

Abstract
Of the numerous inherited diseases known to afflict the pediatric population, spinal muscular atrophy (SMA) is among the most common. It has an incidence of approximately one in 10,000 newborns and a carrier frequency of one in 50. Despite its relatively high incidence, SMA remains somewhat obscure among the many neurodegenerative diseases that affect humans. Nevertheless, the last two decades have witnessed remarkable progress in our understanding of the pathology, underlying biology and especially the molecular genetics of SMA. This has led to a genuine expectation within the scientific community that a robust treatment will be available to patients before the end of the decade. The progress made in our understanding of SMA and, therefore, towards a viable therapy for affected individuals is in large measure a consequence of the simple yet fascinating genetics of the disease. Nevertheless, important questions remain. Addressing these questions promises not only to accelerate the march towards a cure for SMA, but also to uncover novel therapies for related neurodegenerative disorders. This review discusses our current understanding of SMA, considers the challenges ahead, describes existing treatment options and highlights state-of-the-art research being conducted as a means to a better, safer and more effective treatment for the disease.
Keywords: animal models, motor neuron, neurodegeneration, spinal muscular atrophy, survival motor neuron
Spinal muscular atrophy: clinical perspectives
Disease symptoms & clinical course
Spinal muscular atrophy (SMA) is a clinically heterogeneous, autosomal-recessive neuromuscular disorder diagnosed primarily in infants and less frequently in adults. The first documented reports of severe SMA were made in the 1890s at the University of Graz (Austria) and in Heidelberg (Germany), respectively, by the Austrian physician Guido Werdnig and the German clinician Johann Hoffman, each of whom described the neuromuscular phenotype of the disease and the accompanying characteristic loss of the anterior horn cells of the spinal cord [1– 4]. Patients with severe SMA, which is also known as Werdnig–Hoffman disease, typically manifest with weakness during the first 3 months of life and are so debilitated that they fail to ever sit independently, eventually succumbing to the disease before 2 years of age. A much milder form of SMA, with onset after 18 months of age, often during adolescence, and characterized by prolonged ambulation and a normal life expectancy, was described in 1956 and bears the eponymous title of Kugelberg–Welander disease [5]. An intermediate form of the disease with an onset between 6 and 18 months of age was first reported in 1893 at the University of Edinburgh (UK) and described again in depth in the 1960s by Dubowitz [6,7]. Intermediate SMA patients acquire the ability to sit independently but never walk, eventually becoming wheelchair bound with concomitant scoliosis of the spine, the latter condition a result of weak trunk muscles. Although such individuals have a reduced life expectancy, they frequently attain adulthood. A fourth class of patients characterized by disease onset in the fourth to sixth decade of life has also been described [8,9]. The disease in these patients, often referred to as adult-onset SMA, also results in muscle weakness, sometimes rendering affected individuals wheelchair bound. However, life expectancy is not affected.
The four classes of patients referred to above are also frequently described as type I (severe), II (intermediate), III (mild) and IV (adult-onset) SMA based on disease severity (Table 1). This classification has persisted, but only for historical reasons. Today, it is generally accepted that the boundaries between the classes often blur so that SMA patients form a continuum from the truly severe, with onset in utero, to the very mild, with onset during middle or late age.
Table 1.
Classification of spinal muscular atrophy types.
| Type | Eponym | Age at onset | Disease characteristics | Age at death | Proportion of total SMA (%) |
|---|---|---|---|---|---|
| I | Werdnig–Hoffman disease | <6 months | Proximal muscle weakness; patients fail to ever sit | <2 years | 60 |
| II | Dubowitz disease | 6–18 months | Proximal muscle weakness; patients attain ability to sit unassisted, but become wheelchair-bound; scoliosis of spine | >2 years | 27 |
| III | Kugelberg–Welander disease | >18 months | Proximal muscle weakness in early life; patients achieve ability to walk unaided | Normal life expectancy | 12 |
| IV | – | Adulthood | Proximal muscle weakness following active life during early adulthood; eventually may require walking aid | Normal life expectancy | 1 |
SMA: Spinal muscular atrophy.
Although clinical severities vary widely, all SMA patients exhibit a characteristic muscle weakness that affects the proximal muscles more than it does the distal ones, the lower limbs more than the upper ones and intercostal and axial muscle groups more than the diaphragm. It is commonly described as a symmetric condition in the literature, but patients almost always comment on one side or limb being stronger ([10] and personal observations). This asymmetry may contribute to the inevitable scoliosis that complicates the condition, particularly after ambulation is lost. The clinical course of SMA depends on the severity of the disease. Patients in the severe and intermediate regions of the spectrum progress through an early, acute phase of the disease, characterized by actual loss of strength or a period of stasis marked by a failure to gain strength, and therefore an inability to meet normal, rapidly evolving motor milestones. By contrast, milder patients, particularly those with adult-onset SMA, exhibit a gradual weakening of muscles over an extended period of time.
Clinical features of severe SMA are marked by profound hypotonia, a condition reflected in the term ‘floppy infant syndrome’, which is sometimes used to describe the disease. In addition, infants often assume a frog-leg posture owing to weakness of the pelvic muscles, exhibit areflexia of the extremities, fail to suckle and swallow efficiently and develop a bell-shaped torso, a result of breathing that relies primarily on the diaphragm. Tongue atrophy and fasciculations are always evident in severe cases, less common in intermediate forms and rare or absent in the type III and IV phenotypes. In especially severe instances, patients may also be arthrogrypotic. Electrophysiologically, severe SMA is characterized by evidence of profound denervation, suggesting a catastrophic loss of functioning motor units [11,12]. As expected, symptoms of the less severe forms of SMA are not as marked. Proximal muscle weakness in intermediate and mild SMA patients remains a cardinal feature, but in contrast to the severe form of the disease, their electromyographs are characterized by a thinned interference pattern and giant motor units [13], which is a likely consequence of waves of denervation and re-innervation of the muscle by surviving motor neurons. The surviving motor units are increased in both amplitude and duration.
SMA pathology
The pathological hallmarks of SMA are reflective of patient phenotypes and comprise a loss of the motor neurons of the ventral spinal cord and lower brainstem and consequent muscle atrophy (Figure 1). However, a detailed analysis of the cellular pathology, particularly of the spinal cord, has been hampered by the limited availability of suitable patient material. Much of the analysis was carried out on autopsy tissue from severe SMA patients and reflects findings at the end stage of the disease [14]. Analyses of the early pathology of the disease have relied on mouse models and are briefly referred to later. Nevertheless, a few key findings have emerged [14]. Of these, the most striking is a profound loss of large ventral horn spinal motor neurons. Less common is a mispositioning (heterotopy) of these cells in the ventral white matter [15]. The motor neurons that remain often appear swollen or chromatolytic, with accumulations of phosphorylated neurofilaments, vesicles and ribosomes in the cell soma. In rarer instances, such apathology is also observed in the neurons comprising Clarke's column, the dorsal roots and brain structures such as the thalamus. An examination of intramuscular nerves revealed evidence of abnormal beading, as well as Wallerian degeneration.
Figure 1. Pathological hallmarks of spinal muscular atrophy.
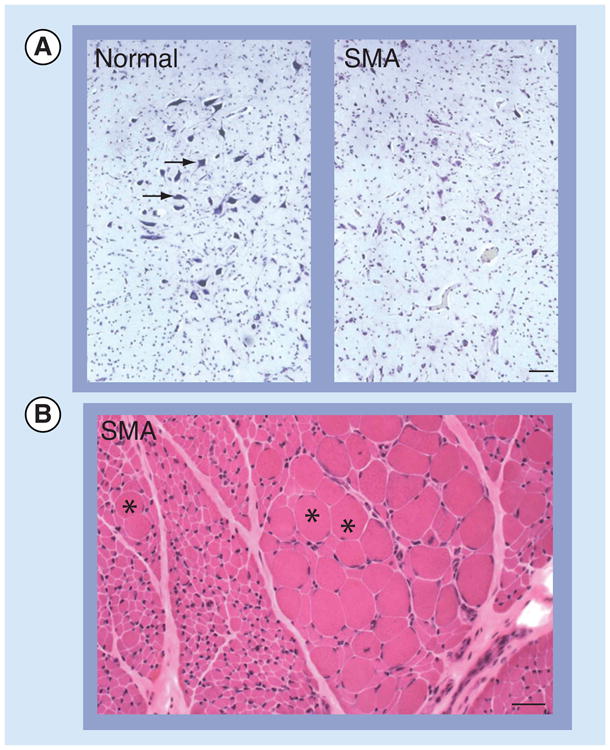
(A) Neurodegeneration in SMA is epitomized by the loss of the large motor neurons (arrows) of the spinal cord and brain stem (e.g., the hypoglossal nucleus [depicted]). Tissue was obtained from autopsy material and stained with cresyl echt violet. Scale bar: 30 μM. (B) Skeletal muscle atrophy, characterized by numerous atrophic muscle fibers interspersed with hypertrophic fibers (asterisks), is a second characteristic feature of the human disease. Tissue was obtained from biopsy material and sections were stained with hematoxylineosin according to standard procedures.
Scale bar: 20 μM.
SMA: Spinal muscular atrophy.
Consistent with the loss of motor neurons, the muscle of SMA patients exhibits evidence of acute (severe/intermediate SMA) or chronic (intermediate/mild SMA) denervation [16]. The muscle from patients at the more severe end of the disease spectrum typically consists of numerous rounded, atrophic fibers interspersed with one or more hypertrophic myofibers (Figure 1). Failure to reinnervate muscle, which is a likely consequence of severely dysfunctional motor neurons, results in the persistence of the normal checkerboard pattern of type I and II fibers. Muscle fibers also appear immature, resembling myotubes with central nuclei, suggesting a primary myopathic condition [17]. By contrast, muscle from patients at the mild end of the disease spectrum exhibits evidence of numerous cycles of denervation and re-innervation, resulting in fiber-type grouping. Transverse sections of muscle from these patients may reveal a mix of atrophic and hypertrophic fibers or, in very mild cases of the disease, normal-sized fibers that have presumably been reinnervated by surviving motor neurons. The muscle from mildly affected patients has also been reported to sometimes exhibit myopathic features, including fiber splitting, central nuclei, basophilia and an increase in endomysial connective tissue [16]. The serum concentrations of creatine kinase are mildly elevated in these mild cases as a laboratory correlate.
More recent data that emerged from a study of SMA model mice and were confirmed in tissue from SMA patients suggest that defects of the neuromuscular synapses are perhaps the earliest pathological hallmarks of the disease [18]. Defects of the pre- as well as the post-synapse have been observed (Figure 2), which is indicative of a disease in which abnormalities of the distal end of the motor unit likely precede those of the motor neuron cell body.
Figure 2. Neuromuscular junction defects are a characteristic feature of spinal muscular atrophy.
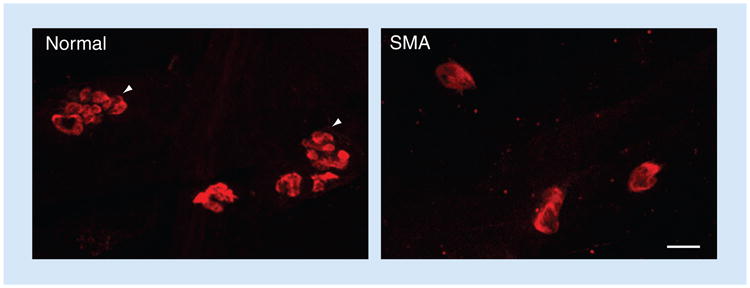
The motor endplates (postsynapse) of the SMA patient are smaller and less complex than those of a control individual (arrowheads). Tissue was obtained from autopsies and stained as whole-mount preparations with fluorescently labeled α-bungarotoxin. Scale bar: 30 μM.
SMA: Spinal muscular atrophy.
Genetic principles of SMA
The genetic defects underlying severe SMA were mapped by linkage analysis to chromosome 5q11.2–13.3 and subsequently shown to also be responsible for the intermediate and mild forms of the disease [19, 20]. In 1995, the defects were shown to lie within a novel gene, SMN1 [21]. Sequence analysis determined that the gene lay within an inverted duplication that also contained an almost identical homolog, SMN2. SMA patients harbor mutations, mostly deletions, in SMN1, but invariably carry one or more copies of the SMN2 gene. In fact, owing to the inherently unstable nature of the SMN locus, humans bear varying numbers of either gene.
The critical difference between the two SMN genes is a single, translationally silent C (SMN1)→T (SMN2) nucleotide change 6 bp inside exon 7 [22,23]. This difference alters the efficiency with which exon 7 is included within the SMN transcript, most likely by creating an exonic splicing suppressor in the SMN2 gene [24]. As a consequence, most of the transcripts from this gene lack exon 7 (SMNΔ7). By contrast, the preponderance of mRNA from SMN1 is full-length. Although both transcripts are translated, the SMNΔ7 protein is unstable and rapidly degraded, resulting in inadequate levels of functional SMN protein from the SMN2 gene (Figure 3). SMA patients therefore express less than normal amounts of the SMN protein [25, 26]. Subsequent work demonstrated that levels of the SMN protein in patients are determined by the SMN2 copy number, which is generally inversely correlated with SMA severity [27, 28]. These findings intuitively make SMN2 an attractive molecular target for the treatment of SMA.
Figure 3. The genetic basis of spinal muscular atrophy, depicting the two major genes involved: SMN1 and SMN2.

SMA patients are either homozygously deleted for or harbor point mutations in SMN1, but they always retain at least one copy of the SMN2 gene.
Complete absence of SMN is embryonic lethal.
ESS: Exonic splicing silencer; Ex: Exon; FL: Full length; SMA: Spinal muscular atrophy.
Adapted with permission from [107].
Functions of the SMN protein: two contrasting views
The 294-amino acid full-length SMN protein is expressed by both SMN genes, even though SMN1 is the predominant contributor to overall levels. Consistent with the expression of the SMN transcripts, the SMN protein is ubiquitously expressed and partitions to both cytoplasm and nucleus. In the nucleus, it concentrates in subnuclear foci that lie adjacent to or colocalize with similar foci that stain positively for coilin, a marker of Cajal bodies [29]. Consequently, the SMN foci were termed ‘Gemini of Cajal bodies’ or simply ‘gems’. Gem numbers in patient fibroblasts frequently correlate with SMA severity [25].
The myriad interactions between SMN and other cellular proteins are indicative of its involvement in multiple biochemical pathways. However, the most widely recognized function of the SMN protein is its role in the biogenesis of spliceosomal U small nuclear ribonuclear protein (snRNP) particles and in the related process of pre-mRNA splicing, which involves an interaction between snRNPs and the nascent, unprocessed RNA. These functions are not carried out by SMN alone, but instead by a complex comprising multimers of SMN bound either directly or indirectly to at least seven additional proteins, the gemins. The complex, referred to as the SMN complex, participates in a coordinated set of steps that involve the export of snRNA molecules into the cytoplasm, the assembly of a heptameric ring of spliceosomal Sm proteins onto the snRNAs by the complex to form an snRNP and the import of the assembled snRNP-bound complex into the nucleus, where it mediates pre-mRNA splicing [30]. The role of the SMN complex in the housekeeping processes of snRNP biogenesis and pre-mRNA splicing has been difficult to reconcile with the selective neuromuscular phenotype observed in SMA patients and animal models. Indeed, in fish and fly models of SMA, studies have uncoupled these two processes from such defining characteristics of the disease as reduced motor activity and defective axon outgrowth [31,32]. While it is possible that genes that are critical for the proper function of the neuromuscular system and therefore selectively expressed in one of its constituent cell types are inappropriately spliced in the absence of adequate SMN protein, only one such gene has been identified (in flies) and shown to contribute to the disease phenotype [33]. The relevance of a perturbation in this gene to the disease in mice and humans is currently not clear. This has prompted speculation of additional SMN functions that might explain the characteristic neuromuscular phenotype of SMA.
Chief among novel SMN functions that might underlie the selective SMA phenotype are those that are important to the health and viability of motor neurons. This has focused attention on the localization and possible functions of the protein in motor axons. Numerous studies have revealed SMN complexes in axons [34]. Interestingly, these complexes lack the spliceosomal Sm proteins, but they do contain some or all of the gemins [35]. Furthermore, SMN in axons has been shown to interact with distinct RNA binding proteins [36–39], suggesting that it may function in assembling a wide variety of RNP particles, some of which could be critical to motor neuron function and thus explaining the early loss of these cells in SMA. Accordingly, it has been suggested that the reported interactions of the SMN protein in the axonal compartment with proteins such as hnRNP-R, HuD and KSRP, which bind and transport β-actin, acetylcholinesterase and p21 transcripts, respectively, may be responsible, at least in part, for the predominantly neuromuscular SMA disease phenotype. Ongoing studies in cell and animal models of SMA continue to refine the putative, physiologically relevant functions of the SMN protein. These have been complemented with findings in human patients that have exploited ‘discordant sibs’ in order to identify genetic modifiers and shed further light on potential SMN functions [40]. This last study, in combination with reports of an increase of RhoA, a regulator of actin dynamics, in SMA mice or SMN-depleted cells, is suggestive of there being a role for the SMN protein in modulating the actin cytoskeleton [41,42]. Assuming that SMN is indeed multifunctional, it would not be surprising if the functions of the protein are disrupted in a hierarchical fashion that is dependent upon precise protein levels. This sequential disruption in SMN functions might very well explain the wide spectrum of phenotypes observed in human patients and mouse models of SMA.
SMA: insights from animal models
Developing an animal model of a human disease is widely used to gain a better understanding of the pathophysiology of the specific disorder. Accordingly, a number of animal models of SMA have been developed since the disease gene was identified in 1995. The collective knowledge gained from these models has not only provided us with greater insights into the mechanisms underlying SMA, but also served as a springboard for the current and emerging treatments for the human condition.
Fish, fly & worm models of SMA
An immediate challenge in developing accurate genetic models of human SMA became apparent with the dual realization that humanity is the only species that harbors an SMN2 gene, and that complete absence of the SMN protein, resulting from homozygous loss of the single SMN gene in nonhumans, is lethal to the organism [43]. Viable animal models of the disease require residual levels of activity from their single SMN gene or the presence of an SMN2-like gene in order to partially compensate for genetic or chemical inactivation of the endogenous locus. In invertebrates, such as the nematode Caenorhabditis elegans and the fruit fly Drosophila melanogaster, the relatively large contribution of maternal SMN to the fertilized oocyte allows partial development of embryos even if their own copy of the SMN gene is inactivated. This is also true of the zebrafish, Danio rerio [44]. However, in each of these organisms, a steady depletion of maternally contributed SMN in Smn−/− embryos results in only a small window of time during which protein concentrations attain disease-relevant levels. This is a limitation of fish, flies and worms, in which the SMN1-equivalent gene is knocked out in an attempt to model SMA. Nevertheless, by exploiting newer technologies, such as RNAi and antisense gene knockdown, or by using novel hypomorphic alleles, much has been learned about the human disease from these model organisms [44].
Mouse models of SMA: further insights into disease pathology
Perhaps the most genetically relevant current models of human SMA are those involving transgenically engineered mice. To circumvent the embryonic-lethal phenotype caused by homozygous inactivation of the murine Smn gene, a human SMN2 transgene was introduced onto the null (Smn−/−) background. Two copies of the SMN2 transgene not only rescued the Smn−/− embryonic lethality, but also resulted in mice with a severe form of SMA [45,46]. As expected, such mice express very low levels of the SMN protein. Conversely, null (Smn−/−) mice harboring eight copies of the SMN2 gene expressed wild-type or greater levels of the SMN protein, appeared asymptomatic and were indistinguishable from healthy, nontransgenic animals [46]. These experiments provide direct evidence of the ability of SMN2 to modify the SMA phenotype, justifying the use of the gene as a molecular target for the treatment of the disease.
In the decade since the first SMN2-expressing SMA model mice were reported, a number of additional SMN transgenes harboring severe as well as mild mutations have been generated to model varying severities of the human disease. The collective analyses of these lines have shed considerable light on the pathophysiology of human SMA. For instance, an inability to reconcile observations of profound early muscle weakness with the persistence of motor neuron cell bodies within the spinal cord prompted an examination of the distal end of the motor unit and led to the discovery of neuromuscular synapse defects in SMA. These defects, which involve nerve terminals (presynapse) as well as the motor endplate (postsynapse), are among the earliest hallmarks of the disease and were subsequently confirmed in material from human patients [18,47]. Parallel experiments uncovered functional, neurotransmission abnormalities that were reflective of the morphological defects of the neuromuscular synapse [18]. Subsequent detailed analyses of the motor neuron perikarya in the model mice revealed that the peripheral synapses were not the only ones to suffer as a consequence of reduced SMN protein. Central synapses onto motor neuron cell bodies are also profoundly affected in SMA mice [48–50]. While this has yet to be confirmed in tissue from human patients, the data suggest that SMA is a disease that likely begins by affecting the synapses, eventually leading to loss of cells within the spinal cord. Although initial studies of the SMA model mice focused on the spinal motor neurons, the relative ease with which the mutant mice can be generated provided investigators with the ability to examine other organ systems as well. These analyses, which have revealed cardiac, brain and muscle defects, suggest that severe SMA may be multi-systemic [51–53]. Indeed, reports in the literature of the most severe SMA patients bear this out [17,54]. Considering the role of the SMN protein in the housekeeping processes of RNP biogenesis and pre-mRNA splicing, such a discovery is not entirely unexpected, although the finding may be limited to a small fraction of the total SMA patient population. SMA model mice have also enabled investigations of the physiological relevance of the known function of the SMN protein to the disease phenotype. In no instance has this been more true than in the case of SMN's role in assembling snRNP particles. These analyses suggest that there is a strict correlation between disease severity and snRNP assembly [55], although studies carried out in fish and fly models that claim to have uncoupled snRNP biogenesis from motor neuronal defects are indicative of additional SMN functions that contribute to the overall SMA phenotype.
Mouse models of SMA: revealing the when & where of SMN
Critical to the success of a treatment involving a disease caused by protein insufficiency is a precise determination of the cellular site(s) of action of the protein and when, during the various stages of life, it is required to prevent disease. The mouse models described above have served as an excellent resource for addressing these questions. Given the neuromuscular nature of the SMA phenotype, initial experiments to determine which tissues require the protein focused on motor neurons and skeletal muscle. Early transgenic experiments suggested that restoring SMN to the muscle of SMA mice provided no benefit, whereas augmenting the protein in nervous tissue was sufficient to rescue the disease phenotype [56]. More recent data have either refined or challenged these conclusions. For instance, more precise targeting strategies have suggested that the SMN protein is important in muscle to prevent disease [57]. Furthermore, despite the motor neuron phenotype observed in SMA, restoring SMN selectively to motor neurons or to all neurons appears not to be sufficient to protect from disease [58–60]. Restoring the protein to both tissues may be sufficient to fully treat SMA, but given the ubiquitous expression of the SMN protein, it will not be surprising if the disease has to be treated systemically. Using model mice harboring inducible alleles engineered to respond in a temporal manner, investigators have also determined when a treatment ought to be administered to be effective. These studies have concluded that although restoring SMN early during the course of the disease is more effective than a treatment initiated later, phenotypic symptoms can be halted, and indeed reversed, even after the onset of profound muscle weakness [61, 62]. The finding is particularly significant for the treatment of symptomatic patients. Collectively, the data exemplify the utility of the SMA models in developing rational therapeutic strategies.
Treatment of SMA
Current therapeutic options
Despite the monogenic nature of the disease and the time that has elapsed since the SMA genes were identified, the currently available treatments for patients afflicted with the disease are palliative at best. Past clinical trials to test the efficacy of promising drugs have mostly involved histone deacetylase inhibitors, a class of compounds that has been shown to increase the expression of a number of genes, including SMN2 [63]. A number of these drugs increase SMN in cultured cells. Some were subsequently demonstrated to provide benefit in mouse models of SMA [64–70]. Three of these compounds – hydroxyurea, phenylbutyrate and valproic acid – were tested in SMA patients, but they had modest, if any, effect [71–74]. Salbutamol, a β-adrenergic agonist, and the neuroprotective agents riluzole and gabapentin have also been tested in patients. The first of these did provide a benefit, but the study did not include placebo-treated patients and must therefore be interpreted with caution [75]. Neither neuroprotective agent conferred any benefit [76–78].
Owing to the disappointing results obtained so far, SMA patients currently have little recourse to effective treatments, while clinicians have been limited to treating secondary complications arising from the disease. Especially insidious in severe SMA, the predominant form of the disease, are complications that are pulmonary in nature. Respiratory failure is the chief cause of mortality in patients at the more severe end of the disease spectrum. This recognition and the ability to intervene using noninvasive ventilation protocols have resulted in a dramatic increase in life expectancy among severely affected SMA patients. Patients born between 1995 and 2006 were reported to exhibit a 70% reduced risk of death compared with those born between 1980 and 1994, over a mean follow-up of approximately 50 months [79]. Of equal concern in many SMA patients are complications that are orthopedic in nature. Scoliosis and contractures are common in patients at the severe end of the disease spectrum; fractures and hip subluxation are often issues in milder patients. In instances of severe scoliosis, surgical intervention is often recommended. In milder SMA patients, physiological fatigue is increasingly being recognized, particularly in ambulatory patients [80,81]. 4-aminopyridine, which has been approved for the treatment of fatigue in patients with multiple sclerosis, is now also being tested in SMA patients for efficacy. Last, but not least, are complications arising from extreme muscle weakness in severely affected SMA infants, and thus the propensity to tire while feeding. Failure to thrive is therefore a common concern, but this can be mitigated by ensuring proper nutrition. In most instances, the placement of a gastrotomy tube within the infant is warranted in order to maintain a state of positive nitrogen balance. Conversely, milder sedentary SMA patients are prone to becoming overweight, owing to an intake of excess calories [82,83]. It is, therefore, recommended that the nutritional status of all SMA patients be carefully monitored.
Emerging treatment strategies
Although the standard of care for SMA patients has vastly improved in the two decades since the SMN genes were identified, it is clear that the available treatments for the disease are far from effective [84]. Nevertheless, progress in developing new therapeutic strategies has been rapid and is, in no small measure, a result of two important factors: first, the monogenic nature of the disease; and second, the availability of a range of disease models. The approaches that are being developed are summarized below.
Pharmacologic approaches
Given the cause of SMA – a deficiency of the SMN protein, the presence of the SMN2 gene in all patients and proof-of-concept studies indicating that this gene can be used to restore SMN as a means to a treatment – it is of little surprise that much of the effort in developing therapeutic strategies has centered on augmenting the SMN protein, either directly or by modulating SMN2 gene expression [21,45,46]. In the last decade, considerable resources have been ploughed into the search for pharmacologic agents that are capable of enhancing the expression of SMN from the SMN2 gene, either via transcriptional activation and/or by altering the splicing pattern of the gene in order to induce more of its transcript to be full length. The effort has involved academic institutions as well as industry and has resulted in a plethora of compounds [62,64–70,85,86]. However, only a handful has been effective in animal models. The most noteworthy include LBH589, a histone deacetylase inhibitor, a C5 quinazoline derivative that targets the DcpS enzyme and a proprietary molecule developed at PTC Therapeutics, Inc. (NJ, USA) [87–89]. The last of these has a major impact on the severe SMA mouse phenotype, enhancing life expectancy from approximately 14 days to more than 6 months [89]. Along with the quinazoline derivative, this compound is being further developed for eventual use in humans.
Small molecule-type drugs often have the distinct advantage of being able to penetrate the BBB, which is a major consideration in the treatment of neurological disorders. However, the knowledge gained from in vitro studies elucidating the elements within SMN2 that are important for the inclusion of exon 7 into the SMN transcripts has spurred the development of short antisense oligonucleotides (ASOs) as a means of enhancing SMN expression from this gene [90]. Targeting one specific element, ISS-N1, within SMN2 is particularly effective in boosting SMN levels from the gene [91]. Indeed, administering an ASO against the element into SMA model mice has been shown to provide as much therapeutic benefit as the most potent small molecules that have been identified so far [92,93]. Based on the promise shown in preclinical studies, these ASOs have been entered into clinical trials and could very well be the basis for the first truly effective treatment of SMA. However, as alluded to earlier, they fail to cross the BBB [94], necessitating direct administration into the subarachnoid space and cerebrospinal fluid of the spinal cord, a somewhat invasive procedure considering the fragility of SMA patients.
A third approach that is used to modulate SMN2 expression has involved the use of aminoglycosides, compounds that are thought to suppress recognition of the first naturally occurring translational stop codon in exon 8 of the SMNΔ7 mRNA and force read-through. The resulting altered transcript is translated into an SMN protein that is relatively stable compared with SMNΔ7 protein and rescues (albeit modestly) the severe SMA phenotype in model mice [95]. Pharmacologic modulators of genes that are distinct from SMN2 have not received nearly as much attention, but may be just as important in the development of safe and effective treatments for SMA. One notable example of such a compound is fasudil, a US FDA-approved ROCK inhibitor that mitigates the SMA phenotype without enhancing SMN levels [96]. Given its reported effect of enhancing the muscle function of SMA model mice, fasudil (or similar compounds) could be combined with ASOs in order to provide benefit to multiple organ systems in affected individuals. Finally, despite their disappointing record so far in US SMA clinical trials [76–78], neuroprotective molecules such as olesoxime, which has been reported to benefit a mouse model of amyotrophic lateral sclerosis [97], may yet be viable therapeutic agents for the treatment of SMA.
Gene- & cell-replacement approaches
A distinct advantage in designing treatments for SMA is the presence of the SMN2 gene, which is a well-validated molecular target in all affected individuals. This precludes having to deliver and replace SMN in the necessary cells of an SMA patient, which is a major challenge, especially in treating neurological disorders. Nevertheless, with the discovery of novel serotypes of adeno-associated virus (AAV), such as AAV9, which not only infect numerous cell types, but also appear to traverse the BBB, there has been renewed interest in applying gene-replacement protocols in the treatment of SMA. Neonatal SMA model mice injected systemically with self-complementing AAV9 (scAAV9) engineered to carry the wild-type SMN gene expressed high levels of SMN protein in multiple tissues and derived remarkable therapeutic benefit, surviving in some instances to 12 months or more with no evidence of muscle weakness [98–100]. Considering the existence of proof-of-concept studies demonstrating the ability of scAAV9 to penetrate the mature BBB and infect adult motor neurons [101], an SMN-replacement strategy to treat SMA would be a promising alternative to pharmacologic approaches. However, based on the design of the vector to be maintained episomally, a drawback to this approach would be the dilution and eventual loss of the therapeutic gene in dividing cells.
In contrast to the pharmacologic and gene-replacement approaches described above, cell-replacement strategies have received less attention. The primary goal of such an approach is to replace the spinal motor neurons that are lost during the course of the disease. Likely sources of the motor neurons would be embryonic stem cells and neural stems cells. Transplantation of stem cell-derived motor neurons has clearly been shown to provide therapeutic benefit in animal models of motor neuron disease (reviewed in [102]). SMA model mice subjected to the treatment not only gained weight and strength, but also survived longer [103]. However, it is unlikely that this benefit involved the establishment of new motor units. Indeed, fewer than 2% of the transplanted cells eventually made connections with muscle. Instead, the benefit is likely to occur through the nourishing effects of the transplanted cells on existing SMA motor neurons. Nevertheless, cell-replacement strategies have exhibited sufficient promise to prompt the clinical community to embark upon clinical trials for a related motor neuron disease, amyotrophic lateral sclerosis, and could be yet another approach to treating SMA.
Conclusion
The progress made by the scientific and patient community towards a treatment for SMA has as much to do with the genetics of the disease as it does with the ability to accurately mimic the condition in model organisms. Restoring SMN by exploiting SMN2 as a target is intuitive and has driven the design of treatments for the disease. The outcome of this collective research is cause for optimism. Small molecules, as well as ASOs, have been shown to provide remarkable therapeutic benefit in model mice. These reagents are currently either being modified for safety or being tested in clinical trials. If the results in mice are any measure of the effects of these molecules in patients, the outcome will be promising. Nevertheless, important questions remain, many of which center on the biology of SMA. For instance, it is still not clear precisely how a deficiency of the SMN protein causes a predominantly motor neuron phenotype. Neither is it certain whether the only relevant function of the protein in ensuring the well-being of a cell is in orchestrating the splicing cascade. Although answers to these questions may not significantly impact treatments centered on restoring SMN, addressing them will surely prove instructive in designing truly effective therapies for SMA.
Future perspective
Since the SMA genes were first identified in 1995, the field has made relatively rapid progress towards the design and, more recently, the implementation of rational therapies for this disorder in humans. This is in large measure a consequence of the relatively simple genetics of the disease, including its recessive pattern of inheritance, the recognition that SMA is caused by insufficient SMN protein and the development of humanized mouse models with which to conduct preclinical work. The relatively severe phenotype of some of the SMA model mice has enabled the community to rapidly and reliably test a variety of therapeutic molecules and related treatment strategies for efficacy. Some of these studies have led to the initiation of human clinical trials using the first truly potent SMN-inducing molecules – ASOs designed to alter the defective splicing of the SMN2 gene (NCT01703988 and NCT01839656 [201,202]). The outcomes of these trials are eagerly anticipated, and the expectation, based on preclinical work and assuming ASOs were administered early in the course of the disease, is that patients will respond favorably. While this development may be a cause for excitement, given the generally disappointing outcomes of past trials with other agents and the caveats inherent in all translational studies, the mood among those of us who have seen the field evolve is more one of guarded optimism. Indeed, the pace of innovation in terms of developing new and complementary treatments has not slowed. For instance, work to develop an orally delivered BBB-penetrating small molecule that is safe, well-tolerated and as potent as the ASOs currently being tested is well underway and constitutes part of the effort to refine current strategies. The implementation of strategies to reverse the disease process and restore function to individuals in advanced stages of the disease would also be a significant development. This will require further investment in basic research.
While the development of promising treatments for SMA has proceeded relatively rapidly, the biology of the disease remains as inscrutable as ever. A few of the most basic unanswered questions are: what is the precise role of SMN in SMA? Are there specific mediators of the SMA phenotype that explain how low SMN precipitates a neuromuscular disease? Does SMN possess more than one function and, if so, does the hierarchical disruption of these functions explain the range of SMA phenotypes? Considering the housekeeping role of SMN in the splicing cascade and the effects of low levels of this protein on the organism, what might the consequences of overexpressing SMN be? Might SMN biology inform our understanding of motor neuron health and disease in general, and might SMN pathways intersect with those that are implicated in other motor neuron diseases (e.g., FUS-, SOD-1- or TDP-43-related amyotrophic lateral sclerosis)? Recent reports suggest that this last question has already gained considerable attention [104–106]. We anticipate that the quest to answer these questions will ensure a continued, robust investment of the scientific community's time in the sort of basic research that resulted in the identification of the SMA genes, the elucidation of a key SMN function and the development of a battery of novel animal models. We would not be surprised if the collective effort invested in such research produces further advances in the treatment of human SMA.
Medscape: Continuing Medical Education Online.
This activity has been planned and implemented in accordance with the Essential Areas and policies of the Accreditation Council for Continuing Medical Education through the joint sponsorship of Medscape, LLC and Future Medicine Ltd. Medscape, LLC is accredited by the ACCME to provide continuing medical education for physicians.
Medscape, LLC designates this journal-based CME activity for a maximum of 1 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at www.medscape.org/journal/fnl; (4) view/print certificate.
Learning Objectives.
Upon completion of this activity, participants should be able to:
Identify the clinical features and pathology of spinal muscular atrophy (SMA)
Describe the biology and molecular genetics of SMA
Outline potential treatment targets for SMA and current management
Executive Summary.
Spinal muscular atrophy (SMA), which was first described in the 1890s, is a clinically heterogeneous disorder that manifests as muscle weakness, usually developing in infancy, in severe, intermediate or mild forms.
Infants exhibiting the most severe form of SMA are profoundly hypotonic and often succumb to the disease in the first 2 years of life.
The pathological hallmarks of SMA include spinal motor neuron loss and attendant skeletal muscle atrophy.
Recent analyses of SMA mouse models and human samples have demonstrated that the neuromuscular synapse is among the first structures to be affected by low SMN protein.
SMA is caused by mutations in SMN1. However, patients always harbor SMN2, an almost identical but functionally deficient copy of the gene.
The best-established functions of the SMN protein are in the biogenesis of spliceosomal U small nuclear ribonuclear protein particles and pre-mRNA splicing.
A number of less well-recognized functions have been inferred from the myriad proteins that SMN is reported to bind.
The precise function of SMN in SMA is still not clear.
Fish, fly and worm models have enhanced our understanding of human SMA, but are limited by the lack of the Smn2 gene in these models, thus resulting in imperfect genetic models of the human disease.
Mouse models of SMA were generated by introducing the human SMN2 gene onto a null murine Smn genetic background. This resulted in a genetic mimic of the human disease.
SMA model mice have provided valuable insights into the natural history of SMA, the cellular sites of action of SMN and the temporal requirements for the protein. They have also facilitated important preclinical work.
A number of US FDA-approved small molecules, many of them histone deacetylase inhibitors, have been tested on SMA patients with modest, if any, therapeutic benefit.
The treatment of SMA symptoms (e.g., respiratory distress, inefficient feeding and scoliosis) has benefited patients, but fails to combat the root cause of the disease.
Emerging disease-modifying treatments exploit the presence of the SMN2 gene in all patients and the ability to induce this gene in order to express enhanced levels of SMN protein. Currently, the most promising of these treatments involves the use of antisense oligonucleotides, potent splice modulators that are presently being tested in patients.
The quest for an SMA treatment has rapidly led to a number of promising therapeutic strategies.
Additional research in order to elucidate the molecular mechanisms underlying human SMA will be important in refining and improving current therapies.
Acknowledgments
The authors thank A Hays and S Kariya for the data presented in Figures 1 & 3, respectively. The authors also apologize to all our colleagues whose important work could not be directly cited.
Footnotes
Financial & competing interests disclosure Editor: Elisa Manzotti, Publisher, Future Science Group. Disclosure: Elisa Manzotti has disclosed no relevant financial relationships.
CME author: Laurie Barclay, MD, Freelance writer and reviewer, Medscape, LLC. Disclosure: Laurie Barclay, MD, has disclosed no relevant financial relationships.
Author & credentials: Umrao R Monani, PhD, Columbia University Medical Center, New York, NY 10032, USA.
Disclosure: Work in the author's laboratory is supported by the the Muscular Dystrophy Association, Spinal Muscular Atrophy Foundation, Spinal Muscular Atrophy-Europe, Families of Spinal Muscular Atrophy, Will Foundation, the Department of Defence and NIH R01NS057482.
Darryl C De Vivo, MD, Columbia University Medical Center, New York, NY 10032, USA.
Disclosure: Work in the author's laboratory is supported by the Colleen Giblin, Spinal Muscular Atrophy and Will Foundations and Milestones for Children.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1•.Werdnig G. Zwei frühinfantile hereditäre Fälle von progressiver Muskelatrophie unter dem Bilde der Dystrophie, aber auf neurotischer Grundlage. Arch Psychiat Nervenkr. 1891;22:437–480. Along with [2], this was the simultaneous first report to describe human spinal muscular atrophy (SMA) [Google Scholar]
- 2•.Hoffmann J. Ueber chronische spinale Muskelatrophie im Kindesalter, auf familiärer Basis. Deutsch Z Nervenheilk. 1893;3:427–470. Along with [1], this was the simultaneous first report to describe human SMA. [Google Scholar]
- 3.Hoffmann J. Weiterer Beitrag zur Lehre von der hereditären progressiven spinalen Muskelatrophie im Kindesalter nebst Bemerkungen über den fortschreitenden Muskelschwund im Allgemeinen. Deutsch Z Nervenheilk. 1897;10:292–320. [Google Scholar]
- 4.Hoffmann J. Dritter Beitrag zur Lehre von der hereditären progressiven spinalen Muskelatrophie im Kindesalter. Deutsch Z Nervenheilk. 1900;18:217–224. [Google Scholar]
- 5.Kugelberg E, Welander L. Heredofamilial juvenile muscular atrophy simulating muscular dystrophy. AMA Arch Neurol Psychiatry. 1956;75:500–509. doi: 10.1001/archneurpsyc.1956.02330230050005. [DOI] [PubMed] [Google Scholar]
- 6.Thompson J, Bruce A. Progressive muscular atrophy in a child with a spinal lesion. Edinb Hosp Rep. 1893;1:372. [Google Scholar]
- 7.Dubowitz V. Infantile muscular atrophy. A prospective study with particular reference to a slowly progressive variety. Brain. 1964;87:707–718. doi: 10.1093/brain/87.4.707. [DOI] [PubMed] [Google Scholar]
- 8.Clermont O, Burlet P, Lefebvre S, et al. SMN gene deletions in adult-onset spinal muscular atrophy. Lancet. 1995;346(8991–8992):1712–1713. doi: 10.1016/s0140-6736(95)92881-2. [DOI] [PubMed] [Google Scholar]
- 9.Brahe C, Servidei S, Zappata S, Ricci E, Tonali P, Neri G. Genetic homogeneity between childhood-onset and adult-onset autosomal recessive spinal muscular atrophy. Lancet. 1995;346(8977):741–742. doi: 10.1016/s0140-6736(95)91507-9. [DOI] [PubMed] [Google Scholar]
- 10.Kang PB, Krishnamoorthy KS, Jones RM, Shapiro FD, Darras BT. Atypical presentations of spinal muscular atrophy type III (Kugelberg–Welander disease) Neuromuscul Disord. 2006;16(8):492–494. doi: 10.1016/j.nmd.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 11.Hausmanowa-Petrusewicz I. Electrophysiological findings in childhood spinal muscular atrophies. Rev Neurol (Paris) 1988;144(11):716–720. [PubMed] [Google Scholar]
- 12.Bromberg MB, Swoboda KJ. Motor unit number estimation in infants and children with spinal muscular atrophy. Muscle Nerve. 2002;25(3):445–447. doi: 10.1002/mus.10050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hausmanowa-Petrusewicz I, Karwańska A. Electromyographic findings in different forms of infantile and juvenile proximal spinal muscular atrophy. Muscle Nerve. 1986;9(1):37–46. doi: 10.1002/mus.880090106. [DOI] [PubMed] [Google Scholar]
- 14•.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3(2):97–110. doi: 10.1006/nbdi.1996.0010. Comprehensive review of the natural history of SMA. [DOI] [PubMed] [Google Scholar]
- 15.Simic G, Mladinov M, Seso Simic D, et al. Abnormal motoneuron migration, differentiation, and axon outgrowth in spinal muscular atrophy. Acta Neuropathol. 2008;115(3):313–326. doi: 10.1007/s00401-007-0327-1. [DOI] [PubMed] [Google Scholar]
- 16.Dubowitz V. Muscle Disorders in Childhood. 2nd. Saunders; PA, USA: 1995. [Google Scholar]
- 17.Nadeau A, D'Anjou G, Debray G, Robitaille Y, Simard LR, Vanasse M. A newborn with spinal muscular atrophy type 0 presenting with a clinicopathological picture suggestive of myotubular myopathy. J Child Neurol. 2007;22(11):1301–1304. doi: 10.1177/0883073807307105. [DOI] [PubMed] [Google Scholar]
- 18••.Kariya S, Park GH, Maeno-Hikichi Y, et al. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17(16):2552–2569. doi: 10.1093/hmg/ddn156. Reported evidence in SMA mouse models and humans of novel, early pathology at the neuromuscular synapse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gilliam TC, Brzustowicz LM, Castilla LH, et al. Genetic homogeneity between acute and chronic forms of spinal muscular atrophy. Nature. 1990;345(6278):823–825. doi: 10.1038/345823a0. [DOI] [PubMed] [Google Scholar]
- 20.Brzustowicz LM, Lehner T, Castilla LH, et al. Genetic mapping of chronic childhood-onset spinal muscular atrophy to chromosome 5q11.2–13.3. Nature. 1990;344(6266):540–541. doi: 10.1038/344540a0. [DOI] [PubMed] [Google Scholar]
- 21••.Lefebvre S, Burglen L, Reboullet S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–165. doi: 10.1016/0092-8674(95)90460-3. Describes the identification of the SMN genes. [DOI] [PubMed] [Google Scholar]
- 22.Monani UR, Lorson CL, Parsons DW, et al. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 23.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci USA. 1999;96(11):6307–6311. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34(4):460–463. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 25.Coovert DD, Le TT, McAndrew PE, et al. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6(8):1205–1214. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 26.Lefebvre S, Burlet P, Liu Q, et al. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 27.McAndrew PE, Parsons DW, Simard LR, et al. Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet. 1997;60(6):1411–1422. doi: 10.1086/515465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldkotter M, Schwarzer V, Wirth R, Wienker TF, Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet. 2002;70(2):358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Dreyfuss G. A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 1996;15(14):3555–3565. [PMC free article] [PubMed] [Google Scholar]
- 30•.Battle DJ, Kasim M, Yong J, et al. The SMN complex: an assembly machine for RNPs. Cold Spring Harb Symp Quant Biol. 2006;71:313–320. doi: 10.1101/sqb.2006.71.001. Concise review of the role of SMN in assembling ribonucleoprotein particles. [DOI] [PubMed] [Google Scholar]
- 31.Carrel TL, McWhorter ML, Workman E, et al. Survival motor neuron function in motor axons is independent of functions required for small nuclear ribonucleoprotein biogenesis. J Neurosci. 2006;26(43):11014–11022. doi: 10.1523/JNEUROSCI.1637-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32•.Praveen K, Wen Y, Matera AG. A Drosophila model of spinal muscular atrophy uncouples snRNP biogenesis functions of survival motor neuron from locomotion and viability defects. Cell Rep. 2012;1(6):624–631. doi: 10.1016/j.celrep.2012.05.014. Along with [33], this report renewed the ongoing debate of the key role of SMN in SMA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33•.Lotti F, Imlach WL, Saieva L, et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151(2):440–454. doi: 10.1016/j.cell.2012.09.012. Along with [32], this report renewed the ongoing debate of the key role of SMN in SMA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Xing L, Rossoll W, Wichterle H, Singer RH, Bassell GJ. Multiprotein complexes of the survival of motor neuron protein SMN with gemins traffic to neuronal processes and growth cones of motor neurons. J Neurosci. 2006;26(33):8622–8632. doi: 10.1523/JNEUROSCI.3967-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rossoll W, Kröning AK, Ohndorf UM, Steegborn C, Jablonka S, Sendtner M. Specific interaction of Smn, the spinal muscular atrophy determining gene product, with hnRNP-R and gry-rbp/hnRNP-Q: a role for Smn in RNA processing in motor axons? Hum Mol Genet. 2002;11(1):93–105. doi: 10.1093/hmg/11.1.93. [DOI] [PubMed] [Google Scholar]
- 37.Akten B, Kye MJ, Hao le T, et al. Interaction of survival of motor neuron (SMN) and HuD proteins with mRNA cpg15 rescues motor neuron axonal deficits. Proc Natl Acad Sci USA. 2011;108(25):10337–10342. doi: 10.1073/pnas.1104928108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tadesse H, Deschênes-Furry J, Boisvenue S, Côté J. KH-type splicing regulatory protein interacts with survival motor neuron protein and is misregulated in spinal muscular atrophy. Hum Mol Genet. 2008;17(4):506–524. doi: 10.1093/hmg/ddm327. [DOI] [PubMed] [Google Scholar]
- 39.Hubers L, Valderrama-Carvajal H, Laframboise J, Timbers J, Sanchez G, Côté J. HuD interacts with survival motor neuron protein and can rescue spinal muscular atrophy-like neuronal defects. Hum Mol Genet. 2011;20(3):553–579. doi: 10.1093/hmg/ddq500. [DOI] [PubMed] [Google Scholar]
- 40.Oprea GE, Krober S, McWhorter ML, et al. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320(5875):524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bowerman M, Beauvais A, Anderson CL, Kothary R. Rho-kinas inactivation prolongs survival of an intermediate SMA mouse model. Hum Mol Genet. 2010;19(8):1468–1478. doi: 10.1093/hmg/ddq021. [DOI] [PubMed] [Google Scholar]
- 42.Nölle A, Zeug A, van Bergeijk J, et al. The spinal muscular atrophy disease protein SMN is linked to the Rho-kinase pathway via profiling. Hum Mol Genet. 2011;20(24):4865–4878. doi: 10.1093/hmg/ddr425. [DOI] [PubMed] [Google Scholar]
- 43.Schrank B, Gotz R, Gunnerson JM, et al. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA. 1997;94(18):9920–9925. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid A, DiDonato CJ. Animal models of spinal muscular atrophy. J Child Neurol. 2007;22:1004–1012. doi: 10.1177/0883073807305667. [DOI] [PubMed] [Google Scholar]
- 45••.Hsieh-Li HM, Chang JG, Jong YJ, et al. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24(1):66–70. doi: 10.1038/71709. Along with [45], these studies described the first mouse models of SMA and provided direct evidence that SMN2 could serve as a viable molecular target in the treatment of SMA. [DOI] [PubMed] [Google Scholar]
- 46••.Monani UR, Sendtner M, Coovert DD, et al. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn-/- mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9(3):333–339. doi: 10.1093/hmg/9.3.333. Along with [44], these studies described the first mouse models of SMA and provided direct evidence that SMN2 could serve as a viable molecular target in the treatment of SMA. [DOI] [PubMed] [Google Scholar]
- 47.Bernal S, Also-Rasso E, Alias L, et al. Synaptic defects in type 1 spinal muscular atrophy in human development. J Pathol. 2013;229(1):49–61. doi: 10.1002/path.4080. [DOI] [PubMed] [Google Scholar]
- 48.Park GH, Maeno-Hikichi Y, Awano T, Landmesser LT, Monani UR. Reduced survival of motor neuron (SMN) protein in motor neuronal progenitors functions cell autonomously to cause spinal muscular atrophy in model mice expressing the human centromeric (SMN2) gene. J Neurosci. 2010;30(36):12005–12019. doi: 10.1523/JNEUROSCI.2208-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ling KK, Lin MY, Zingg B, Feng Z, Ko CP. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE. 2010;5(11):e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mentis GZ, Blivis D, Liu W, et al. Early functional impairment of sensory–motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;69(3):453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19(20):3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wishart TM, Huang JP, Murray LM, et al. SMN deficiency disrupts brain development in a mouse model of severe spinal muscular atrophy. Hum Mol Genet. 2010;19(22):4216–4228. doi: 10.1093/hmg/ddq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mutsaers CA, Wishart TM, Lamont DJ, et al. Reversible molecular pathology of skeletal muscle in spinal muscular atrophy. Hum Mol Genet. 2011;20:4334–4344. doi: 10.1093/hmg/ddr360. [DOI] [PubMed] [Google Scholar]
- 54.Araujo Ade Q, Araujo M, Swoboda KJ. Vascular perfusion abnormalities in infants with spinal muscular atrophy. J Pediatr. 2009;155(2):292–294. doi: 10.1016/j.jpeds.2009.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Workman E, Saieva L, Carrel TL, et al. A SMN missense mutation complements SMN2 restoring snRNPs and rescuing SMA mice. Hum Mol Genet. 2009;18(12):2215–2229. doi: 10.1093/hmg/ddp157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gavrilina TO, McGovern VL, Workman E, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17(8):1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martinez TL, Kong L, Wang X, et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J Neurosci. 2012;32(25):8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gogliotti RG, Quinlan KA, Barlow CB, et al. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory–motor defects are a consequence, not a cause, of motor neuron dysfunction. J Neurosci. 2012;32(11):3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee AJ, Awano T, Park GH, Monani UR. Limited phenotypic effects of selectively augmenting the SMN protein in the neurons of a mouse model of severe spinal muscular atrophy. PLoS ONE. 2012;7(9):e46353. doi: 10.1371/journal.pone.0046353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Le TT, McGovern VL, Alwine IE, et al. Temporal requirement for high SMN expression in SMA mice. Hum Mol Genet. 2011;20:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lutz CM, Kariya S, Patruni S, et al. Post-symptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J Clin Invest. 2011;121(8):3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Avila AM, Burnett BG, Taye AA, et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117(3):659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Echaniz-Laguna A, Bousiges O, Loeffler JP, et al. Histone deacetylase inhibitors: therapeutic agents and research tools for deciphering motor neuron diseases. Curr Med Chem. 2008;15(13):1263–1273. doi: 10.2174/092986708784534974. [DOI] [PubMed] [Google Scholar]
- 64.Sumner CJ, Huynh TN, Markowitz JA, et al. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54(5):647–654. doi: 10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 65.Chang JG, Hsieh-Li HM, Jong YJ, et al. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001;98(17):9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Grzeschik SM, Ganta M, Prior TW, Heavlin WD, Wang CH. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol. 2005;58(2):194–202. doi: 10.1002/ana.20548. [DOI] [PubMed] [Google Scholar]
- 67.Riessland M, Ackermann B, Förster A, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19(8):1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 68.Brahe C, Vitali T, Tiziano FD, et al. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13(2):256–259. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 69.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86(11):1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 70.Chen TH, Chang JG, Yang YH, et al. Randomized, double-blind, placebo-controlled trial of hydroxyurea in spinal muscular atrophy. Neurology. 2010;75(24):2190–2197. doi: 10.1212/WNL.0b013e3182020332. [DOI] [PubMed] [Google Scholar]
- 71.Mercuri E, Bertini E, Messina S, et al. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromusc Disord. 2004;14(2):130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 72.Swoboda KJ, Scott CB, Reyna SP, et al. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS ONE. 2009;4(5):e5268. doi: 10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mercuri E, Bertini E, Messina S, et al. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68(1):51–55. doi: 10.1212/01.wnl.0000249142.82285.d6. [DOI] [PubMed] [Google Scholar]
- 74.Pane M, Staccioli S, Messina S, et al. Daily salbutamol in young patients with SMA type II. Neuromusc Disord. 2008;18(7):536–540. doi: 10.1016/j.nmd.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 75.Russman BS, Iannaccone ST, Samaha FJ. A Phase 1 trial of riluzole in spinal muscular atrophy. Arch Neurol. 2003;60(11):1601–1603. doi: 10.1001/archneur.60.11.1601. [DOI] [PubMed] [Google Scholar]
- 76.Miller RG, Moore DH, Dronsky V, et al. SMA Study Group A placebo-controlled trial of gabapentin in spinal muscular atrophy. J Neurol Sci. 2001;191(1–2):127–131. doi: 10.1016/s0022-510x(01)00632-3. [DOI] [PubMed] [Google Scholar]
- 77.Merlini L, Solari A, Vita G, et al. Role of gabapentin in spinal muscular atrophy: results of a multicenter, randomized Italian study. J Child Neurol. 2003;18(8):537–541. doi: 10.1177/08830738030180080501. [DOI] [PubMed] [Google Scholar]
- 78.Oskoui M, Levy G, Garland CJ, et al. The changing natural history of spinal muscular atrophy type 1. Neurology. 2007;69(20):1931–1936. doi: 10.1212/01.wnl.0000290830.40544.b9. [DOI] [PubMed] [Google Scholar]
- 79.Montes J, Dunaway S, Montgomery MJ, et al. Fatigue leads to gait changes in spinal muscular atrophy. Muscle Nerve. 2011;43(4):485–488. doi: 10.1002/mus.21917. [DOI] [PubMed] [Google Scholar]
- 80.Montes J, McIsaac TL, Dunaway S, et al. Falls and spinal muscular atrophy: exploring cause and prevention. Muscle Nerve. 2013;47(1):118–123. doi: 10.1002/mus.23656. [DOI] [PubMed] [Google Scholar]
- 81.Sproule DM, Montes J, Montgomery M, et al. Increased fat mass and high incidence of overweight despite low body mass index in patients with spinal muscular atrophy. Neuromusc Disord. 2009;19(6):391–396. doi: 10.1016/j.nmd.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sproule DM, Montes J, Dunaway S, et al. Adiposity is increased among high-functioning, non-ambulatory patients with spinal muscular atrophy. Neuromusc Disord. 2010;20(7):448–452. doi: 10.1016/j.nmd.2010.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang CH, Finkel RS, Bertini ES, et al. Participants of the International Conference on SMA Standard of Care Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027–1049. doi: 10.1177/0883073807305788. [DOI] [PubMed] [Google Scholar]
- 84.Andreassi C, Jarecki J, Zhou J, et al. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10(24):2841–2849. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 85.Lunn MR, Root DE, Martino AM, et al. Indoprofen upregulates the survival motor neuron protein through a cyclooxygenase-independent mechanism. Chem Biol. 2004;11(11):1489–1493. doi: 10.1016/j.chembiol.2004.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Garbes L, Riessland M, Hölker I, et al. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009;18(19):3645–3658. doi: 10.1093/hmg/ddp313. [DOI] [PubMed] [Google Scholar]
- 87.Butchbach ME, Singh J, Thorsteinsdóttir M, et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum Mol Genet. 2010;19(3):154–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Naryshkin N, Narasimhan J, Dakka A, et al. Small molecule compounds correct alternative splicing of the SMN2 gene and restore SMN protein expression and function. Neuromusc Dis. 2012;22(9–10):848. [Google Scholar]
- 89.Hua Y, Krainer AR. Antisense-mediated exon inclusion. Methods Mol Biol. 2012;867:307–323. doi: 10.1007/978-1-61779-767-5_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333–1346. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hua Y, Sahashi K, Rigo F, et al. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478(7367):123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Porensky PN, Mitrpant C, McGovern, et al. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum Mol Genet. 2012;21(7):1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mattis VB, Ebert AD, Fosso MY, Chang CW, Lorson CL. Delivery of a read-through inducing compound, TC007, lessens the severity of a spinal muscular atrophy animal model. Hum Mol Genet. 2009;18(20):3906–3913. doi: 10.1093/hmg/ddp333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bowerman M, Murray LM, Boyer JG, Anderson CL, Kothary R. Fasudil improves survival and promotes skeletal muscle development in a mouse model of spinal muscular atrophy. BMC Med. 2012;10:24. doi: 10.1186/1741-7015-10-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96••.Foust KD, Wang X, McGovern VL, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotech. 2010;28(3):271–274. doi: 10.1038/nbt.1610. First proof-of-concept study to demonstrate that restoring SMN to an SMA model can affect near-complete rescue of the disease phenotype. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 97.Sunyach C, Michaud M, Arnoux T, et al. Olesoxime delays muscle denervation, astrogliosis, microglial activation and motoneuron death in an ALS mouse model. Neuropharmacology. 2012;62(7):2346–2352. doi: 10.1016/j.neuropharm.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 98.Valori CF, Ning K, Wyles M, et al. Systemic delivery of scAAV9 expressing SMN prolong survival in a model of spinal muscular atrophy. Sci Transl Med. 2010;2(35):35ra42. doi: 10.1126/scitranslmed.3000830. [DOI] [PubMed] [Google Scholar]
- 99.Dominguez E, Marais T, Chatauret N, et al. Intravenous scAAV9 delivery of a codonoptimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2011;20(4):681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 100.Duque S, Joussemet B, Riviere C, et al. Intravenous administration of self-complementary AAV9 enables transgene delivery to adult motor neurons. Mol Ther. 2009;17(7):1187–1196. doi: 10.1038/mt.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gowing G, Svendsen CN. Stem cell transplantation for motor neuron disease: current approaches and future perspectives. Neurotherapeutics. 2011;8(4):591–606. doi: 10.1007/s13311-011-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Corti S, Nizzardo M, Nardini M, et al. Neural stem cell transplantation can ameliorate the phenotype of a mouse model of spinal muscular atrophy. J Clin Invest. 2008;118(10):3316–3330. doi: 10.1172/JCI35432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kariya S, Re D, Jacquier A, et al. Mutant superoxide dismutase 1 (SOD1), a cause of amyotrophic lateral sclerosis, disrupts the recruitment of SMN, the spinal muscular atrophy protein to nuclear Cajal bodies. Hum Mol Genet. 2012;21(15):3421–3434. doi: 10.1093/hmg/dds174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Groen EJ, Fumoto K, Blokhuis AM, et al. ALS-associated mutations in FUS disrupt the axonal distribution and function of SMN. Hum Mol Genet. 2013;22(18):3690–3704. doi: 10.1093/hmg/ddt222. [DOI] [PubMed] [Google Scholar]
- 105.Tsuiji H, Iguchi Y, Furuya A, et al. Spliceosome integrity is defective in the motor neuron diseases ALS and SMA. EMBO Mol Med. 2013;5(2):221–234. doi: 10.1002/emmm.201202303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yamazaki T, Chen S, Yu Y, et al. FUS–SMN protein interactions link the motor neuron diseases ALS and SMA. Cell Rep. 2012;2(4):799–806. doi: 10.1016/j.celrep.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Monani UR. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron. 2005;48(6):885–896. doi: 10.1016/j.neuron.2005.12.001. [DOI] [PubMed] [Google Scholar]
Websites
- 201.An Open-label Safety, Tolerability and Dose-range Finding Study of Multiple Doses of ISIS SMNRx in Patient With Spinal Muscular Atrophy (SMNRx – CS2) http://clinicaltrials.gov/show/NCT01703988.
- 202.A Study to Assess the Safety and Pharmacokinetics of ISIS SMNRx in Infants With Spinal Muscular Atrophy. http://clinicaltrials.gov/show/NCT01839656.