/− mice infected with 50 cysts i.p. (n = 6/group). Data are from a single experiment representative of three independent experiments. Statistical significance between survival curves was compared using the log rank test.
Susceptibility to T. gondii can be due to an inability to control parasite replication or result from immunopathology. To determine whether death was associated with alterations in parasite replication we evaluated parasite accumulation in the brain 25 days after infection. To our surprise, brains from infected PD-1−/− mice had a ∼2.5-fold higher cyst burden than brains from infected WT mice (Fig. 2A). This suggests that protective immunity is absent or reduced in PD-1−/− mice.
Figure 2.
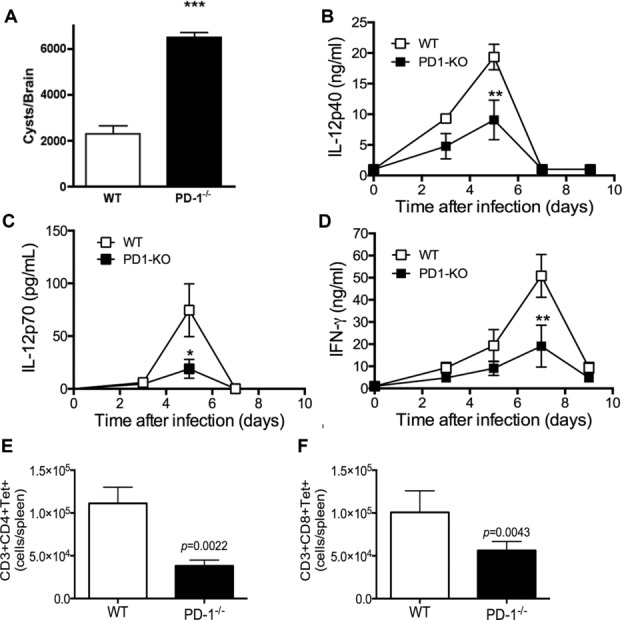
Decreased protective cytokine production and uncontrolled parasite replication in infected PD-1−/− mice. (A) T. gondii brain cysts from infected mice 25 days postinfection. (B–D) Serum level of (B) IL-12p40, (C) IL-12p70, and (D) IFN-γ from infected mice harvested at days 0, 3, 5, and 7 after infection were determined by ELISA. (A–D) Data are shown as mean ± SEM of six mice per group from one experiment representative of three performed. (E, F) Total spleen cells were harvested 7 days after infection from WT (empty bars) or PD-1−/− (filled bars) mice and stained for CD3 and (E) CD4, or (F) CD8, MHCI-GRA4/GRA6 peptide or MHCII-TGME49_0123000 605–619 peptide tetramers. Cells were acquired and analyzed as shown in Supporting Information Fig. 1. Data are shown as mean + SEM of three mice per group from one experiment representative of three performed. *p < 0.05, ***p < 0.001, two-way ANOVA with a Bonferroni posttest.
Type-1 cytokine (IL-12/IFN-γ) production during the acute response to T. gondii is critical for controlling parasite replication 2,3,24. To determine whether increased mortality in PD-1−/− mice was associated with suboptimal cytokine production, we measured serum IL-12p40, IL-12p70, and IFN-γ levels 0, 3, 5, 7, and 9 days after T. gondii infection. Prior to infection, serum cytokine levels were similar between both strains with no detectable differences in the concentrations of serum IL-12p40 (Fig. 2B) (IL-12p70 and IFN-γ were below the LOD). However, PD-1−/− mice had lower serum levels (p < 0.05) of IL-12p40, and IL-12p70 than WT mice 5 days after infection, (Fig. 2B and C) and IFN-γ (Fig. 2D) and parasite-specific CD4+ (Fig. 2E) and CD8+ (Fig. 2F) T cells 7 days after infection. The reduced immune response in infected PD-1−/− mice suggested that their increased parasite burden and death were associated with an inability to develop adequate immunity to infection.
Exogenous IL-12 treatment rescues PD-1−/− mice infected with T. gondii
To determine whether the reduced IL-12 production in PD-1−/− mice could be responsible for their increased mortality and higher parasite burden, WT and PD-1−/− mice were treated with recombinant IL-12 48 and 24 h before T. gondii inoculation. This rescued PD-1−/− mice from mortality (Fig. 3A). rIL-12-also significantly (p < 0.001) reduced brain (Fig. 3B), liver, and spleen (Fig. 3C) parasite burden in PD-1−/− mice to numbers similar to those seen in infected WT mice and normalized serum IFN-γ levels (Fig. 3C). Thus, defective IL-12 induction may mediate the increased susceptibility of PD-1−/− mice to T. gondii infection.
Figure 3.
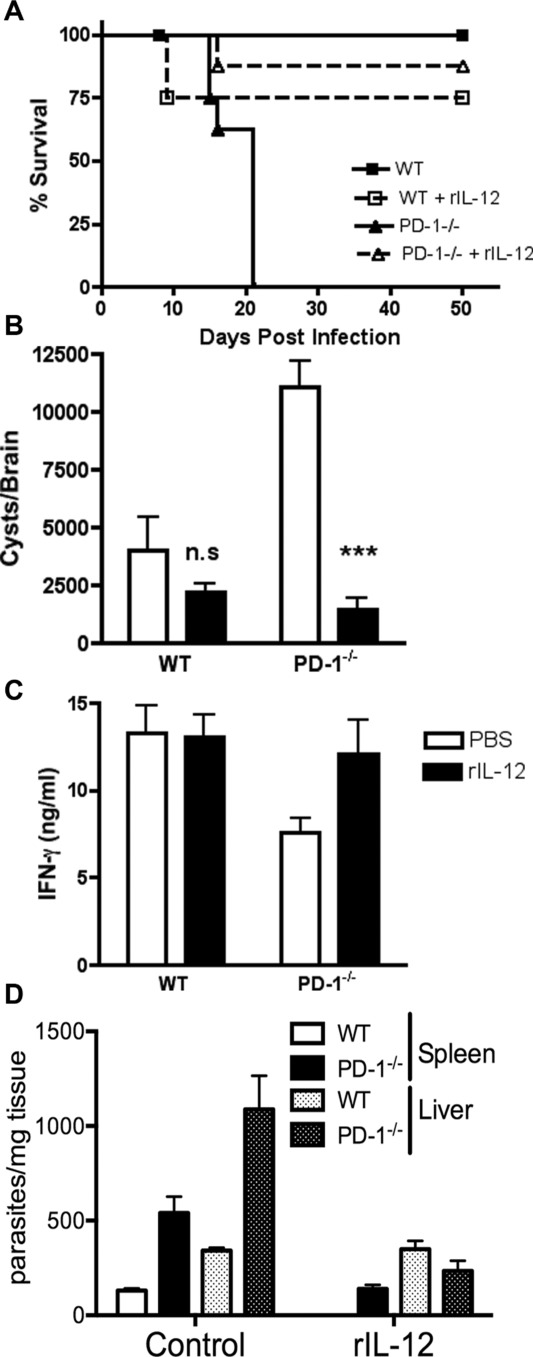
IL-12 treatment rescues PD-1−/− mice during T. gondii infection. WT and PD-1−/− mice were treated with 200 ng of recombinant IL-12 (rIL-12) 48 and 24 h prior to T. gondii infection. (A) WT and PD-1−/− were infected with 50 cysts and monitored daily for survival. (B) Cyst counts from T. gondii-infected mice treated or untreated with IL-12 are shown. (C) Serum levels of IFN-γ were determined in samples harvested 7 days after infection. (D) Total DNA was extracted and PCR performed for T. gondii B1 gene at day 15 after infection. Data are shown as mean + SEM of n = 8 mice/group and are from a single experiment representative of three independent experiments. ***p < 0.001, one–way ANOVA.
IL-12 deficiency in PD-1−/− mice is not due to an intrinsic DC defect
CD11c+ DCs—the primary IL-12 producers after T. gondii infection—are required for protection against this pathogen 1,4,24. CD11c+ DC numbers were similar in WT and PD-1−/− mice (data not shown). To determine whether PD-1−/− CD11c+ DCs respond normally to T. gondii antigen stimulation, mice were injected i.p. with soluble tachyzoite antigen (STAg), which elicits IL-12 production from CD11c+ DCs 4,25. Upon challenge, PD-1−/− mice had lower serum IL-12p70 than their WT counterparts (Fig. 4A). This suggests that in vivo, IL-12 production is attenuated in PD1−/− DCs.
Figure 4.
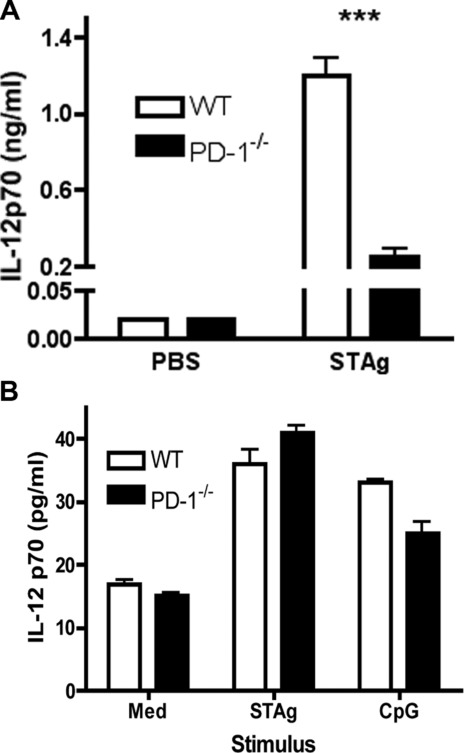
In vivo environment regulates the levels of DC IL-12 production in PD-1−/− mice. (A) IL-12p70 was measured in the serum of WT and PD-1−/−mice challenged with 20 μg of STAg i.p. Data are shown as mean + SEM of n = 4/group. (B) IL-12 p70 was measured in supernatants from WT and PD-1−/− splenic CD11c+ DCs. Data are shown as mean + SEM of n = 3 mice/group. CD11c+ DCs were purified using CD11c-MACS beads. Following purification, cells were stimulated with STAg (10 μg/mL) or CpG (1 μg/mL). Data shown are from single experiments representative of three experiments performed. ***p < 0.001, two-way ANOVA with a Bonferroni posttest.
In order to assess whether the lower in vivo IL-12 production by PD-1−/− DCs is due to a cell-intrinsic defect, splenic CD11c+ cells were purified from naïve WT and PD-1−/− mice using CD11c MACS-beads and stimulated in vitro with STAg or with CpG-oligos. Surprisingly, no detectable differences in IL-12 production following in vitro stimulation was seen in PD-1−/− DCs (Fig. 4B). This similarity in in vitro IL-12 responses by PD-1−/− and WT DCs to TLR stimulation implies that the PD-1−/− in vivo environment inhibits DC IL-12 production, but that this inhibition is lost once DCs are removed from the host. Similarly, “DC paralysis” (unresponsiveness to microbial stimulation in vivo with restored responsiveness in vitro) has been previously observed by us and others 26,27.
Steady-state IL-10 expression is increased in the absence of PD-1 signaling
Because IL-10 has an important counter-regulatory role, including inhibition of DC IL-12 production 28,29 we hypothesized that IL-10 production might be inhibited by PD-1 in naïve mice. Indeed, IL-10 transcript levels in spleen cells from naive PD-1−/− mice were considerably greater than those from age- and sex-matched WT mice, as determined by quantitative RT-PCR (Fig. 5A). To ensure that increased this IL-10 transcript expression correlated with IL-10 protein production, we quantified in vivo IL-10 production with the in vivo cytokine capture assay, which can increase the sensitivity of cytokine detection up to 100-fold 30. Figure 5B shows that the levels of systemic IL-10 protein are significantly (p < 0.05) elevated in naive PD-1−/− mice when compared with those in WT controls (Fig. 5B). Furthermore, systemic IL-10 levels remained significantly (p < 0.01) increased in PD-1−/− mice after T. gondii infection (Fig. 5C).
Figure 5.
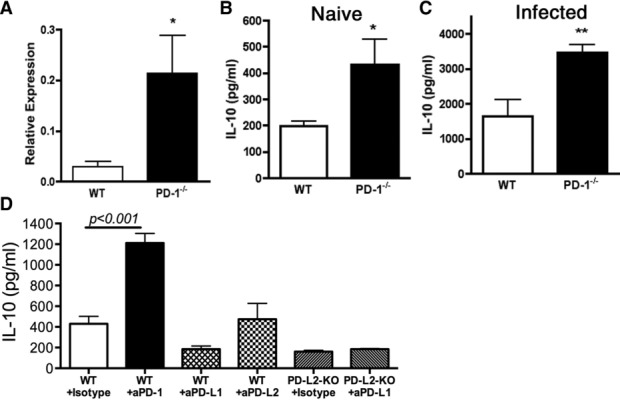
Homeostatic IL-10 production is enhanced in the absence of PD-1. (A) IL-10 mRNA expression was measured by qRT-PCR in total spleen cells from naïve WT and PD-1−/− mice. Data are shown as mean + SEM of n = 6/group. (B) IL-10 was measured in the serum of naïve WT and PD-1−/− mice using in vivo cytokine capture assay (IVCCA) specific for IL-10 and shown as mean + SEM of n = 5–6 mice/group. (C) Serum IL-10 was measured three days after T. gondii infection with 50 cysts i.p. (D) Serum IL-10 was measured, using IVCCA, from naïve WT or PD-L2-deficient mice treated with anti-PD-1, -PD-L1, or -PD-L2 blocking antibodies (250 μg/0.2 mL PBS i.p.) on days 0, 3, 6, 9, and 12. Data are shown as mean + SEM of 4–6 mice and are from single experiments representative of three independent experiments. *p < 0.05, **p < 0.01, **p < 0.001, Stu-dent’s t-test (A–C), or one-way ANOVA (D).
Signaling through PD-1 is triggered by ligation with PD-L1 and PD-L2. To determine the involvement of these PD-1 ligands in inhibition of IL-10 production, we treated WT mice with anti-PD-1 mAb for 12 days; this significantly (p < 0.05) increased serum IL-10 concentrations (Fig. 5D). However, neutralization of PD-L1 significantly (p < 0.01) decreased IL-10 while PD-L2 had no effect. Moreover, anti-PDL1 treatment of PDL2-deficient mice did not change baseline IL-10 levels (Fig. 5D). Notably, treatment with anti-PD-1 mAb for only 3 days did not affect baseline IL-10 (data not shown).
Regulation of T-cell IL-10 is lost in PD-1 deficient mice
IL-10 is produced by T cells, B cells, DCs, macrophages, and other cell types 31. The elevated systemic IL-10 levels seen in PD-1−/− mice suggested that a PD-1-sensitive cell(s) population(s) is (are) the source of this cytokine. To identify this population(s), Vert-X mice (IL-10/eGFP-reporter mice) were crossed with PD-1−/− mice and IL-10-producing cells from naïve mice were identified by flow cytometry. Consistent to our observations above, the frequency as well as in the total number of IL-10+ splenic leukocytes were significantly increased in Vert-X PD-1−/− mice (Fig. 6A and B). Circulating levels of IL-10 in Vert-X PD-1−/− versus VertX PD-1+/+ mice were consistent with the number of IL-10-producing cells (compare Fig. 6A and B with Fig. 6C). As previously described 32, under steady-state conditions naïve WT mice B cells (B220+ CD19+cells) are the major spleen-cell population making IL-10 (approximately 40–50% of GFP+ cells) (Fig. 6D). In PD-1−/− mice the frequency of IL-10+ B cells decreases, to approximately 20% of GFP+ cells, although the total number of IL-10+ B cells is not significantly different between the two genotypes (Fig. 6D and E). These data suggest that PD-1 does not necessarily affect steady-state B-cell-derived IL-10-production. Interestingly, the frequency of CD3+CD4+CD25− and CD3+CD8+ T cells making IL-10 increased fivefold for both populations (Fig. 6D). This increased frequency is corroborated by a 100-fold increase in the total number of IL-10+ cells (Fig. 6E). On the other hand, no significant differences in the frequency or total number of IL-10-producing NK, NKT, and myeloid cells (CD11b+CD11c+, CD11b+CD11c−, and CD11b−CD11c+) were detected (Fig. 6D and E). Elevated IL-10 production in naïve PD1−/− mice was abolished after in vivo depletion of CD4+ or CD8+ cells (Fig. 6F). Conversely, such in vivo depletions allowed for enhanced IL-12p70 production after in vivo STAg challenge, performed as described in Figure 4 (Fig. 6G). Taken together, the data strongly supports CD4+ and CD8+ T cells as the source of elevated IL-10 in PD1-deficient hosts.
Figure 6.
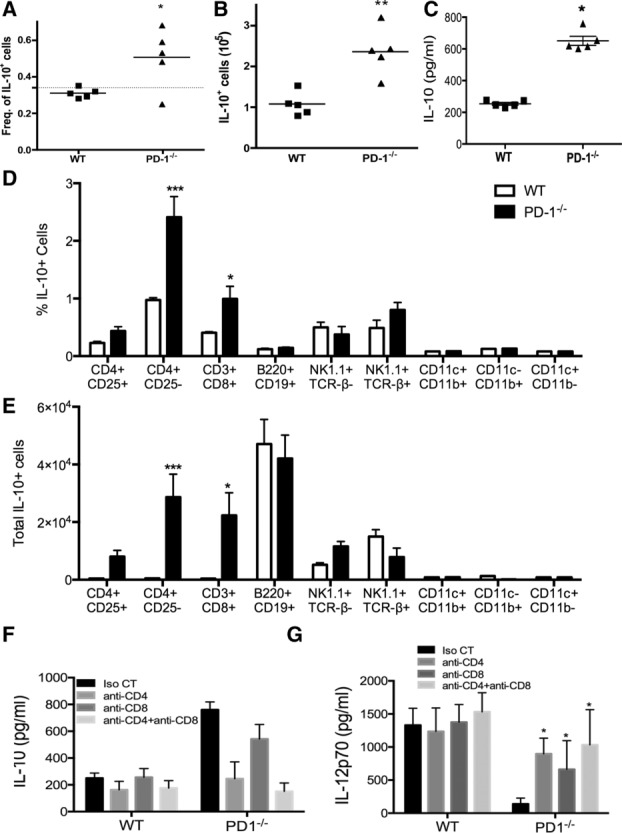
Increased steady-state IL-10 production in PD-1−/− mice is derived from T cells. Splenic leukocytes were harvested from naïve control Vert-X and PD-1−/− Vert-X mice and IL-10 (eGFP) expression was quantified by flow cytometry. The (A) frequency and (B) total number of IL-10+ cells within the spleen were determined by flow cytometry. (C) Serum IL-10 in the same animals as in (A) and (B), was determined by IVCCA. Each symbol represents an individual animal and bar represents the mean. (D) The frequency of IL-10+ cells within various the various lymphocyte subsets within the spleen was measured. (E) The total number of IL-10+ cells within each subset was determined. (F) Serum IL-10 levels in isotype control, anti-CD4 or anti-CD8-treated Vert-X WT or PD1-deficient mice, as determined by IVCCA are shown. (G) Serum IL-12p70 levels after in vivo depletion as described in (F) were determined. (D–G) Data are shown as mean + SEM of four animals and are from single experiments representative of two independent experiments performed. *p < 0.05, **p < 0.01, two-way ANOVA with Bonferroni posttest.
IL-10R neutralization rescues type-1 inflammatory responses in PD-1−/− mice
IL-10 is a potent inhibitor of type-1 response during T. gondii infection as well as during other infections 7,33,34. IL-10-deficient mice are susceptible to T. gondii infection due to an uncontrolled proinflammatory response. To determine the biological relevance of the elevated IL-10 in inhibiting type-1 immunity to infection in PD1–/– mice, WT and PD-1−/− were treated with IL-10R blocking-antibody before infection. As previously shown with IL-10-deficient hosts (30), anti-IL-10R Ab caused increased IL-12p70, and IFN-γ, but not IL-12p40, production in infected WT mice (Fig. 7A–C). Similarly, anti-IL-10R-treated infected-PD-1−/− hosts showed increased IL-12p70/IFN-γ production as compared with that in isotype-control-treated PD-1−/− mice. Moreover, serum IL-12p40 levels were also significantly increased (Fig. 7A–C). Importantly, anti-IL-10R-treated PD-1-deficient hosts showed greatly improved control of brain parasite accumulation (Fig. 7D). However, as shown before with IL-10-deficient hosts 35, anti-IL-10R treatment induced mortality in both WT and PD-1-deficient hosts due to exacerbated proinflammatory reaction (not shown).
Figure 7.
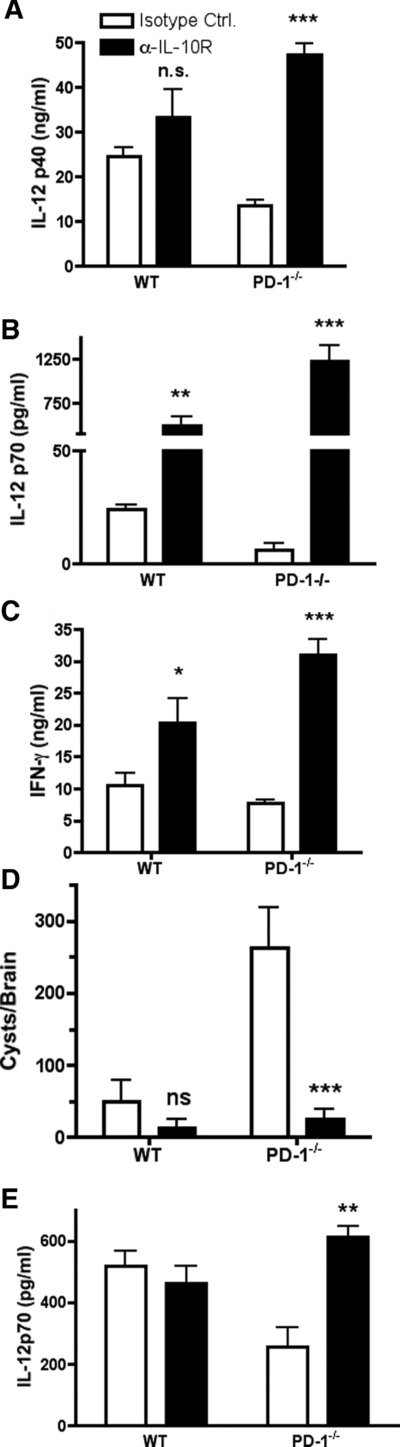
IL-10R neutralization rescues type-1 responses in PD-1−/− mice. IL-10R neutralizing antibody was administered to mice 24 h prior to infection with 50 T. gondii cysts. (A, B) Serum IL-12p40 and IL-12p70 were measured from serum harvested 5 days after infection. (C) IFN-γ was measured in serum harvested 7 days after infection. (D) Cyst counts from brain homogenates obtained 10 days after infection. (E) STAg-induced IL-12 p70 production 6 h after challenge. Data are shown as mean + SEM of triplicate samples and are from single experiments representative of two independent experiments performed. *p < 0.005, **p < 0.01, ***p < 0.001, one-way ANOVA test.
To further establish whether the increased IL-10 levels in naïve PD-1−/− mice are biologically relevant, we evaluated DC IL-12 responses to STAg injection with or without prior anti-IL-10R Ab treatment. IL-10R neutralization induced a two-fold increase in serum IL-12p70 when compared with that in isotype control-treated PD-1−/− mice (Fig. 7E).
In conclusion, the results shown here indicate that in the absence of PD-1, an increase in steady-state IL-10 production modifies the environment by attenuating type-1 cytokine responses to the pathogen as well as to DC cytokine production.
Discussion
PD-1 plays a central role in immune tolerance, however its involvement in modulating inflammation during homeostasis is largely unknown. Use of the experimental T. gondii infection model demonstrates a novel function for PD-1 in supporting the production of protective type-1 inflammatory cytokines, IL-12 and IFN-γ, in response to infection. Also, we demonstrate that the inability to initiate a robust IL-12 response is not due to an intrinsic defect in PD-1−/− DCs but due to the splenic microenvironment, because WT and PD-1−/− DC produce equivalent amounts of IL-12 when stimulated in vitro. Moreover, we establish that type-1 responses are attenuated in the absence of PD-1 due to a systemic increase in IL-10 production in naïve mice. Using IL-10 transcriptional reporter mice (Vert-X), we find an increase in the frequency and total number of IL-10-producing spleen CD4+ and CD8+ T cells. Further, we demonstrate that neutralization of IL-10R activity in PD-1−/− mice rescues production of Type-1 responses to T. gondii as well as STAg challenge. Thus, this study reveals that the absence of PD-1 signaling promotes an increase in IL-10 production, which increases susceptibility to opportunistic infections, including toxoplasmosis.
The inability of PD-1−/− mice to adequately control T. gondii infection provides new insight into the involvement of PD-1 in acute immunity. The requirement of type-1 inflammatory responses for control of T. gondii has been thoroughly characterized 36. Our findings that IL-12 and IFN-γ are significantly lower in T. gondii-infected PD-1−/− mice is intriguing, because a potent type-1 cytokine response is required for protection against several pathogens 2,37,38. T-cell/DC interactions in the lymphoid compartment are required to achieve optimal conditions for development of protective immunity. Further, in vivo DC IL-12 production is influenced by contact-dependent and -independent factors 39. Previous studies of the role of PD-1 in the immune response to infection, which used T-cell deficient mice or immortalized cell lines, did not address the influence of the microenvironment, including DC/T-cell interactions 16,40. Our in vivo assessment of the involvement of PD-1 on IL-12 production suggests that the absence of PD-1 function creates conditions that inhibit development of a type-1 inflammatory response to intracellular microbial infection. The role of PD-1 during T. gondii infection has been previously investigated with neutralizing anti-PD-1 antibodies in WT animals 41,42. Although there are no data on IL-10 production in anti-PD1-treated T. gondii-infected mice, these previously reported findings are in line with a PD-1/PD-L1 counter-regulatory pathway. In contrast, our results indicate that genetic deficiency or antibody-mediated neutralization “prior” to infection can inhibit the development of protective type-1 immunity by increasing IL-10 production. Taking these observations together, we can speculate that there are at least two types of PD-1-dependent interactions, a homeostatic baseline PD-L1/L2-independent mechanism and a PD-L1-dependent pathway that predominates during an ongoing immune response against a pathogen.
Our results reveal that IL-10 production is enhanced when PD-1 activity is absent during steady-state conditions, as well as early after pathogen inoculation. This suppresses IL-12 production by DCs and macrophages in T. gondii-infected mice 7,43,44, even though endogenous IL-10 does not significantly inhibit IL-12 production in response to STAg injection in WT mice 25. Indeed, the observation that IL-10 receptor neutralization and subsequent derepression of IL-12 and IFN-γ production in infected PD-1 deficient mice correct defective parasite elimination demonstrate that the increased steady-state and infection-induced IL-10 production in these mice plays a biologically relevant role in modulating the immune response in vivo.
The mechanism by which PD-1 limits IL-10 production remains unclear. Our results indicate that, as previously observed 45,46, acute neutralization of PD-L1 inhibits IL-10 production, an effect opposite to that of PD-1 neutralization. Moreover, PD-L2 deficiency and PD-L1 neutralization in PD-L2-deficient mice had no detectable effect on baseline IL-10 induction. Interestingly, other reports indicated that PD-L1 neutralization promoted the induction of IL-10 47. The seeming discrepancy between our data and this previously published observation could arise from at least three potential mechanisms, the first of which may be the presence of a biologically active third ligand for PD-1. Second, differences in receptor and ligand expression may have an impact. Baseline PD-L1 expression is very low and is rapidly upregulated by cellular activation. While we followed IL-10 production in the absence of an ongoing immune response, other investigators have studied the role of PD-1 during an ongoing immune responses, which should upregulate ligand/receptor densities on both APCs and T cells. Under such conditions, the effect of PD-1 signaling may switch from limiting to promoting IL-10 production. Finally, the absence of the suppressive activity of PD-1 promotes the development of autoimmune disease, which may trigger increased IL-10 production as a secondary regulatory mechanism. This possibility suggests that combined IL-10 and PD-1 deficiency would promote severe, early development of autoimmunity, in contrast to the late development of autoimmunity in PD-1−/− mice and the relatively mild autoimmunity in IL-10−/− mice 48,49.
PD-1 might also influence the response to T. gondii through other mechanisms. First, PD-1 may suppress protective responses to infection by limiting the activation of effector and memory T cells. Secondly, PD-1/PD-L1 interactions are required for the maturation of DCs and their ability to promote expansion of protective T-cell responses. Finally, PD-1 deficiency can induce significant changes in the gut microbiota 50. Such changes might influence the number of IL-10-producing CD4+ CD25− cells within the gastrointestinal tract. However, data from Kawamoto et al. as well as our experiments do not show significant differences in IL-10 content within lymphocytes from Peyer’s patches or IL-10 producing cells derived from the mesenteric lymph nodes 50.
Steady-state IL-10 is predominantly produced by intestinal leukocytes. These cells, which include Tr1 and Foxp3+ regulatory T cells, release IL-10 that inhibits a pathological immune response to gut flora 51–53. In peripheral tissues other than the small and large intestine, the predominant source of homeostatic IL-10 are B cells, although these cells produce only low levels of this cytokine 32. In contrast, induction of effector T cell IL-10 production requires antigen and APCs 31. However, we did not see increased IL-10 production by gut mucosa or mesenteric lymph node cells from PD-1-deficient hosts (not shown). This suggests that the increased production of IL-10 in the absence of PD-1 is derived from the systemic, rather than the mucosal immune system.
Within the past few years PD-1 has become an immuno-therapeutic target for a variety of illnesses seen in clinics because of its ability to inhibit protective immune responses. PD-1 is upregulated on HIV-specific CD8+ T cells and experimental blockade of PD-1 enhances proliferation and cytokine production 54,55. NK and NKT cells from patients infected with Mycobacterium tuberculosis express elevated levels of PD-1 and in vitro blockade of PD-1 signaling increases NK and NKT cell proliferation 56,57. In contrast, our observations suggest caution in the use of PD-1 antagonists with evaluation of IL-10 levels and susceptibility to opportunistic infections.
Materials and methods
Mice
C57BL/6 mice 6–10 weeks of age were purchased from Jackson Laboratories and housed under specific pathogen-free conditions in the AAALAC-accredited Cincinnati Children’s Hospital Research Foundation animal facility under approved IACUC guidelines. PD-1−/− mice were kindly provided by Tasuku Honjo (Kyoto University, Kyoto, Japan) 58. Vert-X mice (IL-10 reporter) were kindly provided by Christopher Karp (Cincinnati Children’s Hospital Research Foundation) 32. Age (6–10 wk) and sex-matched mice were used in all experiments.
Parasite infection procedures, STAg, and antibody treatment
T. gondii cysts from the avirulent strain ME49 were prepared from brain homogenates obtained from chronically infected C57BL/6 mice and inoculated i.p. (50 cysts/animal unless otherwise indicated). Survival studies were performed following IACUC guidelines. Daily weight was monitored and animals were euthanized when the mice became moribund and severe weight-loss was observed (>20% body weight decline). STAg was prepared as previously described 59. Mice were injected i.p. with STAg (20 μg/0.2 mL/mouse). For in vivo neutralization of IL-10R, mice were injected with anti-IL-10R (clone 1B1.3A) or isotype control antibody (clone GL113) (100 μg/0.2 mL/mouse). For in vivo blockade of PD-1-PD-L signaling, mice received four i.p. injections over 12 days of 250 μg of anti-PD-1 (RMP1–14), -PD-L1 (MIH5), and -PD-L2 (TY25) (PD-1 and PD-L2 blocking antibodies were purchased by Bio X Cell). Prior optimization indicated that 3-day treatment with two injection of anti-PD-1 did not change baseline IL-10 production (data not shown).
Cell purification, culture conditions, and cytokine measurements
Splenocytes and CD11c+ cells were obtained from collagenase-treated spleens from naïve mice. CD11c+ cells were purified using CD11c MicroBeads followed by MACS column purification (Miltenyi Biotech, Gladbach, Germany). Cells were cultured overnight at 37°C and 5% CO2. Supernatants were harvested and stored at –20°C until cytokine analysis. IL-12p40, IL-12p70, and IFN-γ were quantified by ELISA using commercial kits (R&D systems). Samples were analyzed in triplicate. For quantification of IL-10, the in vivo cytokine capture assay was used, as described previously 30. Briefly, mice were injected i.p. with 10 ug of biotin-labeled anti-IL-10. A range of 16–24 h later serum was harvested from mice. Serum was then incubated in microtiter plates previously coated with a mAb against a second epitope of IL-10. The plate coating antibody-IL-10-biotin-antibody complex was then quantitated by adding enzyme-streptavidin complex followed by substrate.
mRNA purification and QRT-PCR
Total RNA was purified from total splenocytes using trizol reagent (Invitrogen). cDNA was prepared from purified RNA using the TaqMan cDNA synthesis kit (Roche). Gene expression was measured using LightCycler 480 software according to the manufacturer’s instructions (Roche). Relative expression was measured using the ΔΔCT-Method.
Flow cytometry
Antibodies to cell surface markers and T. gondii-specific tetramers (NIH Tetramer Core Facility) were used according to directions provided by the manufacturer. For surface staining, cells were labeled in a PBS solution containing 1% FBS + 0.1%NaN3 (FACS buffer) and fixed with 4% methanol-free paraformaldehyde in FACS buffer to minimize loss of eGFP. For tetramer staining, CD3/CD4/CD8 stained cells were incubated with tetramers (TGME49 for CD4+ cells and TGD057 for CD8+ cells) for 1 hr at 37°C 60,61. A LSRII (BD Biosciences) was used for all flow cytometry experiments. FlowJo software (Treestar, Inc.) was used to analyze flow cytometry data. Gating strategies are shown in Supporting Information Fig. 1 and 2.
Statistics
Experimental data are expressed as mean ± SEM. The statistical significance of differences in mean values was determined using Students t test or, when necessary, a two-way ANOVA with a Bonferoni posttest. Survival data are presented as a Kaplan–Meier survival curve and analyzed with a log-rank test. Differences of at least p < 0.05 are considered to be significant.
Acknowledgments
The authors would like to thank Ian Lewkowich and Fred D. Finkelman for critically reading of the manuscript. Christopher L. Karp for generously providing Vert-X-IL-10 reporter mice. Fred D. Finkelman for isotype control antibodies used in experiments. JA is supported by NIH grants AI078969 and AI075038, and DRH is supported by NIH grants RO1GM083204 and RO1 AI095289. CM was supported by a supplement from NIH grant (AI075038) and from the University of Cincinnati Graduate School Yates Fellowship and Scholarship Program.
Glossary
- PD-1
programmed death-1
- STAg
soluble tachyzoite antigen
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Supporting Information
Additional supporting information may be found in the online version of this article at the publisher’s web-site
References
- 1.Mashayekhi M, Sandau MM, Dunay IR, Frickel EM, Khan A, Goldszmid RS, Sher A, et al. CD8α+ dendritic cells are the critical source of interleukin-12 that controls acute infection by Toxoplasma gondii tachyzoites. Immunity. 2011;35:249–259. doi: 10.1016/j.immuni.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yap G, Pesin M, Sher A. Cutting edge: IL-12 is required for the maintenance of IFN-γ production in T cells mediating chronic resistance to the intracellular pathogen, Toxoplasma gondii. J. Immunol. 2000;165:628–631. doi: 10.4049/jimmunol.165.2.628. [DOI] [PubMed] [Google Scholar]
- 3.Scharton-Kersten T, Wynn T, Denkers E, Bala S, Hieny S, Gazzinelli R, Sher A. In the absence of endogenous IFN-gamma, mice develop unimpaired IL-12 responses to Toxoplasma gondii while failing to control acute infection. J. Immunol. 1996;157:4045–4054. [PubMed] [Google Scholar]
- 4.Liu C, Fan Y, Dias A, Esper L, Corn R, Bafica A, Machado F, et al. Cutting edge: dendritic cells are essential for in vivo IL-12 production and development of resistance against Toxoplasma gondii infection in mice. J. Immunol. 2006;177:31–35. doi: 10.4049/jimmunol.177.1.31. [DOI] [PubMed] [Google Scholar]
- 5.Bliss S, Zhang Y, Denkers E. Murine neutrophil stimulation by Toxoplama gondii antigen drives high level production of IFN-gamma-independent IL-12. J. Immunol. 1999;163:2081–2088. [PubMed] [Google Scholar]
- 6.Reis e Sousa C, Hieny S, Scharton-Kersten T, Jankovic D, Charest H, Germain RN, Sher A. In vivo microbial stimulation induces rapid CD40 ligand-indepedent production of interleukin 12 by dendritic cells and their redistribution to T cell areas. J. Exp. Med. 1997;186:1819–1829. doi: 10.1084/jem.186.11.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gazzinelli R, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 8.Gazzinelli R, Wysocka M, Hayashi S, Denkers E, Hieny S, Casper P, Trinchieri G, et al. Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 1994;153:2533–2543. [PubMed] [Google Scholar]
- 9.Neyer L, Gruning G, Fort M, Remington J, Rennick D, Hunter C. Role of interleukin-10 in regulation of T-cell-dependent and T-cell-independent mechanisms of resistance to Toxoplasma gondii. Infect Immun. 1997;65:1675–1682. doi: 10.1128/iai.65.5.1675-1682.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aliberti J, Serhan C, Sher A. Parasite-induced lipoxin A4 is an endogenous regulator of IL-12 production and immunopathology in Toxoplasma gondii infection. J. Exp. Med. 2002;196:1253–1262. doi: 10.1084/jem.20021183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter C, Ellis-Neyer L, Gabriel K, Kennedy M, Grabstein K, Linsley P, Remington J. The role of the CD28/B7 interaction in the regulation of NK cell responses during infection with Toxoplasma gondii. J. Immunol. 1997;158:2285–2293. [PubMed] [Google Scholar]
- 12.Martins G, Tadokoro C, Silva R, Silva J, Rizzo L. CTLA-4 blockage increases resistance to infection with the intracellular protozoan Trypanosoma cruzi. J. Immunol. 2004;172:4893–4901. doi: 10.4049/jimmunol.172.8.4893. [DOI] [PubMed] [Google Scholar]
- 13.Murphy M, Cotterell S, Gorak P, Engwerda C, Kaye P. Blockade of CTLA-4 enhances host resistance to the intracellular pathogen, Leishmania donovani. J. Immunol. 1998;161:4153–4160. [PubMed] [Google Scholar]
- 14.Rowe J, Johanns T, Ertelt J, Lai J, Way S. Cytotoxic T-lymphocyte antigen 4 blockade augments the T-cell response primed by attenuated Listeria monocytogenes resulting in more rapid clearance of virulent bacterial challenge. Immunology. 2009;128:e471–e478. doi: 10.1111/j.1365-2567.2008.03001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat. Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 16.Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, Xu H, et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113:5811–5818. doi: 10.1182/blood-2009-02-203141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang X, Venet F, Wang Y, Lepape A, Yuan Z, Chen Y, Swan R, et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. PNAS. 2009;106:6308–6308. doi: 10.1073/pnas.0809422106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benedict C, Loewendorf A, Garcia Z, Blazar B, Janssen E. Dendritic cell programming by cytomegalovirus stunts naive T cell responses via the PD-L1/PD-1 pathway. J. Immunol. 2008;180:4836–4847. doi: 10.4049/jimmunol.180.7.4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brown KE, Freeman GJ, Wherry EJ, Sharpe AH. Role of PD-1 in regulating acute infections. Curr. Opin. Immunol. 2010;22:397–401. doi: 10.1016/j.coi.2010.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazar-Molnar E, Chen B, Sweeney K, Wang E, Liu W, Lin J, Porcelli S, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. PNAS. 2010;107:13402–13407. doi: 10.1073/pnas.1007394107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barber D, Mayer-Barber K, Feng C, Sharpe A, Sher A. CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 2011;186:1598–1607. doi: 10.4049/jimmunol.1003304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gutierrez F, Marioano F, Oliveir C, Pavanelli W, Guedes P, Silva G, Campanelli A, et al. Regulation of Trypanosoma cruzi-induced myocarditis by programmed death cell receptor 1. Infect Immun. 2011;79:1873–1881. doi: 10.1128/IAI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD-1 inhibits antiviral immunity at the effector phase in the liver. J. Exp. Med. 2003;198:39–50. doi: 10.1084/jem.20022235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldszmid RominaS, Casper P, Rivollier A, White S, Dsutsev A, Hieny S, Kelsall B, et al. NK cell-derived interferon-γ orchestrates cellular dynamics and the differentiation of monocytes into dendritic cells at the site of infection. Immunity. 2012;36:1047–1059. doi: 10.1016/j.immuni.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reis e Sousa C, Yap G, Schulz O, Rogers N, Schito M, Aliberti J, Hieny S, et al. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 1999;11:637–647. doi: 10.1016/s1074-7613(00)80138-7. [DOI] [PubMed] [Google Scholar]
- 26.Reis e Sousa C, Yap G, Schulz O, Rogers N, Schito M, Aliberti J, Hieny S, et al. Paralysis of dendritic cell IL-12 production by microbial products prevents infection-induced immunopathology. Immunity. 1999;11:637–647. doi: 10.1016/s1074-7613(00)80138-7. [DOI] [PubMed] [Google Scholar]
- 27.Aliberti J, Hieny S, Reis e Sousa C, Serhan CN, Sher A. Lipoxin-mediated inhibition of IL-12 production by DCs: a mechanism for regulation of microbial immunity. Nat. Immunol. 2002;3:76–82. doi: 10.1038/ni745. [DOI] [PubMed] [Google Scholar]
- 28.D’Andrea A, Aste-Amezaga M, Valiante N, Ma X, Kubin M, Trinchieri G. Interleukin 10 (IL-10) inhibits human lymphocyte interferon gamma-production by suppressing natural killer cell stimulatory factor/IL-12 synthesis in accessory cells. J. Exp. Med. 1993;178:1041–1048. doi: 10.1084/jem.178.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Aste-Amezaga M, Ma X, Sartori A, Trinchieri G. Molecular mechanisms of the induction of IL-12 and its inhibition by IL-10. J. Immunol. 1998;160:5936–5944. [PubMed] [Google Scholar]
- 30.Finkelman F, Morris S. Development of an assay to measure in vivo cytokine production in the mouse. Int. Immunol. 1999;11:1811–1818. doi: 10.1093/intimm/11.11.1811. [DOI] [PubMed] [Google Scholar]
- 31.Saraiva M, O’Garra A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010;10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 32.Madan R, Demircik F, Surianarayanan S, Allen J, Divanovic S, Trompette A, Yogev N, et al. Nonredundant roles for B cell-derived IL-10 in immune counter-regulation. J. Immunol. 2009;183:2312–2320. doi: 10.4049/jimmunol.0900185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gazzinelli R, Oswald I, James S, Sher A. IL-10 inhibits parasite killing and nitrogen oxide production by IFN-gamma-activated macrophages. J. Immunol. 1992;148:1792–1796. [PubMed] [Google Scholar]
- 34.Hunter C, Subauste C, Van Cleave V, Remington J. Production of gamma interferon by natural killer cells from Toxoplasma gondii-infected SCID mice: regulation by interleukin-10, interleukin-12, and tumor necrosis factor alpha. Infect Immun. 1994;62:2818–2824. doi: 10.1128/iai.62.7.2818-2824.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gazzinelli RT, Wysocka M, Hieny S, Scharton-Kersten T, Cheever A, Kuhn R, Muller W, et al. In the absence of endogenous IL-10, mice acutely infected with Toxoplasma gondii succumb to a lethal immune response dependent on CD4+ T cells and accompanied by overproduction of IL-12, IFN-gamma and TNF-alpha. J. Immunol. 1996;157:798–805. [PubMed] [Google Scholar]
- 36.Denkers E, Gazzinelli R. Regulation and function of T-cell-mediated immunity during Toxoplasma gondii infection. Clin. Microbiol. Rev. 1998;11:569–588. doi: 10.1128/cmr.11.4.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cooper A, Khader S. The role of cytokines in the initiation, expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 2008;226:191–204. doi: 10.1111/j.1600-065X.2008.00702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mocci S, Dalrymple S. The cytokine stew and innate resistance to L. monocytogenes. Immunol. Rev. 1997;158:107–114. doi: 10.1111/j.1600-065x.1997.tb00996.x. [DOI] [PubMed] [Google Scholar]
- 39.Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003;3:133–146. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- 40.Cho H, Choi E, Lee S, Jung K, Seo S, Choi I, Park S, et al. Programmed death-1 receptor negatively regulates LPS-mediated IL-12 production and differentiation of murine macrophage RAW264.7 cells. Immunol. Lett. 2009;127:39–47. doi: 10.1016/j.imlet.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Bhadra R, Gigley JP, Khan IA. PD-1-mediated attrition of polyfunctional memory CD8+ T cells in chronic toxoplasma infection. J. Infect Dis. 2012;206:125–134. doi: 10.1093/infdis/jis304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhadra R, Gigley JP, Weiss LM, Khan IA. Control of toxoplasma reactivation by rescue of dysfunctional CD8+ T-cell response via PD-1-PDL-1 blockade. Proc. Natl. Acad. Sci. USA. 2011;108:9196–9201. doi: 10.1073/pnas.1015298108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Ann. Rev. Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 44.Perona-Wright G, Morhs K, Szaba F, Kummer L, Madan R, Karp C, Johnson L, et al. Systemic but not local infections elicit immunosuppressive IL-10 production by natural killer cells. Cell Host Microbe. 2006;6:503–512. doi: 10.1016/j.chom.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Farina E, Alberoni M, et al. A potential role for the PD1/PD-L1 pathway in the neuroinflammation of Alzheimer’s disease. Neurobiol. Aging. 2012;33:e611–e622. doi: 10.1016/j.neurobiolaging.2011.03.004. 624. [DOI] [PubMed] [Google Scholar]
- 46.Curiel TJ, Wei S, Dong H, Alvarez X, Cheng P, Mottram P, Krzysiek R, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat. Med. 2003;9:562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- 47.Bodhankar S, Chen Y, Vandenbark AA, Murphy SJ, Offner H. PDL1 enhances CNS inflammation and infarct volume following experimental stroke in mice in opposition to PD-1. J. Neuroinflammation. 2013;10:111. doi: 10.1186/1742-2094-10-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 49.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- 50.Kawamoto S, Tran T, Maruya M, Suzuki K, Doi Y, Tsutsui Y, Kato L, et al. The inhibitory receptor PD-1 regulates IgA selection and bacterial composition in the gut. Science. 2012;336:485–489. doi: 10.1126/science.1217718. [DOI] [PubMed] [Google Scholar]
- 51.Maynard C, Harrington L, Janowski K, Oliver J, Zindl C, Rudensky A, Weaver C. Regulatory T cells expressing interleukin 10 develop from Foxp3+ and Foxp3– precursor cells in the absence of interleukin 10. Nat. Immunol. 2007;8:931–941. doi: 10.1038/ni1504. [DOI] [PubMed] [Google Scholar]
- 52.Kamanaka M, Kim S, Wan Y, Sutterwala F, Lara-Tejero M, Galan J, Harhaj E, et al. Expression of interleukin-10 in intestinal lymphocytes detected by an interleukin-10 reporter knock in tiger mouse. Immunity. 2006;25:941–952. doi: 10.1016/j.immuni.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 53.Groux H, O’Garra A, Bigler M, Rouleau M, Antonenko S, de Vries J, Roncarolo M. A CD4+ T-cell subset inhibits antigen-specific T-cell responses and prevents colitis. Nature. 1997;389:737–742. doi: 10.1038/39614. [DOI] [PubMed] [Google Scholar]
- 54.Petrovas C, Casazza JP, Brenchley JM, Price DA, Gostick E, Adams WC, Precopio ML, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J. Exp. Med. 2006;203:2281–2292. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trautmann L, Janbazian L, Chomont N, Said E, Gimmig S, Bessette B, Boulassel M, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006;12:1198–1202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 56.Kee S, Kwon Y, Park Y, Cho Y, Lee S, Kim T, Lee S, et al. Dysfunction of natural killer T cells in patients with active Mycobacterium tuberculosis infection. Infect Immun. 2012;80:2100–2108. doi: 10.1128/IAI.06018-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alvarez I, Pasquinelli V, Jurado J, Abbate E, Musella R, de la Barrera S, Garcia V. Role played by the programmed death-1-programmed death ligand pathway during innate immunity against Mycobacterium tuberculosis. J. Infect Dis. 2010;202:524–532. doi: 10.1086/654932. [DOI] [PubMed] [Google Scholar]
- 58.Nishimura H, Minato N, Nakano T, Honjo T. Immunological studies on PD-1 deficient mice: implication of PD-1 as a negative regulator for B cell responses. Int. Immunol. 1998;10:1563–1572. doi: 10.1093/intimm/10.10.1563. [DOI] [PubMed] [Google Scholar]
- 59.Grunvald E, Chiaramonte M, Hieny S, Wysocka M, Trinchieri G, Vogel S, Gazzinelli R, et al. Biochemical characterization and protein kinase C dependency of monokine-inducing activities of Toxoplasma gondii. Infect Immun. 1996;64:2010–2018. doi: 10.1128/iai.64.6.2010-2018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pollard AM, Skariah S, Mordue DG, Knoll LJ. A transmembrane domain-containing surface protein from Toxoplasma gondii augments replication in activated immune cells and establishment of a chronic infection. Infect Immun. 2009;77:3731–3739. doi: 10.1128/IAI.00450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilson DC, Grotenbreg GM, Liu K, Zhao Y, Frickel EM, Gubbels MJ, Ploegh HL, et al. Differential regulation of effector- and central-memory responses to Toxoplasma gondii infection by IL-12 revealed by tracking of Tgd057-specific CD8+ T cells. PLoS Pathog. 2010;6:e1000815. doi: 10.1371/journal.ppat.1000815. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information may be found in the online version of this article at the publisher’s web-site