

SUMMARY
Diverse environmental cues converge on and are integrated by the mTOR signaling network to control cellular growth and homeostasis. The mammalian Tsc1-Tsc2 GTPase activating protein (GAP) heterodimer is a critical negative regulator of Rheb and mTOR activation. The RalGAPα-RalGAPβ heterodimer shares sequence and structural similarity with Tsc1-Tsc2. Unexpectedly, we observed that C. elegans expresses orthologs for the Rheb and RalA/B GTPases, and for RalGAPα/β but not Tsc1/2. This prompted our investigation to determine whether RalGAPs additionally modulate mTOR signaling. We determined that C. elegans RalGAP loss decreased lifespan, consistent with a Tsc-like function. Additionally, RalGAP suppression in mammalian cells caused RalB-selective activation and Sec5- and exocyst-dependent engagement of mTORC1 and suppression of autophagy. Unexpectedly, we also found that Tsc1-Tsc2 loss activated RalA/B independently of Rheb-mTOR signaling. Finally, RalGAP suppression caused mTORC1-dependent pancreatic tumor cell invasion. Our findings identify an unexpected crosstalk and integration of the Ral and mTOR signaling networks.
INTRODUCTION
Mechanistic target of rapamycin (mTOR) signaling has emerged as a major signaling node that is aberrantly activated in cancer, diabetes, and neurodegenerative disorders (Laplante and Sabatini, 2012). mTOR is an atypical serine/threonine protein kinase that forms two distinct signaling complexes, mTORC1 and mTORC2, that are distinguished primarily by their association with Raptor or Rictor, respectively. Although considerable advances have been made in understanding the signaling mechanisms that regulate mTOR activity, many unresolved and poorly understood issues remain (Wang and Proud, 2011).
mTORC1 activity is regulated by diverse extracellular stimuli that include growth factors and amino acids (Laplante and Sabatini, 2012). A major upstream regulator of mTORC1 is the tuberous sclerosis complex comprised of the Tsc1 (aka hamartin) and Tsc2 (aka tuberin) heterodimer. Tsc1/2 acts as a GTPase activating protein (GAP) for the Rheb small GTPase by converting active Rheb-GTP to inactive Rheb-GDP (Aspuria and Tamanoi, 2004; Inoki et al., 2003). Rheb-GTP associates with and facilitates the localization and activation of mTORC1 at the lysosomal surface in response to nutrients (Inoki et al., 2003). Growth factors such as insulin and insulin-like growth factor are important stimuli of the phosphoinositide 3-kinase (PI3K) and Ras small GTPase pathways (Huang and Manning, 2008; Pollak, 2012), activating the Akt and ERK serine/threonine kinases, respectively, that directly phosphorylate and inactivate Tsc1/2, derepressing Rheb and promoting mTORC1 signaling. Tsc1/2 is also mutationally inactivated or lost in cancers (Laplante and Sabatini, 2012). In contrast, amino acid activation of mTORC1 is independent of Tsc1/2 and instead is mediated through the Rag small GTPases (Sancak et al., 2010). While clinically relevant functions of Tsc1/2 that are independent of Rheb and/or mTORC1 have been described, the mechanisms thereof are unidentified (Neuman and Henske, 2011).
mTORC1 activation regulates diverse cellular processes that include the stimulation of protein synthesis through direct phosphorylation and activation of S6 kinase 1 (S6K) and inactivation of 4E-BP1. mTORC1 also negatively regulates cellular catabolic processes like autophagy, the central degradative process for recycling cellular building blocks. mTORC1 signaling has also been implicated in the aging process, with genetic or pharmacologic suppression of TOR extending lifespan in C. elegans, Drosophila, yeast, and mice (Lapierre and Hansen, 2012; Laplante and Sabatini, 2012). In C. elegans, CeTORC1 inhibition-mediated lifespan extension depends on activation of the transcription factor FOXO/DAF-16. FOXO is also a convergence point with insulin signaling and aging regulation; the C. elegans Insulin Receptor (InsR)/DAF-2 activates a PI3K-PDK-Akt cascade to inhibit FOXO activity, thereby decreasing lifespan.
Although described originally as a tuberin-related protein (TULIP1; (Schwarzbraun et al., 2004), RalGAPα1 and its closely related isoform, RalGAPα2, were independently discovered later as GAPs for the RalA and RalB small GTPases (Chen et al., 2011; Shirakawa et al., 2009). Ral GTPases are best known as regulators of the octameric exocyst complex which controls vesicular transport by tethering secretory vesicles to the plasma membrane prior to fusion (Heider and Munson, 2012). The exocyst also functions independent of exocytosis and aberrant Ral activation has been implicated in cancer growth (Bodemann and White, 2008). In non-cancer cells Ral-exocyst signaling has been implicated in cellular motility (Spiczka and Yeaman, 2008), autophagosome formation (Bodemann et al., 2011), protein sorting (Shipitsin and Feig, 2004), and cytokinesis (Cascone et al., 2008). The RalGAPα1 and α2 GAP catalytic domains share significant sequence identity with the GAP catalytic domain of Tsc2 (26–27% identity). Similar to the requirement for Tsc1 association with Tsc2 for GAP activity, each RalGAPα catalytic subunit requires heterodimerization with a common regulatory RalGAP β subunit (aka RGC1) for their GAP activities (Chen et al., 2011). To date, no other RalGAPs have been described and the embryonic lethality of RalGAPβ knockout mice is consistent with the likelihood that there are no other mammalian RalGAPs (unpublished). C. elegans expresses orthologs of human Ras (LET-60), Rheb (RHEB-1) and Ral (RAL-1), as well as other components of Ral effector (Frische et al., 2007) and TOR signaling (Lapierre and Hansen, 2012; Laplante and Sabatini, 2012). Strikingly, C. elegans expresses RalGAP α and β but not Tsc1 and Tsc2 orthologs. This contrasts with D. melanogaster, which expresses orthologs of Tsc1/2 as well as Rheb, Ral, and RalGAPα/β (Figure 1A). These observations prompted our interest in determining whether RalGAPs may serve as a point of integration of the Ral and mTOR signaling networks.
Figure 1. C. elegans RalGAP and Ral Regulate Lifespan.
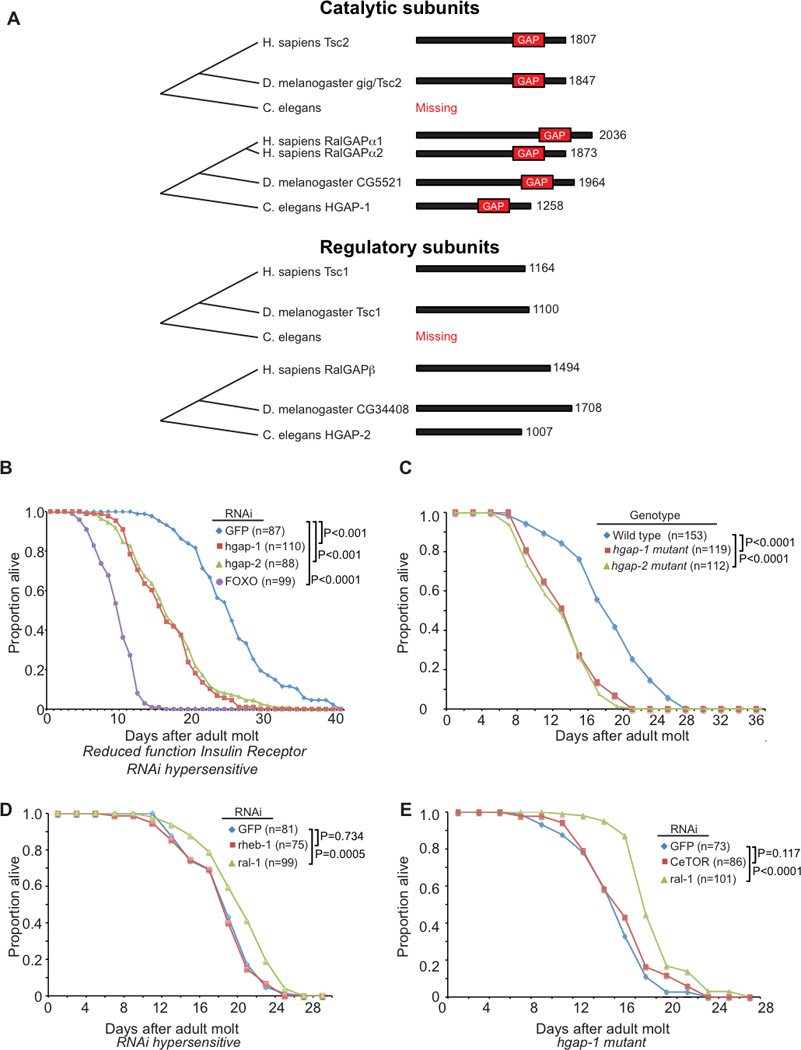
(A) Molecular phylogeny of Ral and Rheb GAPs. Comparison of the domain structure of Rheb and Ral GAP catalytic and regulatory subunits between H. sapiens, D. melanogaster, and C. elegans. Numbers indicate amino acids.
(B) RNAi-mediated knockdown of C. elegans hgap-1 and hgap-2 RalGAP subunits decreases lifespan in daf-2(e1370) reduced function InsR animals (P < 0.0001). daf-16(RNAi) was used as a positive control.
(C) Putative null mutations hgap-1(gk101481) (Y18H1A.3 W1142*) and hgap-2(gk578143) (D2085.5a Q802*) confer reduced lifespan relative to wild-type animals (P < 0.0001).
(D) Bacterially-mediated ral-1(RNAi) (P=0.0005) but not rheb-1(RNAi) (P=0.734) in the eri-1(mg366) RNAi-hypersensitive background extended lifespan.
(E) Bacterially-mediated ral-1(RNAi) increased the lifespan of hgap-1(gk101481); eri-1(mg366) mutant animals to roughly wild-type duration (P = 0.0087; compare to 1C and 1D).
For all experiments, animals were allowed to feed ad libitum with no caloric restrictions Animals in 2B were grown at 25°C, while other experiments were performed at 20°C. P values from log-rank test. See also Figure S1.
In this study we established that GAPs for Ral and Rheb facilitate the integration of Ral and mTOR signaling networks to control aging, autophagy and tumor cell invasion. First, we identified the RalGAP complex, acting through inhibition of RalB, as a negative regulator of mTORC1 signaling through Sec5 and the exocyst complex. Second, we identified Tsc1/2 as an mTORC1-independent negative regulator of Ral. Third, loss of C. elegans RalGAP α or β orthologs phenocopied CeTORC1 activation and decreased lifespan, suggesting that in C. elegans RalGAP substitutes for the Tsc complex. Finally, we showed previously that RalB activation drives pancreatic cancer invasion and metastasis (Lim et al., 2006), and we now showed that RalB-mediated mTORC1 activation is critical for this activity. In summary, our observations establish an unexpected signaling crosstalk between the Ras-Ral and Rheb-mTOR signaling axes.
RESULTS
Aging in C. elegans is Controlled by RalGAP Signaling
The strong GAP domain sequence identity (26–27%) and same heterodimeric complex requirement of the RalGAPs and the Tsc/RhebGAP (Figure 1A) prompted us to examine whether they may exhibit functional similarities. Unexpectedly, we noted that while C. elegans expresses Ral (RAL-1), Rheb (RHEB-1), RalGAP catalytic α (HGAP-1/Y18H1A.3) and regulatory β (HGAP-2/D2085.5) subunit orthologs, notably C. elegans, alone among well-studied model organisms, lacks Tsc1 or Tsc2 orthologs (Figure 1A and Supplemental Table S1). Evolutionary loss of Tsc1/2, otherwise conserved from yeast to humans (Neuman and Henske, 2011), would likely have dire fecundity consequences unless another ancestral protein substituted for this function.
Since Tsc1/2 are known to modulate mTOR signaling to control aging (Lapierre and Hansen, 2012; Laplante and Sabatini, 2012), we determined if RalGAPs could function similarly in C. elegans. Reduced function (rf) of the insulin/IGF receptor ortholog DAF-2 results in doubled lifespan, which in turn is completely suppressed by loss of FOXO/DAF-16 (Kenyon et al., 1993; Kimura et al., 1997; Lin et al., 1997; Ogg et al., 1997). As expected, positive control daf-16(RNAi) reduced daf-2(rf) lifespan. Reduction of RalGAPα/HGAP-1 or RalGAPβ/HGAP-2 function significantly reduced lifespan in daf-2(rf) and wild-type backgrounds (Figures 1B,C and S1B). This result is consistent with the requirement of both subunits for a functional RalGAP (Chen et al., 2011; Shirakawa et al., 2009). Interestingly, we also determined that RAL-1 (RNAi) but not RHEB-1(RNAi) significantly increased C. elegans lifespan (Figure 1D), observations previously described in a whole-genome RNAi screen (Kim and Sun, 2007). Importantly, ral-1 suppression in hgap-1 mutant animals resulted in extended lifespan (Figure 1E), suggesting that hgap-1 acts through RAL-1 to control lifespan. Loss of LET-363/CeTOR results in 3rd stage larval arrest (Jia et al., 2004). hgap-1 (RNAi) and an hgap-2 putative null mutation failed to rescue CeTOR mutant larval arrest, consistent with HGAP-1/2 signaling upstream of CeTOR (Figures S1C and S1D).
RalGAP Signaling Controls mTORC1 Activity
We next pursued further analysis in mammalian cell models where there are well-established assays to access both mTOR and Ral signaling. We isolated and analyzed mouse embryonic fibroblasts (MEFs) isolated from RalGAPβfl/fl conditional knockout embryos. Upon ectopic Cre recombinase expression and RalGAPβ deletion there was a marked decrease in RalGAPβ protein expression and increased RalA/B but not Rheb GTP-loading (Figures 2A and S2A). There was also a large increase in the phosphorylation of the mTORC1 substrate S6K (pS6K(T389); Figure 2A), suggesting an increase in mTORC1 activity. Interestingly, loss of RalGAPβ resulted in increased mTOR kinase activity as measured by in vitro kinase assays, suggesting a crucial role for RalGAPβ in controlling mTOR kinase activity (Figures 2B and S2B). We also observed that overexpression of RalGAPβ with wild-type but not GAP-dead RalGAPα1 resulted in diminished mTOR kinase activity (Figure S2C). RalGAPβ suppression also enhanced mTORC1 signaling that was Raptor dependent and rapamycin treatment ablated pS6K formation in both empty and Cre treated RalGAPβfl/fl MEFs, consistent with a role for mTORC1 in regulating the increase in pS6K upon loss of RalGAPβ (Figures 2C and S2D). We also observed that RalB but not Rheb is necessary for the enhanced pS6K in Cre treated RalGAP βfl/f MEFs indicating that it is RalB but not Rheb that is necessary for mTORC1 activation downstream of RalGAP signaling (Figure 2D).
Figure 2. RalGAPs Regulate mTORC1 Signaling.

(A) RalGAPβ-deficient MEFs have elevated Ral GTPase and mTORC1 activity. RalGAPβfl/fl MEFs were treated with either empty or Cre-expressing adenovirus. Cell lysates were resolved by SDS-PAGE and then analyzed for pS6K and for RalA- and RalB-GTP levels by a GST-Sec5-RBD pulldown assay.
(B) mTOR kinase activity is enhanced in RalGAPβ-deficient MEFs. RalGAPβfl/fl MEFs were treated with empty or Cre-expressing adenovirus. Cells were serum starved overnight and mTOR kinase activity was assessed by in vitro kinase assays measuring S6K(T389) phosphorylation.
(C) Raptor is required for enhanced mTORC1 signaling in response to RalGAPβ-depletion. HEK293T cells stably expressing either NS or RalGAPβ shRNA were transduced with either NS or Raptor shRNA. Cells were selected for shRNA expression, serum-starved, and mTORC1 activity was measured by immunoblotting with the indicated antibodies.
(D) RalB but not Rheb is required for enhanced mTORC1 signaling in RalGAPβ-deficient MEFs. RalGAPβfl/fl MEFs were transfected with the indicated siRNA pools, then treated with either empty or Cre-expressing adenovirus. Cells were serum starved overnight and lysates were immunoblotted with the indicated antibodies.
(E) Depletion of RalGAPβ results in reduced LC3 punctae. RalGAPβfl/fl MEFs grown in serum containing growth medium were analyzed for GFP-LC3 punctae content by confocal microscopy. Images are representative of cells treated with empty (left) or Cre (right) adenovirus-treated RalGAPβfl/fl MEFs.
(F) Quantitation of GFP-LC3 punctae from panel E. Fifteen cells were imaged for each condition and GFP-LC3 punctae were quantitated. p-values were determined by an unpaired Student’s t-test. See also Figure S2.
Enhanced activity of mTORC1 suppresses autophagy (Kim et al., 2011). To determine whether RalGAPβ loss-induced mTORC1 activity was associated with a reduction in autophagy, we utilized two markers of autophagy: formation of the processed form of endogenous LC3 (LC3B-II) and LC3-punctae formation (Klionsky et al., 2012). RalGAPβ depletion resulted in the appearance of fewer GFP-LC3 punctae and a decrease in LC3BII protein levels (Figures 2E, 2F, S2E). Treatment with rapamycin restored LC3B processing, indicating a role for RalGAP upstream of mTOR in autophagy regulation (Figure S2F).
RalB Associates with mTOR and Regulates mTORC1 Signaling in Aging, Autophagy and Tumor Cell Invasion
Rheb-GTP directly engages mTORC1 and promotes mTOR kinase activity and signaling (Laplante and Sabatini, 2012). Tsc1/2 possesses RhebGAP activity to directly antagonize Rheb-mTOR signaling (Tee et al., 2003). Since we found that RalGAP loss activated mTORC1 activity, we investigated whether this was through Ral activation. Since RalGAP did not show GAP activity for Rheb in vitro (Chen et al., 2011; Shirakawa et al., 2009), we assessed the possibility that mTOR is an effector of activated Ral. We determined that mTOR co-precipitated preferentially with constitutively GTP-bound RalB(Q72L) but not the constitutively GDP-bound RalB(S28N) mutant (Figure 3A). Recombinant RalB had a strong interaction with the kinase domain of mTOR (Figure S3A), similar to that reported for Rheb (Long et al., 2005). Consistent with Rheb and RalB interacting with the same region of mTOR, we found that RalB could successfully compete with Rheb for mTOR binding (Figure S3C). These data are consistent with mTOR serving as an effector of activated RalB.
Figure 3. RalB Associates with mTORC1 and Regulates mTORC1 Signaling.

(A) Active, GTP-bound RalB associates with mTOR. HEK293T cells expressing the indicated HA-tagged RalB proteins and Myc-tagged mTOR were subject to anti-HA immunoprecipitation. After SDS-PAGE, association of HA-RalB with Myc-mTOR was determined by western blotting with anti-Myc. Total cell lysates were probed with the indicated antibodies, with anti-vinculin to verify equivalent input of total cellular protein and anti-pS6K to determine mTORC1 activity.
(B) Endogenous RalB associates with mTORC1 upon serum stimulation. HEK293T cells were starved of amino acids, serum, and glucose for 1 h. Cells were then either untreated (−) or treated with serum to a final concentration of 10% (+) for 20 min. Lysates were resolved by SDS-PAGE and subject to immunoprecipitation with anti-RalB antibody. RalB-mTORC1 complex formation was determined by western blotting immune complexes with anti-mTOR and anti-Raptor. Total cell lysates were probed with the indicated antibodies to verify equivalent expression of protein and to determine mTORC1 activity (pS6K).
(C) Raptor is necessary for RalB to associate with mTOR. HEK293T cells stably transduced with the indicated lentiviral shRNA constructs for Raptor and Rictor were subject to immunoprecipitation with an anti-RalB antibody. RalB-mTOR complex formation was determined by western blotting RalB immune complexes with anti-mTOR antibody. Total cell lysates were analyzed with the indicated antibodies to verify equivalent protein expression and protein knockdown.
(D) RalB is necessary for mTORC1 response to serum stimulation. HEK293T cells stably transduced with the indicated lentiviral shRNA constructs for RalA or RalB were starved of amino acids, serum, and glucose for 1 h. Cells were then either untreated (−) or treated with serum to a final concentration of 10% (+). Lysates were analyzed by immunoblotting with the indicated antibodies including pS6K to determine mTORC1 activity. See also Figure S3.
Interestingly, expression of RalB(Q72L) or RalB(S28N) caused an increase or decrease in pS6K levels, respectively (Figure 3A), suggesting that activated RalB association with mTOR could modulate mTORC1 activity. Analyses with RalA, which shares 82% sequence identity with RalB (Feig, 2003; Neel et al., 2011), did not show association with mTOR (Figure S3B). We also determined that endogenous mTOR and Raptor (but not Rictor or other mTORC2 components; Figure S3B) co-immunoprecipitated with endogenous RalB, and this association was enhanced by serum but not amino acid stimulation (Figures 3B and S3E). RalB association with mTOR was dependent on the expression of Raptor but not Rictor (Figure 3C) further indicating a role for RalB in regulating mTORC1 but not mTORC2 signaling. Finally, we found that stable shRNA depletion of RalB but not RalA abrogated serum stimulation of pS6K formation (Figure 3D), indicating the necessity of RalB for serum-stimulated mTOR activation and consistent with the ability of RalB but not RalA to associate with mTOR.
The Tuberous Sclerosis Complex (TSC) Regulates Ral GTPase Signaling
We next addressed whether Tsc1 and Tsc2 could also regulate Ral activation. First, we ectopically expressed the catalytic RalGAPα1 subunit in Tsc2-null (−/−) MEFs and found that RalGAPα1 expression resulted in a decrease in mTORC1 activity, comparable to that seen upon re-expression of Tsc2 (Figure 4A). Examination of Tsc1 or Tsc2 knockout MEFs found that the GTP-bound levels (and total protein) of both RalA and RalB were elevated (Figure 4B). Rapamycin treatment did not result in changes to RalB-GTP levels, indicating mTORC1-independent enhancement in Ral activity (Figure 4C). Knockdown of Rheb1 decreased pS6K (Figure 4D) but not RalB-GTP levels (Figure S4A), further implicating a Rheb-mTOR-independent mechanism for Tsc2 regulation of Ral activity. Despite the ability of active RalB to complex with Tsc2 (Figure S4B), we found that Tsc2 does not possess GAP activity towards Ral. Interestingly, we observed that overexpressed Tsc proteins could heterodimerize with RalGAP proteins, indicating that loss of Tsc expression could possibly reduce RalGAP function (Figures S4C and S4D). To address the necessity of RalB for enhanced mTORC1 activity in Tsc2-null cells, we used stable shRNA expression to deplete RalB or Rheb1. Loss of either RalB or Rheb1 resulted in a similar decrease in mTORC1 activity (Figure 4D).
Figure 4. The Tuberous-Sclerosis Complex (TSC) Regulates Ral GTPase Signaling.

(A) Elevated mTORC1 activity in Tsc2-null MEFs can be rescued by RalGAP expression. Tsc2 wild type (+/+) Tsc2-null (−/−) were transiently transfected with the indicated constructs. Cells were harvested 48 h later and lysates were immunoblotted with the indicated antibodies.
(B) Tsc-deficient cells have elevated Ral GTPase activity. Tsc1-null (−/−) MEFs stably expressing either vector or Tsc1 and Tsc2 wild type (+/+) and null (−/−) MEFs were subject to GST-Sec5-RBD pulldown assays to determine Ral-GTP formation. Pulldowns were analyzed by immunoblotting with anti-RalA or RalB. Total cell lysates were probed with the indicated antibodies to determine protein expression.
(C) Elevated Ral activity in Tsc2-deficient cells is not dependent on mTOR activity. Tsc2 wild type (+/+) or null (−/−) MEFs were treated with 10 nM rapamycin for 16 h. Cells were harvested and subject to GST-Sec5-RBD pulldown assays to determine Ral-GTP formation. Total cell lysates were probed with the indicated antibodies to determine protein expression.
(D) RalB and Rheb1 are necessary for elevated mTORC1 activity associated with Tsc2 deficiency. Tsc2+/+ or Tsc2−/− MEFs were stably transduced with the indicated lentiviral shRNA contructs were starved of serum, glucose, and amino acids for 1 h, then analyzed by immunoblotting with the indicated antibodies.
(E) Tsc2-deficient tumors have elevated RalB activity. Normal and tumor kidney tissue were isolated from Eker rats. Lysates from tissues were subject to GST-Sec5-RBD pulldown assays to determine RalB-GTP formation. Total cell lysates were probed with the indicated antibodies to determine protein expression. *denotes non-specific bands from the pulldown assay. See also Figure S4E.
Lastly we used tissue from the Eker rat, which carries a germline deletion of one Tsc2 allele (Yeung et al., 1995), to determine whether there was altered Ral activity associated with Tsc2 loss in vivo. We found increased total RalB and RalB-GTP levels in renal tumor tissue which had decreased Tsc2 levels (Figures 4E and S4E), suggesting that TSC loss derepresses RalB in vivo.
RalB Regulates Serum-induced mTORC1 Translocation to the Plasma Membrane
We next determined if RalB activation could regulate mTORC1 subcellular localization. We observed a dense perinuclear colocalization of mCherry-tagged RalB with both endogenous mTOR and GFP-Raptor in starved cells (Figures 5A and S5A). Upon serum stimulation RalB plasma membrane localization was enhanced, where it colocalized with both mTOR and GFP-Raptor (Figures 5A and S5A).
Figure 5. RalB Regulates Serum-induced mTORC1 Plasma Membrane Relocalization.

(A) RalB and mTOR relocalize to the plasma membrane after serum stimulation. HEK293T cells were transfected with mCherry-RalB and 48 h later cells were serum starved overnight. Cells were then either untreated or treated with serum to a final concentration of 10% for 10 min and fixed with paraformaldehyde. Endogenous mTOR was stained and colocalization with mCherry-RalB was determined by confocal microscopy. Line scans show the intensity of fluorescence for RalB (red) and mTOR (green) from the nucleus (N) to the plasma membrane (PM).
(B) Serum stimulation results in active RalB accumulation at the plasma membrane. HEK293T cells expressing a RalB biosensor were serum starved overnight. Cells were then either untreated or treated with serum to a final concentration of 10% for 10 min. Live cells were imaged and FRET ratios were determined as described previously (Pertz et al., 2006). Five cells were imaged for each condition.
(C) Loss of RalGAP signaling leads to active RalB accumulation at the plasma membrane. HEK293T cells stably expressing either NS or RalGAPβ shRNA were transfected with a RalB biosensor and serum starved overnight. Live cells were imaged and FRET ratios were determined as in (B). At least 10 cells were imaged for each condition.
(D) Localization of Raptor to Rheb and RalB cellular sites by C-terminal membrane targeting sequences. Wildtype GFP-Raptor (No C) or GFP-Raptor with either Rheb or RalB membrane-targeting regions (Rheb-C or RalB-C) fused to the Raptor C-terminus were expressed in HEK293T cells. Localization of each GFP-Raptor protein was examined by confocal microscopy.
(E) Raptor localization to RalB cellular sites is sufficient for mTORC1 activation. Cells from (D) were serum starved overnight and mTORC1 activity was determined by immunoblotting with the indicated antibodies. For all images scale bars are 20 µm. See also Figure S5.
We next examined whether serum-induced mTOR localization to the plasma membrane was RalB-dependent. In control NS shRNA expressing cells mTOR was enriched in the plasma membrane fraction after serum stimulation (Figure S5B). RalB suppression caused a marked reduction in mTOR recruitment to the plasma membrane fraction demonstrating that RalB is necessary for mTOR plasma membrane recruitment (Figure S5B). mTORC1 is well-established to signal at the lysosome (Sancak et al., 2010). We did observe RalB colocalization with lysosomal marker LAMP2 but this was unchanged upon serum stimulation (Figure S5C).
Since we observed that the active form of RalB preferentially associated with mTOR (Figure 3A), we constructed a FRET-based RalB activity sensor to determine the subcellular localization of active RalB. The biosensor is a unimolecular reporter consisting of CFP and YFP inserted between the Sec5 Ral-binding domain and wild-type RalB. We verified the biosensor detection of active RalB (Figure S5D). We observed that serum stimulation or depletion of RalGAPβ enhanced RalB activation at plasma but not endomembrane compartments (Figures 5B and 5C).
We generated GFP-Raptor mutants with either the Rheb or RalB membrane-targeting regions fused to the Raptor C-terminus to determine if mTORC1 recruitment to the localization of RalB was alone sufficient for activation. As reported previously (Sancak et al., 2010), GFP-Raptor Rheb-C was perinuclear localized, consistent with the lysosomal compartment (Figure 5D), and this was sufficient for mTORC1 activation in serum-starved cells (Figure 5E). GFP-Raptor RalB-C was localized on both internal membranes and the plasma membrane, similarly to what we observed for RalB (Figure 5D). This localization was sufficient for mTORC1 activation in serum-starved cells (Figure 5E) indicating that RalB-dependent plasma membrane translocation is sufficient for mTORC1 activation.
Lastly we constructed GFP-Sec5 Ral binding domain (RBD) fusion proteins bearing different GTPase C-terminal membrane targeting sequences. The isolated RBD can bind and block endogenous Ral-GTP signaling (Bodemann et al., 2011). Addition of the different C-termini allowed us to selectively turn off Ral signaling at different cellular locations. We found that addition of the RalB, Rheb, and H-Ras C-termini showed the localization patterns (Figure S5E) for their respective GTPases. Only expression at the plasma membrane (RalB-C and H-Ras-C) and not the lysosome (Rheb-C) was capable of impairing mTORC1 signaling in response to serum stimulation (Figure S5F). We conclude that the plasma membrane and not the lysosomal subcellular pool of RalB is necessary for proper serum-induced mTORC1 activation.
The Exocyst is the RalB Effector Complex Necessary for mTORC1 Modulation
The best characterized Ral effector proteins are two components of the octomeric exocyst complex involved in vesicle sorting (Sec5 and Exo84), and RalBP1 (aka RLIP76) which has roles in endocytosis and actin reorganization (Neel et al., 2011). To determine their possible association with mTOR, we expressed GFP-tagged Sec5, Exo84, or RalBP1 in HEK293T cells. We then immunoprecipitated the GFP-tagged effector and we found co-precipitation of endogenous mTOR with Sec5 and Exo84 but not RalBP1 (Figure 6A). Next we profiled all eight exocyst subunits and found that each could co-precipitate with mTORC1 to varying degrees (Figure S6A)
Figure 6. The Exocyst is the RalB Effector Responsible for mTORC1 Activity.

(A) The Sec5 and Exo84 components of the exocyst interact with mTOR. HEK293T cells expressing the indicated GFP-tagged Ral effector proteins were subject to GFP immunoprecipitation and SDS-PAGE, and then immunoblotted to assess mTOR co-precipitation.
(B) Sec5 is necessary for RalB to interact with mTOR. HEK293T cells were transiently transfected with the indicated exocyst siRNA pools. Forty-eight h later, cells were subject to immunoprecipitation to isolate endogenous RalB followed by immunoblotting to determine mTOR association. Total cell lysates were analyzed by immunoblotting to determine protein knockdown and to verify protein loading.
(C) Sec5 but not Exo84 is required for the enhanced mTORC1 activity in RalGAPβ-depleted cells. RalGAPβfl/fl MEFs were transfected with the indicated siRNA pools. Cells were then treated with either empty or Cre-expressing adenovirus. Cells were serum starved overnight and lysates were immunoblotted with the indicated antibodies.
(D) Endogenous Sec8 associates with mTOR. HEK293T cells were starved of amino acids, serum, and glucose for 1 h. Cells were then either untreated or treated with 10% serum for 20 min, then subjected to Sec8 or normal mouse IgG immunoprecipitation followed by immunoblotting to determine mTOR co-precipitation. Total cell lysates (Input) were immunoblotted to determine total protein expression.
(E) The exocyst is recruited with mTOR and RalB to the plasma membrane after serum stimulation. HEK293T cells stably expressing mCherry-RalB were serum starved overnight. Cells were then either untreated or treated with 10% serum for 10 min and fixed. Endogenous mTOR (green) and Sec8 (blue) were then immunostained and co-localization with mCherry-RalB (red) was examined by confocal microscopy. Line scans show the intensity of staining from the nucleus (N) to the plasma membrane (PM) for each protein. Scale bars are 20 urn. See also Figure S6.
We used siRNA to deplete cells of either Sec5 or Exo84 to determine if either was necessary for RalB interaction with mTOR. Knockdown of Sec5 but not Exo84 caused a reduction in the coprecipitation of endogenous RalB with immunoprecipitated endogenous mTOR and a reduction in pS6K levels (Figure 6B) implicating Sec5 as the key effector that determines RalB-dependent mTORC1 signaling. Depletion of Sec5 but not Exo84 by siRNA resulted in a reduction of pS6K in RalGAP βfl/fl MEFs upon Cre treatment consistent with a direct role for Sec5 in RalGAPβ–RalB mediated mTORC1 activation (Figure 6C). Overexpression of Sec5 with active RalB enhanced mTORC1 activity while co-expression of active RalB with Exo84 resulted in a decrease in pS6K (Figure S6B) similar to what has been described previously (Bodemann et al., 2011). Using mTOR and Sec5 prepared by in vitro transcription/translation we found that Sec5 could directly interact with mTOR as measured by co-IP (Figure S6C). Finally, using a variant of Sec5 with a T11A mutation that perturbs the Ral-Sec5 association (Fukai et al., 2003), we found that Ral binding is not required for Sec5 to associate with mTORC1 (Figure S6D), suggesting that activated RalB associates with a preformed but inactive Sec5-mTOR complex.
Since there are exocyst-independent functions of Sec5 (Chien et al., 2006), we next determined if RalB-Sec5 engagement of mTOR involved the canonical exocyst complex. We determined immunoprecipitation of endogenous Sec8, a non-Ral effector exocyst core component, also displayed mTOR association independent of the presence of serum (Figure 6D). We also found that upon serum stimulation RalB and endogenous mTOR are found at the plasma membrane together with endogenous Sec8 (Figure 6E). These results suggest that RalB association with mTOR is exocyst-dependent.
RalGAP Suppresses Pancreatic (PDAC) Tumor Cell Invasion
We determined previously that RalA but not RalB is necessary for pancreatic ductal adenocarcinoma (PDAC) anchorage-independent growth in vitro and tumorigenesis in vivo, while RalB is necessary for PDAC invasion and metastasis in vitro and in vivo (Lim et al., 2006). PDAC tumors also have elevated mTOR activity (Falasca et al., 2011). Therefore, we examined PDAC cells to assess whether RalGAP proteins could regulate pancreatic tumor cell growth and invasion. We found expression of both RalGAPα1 and α2 in addition to the regulatory RalGAPβ subunit In PDAC tumor cell lines, (Figure 7A). As expected, we found that stable shRNA knockdown of RalGAPβ enhanced the levels of both GTP-bound RalA and RalB (Figure 7B). This observation was also true for shRNA depletion of RalGAPα1 (Figure S7A). RalGAPβ depletion did not enhance anchorage-dependent or anchorage-independent growth (Figures S4B and S4C), but knockdown of RalGAPβ and RalGAPα1 caused a substantial increase in invasion through Matrigel (Figures 7C and Figure S4A).
Figure 7. RalGAP Signaling Suppresses the Invasive Properties of PDAC Tumor Cells.

(A) PDAC cell lines express all three RalGAP proteins. PDAC tumor cell lines were analyzed by immunoblotting with antibodies for the indicated RalGAP subunits, and for vinculin to verify equivalent loading of total cellular protein.
(B) Loss of RalGAP results in elevated Ral GTPase activity in PDAC cells. PDAC cells were stably transduced with the indicated RalGAPβ and NS shRNAs. Lysates were subject to GST-Sec5-RlBD pulldown assays to determine Ral-GTP formation. Total cellular protein was analyzed with the indicated antibodies to determine protein expression and verify knockdown. Blot analysis for p-actin was used to verify equivalent loading of total cellular protein.
(C) Knockdown of RalGAPβ results in enhanced PDAC cell invasion. PDAC cells from (B) were treated and placed in Matrigel chambers. Numbers represent the average number of invaded cells per well in an assay performed in triplicate (+/− standard error from the mean, S.E.M). p-values were determined by an unpaired Student’s t-test. * denotes p-value less than 0.05 and ** denotes p-value less than 0.001.
(D) Loss of RalGAP results in enhanced mTORC1 signaling in PDAC cells. PDAC cells were stably transduced with the indicated RalGAPβ and NS shRNAs. Lysates were analyzed by immunoblotting with the indicated antibodies. Blot analysis for β-actin was used to verify equivalent loading of total cellular protein.
(E) Enhancement in invasion upon RalGAPβ depletion in PDAC cells is dependent on mTOR activity. PANC-1 cells stably transduced with the indicated RalGAPβ and NS shRNAs were treated with 20 nM rapamycin for 16 h before being placed in Matrigel invasion chambers (left panel). Numbers represent the average number of invaded cells in an assay performed in triplicate (+/− standard error from the mean, S.E.M). p-values were determined by an unpaired Student’s t-test. * denotes p-value less than 0.05 and n.s. (non-significant) denotes p-value greater than 0.05. Blot analysis was done to monitor pS6K levels in cell lines under the indicated conditions (right panel).
Similar to our observations in MEFs, we also observed an increase in mTORC1 activity when PDAC tumor cells were depleted of RalGAPβ (Figure 7D). The increased invasion upon knockdown of RalGAPβ was abolished by rapamycin treatment (Figure 7E), indicating that invasion is driven through mTORC1 signaling.
DISCUSSION
The importance of mTORC1 signaling in cancer is well-established, with two mTORC1 inhibitors approved for cancer treatment (Henske, 2012). Independently, there is considerable evidence for aberrant Ral GTPase activation in driving cancer growth, invasion and metastasis (Bodemann and White, 2008; Neel et al., 2011). Based on the sequence identity and structural heterodimeric complex nature of the GAPs for Ral and Rheb small GTPases, we addressed possible crosstalk between these two signaling networks. We determined that RalGAPs negatively regulate mTORC1 activation via RalB interaction with Sec5 and the exocyst to regulate mTORC1 subcellular localization and activity. RalGAP analysis in C. elegans, where the Tsc1/2 complex is absent, also supported a functional relationship with TOR signaling. Although Ral GTPases have well-validated roles in cancer, the effector functions important for these roles have remained poorly understood (Bodemann and White, 2008; Neel et al., 2011). Our findings suggest that mTORC1 activation is an important consequence of RalB activation. Surprisingly, we also determined that loss of the Tsc1/2 complex increased Ral activation. Although clinically relevant functions of Tsc1/2 independent of Rheb-mTOR signaling have been described, the mechanistic basis for these activities has remained elusive (Neuman and Henske, 2011). We suggest that Ral activation is an important Rheb-independent consequence of Tsc1/2 loss important in disease.
We observed that C. elegans lacks bona fide orthologs of Tsc1 and Tsc2 but does possess a Rheb ortholog and signaling components necessary for mTORC1 signaling found in other organisms. A role for mTORC1 signaling in aging and age-related diseases is well established, but we still have much to learn about the molecular mechanisms involved. Our finding that RalGAP-directed RNAi reduced C. elegans lifespan is consistent with a role for RalGAP repression of Rheb-TORC1. Whether C. elegans RalGAP can act as a GAP for RHEB-1 and whether RAL-1 can activate TOR remain to be determined.
To further implicate RalGAP in regulation of mTOR signaling, we determined that suppression of RalGAPβ caused mTORC1 activation. We established a mechanism whereby activation of RalB but not RalA promotes activation of mTORC1 via Sec5 and the exocyst. In contrast to our findings, a previous study described a mechanism where RalA activation by a RalGEF then facilitated Rheb activation of mTORC1. They also found that growth factor activation of mTORC1 was independent of Ral activation (Maehama et al., 2008; Xu et al., 2011). A second study extended this mechanism and found Rheb-dependent activation of the RalA effector phospholipase D involved in mTORC1 activation by nutrients (Maehama et al., 2008; Xu et al., 2011). Thus, both RalA and RalB can regulate mTORC1 but through distinct upstream stimuli and downstream effectors.
Consistent with our findings, another study observed RalB but not RalA involvement in autophagy (Bodemann et al., 2011). They described a mechanism whereby RalB utilizes two distinct exocyst subcomplexes consisting of either Sec5 or Exo84. Nutrient deprivation promoted RalB-Exo84 complex formation and autophagy. Interestingly, they also found that nutrient deprivation inhibited the RalB-Sec5 association. This is consistent with our finding that the RalB-mTOR complex is both dependent upon Sec5 expression and responsive to the presence of growth factors. Our work further supports and extends their model where RalB can utilize two exocyst subcomplexes: one where the RalB-Sec5 interaction promotes mTOR activation and limits autophagy upon growth factor stimulation and another complex where the RalB-Exo84 complex promotes ULK1 activation to enhance autophagy under nutrient deprived conditions.
Loss of function of either Tsc1 or Tsc2 leads to hyperactive Rheb and chronic mTORC1 activity, stimulating cellular growth and proliferation. Although Rheb is the only bona fide Tsc1/2 substrate, Rheb-independent Tsc1/2 signaling and associated biological properties have been observed. Tsc2 GAP activity has been found to act on both Rab5 (Xiao et al., 1997) and Rap1 (Wienecke et al., 1995), but in vivo characterization has been lacking. We found that loss of either Tsc1 or Tsc2 led to enhanced Ral activity. Since Tsc1/2 displays no RalGAP activity in vitro, it is unclear how Tsc1/2 might be regulating Ral signaling. Nevertheless, we found that tumors deficient in Tsc2 have elevated RalB protein levels and RalB activity, indicating a potential causal role for RalB in Tsc1/2-deficient malignancies. Future efforts to therapeutically target RalB signaling potentially have promise in diseases caused by Tsc1/2 loss.
RalB has been shown to be involved in many important biological processes such as the host immune response (Chien et al., 2006), tumorigenesis (Peschard et al., 2012), tumor cell invasion and metastasis (Bodemann and White, 2008; Neel et al., 2012), control of autophagy (Bodemann et al., 2011) and exocytosis (Martin et al., 2012). Despite controlling diverse biological functions, RalB is only known to interact with a limited number of effector proteins (Martin and Der, 2012). Recently, the exocyst proteins Sec5 and Exo84 have emerged as the most important effectors of RalB. We find that the exocyst engages mTOR and is found to colocalize with active mTOR and RalB at the plasma membrane. We speculate that upon growth factor stimulation, active RalB utilizes the exocyst to traffic mTORC1 to the plasma membrane, where it can actively phosphorylate substrate proteins. This process may play a key role in tumor invasion and metastasis, where RalB and the exocyst have known roles (Bodemann and White, 2008). In fact, we found that in pancreatic tumor cells with elevated RalB activity due to loss of RalGAP function, mTORC1 function is critical, as mTORC1 inhibition with rapamycin decreased invasion in vitro. Our finding that mTORC1 signaling is critical for tumor cells to invade provides a way to potentially inhibit RalB-driven PDAC invasion and metastasis.
In conclusion, we determined that GAPs for the Ral and Rheb small GTPases can facilitate the convergence and interplay of two signaling networks previously considered distinct. Previously, the importance of Tsc1/2-Rheb-mTOR and Ral in disease had stimulated approaches to therapeutically block each pathway. With our finding that these are components of a shared signaling network, combination approaches for blocking Ral or mTOR may be more effective.
EXPERIMENTAL PROCEDURES
Cell Lines and Reagents
Detailed information on cell lines and culture conditions are described in the Extended Experimental Procedures.
Expression Constructs and Antibodies
Detailed information on the source of cDNA and shRNA expression plasmids and antibodies are described in the Extended Experimental Procedures.
Immunofluorescence and Microscopy
HEK293T cells were plated on 0.01% poly-L-lysine (Sigma) coated glass coverslips, fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100, then incubated with primary antibodies, followed by an Alexa Fluor-conjugated secondary antibody (Invitrogen). All images were acquired with sequential scanning with a LSM 710 confocal microscope. Additional information are provided in Extended Experimental Procedures.
C. elegans Aging Assays
Synchronous daf-2(e1370); eri-1(mg366) animals were grown at 15°C to bypass dauer formation. Late L4 animals were picked to RNAi plates at 25°C (day 0) and survival was assessed daily thereafter. Animals that died with eggs hatching within, a common trait of daf-2 mutants grown at 25°C, were removed from plates and not included in the final tally. Other aging assays were performed at 20°C. RNA interference was performed using a standard protocol (Fraser et al., 2000). HT115 bacteria were grown harboring plasmids expressing dsRNA corresponding to gfp (Zand et al., 2011), daf-16 (I-5M24), ral-1 (III-7M13), rheb-1 (III-5C23), let-363 (I-2I02), hgap-1 (I-7B15), and hgap-2 (II-6E14). hgap-1 and hgap-2 nonsense mutations were identified by the Million Mutation Project (Thompson et al., 2013). Additional information is provided in the Extended Experimental Procedures.
Supplementary Material
Highlights.
C. elegans lifespan is controlled by hgap (RalGAP)-Ral-1-CeTOR signaling
RalGAPs negatively regulate mTORC1 signaling by controlling of RalB activity
RalB forms a complex with mTORC1 that is dependent on Sec5 and the exocyst
mTOR inhibition blocks RalB-driven pancreatic tumor cell invasion
ACKNOWLEDGMENTS
We thank Elizabeth Henske for the Tsc-deficient MEFs, Bob Goldstein for helpful discussions, Bob Bagnell for help with FRET, and Jenni Sells for assistance with manuscript preparation. Some C. elegans strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). This work was supported by grants from the National Institutes of Health (CA042978 to C.J.D. and GM085309 to D.J.R.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aspuria PJ, Tamanoi F. The Rheb family of GTP-binding proteins. Cell Signal. 2004;16:1105–1112. doi: 10.1016/j.cellsig.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–267. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodemann BO, White MA. Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer. 2008;8:133–140. doi: 10.1038/nrc2296. [DOI] [PubMed] [Google Scholar]
- Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, White M, Camonis J. Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J. 2008;27:2375–2387. doi: 10.1038/emboj.2008.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XW, Leto D, Xiong T, Yu G, Cheng A, Decker S, Saltiel AR. A Ral GAP complex links PI 3-kinase/Akt signaling to RalA activation in insulin action. Mol Biol Cell. 2011;22:141–152. doi: 10.1091/mbc.E10-08-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, Balakireva MG, Romeo Y, Kopelovich L, Gale M, Jr, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127:157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Falasca M, Selvaggi F, Buus R, Sulpizio S, Edling CE. Targeting phosphoinositide 3-kinase pathways in pancreatic cancer--from molecular signalling to clinical trials. Anticancer Agents Med Chem. 2011;11:455–463. doi: 10.2174/187152011795677382. [DOI] [PubMed] [Google Scholar]
- Feig LA. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 2003;13:419–425. doi: 10.1016/s0962-8924(03)00152-1. [DOI] [PubMed] [Google Scholar]
- Fraser AG, Kamath RS, Zipperlen P, Martinez-Campos M, Sohrmann M, Ahringer J. Functional genomic analysis of C. elegans chromosome I by systematic RNA interference. Nature. 2000;408:325–330. doi: 10.1038/35042517. [DOI] [PubMed] [Google Scholar]
- Frische EW, Pellis-van Berkel W, van Haaften G, Cuppen E, Plasterk RH, Tijsterman M, Bos JL, Zwartkruis FJ. RAP-1 and the RAL-1/exocyst pathway coordinate hypodermal cell organization in Caenorhabditis elegans. Embo J. 2007;26:5083–5092. doi: 10.1038/sj.emboj.7601922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukai S, Matern HT, Jagath JR, Scheller RH, Brunger AT. Structural basis of the interaction between RalA and Sec5, a subunit of the sec6/8 complex. Embo J. 2003;22:3267–3278. doi: 10.1093/emboj/cdg329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heider MR, Munson M. Exorcising the exocyst complex. Traffic. 2012;13:898–907. doi: 10.1111/j.1600-0854.2012.01353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henske E. Getting to the finish line with mTORC1-targeted therapy. J Clin Invest. 2012;122:1970–1972. doi: 10.1172/JCI64227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412:179–190. doi: 10.1042/BJ20080281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Sun H. Functional genomic approach to identify novel genes involved in the regulation of oxidative stress resistance and animal lifespan. Aging Cell. 2007;6:489–503. doi: 10.1111/j.1474-9726.2007.00302.x. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapierre LR, Hansen M. Lessons from C. elegans: signaling pathways for longevity. Trends Endocrinol Metab. 2012;23:637–644. doi: 10.1016/j.tem.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, Der CJ, Counter CM. Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol. 2006;16:2385–2394. doi: 10.1016/j.cub.2006.10.023. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Maehama T, Tanaka M, Nishina H, Murakami M, Kanaho Y, Hanada K. RalA functions as an indispensable signal mediator for the nutrient-sensing system. J Biol Chem. 2008;283:35053–35059. doi: 10.1074/jbc.M805822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TD, Der CJ. Differential involvement of RalA and RalB in colorectal cancer. Small GTPases. 2012;3:126–130. doi: 10.4161/sgtp.19571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TD, Mitin N, Cox AD, Yeh JJ, Der CJ. Phosphorylation by protein kinase Calpha regulates RalB small GTPase protein activation, subcellular localization, and effector utilization. J Biol Chem. 2012;287:14827–14836. doi: 10.1074/jbc.M112.344986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel NF, Martin TD, Stratford JK, Zand TP, Reiner DJ, Der CJ. The RalGEF-Ral effector signaling network: the road less traveled for anti-Ras drug discovery. Genes Cancer. 2011;2:275–287. doi: 10.1177/1947601911407329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neel NF, Rossman KL, Martin TD, Hayes TK, Yeh JJ, Der CJ. The RalB small GTPase mediates formation of invadopodia through a GTPase-activating protein-independent function of the RalBP1/RLIP76 effector. Mol Cell Biol. 2012;32:1374–1386. doi: 10.1128/MCB.06291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuman NA, Henske EP. Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol Med. 2011;3:189–200. doi: 10.1002/emmm.201100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Pertz O, Hodgson L, Klemke RL, Hahn KM. Spatiotemporal dynamics of RhoA activity in migrating cells. Nature. 2006;440:1069–1072. doi: 10.1038/nature04665. [DOI] [PubMed] [Google Scholar]
- Peschard P, McCarthy A, Leblanc-Dominguez V, Yeo M, Guichard S, Stamp G, Marshall CJ. Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr Biol. 2012;22:2063–2068. doi: 10.1016/j.cub.2012.09.013. [DOI] [PubMed] [Google Scholar]
- Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzbraun T, Vincent JB, Schumacher A, Geschwind DH, Oliveira J, Windpassinger C, Ofner L, Ledinegg MK, Kroisel PM, Wagner K, et al. Cloning, genomic structure, and expression profiles of TULIP1 (GARNL1), a brain-expressed candidate gene for 14q13-linked neurological phenotypes, and its murine homologue. Genomics. 2004;84:577–586. doi: 10.1016/j.ygeno.2004.04.013. [DOI] [PubMed] [Google Scholar]
- Shipitsin M, Feig LA. RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol. 2004;24:5746–5756. doi: 10.1128/MCB.24.13.5746-5756.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa R, Fukai S, Kawato M, Higashi T, Kondo H, Ikeda T, Nakayama E, Okawa K, Nureki O, Kimura T, et al. Tuberous sclerosis tumor suppressor complex-like complexes act as GTPase-activating proteins for Ral GTPases. J Biol Chem. 2009;284:21580–21588. doi: 10.1074/jbc.M109.012112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiczka KS, Yeaman C. Ral-regulated interaction between Sec5 and paxillin targets Exocyst to focal complexes during cell migration. J Cell Sci. 2008;121:2880–2891. doi: 10.1242/jcs.031641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Thompson O, Edgley M, Strasbourger P, Flibotte S, Ewing B, Adair R, Au V, Chaudry I, Fernando L, Hutter H, et al. The Million Mutation Project: A new approach to genetics in Caenorhabditis elegans. Genome Res. 2013 doi: 10.1101/gr.157651.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Proud CG. mTORC1 signaling: what we still don't know. J Mol Cell Biol. 2011;3:206–220. doi: 10.1093/jmcb/mjq038. [DOI] [PubMed] [Google Scholar]
- Wienecke R, Konig A, DeClue JE. Identification of tuberin, the tuberous sclerosis-2 product. Tuberin possesses specific Rap1GAP activity. J Biol Chem. 1995;270:16409–16414. doi: 10.1074/jbc.270.27.16409. [DOI] [PubMed] [Google Scholar]
- Xiao GH, Shoarinejad F, Jin F, Golemis EA, Yeung RS. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J Biol Chem. 1997;272:6097–6100. doi: 10.1074/jbc.272.10.6097. [DOI] [PubMed] [Google Scholar]
- Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung RS, Xiao GH, Everitt JI, Jin F, Walker CL. Allelic loss at the tuberous sclerosis 2 locus in spontaneous tumors in the Eker rat. Mol Carcinog. 1995;14:28–36. doi: 10.1002/mc.2940140107. [DOI] [PubMed] [Google Scholar]
- Zand TP, Reiner DJ, Der CJ. Ras effector switching promotes divergent cell fates in C. elegans vulval patterning. Dev Cell. 2011;20:84–96. doi: 10.1016/j.devcel.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.