

Abstract
Significance: During wound healing of the skin, keratinocytes should move over while still adhering to their underlying matrix. Thus, mechanistic insights into the wound-healing process require an understanding of the forms and functions of keratinocyte matrix adhesions, specifically focal contacts and hemidesmosomes, and their components.
Recent Advances: Although the structure and composition of focal contacts and hemidesmosomes are relatively well defined, the functions of their components are only now being delineated using mouse genetic models and knockdown approaches in cell culture systems. Remarkably, both focal contact and hemidesmosomal proteins appear involved in determining the speed and directional migration of epidermal cells by modulating several signal transduction pathways.
Critical Issues: Although many publications are centered on focal contacts, their existence in tissues such as the skin is controversial. Nonetheless, focal contact proteins are central to mechanisms that regulate skin cell motility. Conversely, hemidesmosomes have been identified in intact skin but whether hemidesmosomal components play a positive regulatory function in keratinocyte motility remains debated in the field.
Future Directions: Defective wound healing is a developing problem in the aged, hospitalized and diabetic populations. Hence, deriving new insights into the molecular roles of matrix adhesion proteins in wound healing is a prerequisite to the development of novel therapeutics to enhance tissue repair and regeneration.

Jonathan C.R. Jones, PhD
General Introduction
The skin, comprising an outer epidermal sheet composed of multilayers of keratinocytes and an underlying connective tissue or dermis, is a barrier that protects the body from pathogens and excessive water loss, and which is involved in insulation, temperature regulation, and sensation. In intact skin, keratinocytes in the basal layer of the epidermis abut a basement membrane that is rich in extracellular matrix (ECM) proteins, including laminin, type IV collagen, type VII collagen, nidogens, and perlecan. These matrix components are woven into a complex meshwork that anchors the epidermis to the dermis.
Injuries of the skin, including burns and mechanical trauma, may result in damage to just the keratinocyte layers of the epidermis or damage to both the epidermal and dermal layers. The focus of this review is on the latter type of wounds where the basement membrane is breached and blood vessels are damaged. Under these circumstances, fibronectin is released into the wound bed along with fibrin deposited by platelets.1 These two molecules crosslink to form a plug that provides a provisional matrix for subsequent cellularization of the wound.
Complete epithelialization of a wound by keratinocytes is essential for the restoration of the barrier function of the skin. The epidermal “component” of wound healing comprises three major stages: migration, proliferation, and differentiation of keratinocytes. These processes are initiated, at least in part, as a response to cues from surrounding cells, such as cytokines released from leukocytes and platelets, and from various environmental signals, such as stretch and electrical fields.2,3 The keratinocytes immediately adjacent to the wound proliferate and, at the same time, undamaged keratinocytes at the wound margins migrate over a provisional matrix, rich in matrix molecules, including fibronectin and fibrin, covering the wound bed (Fig. 1).1 Migration of keratinocytes requires that both cell-cell and cell-substratum contacts and the keratinocyte cytoskeleton are remodeled to allow the cells to detach from the intact basement membrane in the unwounded epidermis (Fig. 1).4 These cells then migrate onto the provisional matrix of the wound. As the cells move over the wound, they degrade and remodel the provisional matrix while also depositing new matrix proteins, including laminin-332 (Fig. 1).5,6 After complete epithelialization of the wound, both keratinocytes and fibroblasts contribute to the reformation of the basement membrane. Finally, barrier function is restored as the keratinocytes covering the wound site stratify and differentiate.7
Figure 1.

A schematic of the edge of a wound-healing epidermis. On the left, a keratinocyte in an unwounded region adheres to the laminin-332 (LM332) in the basement membrane (BM) in part through a keratin-associated hemidesmosome (HD) and possibly, an actin-interacting focal contact (FC). The HD is targeted by kinases and proteases (arrow) in cells at the wound margin (middle cell). Cells that leave the BM move over a provisional matrix composed of fibronectin (depicted as fibrils) and laminin-332. Focal contact and hemidesmosomal integrins interact with this matrix and likely regulated the directed migration of the motile keratinocytes. The box indicates the key for α3β1 and α6β4 integrin heterodimers.
The focus of this review is how specific matrix molecules, their receptors, and associated proteins in keratinocytes are involved in epidermal repair after skin insult. In keratinocytes, matrix proteins and their receptors are clustered in two distinct protein complexes: focal contacts (adhesions) and hemidesmosomes (Fig. 2).8 In vivo, at the ultrastructural level, hemidesmosomes link basal keratinocytes to the basement membrane zone and are identified by their characteristic subplasma membrane, cytoplasmic electron dense plaques to which keratin intermediate filaments attach (reviewed in Jones et al.9). In sharp contrast, in vitro, cultured keratinocytes do not assemble bona fide hemidesmosomes, at least as judged electron microscopically. In such cells, hemidesmosomal proteins aggregate into what some have termed stable anchoring complexes (Figs. 3 and 4).10–13 In culture, ex vivo, keratinocytes assemble focal contact structures that serve as the cell surface anchorage site for the actin cytoskeleton (Fig. 4). The tissue equivalent of the focal contact is not obvious in a skin cell at the ultrastructural level although keratinocytes in the basal layer of the epidermis express a wide range of focal contact proteins. That focal contacts and their proteins play important roles in cell migration is undisputed. In contrast, whether hemidesmosomal proteins positively modulate migration is controversial, at least with regard to skin wound healing. We will begin with a brief description of the focal contact and its components. This is not a comprehensive review of focal contact proteins, as reviewing the more than 100 proteins found in focal contacts would be an overwhelming task. Rather, we will focus on a select number of focal contact proteins with known functions in regulating motility.
Figure 2.

Schematics of the protein components of the focal contact (left) and hemidesmosome (right).
Figure 3.
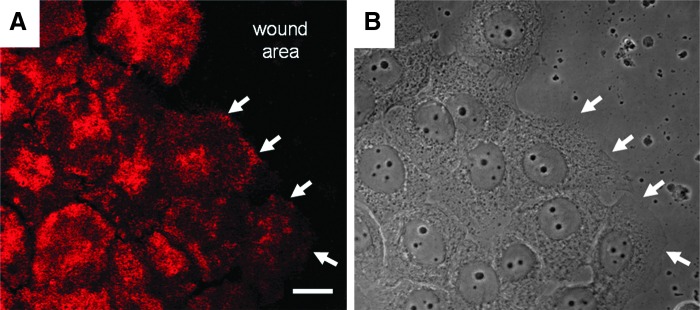
Immortalized human keratinocytes were allowed to attain confluence in vitro and were then scratched wounded. At 6 h after wounding, the cells were prepared for confocal immunofluorescence using antibodies against β4 integrin (A). Cells at the wound edge have begun to move onto the wound site. White arrows indicate β4 integrin staining along the leading lamellipodia of cells at the wound margin. These same areas are marked by arrows in the phase image of the cells in (B). Scale bar, 20 μm.
Figure 4.
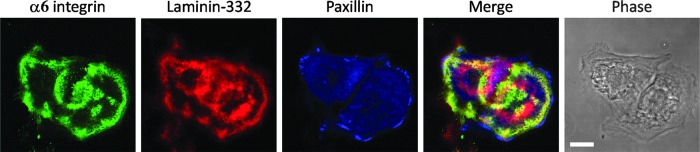
Immortalized human keratinocytes maintained at sub-confluence in vitro were prepared for triple label immunofluorescence using antibodies against α6 integrin, laminin-332, and paxillin as indicated. The merge of the three images indicates that α6 integrin-rich hemidesmosome protein complexes co-distribute with laminin-332, while paxillin focal contacts localize at the very edge of the cells and fail to show clear co-localization with laminin-332. The phase image shows the cell outline. Scale bar, 10 μm.
Discussion of findings and relevant literature
Focal contacts
In vitro, as an individual cell migrates, focal complexes at the cell edge mature into focal contacts, which, in concert with the actin cytoskeleton, generate the traction forces needed to pull the cell forward.4 Focal contacts then disassemble at the cell rear to allow the latter to release from the underlying matrix as the cell advances over its substrate. The migration of cells in tissues is more complex, however, as keratinocytes advance over a wound as a sheet via a process termed collective cell migration.14 Recent studies have begun to uncover the precise mechanisms that regulate collective migration, but they are still incompletely understood.14 With regard to wound healing in the skin, one assumes that focal contacts at the leading edge of the “leader” cells along the migrating tip of the epidermis define sheet polarity, generate traction forces, and guide the migration of “follower” cells.14 In the skin, there is also the complication that there is some multi-layering of cells, one or two cells back from the leading edge of the migrating epidermal tip. Whether these suprabasal layers simply tag along for the ride or play an important role in the wound-healing process remains unknown.
Transmembrane proteins of the focal contact
Integrins
At the core of the focal contact, matrix receptors called integrins link the actin cytoskeleton to various components of the ECM, albeit indirectly (Fig. 2).15 They also mediate both inside-out and outside-in signaling which is central to the role of focal contacts in regulating pathways that determine both the directionality and speed of motile cells.16 Integrins exist as heterodimers of one α and one β subunit. To date, 18 α and 8 β subunits have been identified; they can assemble in 24 specific combinations to form heterodimers in mammals.15 Both subunits contain a large N-terminal extracellular globular head domain (∼1,000 amino acids for α subunits and ∼700 amino acids for β subunits), a transmembrane segment, and a C-terminal cytoplasmic tail that has affinity for cytoskeletal as well as scaffold and signaling proteins.15
Many α subunits contain sites for post-translational processing, such as cleavage into two polypeptides. Cleavage occurs intracellularly and results in two α integrin chains (one heavy and one light) that are covalently linked by a disulfide bridge.17 The heavy chain remains completely extracellular in the mature integrin molecule, whereas the light chain encompasses a single transmembrane region and a short cytoplasmic tail. The β integrin subunits are not cleaved, and their cytoplasmic tails are typically short, around 50 amino acids in length. The one exception to this is the β4 integrin subunit, which has a cytoplasmic tail of more than 1,000 amino acids.15
Each integrin heterodimer binds to a distinct ECM ligand, such as collagen, fibronectin, or laminin. The respective N-terminal head domains of the α and β integrin subunits function cooperatively by binding to specific ligands together.15 Integrins contain multiple sites for interacting with various extracellular ligands; the classic example is the short tripeptide amino-acid sequence of Arg-Gly-Asp (RGD).15 Although this recognition sequence was originally identified as the minimal essential peptide sequence for fibronectin adhesion, it is a common motif that integrins utilize to bind to other cell-adhesive proteins.18 However, laminin-binding integrins, such as α6, utilize different recognition sequences.
During migration and wound healing, cells extend actin-stabilized filopodia to “sample” the terrain.19 Integrins for the ECM substrate become enriched at the filopodia, thereby enhancing matrix adhesion.20 Integrin-ligand interactions also induce conformational changes in the receptor that allow recruitment of various signaling intermediates (see below), which, in turn, activate Rho GTPAses, including Rac. The latter direct cytoskeletal reorganization and the extension of actin-rich lamellipodia.20 Integrin activation of Rac leads to a signaling cascade, resulting in phosphorylation of myosin light chain kinase, which activates myosin II to create actin-myosin contraction forces to pull the cell forward.21 During cell migration, these steps are repeated to generate traction as cells extend lamellipodia and form new points of contact with the ECM.20
A detailed discussion of integrin functions in the intact and wounded epidermis is provided in an excellent recent review.22 For that reason, our discussion on the topic will be brief. In intact skin, keratinocytes express the focal contact-associated integrins α2β1, α9β1, and α3β1.23,24 The functions of these integrins have been examined extensively by the generation of knockout mice. Although the results from such analyses have provided important new mechanistic clues to the molecular pathways regulating wound healing, there are some caveats. Mouse wounds heal primarily by contracture, as their skin is relatively loose. In contrast, human skin is tight and heals primarily by epithelialization. Thus, translating results from in vivo mouse wound healing studies to the human condition may not be valid in every case.
Intriguingly, despite its expression in many tissues, α2β1 integrin-deficient mice are not only viable but display no obvious defects in wound healing relative to their wild-type litter mates.25 Analyses of α9β1 integrin conditional knockout mice demonstrate poor re-epithelialization after wounding. However, this is most likely the result of decreased keratinocyte proliferation, rather than defects in cell migration.24
The role of the laminin-332 receptor α3β1 in cell migration and wound healing is controversial. The wounds of α3β1 integrin-null mouse epidermis exhibit faster wound closure when compared with wild-type skin.26 Moreover, using keratinocytes isolated from the α3β1 integrin null mice, migration assays reveal that the null keratinocytes migrate faster and with greater directional persistence.26,27 These results imply that α3β1 integrin functions as a brake on migration. However, other data argue against this. For example, Choma et al. also analyzed the migratory behavior of α3 integrin-null mouse keratinocytes and have presented evidence that these cells lack persistent migration, stable lamellipodia, and polarity.28 These findings are consistent with studies indicating that α3β1 integrin-laminin-332 ligation promotes keratinocyte polarization and persistent migration.29 Indeed, our recent data, using cultured human keratinocytes which exhibit a loss of α3 integrin expression, indicate that such cells move slower than their wild-type counterparts on laminin-332 rich matrices.30 Thus, together, these studies suggest that α3β1 integrin is a positive regulator of the motility rate of a keratinocyte.28–30 One possible explanation for the discrepancy of these studies is that there are differences in integrin profiles of the α3 integrin null mouse keratinocytes derived in different laboratories. Specifically, Margadant and co-workers have reported an up-regulation of α6 integrin in their α3 integrin-null mouse skin cells.26 Thus, α6β1 integrin could compensate for α3β1 integrin loss in such cells and support migration on laminin-332 matrices.26 However, such up-regulation has not been observed by others and may explain why the latter workers observe motility defects in α3 integrin-null keratinocytes maintained on laminin-332.31 Another possibility comes from recent analyses of the composition of the matrix deposited by mouse and human keratinocytes in vitro. The former assemble a matrix enriched in both fibronectin and laminin-332, whereas the latter deposit a matrix primarily composed of laminin-332 with little, if any, fibronectin.32 Moreover, Hamill et al. demonstrated that fibronectin acts as a brake to laminin-332 induced migration of both mouse and human keratinocytes and that the matrix of α3 integrin-null mouse epidermal cells contains less fibronectin than controls.32 Hence, the differences observed in migration speed of mouse keratinocytes lacking α3 integrin and wild type cells may result from a defect in the deposition of fibronectin by the former cells rather than α3 integrin functioning as a direct brake on migration in the latter cells. Of course, this raises the issue as to how α3 integrin regulates fibronectin deposition and why human and mouse skin cells deposit matrices of a different composition. Interestingly, α3 integrin is found between cells at the leading margin of wounds (Fig. 5A). What function it plays in such sites is unclear. Finally, to complicate the analyses of the functions of α3β1 integrin in wound healing in vivo is the finding that loss of α3β1 integrin in the epidermis of mice impacts wound healing in vivo in animal models via crosstalk with endothelial cells and angiogenic mechanisms.33
Figure 5.
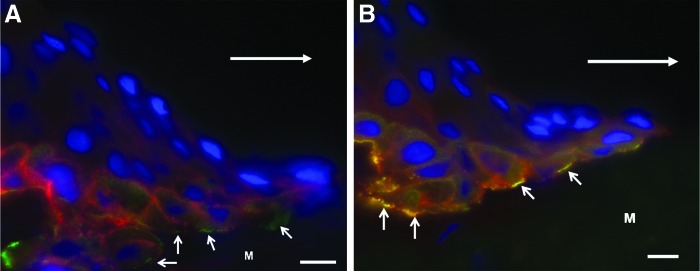
The images show the migrating tongue of keratinocytes in an incisional human wound at 30 h after wounding. The cells were moving toward the right [large arrows in (A) and (B) indicate the direction of movement]. (A) The section was prepared for immunofluorescence using antibodies against α3 integrin (red) and BPAG1e (green). The nuclei are stained with DAPI (blue). α3 integrin (red) is predominantly at sites of cell-to-cell contact with no clear co-distribution with BPAG1e (green) located along the advancing wound margin juxtaposed to the wound matrix (arrows). (B) The section was costained with antibodies against β4 integrin (red) and BPAG1e (green). The nuclei are stained with DAPI (blue). Note that β4 integrin and BPAG1e co-distribute as evidenced by the yellow color along the wound margin (arrows). M, matrix of wound bed. These images were presented in Underwood et al. as Figs. 8C and 5C.104 Scale bar, 10 μm.
On wounding, expression of the fibronectin receptor α5β1 integrin as well as αvβ5 and αvβ6 integrins is upregulated.34 α5β1 integrin likely regulates the migration of keratinocytes over the fibronectin-rich provisional matrix during early stages of wound healing.35 Ablation of αvβ5 or αvβ6 integrin in the epidermis of the mouse has no apparent impact on re-epithelialization of wounds with the possible exception of chronic wounds.36,37 However, whether αvβ6 integrin plays important roles in human skin wound healing remains an open question. Since it is involved in activation of TGF-β and, hence, triggers pathways leading to up-regulation of collagen gene expression, there is considerable interest in its role in the development of scarring and fibrosis in the skin after wounding.38
Syndecans and focal contacts
Cell interaction with matrix at the focal contact is also mediated by members of the syndecan family (Fig. 2).39 Indeed, these transmembrane heparan sulfate proteoglycans often function as integrin co-receptors.40 Each syndecan consists of a conserved transmembrane core and a highly divergent extracellular domain. The ectodomain of a syndecan can interact with a variety of growth factors, cytokines, and ECM proteins, including laminin-332.41 There are four syndecan members expressed in mammals with syndecan-1 and -4 being highly expressed in epithelial tissues, including keratinocytes.41 The heparan sulfate chains of the syndecans bind cytokines and growth factors, which, in turn, influence signaling cascades and matrix protease function that are necessary for wound healing.42,43
That syndecan-1 is involved in wound healing derives from studies of syndecan-1 null mice.44 Closure of wounds in their corneas is impeded as a consequence of epithelial cell migration and proliferation defects.44 The latter may be due to perturbation in the expression and localization of α9 integrin.24,44,45 In addition, Stepp et al. have demonstrated that syndecan-1 null keratinocytes exhibit enhanced adhesion with a corresponding decrease in the rate of their migration.46 This appears due to changes in either the organization of their laminin-332 matrix and/or by an indirect increased activation of adhesion-promoting α6β4 integrin complexes.46
Syndecan-4 binds the G domain of the α3 subunit of laminin-332.47 Addition of a synthetic peptide corresponding to the syndecan-4 binding region to cultured primary human keratinocytes promotes their migration apparently by activating the β1 integrin subunit.47 A role for syndecan-4 in regulating wound healing is also indicated in studies of syndecan-4 null mice that exhibit delayed wound repair. However, this wound-healing defect may be due to motility defects in fibroblasts rather than due to defects in keratinocyte migration.48
Interestingly, the extracellular domains of syndecans are constitutively shed, a process that is up-regulated in inflammation, wound healing, and some tumors.40 Specifically, the shed ectodomains of syndecan-1 and -4 are elevated in injured tissue as well as wound fluid where they likely influence signal transduction, protease activities and modulate the functions of intact syndecans by competing for interactions with growth factors or cytokines.49–51
Cytoplasmic proteins of the focal contact
Kindlins and talin
Proteins of the kindlin family are found in focal contacts where they regulate the function of β integrin subunits (Fig. 2). There are three members of the kindlin family of focal contact proteins: kindlin-1, mainly in epithelial cells; kindlin-2, expressed in most tissues; and kindlin-3, which is expressed in cells of the hematopoietic system.52
In humans, mutations in kindlin-1 are known to cause Kindler syndrome, a rare skin disease that is characterized by fragile skin with trauma-induced microblisters, poikiloderma, cutaneous atrophy, and photosensitivity in adulthood.53
All three kindlin proteins share a high degree of sequence similarity and domain structure. Each contains a pleckstrin homology domain and a 4.1 protein, ezrin, radixin, and moesin (FERM) domain. The FERM domain consists of three subdomains, one of which encompasses an integrin binding site.52 The FERM domain of the kindlins is similar to that found in talin, especially within the integrin-binding region.54 Indeed, it has been shown the both kindlin-1 and -2 directly bind the cytoplasmic tails of β1 and β3 integrin, where they serve as adaptor proteins.55
To distinguish the functions of kindlin-1 and -2, He et al. developed keratinocytes deficient in one or both of the kindlins. Loss of both kindlin-1 and -2 results in more severe changes in cell morphology than loss of one or the other.56 In addition, these proteins compensate for each other with regard to activation of β1 integrin and in keratinocyte adhesion to a substrate. In a scratch wound assay, keratinocytes deficient in both kindlins are unable to migrate. Kindlin-1 deficient cells move but heal a wound more slowly than their wild-type counterparts.56 In contrast, kindlin-2 deficient keratinocytes demonstrate an even more marked defect in migration in a scratch assay compared with kindlin-1 knockdown cells. Both kindlin-2 and the double-deficient cells also exhibit a loss in an ability to migrate directionally.56
Analyses of keratinocytes from patients suffering from Kindler syndrome have demonstrated that they have an abnormal, irregular shape, appearing more fibroblastic in nature, and have lost the ability to form colonies.57 When grown in culture, these cells show aberrant localization and decreased expression of α6β4 integrin and collagen XVII, but surprisingly, an increase in the level of laminin-332.57 These results, as well as immunoprecipitation data and structural modeling, have led to the speculation that kindlin-1 may associate with the hemidesmosome.58 Indeed, kindlins are candidate mediators of the crosstalk between hemidesmosomes and focal contacts (see below).8
Similar to the kindlins, talin is an integrin-activating and actin-binding protein. The precise functions of talin in wound healing of the skin and the migration of keratinocytes have not been precisely elucidated. Nonetheless, since talin is expressed by cultured keratinocytes and localizes to the epidermal-dermal junction, it likely plays roles in wound repair and merits some discussion.59 Talin is composed of an N-terminal globular head and a larger C-terminal rod. The head region contains an FERM domain that is composed of three subdomains, F1, F2, and F3.54 The F3 subdomain binds to and activates the β3 integrin tail, while a more extensive head fragment is necessary for activation of β1 integrins.54 The talin rod domain contains binding sites for actin, vinculin, and β integrin tails. Thus, talin regulates both integrin affinity for its extracellular matrix ligand and serves as a linker between ligand bound integrins and the actin cytoskeleton.60
The talin molecule is cleaved between the head and tail domain by calpain.61 The cleavage of talin plays an important role in focal contact dynamics and, thus, cell migration. Indeed, loss of expression of calpain has been implicated in defects in fibroblast migration.62 In Chinese hamster ovary cells, it has been shown that the talin head is phosphorylated by Cdk5, blocking its degradation and leading to stabilization of focal contacts.63 If talin is not phosphorylated, it binds Smurf1 and is ubiquitinated, and marked for degradation, thus promoting focal contact disassembly.63 In migrating cells, talin is a regulator that is involved in the balance between maintenance of stable focal contacts which facilitate development of traction forces and the disassembly of adhesion structures in order to permit the cell to move forward. Intriguingly, talin interacts directly with the Rac exchange factor Tiam 1, thereby regulating Rac1 activity and polarized cell migration in certain cells.64 In this regard, Tiam1 has been reported to be involved in laminin-332 deposition that is regulated by α3β1 integrin and keratinocyte migration.65
Paxillin
Paxillin is an adaptor protein that is localized to focal contacts and has roles in cell adhesion and cell migration (Fig. 2).66 It contains multiple docking sites for a variety of proteins, including β integrins, actopaxin, GTPase activating proteins, and vinculin, as well as tyrosine kinases such as focal adhesion kinase (FAK) and integrin-linked kinase (ILK).66 Paxillin localizes near the leading edge of migrating cells and is believed to be a very early component of the assembling focal contact.67 In addition to recruiting various kinases to focal contacts, the phosphorylation of paxillin by FAK, c-Jun amino-terminal kinase (JNK), extracellular signal-regulated kinase, and p38 mitogen-activated protein kinase (MAPK) regulates its activity and localization.68,69
In nonepithelial cells and tumor cells, paxillin regulates motility by its association with various signaling pathways as well as its effect on focal contact remodeling and lamellipodial stability.66,70 In wounded epithelial tissues, epidermal growth factor (EGF) activates JNK, which, in turn, phosphorylates paxillin on residue serine 178.71 This phosphorylation event is required for additional FAK-mediated phosphorylation at residues tyrosine 31 and 118, which is necessary for corneal keratinocyte motility.72
FAK and ILK
FAK is a nonreceptor tyrosine kinase that is localized to focal contacts and which is involved in multiple signaling cascades and integrin-mediated signaling pathways (Fig. 2).73 FAK is composed of a kinase domain that is flanked on the N-terminal side by a FERM domain and on the C-terminal end by two proline-rich docking sites and a focal contact targeting sequence.74 Integrin/ECM ligation activates FAK, inducing a conformational change and resulting in autophosphorylation and interaction with and activation of Src family kinases.74 This process then leads to phosphorylation of multiple downstream targets that drive cell migration. The importance of FAK in keratinocyte migration is indicated by its localization at the epidermal-dermal junction in partial-thickness burn wounds, a site corresponding to actively migrating keratinocytes covering a wound.75 Moreover, studies in cultured keratinocytes have demonstrated an increase in phosphorylation of FAK in migrating cells compared with nonmotile cells.76 Finally, when FAK expression is knocked down or ablated, keratinocyte migration is significantly reduced compared with controls, with focal contacts in the knockdown cells being less dynamic and exhibiting defective disassembly properties.76,77 Support for the importance of FAK in keratinocyte migration comes from analyses of human and mouse skin explants. FAK is up-regulated at the front of migrating keratinocytes, and surprisingly, also within the explant itself.78 Addition of a tyrosine kinase inhibitor results in decreased keratinocyte migration that is relieved by removal of the inhibitor.78 Moreover, keratinocytes emerging from FAK-null mouse skin explants show loss of directed migration that is due, most likely, to their aberrant actin cytoskeleton organizations when compared with wild-type cells.77
ILK binds the cytoplasmic tails of β1, β2, and β3 integrins, linking them to the actin cytoskeleton and serves as an adaptor/scaffolding protein for signal transduction.79 ILK has been linked to phosphorylation of Akt and it is activated by substrate adhesion in a PI3 kinase-dependent manner.80 Akt phosphorylation is known to regulate the motility of a variety of cells.81 Therefore, one might assume that Akt phosphorylation plays a role in keratinocyte migration. In support of this, ILK knockout mice display impaired migration of keratinocytes in the hair follicle.82 In addition, keratinocytes isolated from these mice exhibit unstable lamellipodia, a lack of persistent migration, and failure to close scratch wounds efficiently.82
Alpha actinins
The actinins are members of a large family of actin-bundling proteins, including fimbrin, spectrin, and dystrophin, which share similar actin-binding domains and an EF hand calcium regulatory domain.83 There are four major isoforms of actinin, including two muscle-specific isoforms, actinin-2 and actinin-3, and two nonmuscle isoforms, actinin-1 and actinin-4.84–86 Though actinin-1 and actinin-4 share about 87% protein sequence homology, they show differences in subcellular localization and function, depending on cell type. Actinin-1 localizes exclusively in the cytoplasm, where it is found along actin stress fibers and is required to crosslink linear dorsal stress fibers.87,88 It associates with the cytoplasmic domain of β1 integrin and, when phosphorylated, it can interact with both FAK and Src, where it enhances signaling that is mediated by focal contacts and increases integrin-mediated adhesion (Fig. 2).
In contrast to actinin-1, actinin-4 is concentrated at the leading edge of migrating cells as well as localizing with actin stress fibers and cell-cell contacts.85,89,90 A role for actinin-4 in skin cell migration comes from recent studies using a knockdown approach. Actinin-4 deficient cultured human keratinocytes have defects in their lamellipodial dynamics, an inability to migrate progressively, and a decrease in the activation state of the actin severing protein cofilin.91 These defects are rescued by plating the actinin-4 knockdown keratinocytes onto the laminin-332-rich matrix laid down by wild-type keratinocytes, implying a role for actinin-4 in both signaling required for directed migration and the deposition of a laminin matrix.91
Flii
Flii, a member of the gelsolin family, is an actin remodeling protein that interacts with the focal contacts protein talin through its leucine-rich repeat domain.92,93 Moreover, it appears to function as a negative regulator of wound healing in mouse skin (Fig. 2).92,93 Specifically, overexpression of Flii in mice leads to impaired wound healing with larger wound beds, while mice with decreased expression of Flii show enhanced wound healing and migration.92 Though the mechanism for Flii function in wound healing is not fully understood, it may modulate the migration of skin cells via its effects on the pool of talin that is available for binding to and activating integrins.94
Hemidesmosomes
Unlike fibroblasts and many epithelial cells, keratinocytes in intact skin and cornea assemble matrix adhesion attachment devices termed hemidesmosomes. The core of the hemidesmosome is composed of α6β4 integrin, which, via an interaction with collagen XVII (BPAG2, BP180), a type II membrane protein, and the plakin family members BPAG1e (BP230) and plectin, mediates cell surface interaction of the keratin cytoskeleton. α6β4 integrin and the extracellular collagen-like domain of collagen XVII bind to the matrix molecule laminin-332 in the basement membrane8,9,95 (Fig. 2). Within the complex itself, β4 integrin has binding sites for BPAG1e and plectin as well as collagen XVII. α6 integrin also binds collagen XVII.96 Moreover, there are a number of accessory proteins that have been reported to interact with the hemidesmosomes, including CD151.97 It should be noted that there is far less molecular complexity to the hemidesmosome than the focal contact, at least with regard to the number of proteins associated with it (Fig. 2).
Hemidesmosomes are important for stable adherence of the epidermis to the dermis. Evidence for this comes from studies of numerous skin diseases where loss of hemidesmosomes results in blistering due to dysadhesion of the epidermis at or close to the basement membrane zone.8 The molecular defects in these diseases will be discussed next. Nevertheless, in wound healing, undamaged adherent keratinocytes at the margin of the wound release themselves from the constraints of the basement membrane zone, at least in part, by disassembling their hemidesmosomes (Fig. 1). Bona fide hemidesmosomes are only assembled after complete epithelialization of the wound bed. Such observations have led to the idea that hemidesmosomal proteins are not involved in the wound-healing process per se. Rather, hemidesmosomal proteins might be thought of as restricting motility over the ECM. However, recent studies from own lab argue otherwise and we have provided evidence for the role of several hemidesmosomal proteins in determining the motility of keratinocytes as detailed next.
Transmembrane components of the hemidesmosome
α6β4 integrin
α6β4 integrin binds laminin-332 in the ECM (Figs. 3 and 4).98 The β4 integrin subunit is unique among its β integrin relatives in that its cytoplasmic is more than 1,000 amino acids long.15 This allows it to interact with a number of high molecular cytoskeleton linker proteins, namely BPAG1e and plectin, which will be described next. BPAG1e and plectin bind the keratin cytoskeleton and, hence, α6β4 integrin possesses the ability to anchor keratin filaments to the cell surface.9,95 It is well established from both antibody inhibition studies and analyses of knockout mice that ligation of α6β4 integrin by laminin-332 nucleates hemidesmosome assembly.99,100 In addition, the long tail of β4 integrin interacts with a number of signaling intermediates although whether α6β4 integrin is capable of signaling when it resides within the hemidesmosome is not clear.101 Rather, α6β4 integrin more likely signals to various pathways when not incorporated into the hemidesmosome proper.
On skin wounding, EGF is released into the wound and induces the Src kinase family member Fyn to phosphorylate β4 integrin in those hemidesmosomes in the keratinocytes in the undamaged margins of the wound.102 This process may involve the interaction between syndecans and α6β4 integrin.103 Indeed, it has been suggested that syndecans may function to bring the cytoplasmic domain of the β4 integrin in close proximity to membranes to facilitate its phosphorylation.103 Regardless, phosphorylation of the β4 integrin cytoplasmic tail results in disassembly of hemidesmsomes, allowing keratinocytes to escape the constraints of the basement membrane.
Loss of expression of α6β4 integrin is the underlying cause of the blistering skin disease junctional epidermolysis bullosa (JEB) with pyloric atresia.8 This loss not only blocks hemidesmosome assembly but also results in dysadhesion of epidermal cells from the basement membrane. Indeed, this consequence has been used to suggest that the primary function of α6β4 integrin in the skin is to maintain epidermal-dermal adherence and that it is a stable anchor.22 Moreover, antibody blockade studies and analyses of motility of mouse keratinocytes deficient in β4 integrin expression have also suggested that α6β4 integrin is a stable anchoring integrin.99,100 However, there is now emerging evidence for the importance of α6β4 integrin in determining directed cell movement after wounding. First, α6β4 integrin localizes to the leading front of the cells migrating into wound sites in vitro and in tissue (Figs. 3 and 4B).104 Moreover, Pullar and associates explored the directed migration of primary keratinocytes exposed to an electrical field, an early signal in the wound-healing process.3 They compared migration of wild-type keratinocytes and keratinocytes lacking β4 integrin, isolated from a patient suffering from JEB with pyloric atresia, when exposed to an electrical field. Both the presence of laminin 332 and a functional β4 integrin cytoplasmic tail were shown to be necessary for directed migration in this in vitro model. Moreover, such migration requires activation of Rac1, a finding that is consistent with several other studies.3,105,106
Our own studies have also provided support for the idea that α6β4 integrin is required for directed migration. Wild-type keratinocytes move in a directed fashion along a linear track of laminin 332 matrix, whereas β4 integrin-deficient keratinocytes move primarily in circles and lay down a laminin 332 matrix that is also in a circular pattern. Interestingly, the motility defect in the β4 integrin-deficient keratinocytes can be corrected by either re-expressing wild-type β4 integrin or by plating the β4 integrin-deficient keratinocytes onto a matrix laid down by wild-type keratinocytes.106 These results suggest that migration defect seen in the β4 integrin-deficient keratinocytes is not due to a problem in the motility machinery, but rather is a consequence of an aberrant matrix deposition.
Rac1 immunoprecipates with α6β4 integrin in motile keratinocytes.106 Moreover, Rac1 signals to activate cofilin, an actin severing protein that is involved in actin cytoskeletal remodeling in such cells.106 Cofilin is activated by dephosphorylation, and our data indicate that members of the slingshot family of phosphatases are involved in this activation in migrating skin cells.107 Slingshot activity, in turn, is regulated through its interaction with 14-3-3 binding proteins. When slingshot is phosphorylated, it interacts with 14-3-3 protein, leading to inactivation by sequestering the complex in the cytoplasm.108,109 Intriguingly, α6β4 integrin binds 14-3-3 protein when the β4 integrin cytoplasmic tail is phosphorylated.110 Thus, we speculate that this places 14-3-3 protein in a key position to regulate Rac1-mediated cell migration in keratinocytes. Specifically, inactive, phosphorylated slingshot binds 14-3-3, but on receipt of migratory signals, β4 integrin becomes phosphorylated, allowing it to bind 14-3-3 to set up a scaffold for further signaling through Rac1.111 In the absence of β4 integrin, this signal pathway is compromised, leading to inactivation of both Rac1 and cofilin and perturbation in the actin remodeling machinery at the leading front of the cells. This is manifest in lamellipodial defects, in both their stability and formation, and a pronounced deficiency in directed migration111
Collagen XVII
A second transmembrane component of the hemidesmosomes is a 180 kD type II trimeric protein with a cytoplasmic amino terminus and a long extracellular tail composed of collagen repeats, termed collagen XVII (Fig. 2).112,113 This protein was first identified using autoantibodies in the serum of patients afflicted with the disease bullous pemphigoid (BP).114 Hence, it is sometimes termed BP180. Mutations in collagen XVII result in a nonlethal variant of a JEB-related blistering disease, characterized by decreased epidermal adhesion and skin fragility.8 Collagen XVII has been shown to contain multiple binding sites for hemidesmosomal proteins, including BPAG1e and β4 integrin in the cytoplasm, and interacts with laminin 332 via its collagen tail and α6 integrin via its NC16A domain.95,96 Collagen XVII is also able to link β4 integrin and BPAG1e when the β4 integrin binding site for BPAG1e has been deleted.115
The role of collagen XVII in migration is controversial. Keratinocytes derived from patients lacking the protein show enhanced migration over controls.116 However, studies using HaCat cells reveal that knockdown of collagen XVII expression using siRNA results in a decrease in migration in single-cell motility assays.117 To further complicate matters, keratinocytes isolated from a GABEB patient display an increase in motility in scratch wounds and cell scattering assays.116 To add to the complexity, collagen XVII is also proteolytically processed, releasing the extracellular region into the basement membrane region.118 One study has shown that this shed domain functions as an inhibitor of migration in a scratch wound assay, while other groups have demonstrated that the collagen XVII ectodomain is involved in both transmigration of squamous cell carcinomas and invasion of extravillous trophoblasts in the placenta.119,120
Our recent work using lentiviral-mediated shRNA knockdown of collagen XVII in cultured keratinocytes has shown that the depleted cells have impaired lamellipodia dynamics, loss of directed migration, and reduced Rac1 activity. Although surface expression of β4 and α3 integrins is unchanged in the knockdown cells, loss of expression of collagen XVII results in a decreased expression of BPAG1e.115 This indicates a role for collagen XVII in assembly of a α6β4 integrin/BPAG1e signaling complex in motile skin cells.
CD151
CD151 is a member of the tetraspanin family of transmembrane proteins and is found in association with hemidesmosomes (Fig. 2).97 All the tetraspanins share a similar structure of series of four passes through the cell membrane with one large extracellular loop that can interact with α integrin subunits and one small extracellular loop. The amino and carboxy termini are in the cytoplasm, and the carboxy tail is involved in signaling.121 These proteins are involved in cell adhesion, migration, and signaling.122 Tetraspanins may function as scaffolds or organizers of larger complexes of proteins, linking them to signaling enzymes and to each other. This complex has been termed a tetraspanin “web”123 or tetraspanin-enriched microdomain.124
In mouse skin, CD151 expression is up-regulated at the migrating front of the epithelium at the wound edge.125 Moreover, using blocking antibodies, tetraspanin CD151, along with its relative CD9 and CD81, is implicated in keratinocyte wound healing in scratch wound assays.126 Skin explant cultures from punch biopsies of neonatal skin from CD151 knockout mice also demonstrate defects in keratinocyte motility.127 In addition, in vivo studies from the CD151 knockout mice reveal wound-healing defects.125 The mechanisms involved in the integrin-tetraspanin interaction during keratinocyte migration are not well understood. One possibility is that CD151 plays a crucial role in integrin trafficking.128 During migration, some integrins are endocytosed from the rear end of the cell and recycled to the leading front.129 Interestingly, the cytoplasmic tail of CD151 contains an endocytosis/sorting motif. When residues of this motif are mutated, endocytosis of the CD151-associated integrins is compromised, and cell motility is defective as measured by scratch wound assays.128 Moreover, knockdown of CD151 expression using RNAi technology results in defects in cell migration on laminin-332, as well as on collagen and fibronectin.130 These authors propose a role for CD151 in regulating internalization and recycling of α3β1 integrin during cell migration and perhaps influencing the role of α3β1 integrin in maintaining polarization of migrating cells during migration by Rac1 activation.130
Cytoplasmic components of the hemidesmosome
BPAG1e
In the skin, BPAG1e (BP230) was originally identified as a 230 kD component of the plaque of the hemidesmosome using autoantibodies present in the serum of patients afflicted with BP.114 It is a plakin family member sharing homology with desmoplakins and plectin.131 Several alternatively spliced proteins are transcribed from the BPAG1 gene and have distinctive distribution across epithelial, neuronal, and muscle tissues.132 BPAG1e, the epithelial form, is located within the intracellular hemidesomsomal plaque on the basal surface (Fig. 2).9,114 Mice lacking BPAG1e have poorly formed hemidesmosomes, lacking keratin cytoskeleton connection, and a fragile skin prone to trauma-induced wounds and delayed wound healing.133
A role for BPAG1e in wound healing is suggested by the finding that wounds in the skin of the knockout mouse heal more slowly than those in wild-type animals and is supported by the link between an increase in BPAG1e expression and invasive squamous cell carcinomas.133,134 Moreover, on wounding skin, both β4 integrin and BPAG1e colocalize at the leading edge of the migrating keratinocytes (Fig. 5B).135 More direct evidence for a role for BPAG1e in keratinocyte migration derives from studies of keratinocytes that are depleted of BPAG1e.135 They have defects in front–rear polarity, exhibit a significant reduction in lamellipodial stability, and display aberrant motility with loss of directionality.135 Moreover, the amount of Rac1 immunoprecipitated with α6β4 integrin is reduced in BPAG1e-deficient keratinocytes compared with that in control cells.135 Taken together, these results strongly suggest a role for BPAG1e in the formation of a signaling complex with Rac1 that mediates α6β4 integrin-regulated directed migration.
Plectin
Plectin is a high-molecular-mass (over 600 kDa) protein that also indirectly binds to α6 integrin within hemidesmosomes (Fig. 2).95 This plakin family member can be found in both focal contacts and the plaques of hemidesmosomes via a direct interaction with β4 integrin.131 Plectin cooperates in some way with BPAG1e in connecting keratin filaments to the basal plasma membrane at the site of the hemidesmosome. Its N-terminal globular head region contains binding sites for the cytoplasmic tail of β4 integrin and actin, while the C-terminal tail associates with keratin.136 Structural studies indicate that plectin does not interact with β4 integrin and actin at the same time.136
Plectin plays a general role in the reinforcement of mechanically stressed cells such as those in the skin.137 Moreover, patients with mutations within the PLEC gene often have either no plectin or a truncated form and display a trauma-induced blistering phenotype within the epidermal basal layer.138 Fewer hemidesmosomes are formed, compromising anchorage and resulting in blister formation. Plectin-null keratinocytes migrate with higher velocities than their wild-type counterparts indicating that plectin plays a role in stabilization matrix attachments to their underlying matrix.139
Matrix, focal contacts, and hemidesmosomes
During wound healing, migrating keratinocytes move over or under a complex of matrix proteins composed of fibronectin and fibrin (the clot) and over the collagen present in the dermis.1 In addition, the migrating skin cells lose their connection to the laminins in the intact basement membrane and as they move, they deposit a fresh matrix rich in laminin-332.6 We will discuss fibronectin and laminin-332, focusing on our recent data indicating a complex interplay between the functions of fibronectin and laminin-332 in keratinocyte migration.30,32,140
Fibronectin is a large dimeric glycoprotein containing two similar polypeptide subunits of about 250 kDa linked by disulfide bonds (reviewed in Singh et al.141). It is a component of blood plasma and is also secreted by fibroblasts and keratinocytes. The fibronectin molecule contains three distinct modules termed type I, II, and III fibronectin repeats. These modules are arranged along the fibronectin monomer and contain various matrix and integrin-interacting domains, with the most famous one being the arginine-glycine-aspartic acid (RGD) motif, which interacts with the focal contact-associated α5β1 integrin to promote cell attachment and migration of keratinocytes.15,35
Laminin-332 is a major component of the epidermal basement membrane. The structure and functions of laminin-332 have been extensively described in a recent review and so, we will only briefly discuss laminin-332 structure here.98 It is a heterotrimer that is composed of α3, β3, and γ2 subunits. In intact skin, laminin-332 protein is the ligand for the integrin that is associated with the hemidesmosome and provides firm anchorage to the underlying matrix (see above).9 The importance of this component of the basement membrane of skin in maintaining the interaction of the epidermis and the dermis is highlighted by the effects of mutations in the subunits of laminin-332, the genetic defect that is the underlying cause of blistering in a variant of JEB.8 Loss of laminin-332 expression in the skin of these patients leads to dysadhesion of the epidermis at the level of the basement membrane.8
Laminin-332 is an atypical laminin with short β and γ subunit side arms compared with the comparable side arms in laminin-111.98 Moreover, both the α3 subunit and the γ2 laminin subunits are subject to proteolytic processing after secretion.142,143 In the case of the α3 subunit, this processing occurs in the C-terminus such that the mature molecule is missing domains 4 and 5 of its G domain.142 There is also evidence of processing occurring in the N-terminus of the α3 subunit, while the γ2 subunit undergoes an N-terminal cleavage.98 During tissue remodeling, additional cleavage of the γ2 subunit appears important in allowing cells to break their connection with the basement membrane. In this regard, syndecan-1 interacts with the short arm of the γ2 chain.144 This interaction, in turn, suppresses EGF-stimulated phosphorylation of the β4 integrin subunit, inhibits hemidesmosome disassembly, and promotes migration into the wound site.144
In addition to its role in stabling anchoring the keratinocytes in the basal layer of the epidermis to the dermis, laminin-332 is a crucial mediator of the migration of keratinocytes. Laminin-332 is not only localized to the wound edge of the epidermis, but it also supports the migration of cells in vitro.6 Moreover, live cell imaging of keratinocytes expressing a tagged subunit of the laminin-332 protein demonstrates that migrating cells deposit laminin-332 in linear arrays or tracks along which they move.106,145
Laminin-332 containing an unprocessed γ2 subunit has also been reported to drive the hypermotility of keratinocytes.146 Moreover, there is evidence that matrices rich in laminin-332 containing uncleaved α3 subunits support migration and that they do so in an α3β1 integrin-dependent manner.6 In addition, several heparin-binding domains have been identified in the G4 and 5 domains of the unprocessed α3 laminin, suggesting that heparan sulfate proteoglycans could interact with these regions.47,147,148 Moreover, a synthetic peptide encompassing the syndecan-binding domain of α3 laminin LG4 domain has been shown to promote keratinocyte migration by its interaction with syndecan-4, induction of conformational changes, and activation of β1 integrin. Interestingly, inhibition of p38MAPK partially blocked the ability of keratinocytes to migrate in the presence of the soluble peptide, implying syndecan-4 ligation to laminin-332 containing an unprocessed α3 activates a migratory signaling cascade involving p38MAPK.47 Further support for a syndecan-dependent motility inducing role for the G4 and 5 domains of the α3 laminin subunit derives from the finding that mutations perturbing syndecan-4 binding inhibit keratinocyte motility.147
Interestingly, the ability of laminin-332 to sustain migration appears fine-tuned by fibronectin. Specifically, addition of fibronectin to laminin-332 deposited matrix leads to a reduction in speed of the motile cultured keratinocytes, while enhancing their adhesion to substrate.32 We speculate that fibronectin acts to prevent detachment of keratinocytes by actively migrating over the wound bed in a laminin-332 dependent fashion.32
Crosstalk between focal contacts, hemidesmosomes, and their proteins
Although we have described focal contacts and hemidesmosomes in separate sections, their functions in adhesion and migration are closely interlinked.12 Moreover, although they share only one component, namely plectin, it is becoming increasingly clear that there is crosstalk between the proteins in focal contacts and hemidesmosomes. We have already mentioned data suggesting that the focal contact proteins kindlin and syndecan regulate hemidesmosome protein localization and possibly hemidesmosome protein functions. In addition, the focal contact protein Flii may be involved in this phenomenon, as mice overexpressing Flii have poorly formed hemidesmosomes and more fragile skin compared with wild-type mice.93 Another example of crosstalk is the regulation of translation of the α3 integrin subunit by α6β4 integrin-driven activation of the Akt-mTOR pathway in an initiation factor 4E binding protein 1-dependent manner.30 Moreover, it was also recently shown that α3 integrin subsequently regulates the transcription of α2 integrin via an unknown mechanism.30
Future Directions
Further work on the exact manner via which α6β4 integrin and the BP antigens regulate signaling that determines motile behavior is clearly needed. Moreover, their role in the context of the complex skin tissue environment also needs elucidation. Knockout mice have proved useful in dissecting out the functions of a number of proteins in wound healing, but mice are problematic for testing the activities of proteins that are involved in epithelialization. This is because mice wounds heal by contracture rather than epithelialization, the process via which human skin wounds repair. Thus, technical improvement in animal models such as the use of splints that prevent contracture and the use of three-dimensional culture systems may circumvent this problem in the future.149
The hemidesmosome is associated with keratin, but its components, most notably α6β4 integrin and plectin, also show association with the actin cytoskeleton.22 The mechanisms underlying the regulation of such associations that appear central in their role in migration still require elucidation. Moreover, intriguingly, a role for the keratin cytoskeleton in wound healing is also indicated. Specifically, the wound-induced keratin 6 negatively regulates Src kinase and the migratory behavior of keratinocytes.150 However, more work in this area is still needed to clarify whether keratins play a positive role in motility.
Finally, a better understanding of focal contact/hemidesmosome protein crosstalk should provide novel clues to the molecular mechanisms that regulate both adhesion and migration in skin cells. Such studies may reveal new strategies for the treatment of not only blistering skin diseases but also the treatment of chronic wounds and other defects in the repair of skin after tissue damage.
Take-Home Messages.
• Focal contacts and hemidesmosomes and/or their components play critical roles in wound repair.
• Focal contact proteins regulate the traction forces and signals that are necessary for forward movements of cells as they migrate over a wound.
• Although the hemidesmosome is disassembled to allow keratinocytes to move onto the wound bed and/or over the provisional wound matrix, hemidesmosome proteins play important regulatory and signaling roles in determining aspects of the motile behavior of skin cells.
• Crosstalk between focal contact and hemidesmosome proteins appears central to the molecular mechanisms and fine-tuning of the repair response.
Abbreviations and Acronyms
- BP
bullous pemphigoid
- BPAG
bullous pemphigoid antigen
- ECM
extracellular matrix
- EGF
epidermal growth factor
- FAK
focal adhesion kinase
- FERM
4.1 protein, ezrin, radixin, moesin
- Flii
Protein flightless-1 homolog
- ILK
integrin-linked kinase
- JEB
junctional epidermolysis bullosa
- JNK
c-Jun amino-terminal protein kinase
- MAPK
mitogen-activated protein kinase
Acknowledgments and Funding Sources
The research in the Jones lab is supported by grants from the NIH (RO1 AR054184 and RO1 HL092963). They are very grateful to Dr. John Olerud, Dr. Robert Underwood, and their coauthors for providing original micrographs from their publication for the generation of Fig. 5.104
Author Disclosure and Ghostwriting
The authors disclose no commercial associations or known conflicts of interest in connection with this work. No ghostwriters were used to write this article.
About the Authors
The authors are members of the Department of Cell and Molecular Biology at the Feinberg School of Medicine at Northwestern University in Chicago. Their research centers on the functions of matrix receptors and matrix in both the adhesion and motility of keratinocytes. Jonathan C.R. Jones, PhD, is Professor of Cell and Molecular Biology and Dermatology.
References
- 1.Clark RA: Fibronectin matrix deposition and fibronectin receptor expression in healing and normal skin. J Invest Dermatol 1990; 94:128S. [DOI] [PubMed] [Google Scholar]
- 2.Kippenberger S, Bernd A, Loitsch S, et al. : Signaling of mechanical stretch in human keratinocytes via MAP kinases. J Invest Derm 2000; 114:408. [DOI] [PubMed] [Google Scholar]
- 3.Pullar CE, Baier BS, Kariya Y, et al. : β4 Integrin and epidermal growth factor coordinately regulate electric field-mediated directional migration via Rac1. Mol Biol Cell 2006; 17:4925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lauffenburger DA. and Horwitz AF: Cell migration: a physically integrated molecular process. Cell 1996; 84:359. [DOI] [PubMed] [Google Scholar]
- 5.Nguyen BP, Gil SG, and Carter WG: Deposition of laminin 5 by keratinocytes regulates integrin adhesion and signaling. J Biol Chem 2000; 275:31896. [DOI] [PubMed] [Google Scholar]
- 6.Goldfinger LE, Hopkinson SB, deHart GW, Collawn S, Couchman JR, and Jones JCR. The α3 laminin subunit, α6β4 and α3β1 integrin coordinately regulate wound healing in cultured epithelial cells and in the skin. J Cell Sci 1999; 112:2615. [DOI] [PubMed] [Google Scholar]
- 7.Sivamani RK, Garcia MS, and Isseroff RR: Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front Biosci 2007; 12:2849. [DOI] [PubMed] [Google Scholar]
- 8.Tsuruta D, Hashimoto T, Hamill KJ, and Jones JCR: Hemidesmosomes and focal contact proteins: functions and cross-talk in keratinocytes, bullous diseases and wound healing. J Derm Sci 2011; 62:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones JCR, Hopkinson SB, and Goldfinger LE: Structure and assembly of hemidesmosomes. BioEssays 1998; 20:488. [DOI] [PubMed] [Google Scholar]
- 10.Carter WG, Kaur P, Gil SG, Gahr PJ, and Wayner EA: Distinct functions for integrins alpha 3 beta 1 in focal adhesions and alpha 6 beta 4/bullous pemphigoid antigen in a new stable anchoring contact (SAC) of keratinocytes: relation to hemidesmosomes. J Cell Biol 1990; 111:3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hamill KJ, Paller AS, and Jones JC: Adhesion and migration, the diverse functions of the laminin alpha3 subunit. Dermatol Clin 2010; 28:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ozawa T, Tsuruta D, Jones JCR, et al. : Dynamic relationship of focal contacts and hemidesmosome protein complexes in live cells. J Invest Dermatol 2010; 130:1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsuruta D, Hopkinson SB, and Jones JC: Hemidesmosome protein dynamics in live epithelial cells. Cell Mot Cytoskel 2003; 54:122. [DOI] [PubMed] [Google Scholar]
- 14.Friedl P. and Gilmour D: Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 2009; 10:445. [DOI] [PubMed] [Google Scholar]
- 15.Hynes RO: Integrins: bidirectional, allosteric signaling machines. Cell 2002; 110:673. [DOI] [PubMed] [Google Scholar]
- 16.Huveneers S. and Danen EHJ: Adhesion signaling–crosstalk between integrins, Src and Rho. J Cell Sci 2009; 122:1059. [DOI] [PubMed] [Google Scholar]
- 17.deMelker AA. and Sonnenberg A: Integrins: Alternative splicing as a mechanism to regulate ligand binding and integrin signaling events. Bioessays 1999; 21:499. [DOI] [PubMed] [Google Scholar]
- 18.Pfaff M: Recognition sites of integrins within their ligands. In: Integrin-Ligand Interactions, edited by Eble JA. and Kühn K. Vol. xxi Austin, TX: R.G. Landers Company, 1997, pp. 101–122 [Google Scholar]
- 19.Defilippi P, Olivo C, Venturino M, Dolce L, Silengo L, and Tarone G: Actin cytoskeleton organization in response to integrin-mediated adhesion. Microsc Res Tech 1999; 47:67. [DOI] [PubMed] [Google Scholar]
- 20.Huttenlocher A. and Horwitz AR: Integrins in cell migration. Cold Spring Harb Perspect Biol 2011; 3:a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klemke RL, Cai S, Giannini AL, Gallagher PJ, Lanerolle PD, and Cheresh DA: Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 1997; 137:481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margadant C, Charafeddine RA, and Sonnenberg A: Unique and redundant functions of integrins in the epidermis. FASEB J 2010; 24:4133. [DOI] [PubMed] [Google Scholar]
- 23.Grose R, Hutter C, Bloch W, et al. : A crucial role of β1 integrins for keratinocyte migration in vitro and during cutaneous wound repair. Development 2002; 129:2303. [DOI] [PubMed] [Google Scholar]
- 24.Singh P, Chen C, Pal-Ghosh S, Stepp MA, Sheppard D, and Van De Water L: Loss of integrin α9β1 results in defects in proliferation, causing poor re-epithelialization during cutaneous wound healing. J Invest Dermatol 2008; 129:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen J, Diacovo TG, Grenache DG, Santoro SA, and Zutter MM: The α2 integrin subunit-deficient mouse: a multifaceted phenotype including defects of branching morphogenesis and hemostasis. Am J Path 2002; 161:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Margadant C, Raymond K, Kreft M, Sachs N, Janssen H, and Sonnenberg A: Integrin α3β1 inhibits directional migration and wound re-epithelialization in the skin. J Cell Sci 2009; 122:278. [DOI] [PubMed] [Google Scholar]
- 27.deHart GW, Healy KE, and Jones JCR: The role of α3β1 integrin in determining the supramolecular organization of laminin-5 in the extracellular matrix of keratinocytes. Exp Cell Res 2003; 283:67. [DOI] [PubMed] [Google Scholar]
- 28.Choma DP, Pumiglia K, and DiPersio CM: Integrin α3β1 directs the stabilization of a polarized lamellipodium in epithelial cells through activation of Rac1. J Cell Sci 2004; 117:3947. [DOI] [PubMed] [Google Scholar]
- 29.Frank DE. and Carter WG: Laminin 5 deposition regulates keratinocyte polarization and persistent migration. J Cell Sci 2004; 117:1351. [DOI] [PubMed] [Google Scholar]
- 30.Kligys KR, Wu Y, Hopkinson SB, Kaur S, Platanias LC, and Jones JCR: α6β4 integrin, a master regulator of expression of integrins in human keratinocytes. J Biol Chem 2012; 287:17975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DiPersio CM, Hodivala-Dilke KM, Jaenisch R, Kreidberg JA, and Hynes RO: α3β1 integrin is required for normal development of the epidermal basement membrane. J Cell Biol 1997; 137:729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hamill KJ, Hopkinson SB, Hoover P, Todorovic V, Green KJ, and Jones JC: Fibronectin expression determines skin cell motile behavior. J Invest Dermatol 2012; 132:448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitchell K, Szekeres C, Milano V, et al. : α3β1 integrin in epidermis promotes wound angiogenesis and keratinocyte-to-endothelial-cell crosstalk through the induction of MRP3. J Cell Sci 2009; 122:1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Juhasz I, Murphy GF, Yan HC, Herlyn M, and Albelda SM: Regulation of extracellular matrix proteins and integrin cell substratum adhesion receptors on epithelium during cutaneous human wound healing in vivo. Am J Path 1993; 143:1458. [PMC free article] [PubMed] [Google Scholar]
- 35.Kim JP, Zhang J, Chen JD, Wynn KC, Kramer RH, and Woodley DT: Mechanisms of human keratinocyte migration on fibronectin: unique roles of RGD site and integrins. J Cell Physiol 1992; 151:443. [DOI] [PubMed] [Google Scholar]
- 36.AlDahlawi S, Eslami A, Häkkinen L, and Larjava HS: The αvβ6 integrin plays a role in compromised epidermal wound healing. Wound Repair Regen 2006; 14:289. [DOI] [PubMed] [Google Scholar]
- 37.Huang X, Griffiths M, Wu J, Farese RV, and Sheppard D: Normal development, wound healing, and adenovirus susceptibility in β5-deficient mice. Mol Cell Biol 2000; 20:755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Munger JS, Huang XZ, Kawakatsu H, et al. : The integrin αvβ6 binds and activates latent TGF-β1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999; 96:319. [DOI] [PubMed] [Google Scholar]
- 39.Kim CW, Goldberger OA, Gallo RL, and Bernfield M: Members of the syndecan family of heparan sulfate proteoglycans are expressed in distinct cell-, tissue-, and development-specific patterns. Mol Biol Cell 1994; 5:797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan MR, Humphries MJ, and Bass MD: Synergistic control of cell adhesion by integrins and syndecans. Nat Rev Mol Cell Biol 2007; 8:957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zimmerman P. and David G: The syndecans, tuners of transmembrane signaling. FASEB J 1999; 13:91. [DOI] [PubMed] [Google Scholar]
- 42.Charnaux N, Brule S, Chaigneau T, et al. : RANTES (CCL5) induces a CCR5-dependent accelerated shedding of syndecan-1 (CD138) and syndecan-4 from HeLa cells and forms complexes with the shed ectodomains of these proteoglycans as well as with those of CD44. Glycobiology 2005; 15:119. [DOI] [PubMed] [Google Scholar]
- 43.Li Q, Park PW, Wilson CL, and Parks WC: Matrilysin shedding of syndecan-1 regulates chemokine mobilization and transepithelial efflux of neutrophils in acute lung injury. Cell 2002; 111:635. [DOI] [PubMed] [Google Scholar]
- 44.Stepp MA, Gibson HE, Gala PH, et al. : Defects in keratinocyte activation during wound healing in the syndecan-1-deficient mouse. J Cell Sci 2002; 115:4517. [DOI] [PubMed] [Google Scholar]
- 45.Nakayama Y, Kon S, Kurotaki D, Morimoto J, Matsui Y, and Uede T: Blockade of interaction of α9 integrin with its ligand hinders the formation of granulation in cutaneous wound healing. Lab Invest 2010; 90:881. [DOI] [PubMed] [Google Scholar]
- 46.Stepp MA, Liu Y, Pal-Ghosh S, et al. : Reduced migration, altered matrix and enhanced TGF1 signaling are signatures of mouse keratinocytes lacking Sdc1. J Cell Sci 2007; 120:2851. [DOI] [PubMed] [Google Scholar]
- 47.Araki E, Momota Y, Togo T, et al. : Clustering of syndecan-4 and integrin β1 by laminin α3 chain-derived peptide promotes keratinocyte migration. Mol Biol Cell 2009; 20:3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Echtermeyer F, Streit M, Wilcox-Adelman S, et al. : Delayed wound repair and impaired angiogenesis in mice lacking syndecan-4. J Clin Invest 2001; 107:R9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fitzgerald ML, Wang Z, Park PW, Murphy G, and Bernfield M: Shedding of syndecan-1 and -4 ectodomains is regulated by multiple signaling pathways and mediated by a Timp-3–sensitive metalloproteinase. J Cell Biol 2000; 148:811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kainulainen V, Wang H, Schick C, and Bernfield M: Syndecans, heparan sulfate proteoglycans maintain the proteolytic balance of acute wound fluids. J Biol Chem 1998; 273:11563. [DOI] [PubMed] [Google Scholar]
- 51.Subramanian SV, Fitzgerald ML, and Bernfield M: Regulated shedding of syndecan-1 and -4 ectodomains by rhrombin and growth factor receptor activation. J Biol Chem 1997; 272:14713. [DOI] [PubMed] [Google Scholar]
- 52.Meves A, Stremmel C, Gottschalk K, and Fässler R: The kindlin protein family: new members to the club of focal adhesion proteins. Trends Cell Biol 2009; 19:504. [DOI] [PubMed] [Google Scholar]
- 53.Jobard F, Bouadjar B, Caux F, et al. : Identification of mutations in a new gene encoding a FERM family protein with a pleckstrin homology domain in Kindler syndrome. Hum Mol Genet 2003; 12:925. [DOI] [PubMed] [Google Scholar]
- 54.Calderwood DA, Yan B, de Pereda JM, et al. : The phosphotyrosine binding-like domain of talin activates integrins. J Biol Chem 2002; 277:21749. [DOI] [PubMed] [Google Scholar]
- 55.Harburger DS, Bouaouina M, and Calderwood DA: Kindlin-1 and -2 directly bind the C-terminal region of β integrin cytoplasmic tails and exert integrin-specific activation effects. J Biol Chem 2009; 284:11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.He Y, Esser P, Heinemann A, Bruckner-Tuderman L, and Has C: Kindlin-1 and -2 have overlapping functions in epithelial cells: implications for phenotype modification. Am J Path 2011; 178:975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Qu H, Wen T, Pesch M, and Aumailley M: Partial loss of epithelial phenotype in kindlin-1–deficient keratinocytes. Am J Path 2012; 180:1581. [DOI] [PubMed] [Google Scholar]
- 58.Larjave H, Plow EF, and Wu C: Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. ENBP Rep 2008; 9:1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaiser HW, Ness W, Offers M, O'Keefe EJ, and Kreysel HW: Talin: adherens junction protein is localized at the epidermal-dermal interface in skin. J Invest Dermatol 1993; 101:789. [DOI] [PubMed] [Google Scholar]
- 60.Zhang X, Jiang G, Cai Y, Monkley SJ, Critchley DR, and Sheetz MP: Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol 2008; 10:1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franco SJ, Rodgers MA, Perrin BJ, et al. : Calpain-mediated proteolysis of talin regulates adhesion dynamics. Nat Cell Biol 2004; 6:977. [DOI] [PubMed] [Google Scholar]
- 62.Dourdin N, Bhatt AK, Dutt P, et al. : Reduced cell migration and disruption of the actin cytoskeleton in calpain-deficient embryonic fibroblasts. J Biol Chem 2001; 276:48382. [DOI] [PubMed] [Google Scholar]
- 63.Huang C, Rajfur Z, Yousefi N, Chen Z, Jacobson K, and Ginsberg MH: Talin phosphorylation by Cdk5 regulates Smurf1-mediated talin head ubiquitylation and cell migration. Nat Cell Biol 2009; 11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang S, Watanabe T, Matsuzawa K, et al. : Tiam1 interaction with the PAR complex promotes talin-mediated Rac1 activation during polarized cell migration. J Cell Biol 2012; 199:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamelers IHL, Olivo C, Mertens AEE, et al. : The Rac activator Tiam1 is required for α3β1-mediated laminin-5 deposition, cell spreading, and cell migration. J Cell Biol 2005; 171:871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brown MC. and Turner CE: Paxillin: adapting to change. Physiol Rev 2004; 84:1315. [DOI] [PubMed] [Google Scholar]
- 67.Laukaitis CM, Webb DJ, Donais K, and Horwitz AF: Differential dynamics of α5 integrin, paxillin, and α-actinin during formation and assembly of adhesions in migrating cells. J Cell Biol 2001; 153:1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaller MD. and Parsons JT: pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high-affinity binding site for Crk. Mol Cell Biol 1995; 15:2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Webb DJ, Schroeder MJ, Brame CJ, et al. : Paxillin phosphorylation sites mapped by mass spectrometry. J Cell Sci 2005; 118:4925. [DOI] [PubMed] [Google Scholar]
- 70.Sero JE, Thodeti CK, Mammoto A, Bakal C, Thomas S, and Ingber DE: Paxillin mediates sensing of physical cues and regulates directional cell motility by controlling lamellipodia positioning. PLoS One 2011; 6:e28303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang C, Rajfur Z, Borchers C, Schaller MD, and Jacobson K: JNK phosphorylates paxillin and regulates cell migration. Nature 2003; 424:219. [DOI] [PubMed] [Google Scholar]
- 72.Huang Z, Yan D-P, and Ge B-X: JNK regulates cell migration through promotion of tyrosine phosphorylation of paxillin. Cell Signal 2008; 20:2002. [DOI] [PubMed] [Google Scholar]
- 73.Parsons JT: Focal adhesion kinase: the first ten years. J Cell Sci 2003; 116:1409. [DOI] [PubMed] [Google Scholar]
- 74.Lietha D, Cai X, Ceccarelli DFJ, Li Y, Schaller MD, and Eck MJ: Structural basis for the autoinhibition of focal adhesion kinase. Cell 2007; 129:1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gates R, King L, Jr, Hanks S, and Nanney L: Potential role for focal adhesion kinase in migrating and proliferating keratinocytes near epidermal wounds and in culture. Cell Growth Differ 1994; 5:891. [PubMed] [Google Scholar]
- 76.Yurko MA, O'Toole EA, and Woodley DT: Phosphorylation of focal adhesion kinase (pp125FAK) is increased in human keratinocytes induced to migrate by extracellular matrices. J Cell Physiol 2001; 188:24. [DOI] [PubMed] [Google Scholar]
- 77.Schober M, Raghavan S, Nikolova M, et al. : Focal adhesion kinase modulates tension signaling to control actin and focal adhesion dynamics. J Cell Biol 2007; 176:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim LT, Wu J, and Turnage RH: FAK Induction in keratinocytes in an in vitro model of reepithelialization. J Surg Res 2001; 96:167. [DOI] [PubMed] [Google Scholar]
- 79.Brakebusch C. and Fassler R: The integrin-actin connection, an eternal love affair. EMBO J 2003; 22:2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hannigan G, Troussard AA, and Dedhar S: Integrin-linked kinase: a cancer therapeutic target unique among its ILK. Nat Rev Cancer 2005; 5:51. [DOI] [PubMed] [Google Scholar]
- 81.Xue G. and Hemmings BA: PKB/Akt–dependent regulation of cell motility. J Nat Cancer Inst 2013; 105:393. [DOI] [PubMed] [Google Scholar]
- 82.Lorenz K, Grashoff C, Torka R, et al. : Integrin-linked kinase is required for epidermal and hair follicle morphogenesis. J Cell Biol 2007; 177:501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Matsudaira P: Modular organization of actin crosslinnking proteins. Trends Biochem Sci 1991; 16:87. [DOI] [PubMed] [Google Scholar]
- 84.Beggs AH, Byers TJ, Knoll JH, Boyce FM, Bruns GA, and Kunkel LM: Cloning and characterization of two human skeletal muscle alpha-actinin genes located on chromosomes 1 and 11. J Biol Chem 1992; 267:9281. [PubMed] [Google Scholar]
- 85.Honda K, Yamada T, Endo R, et al. : Actinin-4, a novel actin-bundling protein associated with cell motility and cancer invasion. J Cell Biol 1998; 140:1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Millake DB, Blanchard AD, Paatel B, and Critchley DR: The cDNA sequence of a human placental alpha-actinin. Nucl Acids Res 1989; 17:6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Otey CA, Pavalko FM, and Burridge K: An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol 1990; 111:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vallenius T, Luukko K, and Makela TP: CLP-36 PDZ-LIM protein associates with nonmuscle alpha-actinin-1 and alpha-actinin-4. J Biol Chem 2000; 275:11100. [DOI] [PubMed] [Google Scholar]
- 89.Araki N, Hatae T, Yamada T, and Hirohashi S: Actinin-4 is preferentially involved in circular ruffling and macropinocytosis in mouse macrophages: analysis by fluorescence ratio imaging. J Cell Sci 2000; 130:67. [DOI] [PubMed] [Google Scholar]
- 90.Honda K, Yamada T, Hayashida Y, et al. : Actinin-4 increases cell motility and promotes lymph node metastasis of colorectal cancer. Gastroenterology 2005; 128:51. [DOI] [PubMed] [Google Scholar]
- 91.Hamill KJ, Hopkinson SB, Skalli O, and Jones JCR: Actinin-4 in keratinocytes regulates motility via an effect on lamellipodia stability and matrix adhesions. FASEB J 2013; 27:546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cowin AJ, Adams DH, Strudwick XL, et al. : Flightless I deficiency enhances wound repair by increasing cell migration and proliferation. J Path 2007; 211:572. [DOI] [PubMed] [Google Scholar]
- 93.Kopecki Z, Arkell R, Powell BC, and Cowin AJ: Flightless I regulates hemidesmosome formation and integrin-mediated cellular adhesion and migration during wound repair. J Invest Dermatol 2009; 129:2031. [DOI] [PubMed] [Google Scholar]
- 94.Kligys K. and Jones JCR: Flii control: balancing migration and adhesion. J Invest Dermatol 2009; 129:1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Borradori L. and Sonnenberg A: Structure and function of hemidesmosomes: more than simple adhesion complexes. J Invest Dermatol 1999; 112:411. [DOI] [PubMed] [Google Scholar]
- 96.Hopkinson SB, Findlay K, and Jones JCR: Interaction of BP180 (type XVII collagen) and α6 integrin subunit is necessary for stabilization of hemidesmosome structure. J Invest Dermatol 1998; 111:1015. [DOI] [PubMed] [Google Scholar]
- 97.Sterk LMT, Geuijen CAW, Oomen LCJM, Calafat J, Janssen H, and Sonnenberg A: The tetraspan molecule CD151, a novel constituent of hemidesmosomes, associates with the integrin α6β4 and may regulate the spatial organization of hemidesmosomes. J Cell Biol 2000; 149:969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rousselle P. and Beck K: Laminin 332 processing impacts cellular behavior. Cell Adh Migr 2013; 7:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dowling J, Yu Q-C, and Fuchs E: β4 integrin is required for hemidesmosome formation, cell adhesion and cell survival. J Cell Biol 1996; 134:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Jones JCR, Kurpakus MA, Cooper HM, and Quaranta V: A function for the integrin α6β4 in the hemidesmosome. Cell Regul 1991; 2:427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Mainiero F, Pepe A, Wary KK, et al. : Signal transduction by the α6β4 integrin: distinct β4 subunit sites mediate recruitment of Shc/Grb2 and association with the cytoskeleton of hemidesmosomes. EMBO J 1995; 14:4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mariotti A, Kedeshian PA, Dans M, Curatola AM, Gagnoux-Palacios L, and Giancotti FG: EGF-R signaling through Fyn kinase disrupts the function of integrin α6β4 at hemidesmosomes: role in epithelial cell migration and carcinoma invasion. J Cell Biol 2001; 155:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang H, Leavitt L, Ramaswamy R, and Rapraeger AC: Interaction of syndecan and α6β4 integrin cytoplasmic domains: regulation of ErbB2-mediated integrin activation. J Biol Chem 2010; 285:13569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Underwood RA, Carter WG, Usui ML, and Olerud JE: Ultrastructural localization of integrin subunits β4 and α3 within the migrating epithelial tongue of in vivo human wounds. J Histochem Cytochem 2009; 57:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Russell AJ, Fincher EF, Millman L, et al. : α6β4 integrin regulates keratinocyte chemotaxis through differential GTPase activation and antagonism of α3β1 integrin. J Cell Sci 2003; 116:3543. [DOI] [PubMed] [Google Scholar]
- 106.Sehgal BU, Debiase P, Matzno S, et al. : Integrin β4 regulates migratory behavior of keratinocytes by determining laminin-332 (laminin-5) matrix organization. J Biol Chem 2006; 281:35487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kligys K, Claiborne JN, DeBiase P, Hopkinson SB, Mizuno K, and Jones JCR: The Slingshot family of phosphatases mediates Rac1 regulation of cofilin phosphorylation, laminin-332 organization and motility behavior of keratinocytes. J Biol Chem 2007; 282:32520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nagata-Ohashi K, Ohta Y, Goto K, et al. : A pathway of neuregulin-induced activation of cofilin-phosphatase slingshot and cofilin in lamellipodia. J Cell Biol 2004; 165:465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Soosairajah J, Maiti S, Wiggan ON, et al. : Interplay between components of a novel LIM kinase-slingshot phosphatase complex regulates cofilin. EMBO J 2005; 24:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Santoro MM, Gaudino G, and Marchisio PC: The MSP receptor regulates α6β4 and α3β1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell 2003; 5:257. [DOI] [PubMed] [Google Scholar]
- 111.Kligys K, Yun J, Yu D, and Jones JCR: 14-3-3ζ/τ heterodimers regulate slingshot activity in migrating keratinocytes. Biochem Biophys Res Comm 2009; 383:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Giudice GJ, Emery DJ, and Diaz LA: Cloning and primary structural analysis of the bullous pemphigoid autoantigen, BP-180. J Invest Dermatol 1992; 99:243. [DOI] [PubMed] [Google Scholar]
- 113.Hopkinson SB, Riddelle KS, and Jones JCR: The cytoplasmic domain of the 180kD bullous pemphigoid antigen, a hemidesmosomal component: molecular and cell biologic characterization. J Invest Dermatol 1992; 99:264. [DOI] [PubMed] [Google Scholar]
- 114.Klatte DH, Kurpakus MA, Grelling KA, and Jones JCR: Immunochemical characterization of three components of the hemidesmosome and their expression in cultured epithelial cells. J Cell Biol 1989; 109:3377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hamill KJ, Hopkinson SB, Jonkman MF, and Jones JC: Type XVII collagen regulates lamellipod stability, cell motility, and signaling to Rac1 by targeting bullous pemphigoid antigen 1e to α6β4 integrin. J Biol Chem 2011; 286:26768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tasanen K, Tunggal L, Chometon G, Bruckner-Tuderman L, and Aumailley M: Keratinocytes from patients lacking collagen XVII display a migratory phenotype. Am J Pathol 2004; 164:2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Qiao H, Shibaki A, Long HA, et al. : Collagen XVII participates in keratinocyte adhesion to collagen IV, and in p38MAPK-dependent migration and cell signaling. J Invest Dermatol 2009; 129:2288. [DOI] [PubMed] [Google Scholar]
- 118.Franzke C-W, Tasanen K, Borradori L, Huotari V, and Bruckner-Tuderman L: Shedding of collagen XVII/BP180: Structural motifs influence cleavage from cell surface. J Biol Chem 2004; 279:24521. [DOI] [PubMed] [Google Scholar]
- 119.Huilaja L, Hurskainen T, Autio-Harmainen H, et al. : Pemphigoid gestationis autoantigen, transmembrane collagen XVII, promotes the migration of cytotrophoblastic cells of placenta and is a structural component of fetal membranes. Matrix Biol 2008; 27:190. [DOI] [PubMed] [Google Scholar]
- 120.Parikka M, Nissinen I T K, et al. : Collagen XVII promotes integrin-mediated squamous cell carcinoma transmigration-A novel role for alpha II(b) integrin and tirofiban. Exp Cell Res 2006; 312:1431. [DOI] [PubMed] [Google Scholar]
- 121.Yanez-Mo M, Mittelbrunn M, and Sanchez-Madrid F: Tetraspanins and intercellular interactions. Microcirculation 2001; 8:153. [DOI] [PubMed] [Google Scholar]
- 122.Berditchevski F: Complexes of tetraspanins with integrins: more than meets the eye. J Cell Sci 2001; 114:4143. [DOI] [PubMed] [Google Scholar]
- 123.Boucheix C. and Rubenstein E: Tetraspanins. Cell Mol Life Sci 2001; 58:1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hemler ME: Tetraspanin functions and associated microdomains. Nat Rev Mol Cell Biol 2005; 6:801. [DOI] [PubMed] [Google Scholar]
- 125.Cowin AJ, Adams D, Geary SM, Wright MD, Jones JCR, and Ashman LK: Wound healing is defective in mice lacking tetraspanin CD151. J Invest Dermatol 2006; 126:680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Penas P, Garci-Diez A, Sanchez-Madrid F, and Yaniez-Mo M: Tetraspanins are localized at motility-related structures and involved in normal human keratinocyte wound healing migration. J Invest Dermatol 2000; 114:1126. [DOI] [PubMed] [Google Scholar]
- 127.Wright MD, Geary SM, Fitter S, et al. : Characterization of mice lacking the tetraspanin superfamily member CD151. Mol Cell Biol 2004; 24:5978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu L, He B, Liu W, Zhou D, Cox JV, and Zhang XA: Tetraspanin CD151 promotes cell migration by regulating integrin trafficking. J Biol Chem 2007; 282:31631. [DOI] [PubMed] [Google Scholar]
- 129.Regen CM. and Horwitz AF: Dynamics of beta 1 integrin-mediated adhesive contacts in motile fibroblasts. J Cell Biol 1992; 119:1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Winterwood NE, Varzavand A, Meland MN, Ashman LK, and Stipp CS: A critical role for tetraspanin CD151 in α3β1 and α6β4 integrin–dependent tumor cell functions on laminin-5. Mol Biol Cell 2006; 17:2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rurhberg C. and Watt FM: The plakin family: versatile organizers of cytoskeletal architecture. Curr Opin Genet Dev 1997; 7:392. [DOI] [PubMed] [Google Scholar]
- 132.Leung CL, Zheng M, Prater SM, and Liem RKH: The BPAG1 locus: alternative splicing produces multiple isoforms with distinct cytoskeletal linker domains, including predominant isoforms in neurons and muscles. J Cell Biol 2001; 154:691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Guo L, Degenstein L, Dowling J, et al. : Gene targeting of BPAG1: abnormalities in mechanical strength and cell migration in stratified epithelia and neurologic degeneration. Cell 1995; 81:233. [DOI] [PubMed] [Google Scholar]
- 134.Herold-Mende C, Kartenbeck J, Tomakidi P, and Bosch F: Metastatic growth of squamous cell carcinomas is correlated with upregulation and redistribution of hemidesmosomal components. Cell Tissue Res 2001; 306:399. [DOI] [PubMed] [Google Scholar]
- 135.Hamill KJ, Hopkinson SB, DeBiase P, and Jones JCR: BPAG1e maintains keratinocyte polarity through β4 integrin-mediated modulation of Rac 1 and cofilin activities. Mol Biol Cell 2009; 202954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Koster S, van Wilpe I, Kuikman , Litjens SHM, and Sonnenberg A: Role of binding of plectin to the integrin beta4 subunit in the assembly of hemidesmosomes. Mol Biol Cell 2004; 15:1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wiche G: Role of plectin in cytoskeleton organization and dynamics. J Cell Sci 1998; 111:2477. [DOI] [PubMed] [Google Scholar]
- 138.McLean WHI, Pulkkinen L, Smith FJD, et al. : Loss of plectin causes epidermolysis bullosa with muscular dystrophy: cDNA cloning and genomic organization. Genes Dev 1996; 10:1724. [DOI] [PubMed] [Google Scholar]
- 139.Osmanagic-Myers S, Gregor M, Walko G, Burgstaller G, Reipert S, and Wiche G: Plectin-controlled keratin cytoarchitecture affects MAP kinases involved in cellular stress response and migration. J Cell Biol 2006; 174:557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kligys K, Hamill KJ, and Jones JCR: Hemidesmosomes and their components: adhesion versus signaling. In Health and Disease, edited by LaFlamme SE. and Kowalczyk AP. Weinheim, Germany: Wiley-VCH Verlag GmbH, 2008, pp. 109–133 [Google Scholar]
- 141.Singh P, Carraher C, and Schwarzbauer JE: Assembly of fibronectin extracellular matrix. Ann Rev Cell Dev Biol 2010; 26:397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goldfinger LE, Stack MS, and Jones JCR: Processing of laminin-5 and its functional consequences: role of plasmin and tissue-type plasminogen activator. J Cell Biol 1998; 141:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marinkovich MP, Lunstrum GP, and Burgeson RE: The anchoring filament protein kalinin is synthesized and secreted as a high molecular weight precursor. J Biol Chem 1992; 267:17900. [PubMed] [Google Scholar]
- 144.Ogawa T, Tsubota Y, Hashimoto J, Kariya Y, and Miyazaki K: The short arm of laminin γ2 chain of laminin-5 (laminin-332) binds syndecan-1 and regulates cellular adhesion and migration by suppressing phosphorylation of integrin β4 chain. Mol Biol Cell 2007; 18:1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hopkinson SB, DeBiase PJ, Kligys K, Hamill K, and Jones JCR: Fluorescently tagged laminin subunits facilitate analyses of the properties, assembly and processing of laminins in live and fixed lung epithelial cells and keratinocytes. Matrix Biol 2008; 27:640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Natarajan E, Omobono JD, II, Guo Z, et al. : A keratinocyte hypermotility/growth arrest response involving laminin 5 and p16INK4A activated in wound healing and senescence. Am J Pathol 2006; 168:1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carulli S, Beck K, Dayan G, Boulesteix S, Lortat-Jacob H, and Rousselle P: Cell surface proteoglycans syndecan-1 and -4 bind overlapping but distinct sites in laminin α3 LG45 protein domain. J Biol Chem 2012; 287:12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Utani A, Nomizu M, Matsuura H, et al. : A unique sequence of the laminin α3 G domain binds to heparin and promotes cell adhesion through syndecan-2 and -4. J Biol Chem 2001; 276:28779. [DOI] [PubMed] [Google Scholar]
- 149.Galiano RD, Michaels V J, Dobryansky VM, Levine JP, and Gurtner GC: Quantitative and reproducible murine model of excisional wound healing. Wound Repair Regen 2004; 12:485. [DOI] [PubMed] [Google Scholar]
- 150.Rotty JD. and Coulombe PA: A wound-induced keratin inhibits Src activity during keratinocyte migration and tissue repair. J Cell Biol 2012; 197:381. [DOI] [PMC free article] [PubMed] [Google Scholar]