

Abstract
Recent advances in genome sequencing technologies provide unprecedented opportunities to characterize individual genomic landscapes and identify mutations relevant for diagnosis and therapy. Specifically, whole-exome sequencing using next-generation sequencing (NGS) technologies is gaining popularity in the human genetics community due to the moderate costs, manageable data amounts and straightforward interpretation of analysis results. While whole-exome and, in the near future, whole-genome sequencing are becoming commodities, data analysis still poses significant challenges and led to the development of a plethora of tools supporting specific parts of the analysis workflow or providing a complete solution. Here, we surveyed 205 tools for whole-genome/whole-exome sequencing data analysis supporting five distinct analytical steps: quality assessment, alignment, variant identification, variant annotation and visualization. We report an overview of the functionality, features and specific requirements of the individual tools. We then selected 32 programs for variant identification, variant annotation and visualization, which were subjected to hands-on evaluation using four data sets: one set of exome data from two patients with a rare disease for testing identification of germline mutations, two cancer data sets for testing variant callers for somatic mutations, copy number variations and structural variations, and one semi-synthetic data set for testing identification of copy number variations. Our comprehensive survey and evaluation of NGS tools provides a valuable guideline for human geneticists working on Mendelian disorders, complex diseases and cancers.
Keywords: Mendelian disorders, cancer, variants, bioinformatics tools, next-generation sequencing
INTRODUCTION
Recent advances in genome sequencing technologies are rapidly changing the research and routine work of human geneticists. Due to the brisk decline of costs per base pair, next-generation sequencing (NGS) is now affordable even for small- to mid-sized laboratories. Whole-genome sequencing and whole-exome sequencing have proven to be valuable methods for the discovery of the genetic causes of rare and complex diseases [1]. Although cheaper than Sanger sequencing, whole-genome sequencing remains expensive on a grand scale. Over and above, one sequencing run provides enormous amounts of data and poses considerable challenges for the analysis and interpretation. In contrast, whole-exome sequencing is becoming a popular approach to bridge the gap between genome-wide comprehensiveness and cost-control, by capturing and sequencing the ∼1% of the human genome that codes for protein sequences [2, 3]. Furthermore, novel, non-optical technologies [4] are developing fast, and soon devices will be available that can sequence up to 10 Gbases at a fraction of the current costs. Thus, it is expected that by the end of the year 2012 whole-exome sequencing can be performed at a reagent cost per sample in the range of 400–500€ [5].
Whole-exome sequencing has already been used for identifying the molecular defects of single gene disorders [6, 7], for elucidating some genetically heterogeneous disorders [8, 9] and for improving the accuracy of diagnosis of patients [10, 11]. The amount of both raw and processed data for whole-exome sequencing is orders of magnitude smaller than for whole-genome sequencing. Nevertheless, each sequencing run identifies a wealth of simple nucleotide variations (SNVs), including single-nucleotide polymorphisms (SNPs) and small insertions and deletions (INDELs). On average, whole-exome sequencing identifies 12 000 variants in coding regions [12], of which ∼90% are found in publicly available databases [13]. In comparison, ∼5 million variants, including 144 000 new variants, are reported on average by whole-genome sequencing [14].
Not surprisingly, initiatives have been started using whole-exome sequencing to explore all Mendelian disorders, which will improve the functional annotation of the human genome and will provide new insights into mechanisms of disease development [15]. As of to date, OMIM [16], a catalog of Mendelian disorders, lists >3000 disorders where the molecular basis has been reported. Still, >3500 disorders are listed where the genetic cause remains unknown and has yet to be identified [17].
Other main fields of interest for human geneticists are complex diseases and cancer. For example, the usage of NGS has led to the identification of driver mutations for specific types of cancer [18, 19], which are often reported to be structural or non-coding [20]. NGS paved the way for the fundamental understanding of mutated genes in cancer cells, affected pathways, and how these data inform our models and knowledge of cancer biology [21]. A recent study with regard to tumor vaccination showed a proof-of-concept in which somatic mutations are first detected using NGS. Subsequently the immunogenicity of these mutations is defined, and finally, mutations are tested for their capability to elicit T-cell immunogenicity [22]. Thus, tailored vaccine concepts based on the genome-wide discovery of cancer-specific mutations and individualized therapy seem technically feasible.
The current bottleneck of whole-genome and whole-exome sequencing projects is not the sequencing of the DNA itself but lies in the structured way of data management and the sophisticated computational analysis of the experimental data [23]. In order to get meaningful biological results, each step of the analysis workflow needs to be carefully considered, and specific tools need to be used for certain experimental setups.
The complete NGS data analysis process is complex, includes multiple analysis steps, is dependent on a multitude of programs and databases and involves handling large amounts of heterogeneous data. It is not surprising that due to the enormous success of NGS projects, a flood of tools has been created to support specific parts of the analysis workflow. It is apparent that the appropriate choice of tools is a non-trivial task, especially for inexperienced users. Therefore, a number of review articles were recently published (e.g. [24–28]) to facilitate the choice of the most suitable tool for a particular application. However, these articles review only selected components of the NGS data analysis pipeline such as mapping and assembly [24], sequence alignments [26], algorithms for SNP and genotype calling [25] or detection of structural variations (SVs) and copy number variations (CNVs) [27]. To the best of our knowledge, a comprehensive review covering all individual analysis steps has not been reported yet. Such a review would be tremendously helpful for researchers planning a NGS project since it would provide a rich resource to guide the assembly of an analytical pipeline for a particular application or alternatively select a fully integrated pipeline. In addition, by covering multiple steps of the workflow it addresses issues such as data handling and tool compatibility, which are neglected when only individual components are reviewed. We therefore initiated this study to survey existing tools spanning the complete analysis workflow and compile a comprehensive list of programs for variant analysis of NGS data. Additionally, we wanted to test a particular scenario typical for a human geneticist, i.e. applying tools in the context of mutation discovery on a Mendelian disorder or on a cancer data set. This type of hands-on evaluation of NGS software using real data sets rather than benchmarking the performance of a bioinformatics pipeline provides additional criteria for making a decision to select the appropriate tools.
We surveyed 205 tools for whole-genome/whole-exome sequencing data analysis supporting five distinct analytical steps (Figure 1): quality assessment, alignment, variant identification, variant annotation and visualization. We report an overview of the functionality, features and specific requirements of the individual tools. We then selected 32 programs for variant identification, variant annotation and visualization, which were subjected to hands-on evaluation using four data sets: one set of whole-exome data from two patients with a rare disease for testing identification of germline mutations, two cancer data sets for testing variant callers for somatic mutations, CNVs and SVs, and one semi-synthetic data set for testing identification of CNVs. Commercial tools such as Avadis NGS (www.avadis-ngs.com), CLC Genomics Workbench (www.clcbio.com), DNAnexus (dnanexus.com), Ingenuity Pathways Analysis (www.ingenuity.com), NextGENe (www.softgenetics.com/NextGENe.html), Partek Genomics Suite (www.partek.com) or SNP and Variation Suite (www.goldenhelix.com) were not part of this survey.
Figure 1:
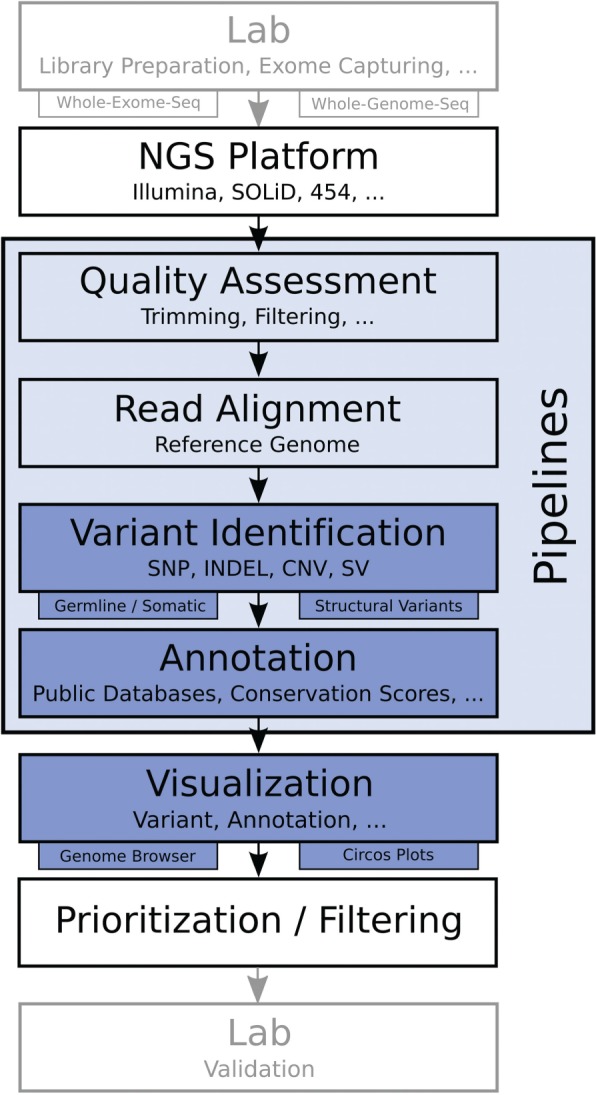
Basic workflow for whole-exome and whole-genome sequencing projects. After library preparation, samples are sequenced on a certain platform. The next steps are quality assessment and read alignment against a reference genome, followed by variant identification. Detected mutations are then annotated to infer the biological relevance and results can be displayed using dedicated tools. The found mutations can further be prioritized and filtered, followed by validation of the generated results in the lab.
The article is structured as follows. We first introduce the main application fields for NGS in human genetics, namely, Mendelian diseases, i.e. single gene disorders as a result of a single mutated gene like cystic fibrosis or sickle cell anemia, complex diseases, i.e. polygenic diseases associated with the effects of multiple genes in combination with other factors like diabetes or hypertension and cancer. Next, we describe NGS platforms and the NGS data analysis workflow and review available tools for the distinct analysis steps. Then we explain the scope and the method used to evaluate the tools and report hands-on experience of the selected programs. Finally, we make general recommendations for tool selection and prioritization of candidate genes.
APPLICATION OF NEXT-GENERATION GENOME SEQUENCING IN HUMAN GENETICS
We considered three common scenarios for human geneticists using NGS data: (i) identification of causative genes in Mendelian disorders (germline mutations), (ii) identification of candidate genes in complex diseases for further functional studies and (iii) identification of constitutional mutations as well as driver and passenger genes in cancer (somatic mutations).
Mendelian disorders
Traditionally, the main approach to elucidate causes of Mendelian disorders has been positional cloning based on linkage analysis [29]. The resulting genomic interval should normally contain <300 candidate genes which are further investigated by Sanger sequencing [13]. This study design is only applicable for familial diseases where an appropriate sample size is available [30] and is not suitable for the identification of de novo dominant mutations.
Currently, OMIM lists ∼3500 Mendelian disorders with unknown genetic causes [17]. Whole-exome sequencing is a powerful tool that has not only revolutionized comprehensive candidate gene sequencing in traditional positional cloning studies but also allowed identification of autosomal recessive disease genes in single patients from non-consanguineous families (e.g. [6, 7, 31]) as well as de novo dominant mutations. As whole-exome sequencing identifies a vast amount of variants, sophisticated filtering approaches are needed to reduce the number of genes for further investigation. Furthermore, as current capturing methods cannot evenly capture exonic regions [32], potentially interesting mutations in these regions are possibly neglected.
Whole-genome sequencing provides a complete view of the human genome, including point mutations in distant enhancers and other regulatory elements which have been previously associated with hereditary diseases [33]. As the cost per sequenced base will likely drop in the future, whole-genome sequencing will presumably replace whole-exome sequencing.
Complex diseases
The genetics of complex phenotypes have been investigated for decades through association studies with candidate genes that, based on pathophysiological considerations, were suspected to be involved in the development of the phenotype [34]. This approach was severely hampered by relying on sometimes unfounded functional hypotheses as well as by applying wrong statistical assumptions. An alternative to this candidate gene approach are genome-wide association studies (GWASs), which have become more feasible through the advancement of high-throughput genotyping technologies. GWASs are based on the principle of linkage disequilibrium—the non-random association between alleles at different loci—at the population level [35]. The development of SNP arrays, which can genotype many markers in a single assay in conjunction with biobanks of either population cohorts or case–control samples, facilitated the ability to conduct GWAS. This unbiased survey of many genes and variants robustly identified associations between 1300 loci and 200 diseases or traits [36].
Genetic studies of complex phenotypes are based on either ‘common disease–common variant’ or ‘common disease–rare variant’ hypotheses. GWAS primarily test the ‘common disease–common variant’ hypothesis, where complex phenotypes are the result of cumulative effects of a large number of common variants. In contrast, the ‘common disease–rare variant’ hypothesis posits that multiple rare variants with large effect sizes are the main determinants of heritability of the disease [34]. The field is now shifting toward the study of lower frequencies of rare variants [37], which can only be empowered by NGS and sophisticated bioinformatics approaches [38].
Defining the genetic basis of complex diseases using NGS can be performed by the following: (i) whole-genome, (ii) whole-exome and (iii) targeted subgenomic sequencing. Whole-genome and whole-exome sequencing have been successfully utilized to identify the genes responsible for complex hereditary diseases [39, 40], where whole-genome sequencing enables testing of both mentioned hypotheses.
Finally, NGS can also be used to identify trait loci by re-sequencing candidate genes in a large number of patients and controls as demonstrated for Type 1 diabetes [41]. This targeted subgenomic sequencing is likely to be supplanted by whole-exome (or whole-genome) sequencing in the near future. The challenge is now to utilize sequencing to enable the discovery of novel genes that contribute to the studied diseases. Given the vast number of genetic and non-genetic etiological factors of complex diseases, the ultimate approach will require exploiting biological and clinical data, and integration of additional data sets including RNA sequencing data, proteomics data and metabolomics data.
Somatic mutations
For human geneticists, there is an important distinction between constitutional and somatic mutations. Constitutional mutations, which have been inherited from the parents, are present in all cells of the body and may increase the susceptibility of an individual to be diagnosed with cancer [42]. New methods have led to the identification of novel genetic components which may improve assessment of cancer predisposition [43]. Indeed multiple cancer susceptibility loci have been identified, which indicate that there may be a significant number of common alleles that contribute to the heritability of a specific cancer. However, each of these loci confers only a small contribution to the risk for cancer [44]. For example, 22 common breast cancer susceptibility loci have been reported which explain only ∼8% of the disease’s heritability [45]. For this reason, it is daunting to transfer these common alleles, which on their own have only a minor impact on disease risk, into a clinical test [43]. In contrast, according to the ‘common disease–rare variant’ hypothesis [46], constitutional high-penetrance mutations in specific genes may cause a high risk of developing particular types of cancer. Whole-exome sequencing has led to the identification of high-penetrance mutations in other genes that are only relevant for a small proportion of families [47].
There is considerable interest in the exploitation of somatic mutations as a tool used to improve the detection of disease and, ultimately, to allow individualized treatment leading to better outcomes. The aim is to give patients a drug tailored to the genetic makeup of their tumor. The most famous example is the Bcr-Abl tyrosine kinase inhibitor imatinib, which represents a major therapeutic advance over conventional therapy in patients with Philadelphia chromosome positive chronic myelogenous leukemia. Here, >90% of patients obtained complete hematologic response and 70–80% of patients achieved a complete cytogenetic response [48]. In colorectal cancer, the paradigms are ‘KRAS’ mutations in exon 2 (codons 12 and 13), which have been established as a predictive marker for treatment with epidermal growth factor receptor inhibitors [49]. Therefore, it has become increasingly urgent for the clinical oncologist to have access to accurate and sensitive methods for the detection of such predictive biomarkers.
NGS VARIANT ANALYSIS WORKFLOW
NGS platforms
NGS instruments provide higher throughput at an unprecedented speed by sequencing millions of short DNA fragments in parallel [50, 51]. Currently, the three most commonly used platforms are Roche 454 (introduced in 2005), Illumina (launched in 2006) and ABI SOLiD (followed in 2008). All three platforms sequence DNA by measuring and analyzing signals, which are emitted during the creation of the second DNA strand, but differ in how the second strand is generated.
In order to produce detectable signals, template DNA is fragmented into small pieces, amplified and immobilized on a glass slide before sequencing. Roche 454 implements pyrosequencing, which measures released pyrophosphates allowing the analysis of read fragments up to a few hundred base pairs. Since this technique infers the number of incorporated nucleotides from the signal’s intensity, the system experiences problems when homopolymer stretches longer than 8 bp are sequenced [52]. This complicates identification of small insertions and deletions. Illumina applies a sequencing-by-synthesis approach where only 1 nt per sequencing cycle is incorporated using reversible dye terminators. Thereby, it avoids homopolymer calling problems at the cost of being capable of sequencing only shorter fragments. ABI SOLiD analyzes DNA by ligating fluorescently labeled di-base probes to the first strand, requiring reading each base twice. Due to the nature of this approach, identified calls are not stored in nucleotide but in color space—a property that needs to be considered in downstream analyses.
Depending on library preparation and sequencing technology, it is possible to sequence reads that are of a known chromosomal distance [26]. These so-called paired-end or mate-pair reads provide additional information which can be used for enhancing mapping accuracy and identifying structural rearrangements [53].
After completing laboratory work and the actual sequencing, the researcher is confronted with a huge amount of raw data. The analysis of the data can be decomposed into five distinct steps (Figure 1): (i) quality assessment of the raw data, (ii) read alignment to a reference genome, (iii) variant identification, (iv) annotation of the variants and (v) data visualization. In the following paragraphs, we briefly explain each of the steps and review available software tools. The initial list of analysis tools was acquired by performing multiple PubMed searches. Furthermore, we conducted additional Internet searches to identify tools not indexed by PubMed. An overview of the surveyed tools is given in Supplementary Tables (see also http://icbi.at/ngs_survey).
Quality assessment
The first analysis step after completing the sequencing run is to evaluate the quality of raw reads and to remove, trim or correct reads that do not meet the defined standards. Raw data generated by sequencing platforms are compromised by sequence artifacts such as base calling errors, INDELs, poor quality reads and adaptor contamination [54]. These errors are quite common in sequencing data, and platforms are susceptible to a wide range of chemistry and instrument failures [55, 56]. As many downstream analysis tools are not capable of checking for low-quality reads, it is necessary to perform filtering and trimming tasks in advance to avoid drawing of wrong biological conclusions. Generally, these steps include visualization of base quality scores and nucleotide distributions, trimming of reads and read filtering based on base quality score and sequence properties such as primer contaminations, N content and GC bias.
Several tools have been developed to perform the various stages of quality assessment (Supplementary Table S1 shows 11 selected tools). The stand-alone tools NGSQC Toolkit [54] and PRINSEQ [57] are able to handle FASTQ and 454 (SFF) files, produce summary reports and are capable of filtering and trimming reads. FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc) is compatible with all main sequencing platforms and outputs summary graphs and tables to quickly assess the data quality. The tool ContEst [58] can be used to estimate the amount of cross-sample contamination in NGS data. Galaxy offers an integrated tool [59] that creates FASTQ summary statistics and performs flexible trimming and filtering tasks. htSeqTools [60] and SolexaQA [55] include quality assessment, processing and visualization functionality. In addition, software tools have been published that support only the Illumina platform (i.e. FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit), PIQA [61] and TileQC [62]) or provide certain specialized functionality (i.e. TagCleaner [63]).
Alignment
After reads have been processed to meet a certain quality standard, they are usually aligned to an existing reference genome [25]. Currently, there are two main sources for the human reference genome assembly: the University of Santa Cruz (UCSC), which is also hosting the central repository for ENCODE data [64] and the Genome Reference Consortium (GRC), which focuses on creating reference assemblies [65]. Both resources provide several versions of the human genome. UCSC offers versions hg18 and hg19 while GRC provides GRCh36 and GRCh37. Together these are the most widely used reference genomes. UCSC (hg) and GRC (GRCh) human assemblies are identical [66] but differ with regards to their nomenclature (e.g. UCSC uses a ‘chr’ prefix).
Over the last years, many alignment programs have been developed [67] to efficiently process millions of short reads and include, among others, Bowtie/Bowtie2 [68, 69], BWA [70, 71], MAQ [72], mrFAST [73], Novoalign (http://novocraft.com), SOAP [74], SSAHA2 [75], Stampy [76] and YOABS [77] (Supplementary Table S2). Since reviews about characteristics and properties of alignment methods have been published elsewhere [26, 67, 78], we do not report a comprehensive list but rather a shorter list of 17 commonly used tools and refer to the literature for an in-depth review of the methods and the corresponding tools.
The sequencing technologies are constantly pushing the lengths of generated reads—requiring new and improved algorithms. First-generation short-read aligners were often optimized for ungapped alignment whereas today’s programs can deal with longer read lengths and gaps. The majority of currently available long-read alignment algorithms may be classified as either using hash table indexing, like in BLAT [79] or in SSAHA2 or using some sort of compressed tree indexing based on the Burrows–Wheeler transform [80]. Most alignment algorithms follow the seed-and-extend paradigm, where one or more of so-called seeds are searched followed by an extension to cover the whole query sequence [26].
Additionally to the selection of the alignment program, three issues are noteworthy. First, to overcome the problem of ambiguity when mapping short reads to a reference genome, paired-end reads have proven to be a valuable solution and are highly recommended, if not even a requirement for whole-exome sequencing and whole-genome sequencing [81]. Second, reads that can only be mapped with many mismatches should not be considered and as a consequence, mutations that are only backed by such reads should be discarded from further analysis. And third, as current NGS technologies incorporate PCR steps in their library preparations, multiple reads originating from only one template might be sequenced, thereby interfering with variant calling statistics. For that reason, it is common practice to remove PCR duplicates after alignment in whole-genome and whole-exome sequencing studies.
Variant identification
A crucial part of next-generation genome sequencing data analysis is the identification of variants. The choice of applied strategies for genotype-calling, somatic mutation identification and SV exploration is ultimately related to the data usage. It is of utmost importance to first carefully design the study as it ultimately affects analysis and testing strategies [82]. In addition, sequence coverage is an important factor in variant identification as called mutations should be supported by several reads [83]. Finally, appropriate tools for analyzing NGS data [25] have to be thoroughly evaluated.
Tools for genome-wide variant identification can be grouped into four categories: (i) germline callers, (ii) somatic callers, (iii) CNV identification and (iv) SV identification. The detection of germline mutations is a central part for finding causes of rare diseases. Cancer studies focus on the identification of somatic mutations by comparing sequencing results of tumor/normal pairs from one subject. The tools for the identification of large structural modifications can be divided into those which find CNVs and those which find other SVs such as inversions, translocations or large INDELs. CNVs are currently the only SVs that can be detected in both whole-genome and whole-exome sequencing studies. Therefore, dedicated tools have been designed that consider the properties of exome capturing [84].
We surveyed 63 tools for variant identification and provide a list for each of the four categories including input/output formats, supported platform, types of detectable variants and usage notes (Supplementary Tables S3–S6).
Variant annotation
With the large amount of data produced by NGS experiments, the possibility of predicting the functional impact of variants in an automated fashion is becoming increasingly important. Computer-aided annotation enables research groups to filter and prioritize potential disease-causing mutations for further analysis. The available tools implement different methods for variant annotation. Most of them focus on the annotation of SNPs, since they can be easily identified and analyzed. INDELs are also covered by some tools, whereas annotation of structural variants is limited to CNVs and only performed by recently developed applications.
The most common form of annotation is to provide database links to various public variant databases such as dbSNP. In terms of functional prediction of the variants, the tools employ different approaches, ranging from simple sequence-based analysis over region-based analysis to the evaluation of the structural impact on proteins. The result of the functional analysis is a classification into accepted and deleterious mutations. The programs often have more fine-grained risk classes or scores reflecting the likelihood of a deleterious effect.
Many annotation tools are provided as web applications, which have the advantage that there is no need for installing or maintaining a local copy. Usually, applications using web interfaces are easy to use and self-explanatory. However, users are dependent on the availability and the performance of the provided services. Another issue is that many of the online tools do not support batch submission of variants, thus making them only viable for manual analysis of a small set of variants. Moreover, legal issues might arise since most services do not guarantee data confidentiality. On the other hand, offline tools usually provide more flexibility and are not dependent on the availability of any specific web-service, but require the user to have a certain degree of IT skills. The inspected annotation tools (74) with their respective input/output formats, supported variants, type [graphical user interface (GUI), client or web tools] and usage notes are given in Supplementary Table S7.
Visualization
An important and challenging step in every NGS data analysis workflow is the validation and visualization of the generated results. Visual representation of data can be tremendously useful for the interpretation of obtained results. Therefore, NGS visualization tools should support users by displaying aligned reads, mapping quality and identified mutations combined with annotations from various public resources. In addition, the tools should be user-friendly, intuitive and responsive.
Visualization tools for genomic data can be divided into three different types [85]: (i) finishing tools supporting the interpretation of sequence data of de novo or re-sequencing experiments, (ii) genome browsers that allow users to browse mapped experimental data in combination with different types of annotation and (iii) comparative viewers that facilitate the comparison of sequences from multiple organisms or individuals. In addition to genome browsers, software suites have been published which enable visualization of identified CNVs and SVs [86–88], showing a global picture of genomic rearrangements and SVs. A list of 38 surveyed genome browsers and two CNV/SV visualization tools can be seen in Supplementary Tables S8 and S9.
Genome browsers can be divided into two major types: web-based applications running on a dedicated web server [89] and stand-alone tools, where most of them use a GUI. As all genome browsers with a GUI are implemented in Java, they can be used on platforms such as Windows, Mac and Linux systems.
The main advantage of web-based genome browsers is the support of a variety of annotations. The user can browse reference genomes as well as different types of genomic annotations derived from a variety of public databases. Furthermore, users do not need to install new applications with numerous dependencies, and computational intensive calculations are performed on the server. A drawback of the web-based genome browsers is the necessity of uploading the data to a remote server, which poses security and legal issues.
Stand-alone genome browsers offer interactive browsing and zooming features, which are missing in some web-based genome browsers. Furthermore, they do not require uploading the data to websites. Shortcomings include the need to download annotation files and the user’s responsibility to keep annotations up-to-date. In addition, complex calculations have to be performed by regular desktop PCs, which may not be powerful enough to deal with this workload.
When interpreting aligned sequences using a genome browser, it is recommended to consider several important aspects [90]. Reads, which could be mapped with many mismatches should not be trusted and mutations, which are only backed by a small fraction of reads should be discarded. Moreover, reads should only be trusted for further processing if they align at a unique starting position.
Analytical pipelines and workflow systems
As can be seen from this review, the scientific community has access to a plethora of tools for NGS analysis. Combining these methods for analysis to obtain biologically meaningful results is still a challenging task even for experienced users. A viable alternative is the use of complete analytical pipelines capable of analyzing all steps starting from raw sequences to a set of identified and annotated variants.
The analytical pipelines in general have a predefined order of analysis steps and built-in algorithms that cannot be easily modified and/or replaced. In contrast to pipelines, workflow management systems offer the flexibility to arrange a specific order of analytical steps and execute a series of data manipulation or analysis steps. Most existing systems provide GUI allowing the user to build and modify complex workflows with little or no programming expertise.
We identified 13 published pipelines and 12 available workflow systems suitable for NGS analysis, which process data from various platforms using different tools (see Supplementary Tables S10 and S11, respectively).
SCOPE AND METHOD OF EVALUATION OF NGS TOOLS
The wealth of available tools reflects the importance of NGS and the tremendous dynamic of the field of data analysis. Of the five distinct analytical steps, quality assessment and read alignment can be considered as matured and robust. Variant identification, variant annotation and visualization methods are essential for the detection of relevant variants and are still being developed. We therefore assessed selected tools for the latter three categories by installing and testing them.
Criteria for tool selection
We considered only published, freely available and constantly maintained tools for in-depth evaluation. To be classified as maintained, the tool had to be updated within the last year (cutoff date September 2011) since the analysis of NGS data is a fast-evolving field and tools need to be constantly adapted. A further requirement for tool selection was the support of accepted standard input and output formats. Additionally, due to the heterogeneity of tools available for different parts of the analysis workflow, we specified distinct selection criteria for each category as described below.
Variant identification
Germline and somatic variant callers were selected if they were able to: (i) use Sequence Alignment/Map (SAM) [91], Binary Alignment/Map (BAM) or pileup format as input and (ii) provide output results in the variant call format (VCF). Tools that determine CNVs and SVs were selected when they accepted SAM/BAM as input format. Table 1 lists all tools selected for testing.
Table 1:
Variant identification
| Name | OS | BAM/SAM input | Other inputs | Output | Identifies | Data set | Resulta |
|---|---|---|---|---|---|---|---|
| Germline callers | |||||||
| CRISP | Lin | Yes | – | VCF | SNP, INDEL | KTS | 24 034 SNPs, 259 INDELs |
| GATK (UnifiedGenotyper) | Lin | Yes | – | VCF | SNP, INDEL | KTS | 49 476 SNPs, 1959 INDELs |
| SAMtools | Lin | Yes | FASTA | VCF | SNP, INDEL | KTS | 21 852 SNPs, 332 INDELs |
| SNVer | Lin, Mac, Win | Yes | – | VCF | SNP, INDEL | KTS | 22 105 SNPs, 234 INDELs |
| VarScan 2 | Lin, Mac, Win | No | pileup/mpileup | VCF, VarScan CSV | SNP, INDEL | KTS | 34 984 SNPs, 1896 INDELs |
| Somatic callers | |||||||
| GATK (SomaticIndelDetector) | Lin | Yes | – | VCF | INDEL | WES | 151 INDELs |
| SAMtools | Lin | Yes | FASTA | BCF | SNP, INDEL | WES | Canceledb |
| SomaticSniper | Lin | Yes | – | VCF, somatic sniper output | SNP, INDEL | WES | 6926 SNPs |
| VarScan 2 | Lin, Mac, Win | No | pileup/mpileup | VCF, VarScan CSV | SNP, INDEL, CNV | WES | 1685 SNPs, 324 INDELs |
| CNV identification tools | |||||||
| CNVnator | Lin | Yes | FASTA | CSV | CNV | cnv_sim | 39 CNVs |
| RDXplorer | Lin, Mac | Yes | FASTA | CSV | CNV | cnv_sim | 4 CNVsc |
| CONTRA | Lin, Mac | Yes | FASTA | VCF, CSV | CNV | WES | 3 CNVs |
| ExomeCNV | Lin, Mac, Win | Yes | pileup + BED + FASTA | CSV | CNV, LOH | WES | 137 CNVs |
| SV identification tools | |||||||
| BreakDancer | Lin, Mac | Yes | config file | CSV, BED | INDEL, INV, TRANS, CNV | WGS (tumor + normal) | 6219 DELs, 0 INSs, 7 INVs, 17 303 ITX, 5037 CTX |
| Breakpointer | Lin | Yes | – | GFF | INDEL | WGS (tumor) | d |
| CLEVER | Lin | Yes | FASTA | CLEVER format | INDEL | WGS (tumor) | d |
| GASVPro (GASVPro-HQ) | Lin, Mac | Yes | – | clusters file | INDEL, INV, TRANS | WGS (tumor) | 2529 DELs, 207 INVs |
| SVMerge | Lin | Yes | FASTA | BED | INDEL, INV, CNV | – | Abortede |
Four different types of tools for variant identification can be distinguished: germline callers, somatic callers, CNV identification and SV identification tools. Listed are the results of the tested applications (4, 2, 3 and 5, respectively). All surveyed applications are listed in Supplementary Tables S3–S6. aSNVs are counted based on their position but in a sequence independent manner. bSomatic mutation calling with SAMtools was canceled due to unclear definition of tumor and normal files. Furthermore, we were not able to find the CLR field in the resulting vcf file, which should hold the Phred-log ratio between the likelihood by treating the two samples independently, and the likelihood by requiring the genotype to be identical. cFor RDXplorer the filtered result data set was used. dCLEVER and Breakpointer created result files with >2.6 million lines, which need to be further processed. eInstallation was aborted due to unreasonable dependencies. OS, operating system; Lin, Linux; Mac, Mac OS X; Win, Windows; BAM, Binary SAM; BED, Browser Extensible Data, a text-based file format; CSV, comma separated values; FASTA, text-based format for representing nucleotide sequences; GFF, general feature format; mpileup, multisample pileup; pileup, text-based format representing base-pair information at each chromosomal position; SAM, Sequence Alignment/Map; VCF, Variant Call Format; CNV, copy number variation; CTX, inter-chromosomal translocation; DEL, deletion; INDEL, insertion/deletion; INS, insertion; INV, inversions; ITX, intra-chromosomal translocation; LOH, loss of heterozygosity; SNP, single-nucleotide polymorphism; SNV, simple nucleotide variant; SV, structural variant; TRANS, translocations.
Variant annotation
For further consideration, annotation programs were required to: (i) accept VCF as input format (including tools that offer converters) and (ii) integrate results from other software (Table 2). Furthermore, results had to be reported in an output file, and web-based tools needed to provide a batch submission system.
Table 2:
Variant annotation
| Name | OS | Input | Output | SNP | INDEL | CNV | GUI | CLI | Web | Function/Location Parameters | DB IDs | Number of scores |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ANNOVAR | Lin, Mac, Win, web interface | VCF, pileup, CompleteGenomics, GFF3-SOLiD, SOAPsnp, MAQ, CASAVA | TXT | Yes | Yes | Yes | No | Yes | No | 9 (func) + 11(exonic-func) | Yes | GERP++ conservation, LRT, MutationTaster, PhyloP conservation, PolyPhen, SIFT |
| AnnTools | Lin, Mac | VCF, pileup, TXT | VCF | Yes | Yes | Yes | No | Yes | No | 5 (position) + 4 (functional class) | Yes | – |
| NGS–SNP | Lin, Mac | VCF, pileup, MAQ, diBayes, TXT | TXT | Yes | No | No | No | Yes | No | 17 | Yes | Condel, PolyPhen, SIFT |
| SeattleSeq | web interface | VCF, MAQ, CASAVA, GATK BED, custom | VCF, SeattleSeq | Yes | Yes | No | No | No | Yes | 11(dbSNP) + 5 (GVS) | Yes | GERP, Grantham, phastCons, PolyPhen |
| snpEff | Lin, Mac, Win | VCF, pileup/TXT (deprecated) | VCF, TXT, HTML overview | Yes | Yes | No | No | Yes | No | 34 | Yes | – |
| SVA | Lin | VCF, SV.events file, BCO | CSV | Yes | Yes | Yes | Yes | Yes | No | 17 (SNP), 17 (INDEL), 10 (CNV) | Yes | – |
| VARIANT | web interface | VCF, GFF2, BED | web report, TXT | Yes | Yes | No | No | Yes | Yes | 26 | Yes | – |
| VEP | Lin, web interface | VCF, pileup, HGVS, TXT, variant identifiers | TXT | Yes | Yes | No | No | Yes | Limited | 28 | Yes | Condel, PolyPhen, SIFT |
Tools for annotation of different variants are displayed. Some of the listed applications are available via web, whereas others have to be installed locally and can be accessed via a command-line interface. These reviewed applications calculate different scores and use public databases for annotation. Each mutation will end up with several annotations from each single tool. Annotation tools that were not tested are listed in Supplementary Table S7. OS, operating system; Lin, Linux; Mac, Mac OS X; Win, Windows; CLI, command line interface; CNV, copy number variation; GUI, graphical user interface; INDEL, insertion/deletion; SNP, single-nucleotide polymorphism; ASM.tsv, Complete Genomics’ text-based genotyping-calling format; BCO, binary format coverage and quality score file including: consensus quality, SNP quality, RMS mapping quality, read depth; BED, Browser Extensible Data, a text-based file format; CASAVA, genotype-calling output format of Illumina’s CASAVA (Consensus Assessment of Sequence and Variation) software; CSV, Comma Separated Values; GFF2, Generic Feature Format version 2; GFF3, Generic Feature Format version 3; HGVS, nomenclature for the description of sequence variants by HGVS (Human Genome Variation Society); MAQ, genotype-calling output format of Maq (Mapping and Assembly with Qualities); variant identifiers, e.g. dbSNP rsIDs or any synonym for a variant present in the Ensembl Variation database; VCF, variant call format; pileup, text-based format representing base-pair information at each chromosomal position; SOAPsnp, genotype-calling format from the SOAPsnp component of the Short Oligonucleotide Analysis Package (SOAP); SV.events file, ERDS (Estimation by Read Depth with SNVs) output, each row in the.events file corresponds to a CNV.
Visualization
Selection criteria were the availability of a GUI and the support of VCF, SAM and BAM, as these formats are considered a de facto standard in the field. We did not consider finishing tools, as they are primarily designed for de novo sequencing projects as well as comparative viewers and genome browsers that only supported viewing of aligned reads.
Analysis pipelines and workflow systems
The quality assessment step is NGS platform specific and it is not necessarily a part of an analytical pipeline or workflow system. Therefore, pipelines and workflow systems were selected for close inspection if they cover the analysis steps of read alignment, variant detection and variant annotation.
Test data sets and hardware
In order to test the software suites, we selected four data sets. The first data set (KTS) contains two samples and is a paired-end whole-exome sequencing run used for the elucidation of the Kohlschütter–Tönz syndrome [92]. Reads were aligned to the UCSC reference human genome assembly (hg19) using the sequence alignment software BWA version 0.5.10 [71] using the default parameters.
For the second data set, we randomly selected one of the simulated artificial tumor data sets published by [93] (csv_sim). This data set (14.7 million reads) is based on chromosome 22 (UCSC reference human genome assembly hg18), where 42 CNVs with set sizes of 100, 500 bp, 1, 5, 10, 50 or 100 kb were simulated. The copy number of the CNV segments had been chosen to represent either a homo- or heterozygous deletion or up to a four-copy gene gain.
The third data set is an example of a whole-exome sequencing study containing paired-end data from germline and tumor samples derived from a 42-year-old Hispanic female with mast-cell leukemia (WES) (SRP008740 [94]). Tumor-genomic DNA originated from bone marrow biopsies, whereas the germline sample was derived from saliva of the same patient. Both samples were aligned to UCSC reference human genome assembly (hg19) using BWA version 0.5.10.
For testing SVs, a whole-genome sequencing data from a liver metastatic lung cancer samples obtained from lung adenocarcinoma patients (ERP001071, [95]) was selected (WGS). We randomly chose one tumor/normal pair (subject AK55) sequenced on an Illumina HiSeq 2000 of the available data sets. To enable comparison with the study’s results, read alignment was performed to UCSC reference human genome assembly (hg19) using the alignment program GSNAP [96] version 2012-07-20 and default parameters.
All tools have been tested with default parameters on the same server system to ensure comparable results between the test runs. This system consists of an HP Proliant DL580 G7 server, equipped with four Intel E7-4870 CPUs and 512 GB of main memory. This results in 40 CPU cores at a tact rate of 2.4 GHz each and 12.8 GB of memory per core. Data storage was provided by a hybrid system of both 900 GB of directly attached high-performance hard drives and 30 TB of network attached storage via a 6 GB/s fiber channel link. On the software side, CentOS 6.2 was used with all packages on their latest versions. All Python tools have been tested with a manually compiled version of Python 2.7.3. R scripts were executed with R version 2.15.1 [97].
EVALUATION RESULTS
In this section, we present the evaluation results for the three essential NGS data analysis steps (Figure 1): (i) variant identification, (ii) variant annotation and (iii) visualization. Additionally, we closely describe existing pipelines integrating read alignments, variant identification and annotation. We were able to install and run all selected tools (in total 32 programs). It is noteworthy that the installation and maintenance of the majority of the tools and/or pipelines require certain degree of IT expertise, which is usually provided by the local IT department. Below we briefly describe the tools and provide additional information of the software in Supplementary Information I1.
Variant identification
The evaluated tools for variant identification were divided into four groups (Table 1): germline callers (five tools), somatic callers (four tools), CNV identification tools (four tools) and SV identification tools (five tools).
Germline callers
‘CRISP’ [98] is a tool for identifying SNPs and INDELs from pooled NGS data. It is intended to detect both rare and common variants. Furthermore, it has been specifically designed to detect variants from pooled data and should not be used for analysis of single samples.
‘GATK’ [99] is a software library that provides a suite of tools for working with human data, including depth of coverage analyzers, a quality score recalibrator, a local realigner and a SNP/INDEL caller. The authors offer extensive documentation and an informative wiki system. In addition to calling germline variants, GATK can be used to identify somatic mutations.
‘SAMtools’ [91] is a versatile collection of tools for manipulating SAM and BAM files. It contains a subset of commands called BCFtools. BCFtools has also the ability to call SNPs and short INDELs from a single alignment file. Furthermore, SAMtools can be used to call somatic mutations from a pair of samples.
‘SNVer’ [100] is an operating system independent statistical tool for the identification of SNPs as well as INDELs, in both pooled and individual NGS data. Recently, SNVerGUI [101] has been published, which provides a GUI version of SNVer tool.
‘VarScan 2’ [102] is a platform-independent program that can be used to identify germline variants, as well as shared and private variants. In addition to germline variants, ‘VarScan 2’ is able to identify somatic mutations and somatic CNVs. Germline variants are called using a heuristic method as well as a statistical test based on the number of aligned reads supporting each allele.
CRISP, GATK, SAMtools, SNVer and VarScan 2 were tested with the KTS data set. The tools identified ∼24 000, 49 000, 22 000, 22 000 and 34 000 SNPs as well as 259, 1959, 234, 332 and 1896 INDELs, respectively. Figure 2A depicts the overlap of identified variants in a Venn diagram. More than 13 000 of identified SNPs are overlapping between all used tools, in contrast to INDELs where none is shared with all applications. CRISP reports the lowest number of non-overlapping SNPs (8), whereas >23% of SNPs are only identified by GATK. VarScan 2 and GATK share the largest number of overlapping INDELs (∼57%) in contrast to SNVer, where 99% of reported INDELs are not identified by any other tool. In summary, tools differ widely regarding called INDELs and show larger agreement in identified SNPs. It is however noteworthy that all INDELs called by CRISP were also identified by GATK (96% by VarScan2).
Figure 2:

Venn diagrams showing the number of identified variants for tested germline (A), somatic (B), CNV (C) and exome CNV (D) tools. The depicted numbers in (A) and (B) report identified SNPs and INDELs. Venn diagram (C) shows the overlap between known (cnv_sim) and predicted CNVs. Figure (D) illustrates the overlap between CONTRA and ExomeCNV. The intersection numbers were adjusted to reflect that 10 CNVs detected by CONTRA are located within 3 CNVs reported by ExomeCNV.
Somatic callers
In addition to GATK, SAMtools and VarScan 2, we evaluated another somatic caller: ‘SomaticSniper’. ‘SomaticSniper’ [103] is a command-line application to identify SNPs that are different between tumor/normal pairs. It calculates a somatic score that predicts the probability that tumor and normal genotypes are different.
GATK, SAMtools, SomaticSniper and VarScan 2 were tested using the whole-exome tumor data set. As SAMtools does not clearly specify how to define the input for tumor and normal data sets and the generated result file lacked information about the somatic score, SAMtools was not further considered for calling somatic mutations. GATK is only capable of calling INDELs and reported 151 mutations. SomaticSniper could identify 6926 SNPs and VarScan 2 was able to detect 1685 SNPs and 324 INDELs. Compared with germline results, the agreement of the identified variants is small (depicted in Figure 2B).
Copy number variations identification
We tested four tools for CNV identification: (i) ‘CNVnator’ [104], a CNV discovery and genotyping tool for whole-genome sequencing data which uses read-depth analysis based on mean shift; (ii) CONTRA [105], a tool for detecting CNVs in whole-exome data. The application calls copy number gains and losses for each specified target region; (iii) ‘ExomeCNV’ [84], a R package for the identification of CNVs and loss of heterozygosity from whole-exome sequencing data. The tool works best when paired samples (i.e. tumor/normal pairs) are available and (iv) ‘RDXplorer’ [106], a tool for detecting CNVs in human whole-genome sequencing data. It uses read depth coverage and detects CNV based on the event-wise testing algorithm. It should be noted that due to numerous dependencies, the installation of this tool is challenging for non-experienced users.
CNVnator and RDXplorer were tested using the simulated CNV data set with known number of CNVs (42). CNVnator was able to call 39 CNVs; RDXplorer identified 4 CNVs (filtered results). This results in a precision and recall of 0.74 and 0.69, as well as 1 and 0.1 for CNVnator and RDXplorer, respectively. Figure 2C depicts the agreement between known (cnv_sim) and predicted CNVs.
As CONTRA and ExomeCNV were designed for the analysis of whole-exome sequencing data, the ‘WES’ data set was used for testing. ExomeCNV identified 137 CNVs, whereas CONTRA was able to call 8940 CNVs using a P-value ≤ 0.05. Three out of those were reported as significant (adjusted P-value ≤ 0.05), whereby the usage of a less-conservative threshold (adjusted P-value ≤ 0.1) resulted in 11 CNVs. Out of the remaining 11, 10 CNVs were located within 3 CNVs reported by ExomeCNV. The direction (gain/loss) reported by both tools was the same for all of the overlapping variants.
Structural variants identification
‘BreakDancer’ [107] predicts five types of structural variants: insertions, deletions, inversions, inter- and intra-chromosomal translocations.
‘Breakpointer’ [108] is a command-line tool especially designed for the location of potential intra-chromosomal sequence breakpoints from single end reads. It uses a heuristic method to identify local mapping signatures created by INDELs longer than the read’s length or other SVs.
‘CLEVER’ [109] identifies SVs in genomes from paired-end sequencing reads. It uses an insert size-based approach, which takes all reads into account. The tool offers an intuitive script with default parameters to facilitate usability.
‘GASVPro’ [110] represents the probabilistic version of the original GASV algorithm [111] and detects SVs from paired-end data.
‘SVMerge’ [112] is a software suite that integrates results from several different SV callers and performs subsequent validation and refinement of identified breakpoints. Unfortunately, the software suite is not provided as a ready-to-use virtual box or cloud implementation, which would enhance the usability.
BreakDancer was tested with the WGS tumor/normal data set and identified 6219 deletions, 7 inversions, 17 303 intra- and 5037 inter-chromosomal translocations. All other inspected SV callers were applied on the WGS tumor data set only. Breakpointer determined 2.7 million possible intra-chromosomal breakpoints in total. CLEVER produced 2.6 million entries. ‘GASVPro-HQ’ identified 3941 raw deletions and 407 raw inversions, which were automatically reduced to 2529 and 207, respectively. Although GASVPro commands offer to call inter-chromosomal variations, the feature is not yet supported by its pipeline scripts. Testing of SVMerge was aborted, as installation dependencies are likely beyond the IT skill levels of many bench scientists.
In summary, all selected tools for variant identification were successfully installed and tested with the appropriate data sets. As can be seen in Figure 2 and Table 1, there were considerable differences in the obtained results. The agreement between the callers was larger for SNPs compared with INDELS and larger for germline than for somatic mutations, respectively. Furthermore, the results show even bigger discrepancy for the identified CNVs and SVs (Figure 2 and Table 1).
There are several possible explanations for this observation. First, there are a number of parameters and thresholds that can be adjusted for the specific tools and hence, influence the outcome. Early methods for genotype calling are based on fixed cut-offs whereas recent methods such as GATK or SAMtools provide measures of statistical uncertainty when calling genotypes and hence, might improve the accuracy (see also excellent review on genotype and SNP calling by Nielsen et al. [25]). Second, the underlying statistical models are divergent and harbor different assumptions, which are often difficult or impossible to evaluate experimentally. For example, the ploidy, the degree of tumor heterogeneity and the percentage of normal cells in the sample are in most cancer studies difficult to estimate. Thus, using a statistical model based on a diploid genome would in this case inevitably lead to erroneous results. Third, in order to assess the performance of newly developed tools, the authors often rely on simulated data to benchmark their method. However, due to the complexity of the underlying error model and the varying properties of NGS data, simulated data may be neglecting certain error cases and make assumptions that are not valid. Furthermore, the use of simulated data for the development of a new tool might tailor the method to specific data sets and hence, severely hamper the performance when using real NGS data. Further studies are required in order to benchmark the performance of the variant callers either by using an exome or genome-scale size simulation data set or a large number of experimentally evaluated variants by Sanger re-sequencing as shown recently [113]. We also strongly believe that further theoretical studies are necessary to develop new and/or improved variant callers. As the number of sequenced genomes/exomes is steadily increasing, specific statistical models will become available for certain diseases or cohorts.
Variant annotation
Below we describe the evaluation results of the eight selected tools (see also Table 2).
‘ANNOVAR’ [114] is a command-line tool for up to date functional annotation of various genomes, supporting SNPs, INDELs, block substitutions as well as CNVs. The tool provides a wide variety of different annotation techniques, organized in the categories gene-based, region-based and filter-based annotation. The tool depends on several databases, which need to be downloaded individually. This approach ensures that the correct database version is used and the download of large unnecessary data sets is avoided.
‘AnnTools’ [115] is a command-line tool for analyzing SNPs, INDELs and CNVs found in both coding and non-coding regions. The program relies on 15 different widely used data sources such as dbSNP, which are regularly updated. A database update tool is provided to help keep the local database up-to-date.
‘NGS–SNP’ [116] is a collection of Perl scripts for the annotation of SNVs using the Ensembl database as a reference. The program uses the online version of the Ensembl database, which has the advantage that the reference database is always up-to-date. In comparison to other tools, NGS–SNP took several days to complete the annotation process, which is likely due to the latency of querying the online database during the tool’s execution.
The ‘SeattleSeq’ Annotation server (http://snp.gs.washington.edu/SeattleSeqAnnotation) provides a web application for annotating human SNPs and INDELs. In contrast to most other web-based annotation services, SeattleSeq provides the possibility to directly upload input files in various formats for batch analysis of multiple variants. The analysis of both hg18/GRCh36 and hg19/GRCh37 data is supported by providing a separate input page for each genome version. As variant annotation is performed on a remote server, the tool might be interesting for research groups without dedicated hardware for data analysis.
‘Sequence variant analyzer (SVA)’ [117] is a stand-alone tool with a GUI dedicated to annotating and visualizing variants identified by NGS experiments. The tool includes its own genome browser and supports annotation of SNPs, INDELs and CNVs. Setting up the program and writing the project configuration file might be difficult without IT expertise. As the hardware requirements are rather demanding, the use of a powerful workstation is recommended (>48 GB of RAM; >1 TB free hard disk; quad core processor). The program could not correctly annotate VCF files, which were using a UCSC hg reference version.
‘snpEff’ [118] is a popular variant annotation, which has also been integrated within Galaxy and GATK. In addition to SNPs, the tool also supports INDELs and multiple-nucleotide polymorphisms. snpEff identifies various different effects, which are categorized into four classes (high, moderate, low and modifier) by their functional impact.
‘VARIANT’ [119] can detect the functional properties of SNVs in coding as well as non-coding regions. The program can be used via a web interface, as a command-line tool or as a web service. Since the command-line tool also makes use of the remote VARIANT database, the tasks of maintaining and searching of databases are provided by the authors. Therefore, the command-line version of the tool is usable without profound IT expertise and can be executed on regular office PCs. The web interface features anonymous usage and allows creation of personal accounts, which enables users to view their uploaded input files and analysis results once they log in.
‘Variant effect predictor (VEP)’ [120] is Ensembl’s own functional annotation tool, formerly known as SNP effect predictor. The tool can be used either by a web interface, as command-line tool or via a Perl API. The web interface version is aimed at users analyzing smaller sets of variants, as it is only capable of processing 750 variants per file.
In summary, all annotation tools provide a set of general (e.g. mutation introduces a new stop codon) or detailed (e.g. mutation hits an exon) attributes for each identified mutation. These properties can be used to assess the potential impact of each mutation. All tested applications provide links to one or more public databases of known mutations. Four of the tested tools provide prediction scores reflecting their estimated deleterious impact. ANNOVAR uses six different scores: GERP++ [121], LRT [122], MutationTaster [123], PolyPhen [124], PhyloP conservation [125] and SIFT [126]. SeattleSeq supplies four scores: GERP [127], Grantham [128], phastCons [129] and PolyPhen. NGS–SNP and VEP provide three scores: Condel [130], PolyPhen and SIFT. These scores are computed based on various different approaches, such as sequence homology, evolutionary conservation, protein structure or statistical prediction based on known mutations. Due to the possibility of precalculating several scores (GERP, PolyPhen and SIFT) for every position in the genome, tools such as ANNOVAR or SeattleSeq make use of databases to look up the requested annotation. Hence, the annotation process itself is very fast as no on the fly calculation is needed and similar results are reported. It should be noted that results are often difficult to interpret for inexperienced users. Furthermore, among the tested tools, only stand-alone tools (ANNOVAR, AnnTools and SVA) were able to annotate identified CNVs.
Visualization
All of the evaluated genome browsers have in common that they are capable of displaying numerous 1D tracks which contain information about the reference genome, the transcriptome, aligned reads, found mutations, annotations collected from public data sources or other data types important for the correct interpretation of results [131]. The two types of genome browsers, namely web-based applications and stand-alone tools, as well as CNV/SV visualization tools are given in Table 3.
Table 3:
Visualization
| Name | OS | BAM/ SAM | VCF | Other formats | Annotation |
|---|---|---|---|---|---|
| Web-based genome browsers | |||||
| Ensembl Genome Browser | web interface | Yes | Yes | BED, bedGraph, GFF, GTF, PSL, WIG, BAM, bigWig | Yes |
| UCSC Genome Browser | web interface | Yes | Yes | BED, bigBed, bedGraph, GFF, GTF, WIG, bigWig, MAF, SNP, PSL | Yes |
| VEGA Genome Browser | web interface | Yes | Yes | BED, bedGraph, bigBed, bigWig, GBrowse, GFF, GTF, PSL, WIG | Yes |
| Stand-alone genome browsers | |||||
| Artemis | Lin, Mac, Win | Yes | Yes | BCF, FASTA | Yes |
| Integrative Genomics Viewer (IGV) | Lin, Mac, Win | Yes | Yes | SNP, GFF, BED, IGV, TAB, WIG, (>30 formats) | Yes |
| Savant | Lin, Mac, Win | Yes | Yes | FASTA, BED, GFF, WIG, TAB | Yes |
| CNV and SV visualization | |||||
| Circos | Lin, Mac, Win, web interface | No | No | GFF, CSV | Yes |
This table holds genome browsers as well as tools producing circos plots, whereby genome browsers were split into web-based applications, accessible using a web browser and stand-alone tools with a graphical user interface. All genome browsers use tracks to display different features like reference genome, annotations or experimental data. Further visualization tools can be found in Supplementary Tables S8 and S9. OS, operating system; Lin, Linux; Mac, Mac OS X; Win, Windows; BAM, Binary SAM; BCF, Binary VCF; BED, Browser Extensible Data, a text-based file format; bedGraph, file format allowing the display of continuous-valued data in track format; bigBed, compressed, binary-indexed BED file; bigWig, compressed, binary indexed WIG file; FASTA, text-based format for representing nucleotide sequences; GBrowse, Gbrowse proprietary format; GFF, General Feature Format; GTF, Gene Transfer Format; IGV, Integrative Genomics Viewer format; MAF, Multiple Alignment Format; PSL, pattern space layout; SAM, Sequence Alignment/Map; SNP, Personal Genome SNP format; TAB, tab-delimited file; VCF, Variant Call Format; WIG, Wiggle Track Format.
Web-based applications
‘The Ensembl Genome Browser’ [132] provides a variety of reference genomes in combination with numerous local annotation sets and external resources. This genome browser is able to display continuative information when searching for a specific entity. Users can add their own tracks either by uploading the data file (restricted to 5 MB) or by specifying a remote URL or the distributed annotation system (DAS). It is not necessary to create an Ensembl account to attach or upload data, but it can be used to save specific information for later use.
‘The University of Santa Cruz (UCSC) Genome Browser’ [133] offers several different annotations, including phenotype and disease annotations. User-specific data can either be uploaded or remotely hosted and provided by an URL. The UCSC database comprises almost 1800 annotation tracks for the human genome GRCh37/hg19.
‘The Vertebrate Genome Annotation (Vega) Genome Browser’ [134] is built upon code of the Ensembl Genome Browser. Data available in Vega is based on a freezed Ensemble release that has undergone manual annotation and curation by the Havana group at the Welcome Trust Sanger Institute. The genome browser offers standardized gene classification encompassing pseudogenes, non-coding transcripts and PolyA sites. Vega contains annotations from different species for comparative analysis, but currently human chromosomes 18 and 19 are still outstanding.
In general, web-based genome browsers provide different reference genomes already integrated with a variety of annotation tracks. A drawback is the required data handling, as the user has to provide URLs for external files, which requires uploading large amount of data to either a local web server or a remote location. In addition, files need to be packed, sorted and indexed before they can be used.
Stand-alone applications
‘Artemis’ [135] incorporates BamView [136] for viewing aligned reads and is able to display different properties of a loaded sequence. It features two sequence windows, which can be used to display the same sequence at different zoom levels. Artemis allows filtering of variants based on different properties in real time and supports the export of calculated properties such as SNP density and read counts.
‘The Integrative Genomics Viewer (IGV)’ [137] supports loading of tracks, reference genome as well as annotation data from local or remote data sources. Furthermore, the tool uses the DAS system and is capable of handling >30 different file formats. IGV is also capable of displaying sample metadata (e.g. gender and age) as a heatmap, which can be shown adjacent to the sample’s corresponding track. The IGV tool package provides functionality for tiling, counting, sorting and indexing of several file formats. In addition to the stand-alone GUI version, IGV can be launched and controlled using scripts enabling the generation of different image snapshots at once.
‘Sequence Annotation and Visualization and ANalysis Tool (Savant)’ [138] uses a modular docking framework, where each module appears as a separate window. Local as well as remote data sets can be loaded into Savant, and it supports simple file formatting functionality (e.g. VCF zipping and indexing).
All tested stand-alone genome browsers are implemented in Java, and therefore run on all operating systems with an installed JVM. The applications are able to load different reference genomes and combine them with tracks holding experimental data or annotations. The stand-alone genome browsers showed excellent performance for zooming and panning and were able to visualize SNPs and INDELs. Moreover, they generally feature a rich user interface and sometimes support the inclusion of custom analytical modules using a plug-in architecture (Savant). Web-based genome browsers facilitate displaying of data in context with already existing annotations and data tracks without the need for downloading or installing additional information. Moreover, due to their universal accessibility, they support collaboration within or between different institutions. Thus, given the above characteristics, the selection of a genome browser for prospective users and for specific criteria is straightforward.
CNV and SV visualization
‘Circos’ is a widely used command-line tool written in Perl for the visualization of similarities and differences of genomes. Although it is capable of visualizing arbitrary data, it is clearly intended for multi-sample genomic data. Extensive documentation and tutorials are provided which are helpful to correctly set up all configuration files. In addition to the simple command-line interface, an online version of the tool is available allowing the upload of input data and configuration files using a simple web interface. Circos is very flexible and allows users to display data as scatter, line and histogram plots, as well as heat maps, tiles, connectors and text. Due to this flexibility, the learning curve for new users is steep and it might take some time to explore all available options.
Analysis pipelines and workflow systems
We evaluated three analytical pipelines (‘HugeSeq’, ‘SIMPLEX’ and ‘TREAT’) and three workflow systems (‘Galaxy’, ‘LONI’ and ‘Taverna’).
‘HugeSeq’ [139] is a fully integrated pipeline for NGS analysis from aligning reads to the identification and annotation of variants (SNPs and INDELs for whole-genome and whole-exome sequencing data as well as CNVs and SVs for whole-genome data only). It consists of three main parts: (i) preparing and aligning reads, (ii) combining and sorting reads for parallel processing of variant calling and (iii) variant calling and annotating. The pipeline accepts as an input reads in FASTA or FASTQ format and outputs identified variants in VCF format, and SVs and CNVs in GFF format. Identified variants are further processed with ANNOVAR to include additional annotations.
‘SIMPLEX’ [140] is an autonomous analysis pipeline for the analysis of NGS exome data, covering the workflow from sequence alignment to SNP/INDEL identification and variant annotation. It supports input from various sequencing platforms and exposes all available parameters for customized usage. It outputs summary reports and annotates detected variants with additional information for discrimination of silent mutations from variants that are potentially causing diseases. SIMPLEX is provided as a ready-to-use virtual machine and can be used in the Amazon EC2 Cloud.
‘TREAT’ [141] is a pipeline where each of the three modules (alignment, variant calling and variant annotation) can be used separately or as an integrated version for an end-to-end analysis. It provides a rich set of annotations, HTML summary report and variant reports in Excel format. TREAT can be downloaded for local use (requires large main memory) or launched on the Amazon EC2 Cloud. The pipeline currently provides only the human reference genome hg18.
‘Galaxy’ [142] is a web-based platform where the user can perform, reproduce and share complete analyses. Pipelines are represented as a history of user actions, which can be stored as a dedicated workflow. It contains scripts for over a 100 analysis tools and users can add new tools (requiring basic informatics skills) and share all analysis steps and pipelines. Workflows are stored directly in a dedicated database, and jobs can be distributed onto a high-performance computing infrastructure. Researchers can use the online version, install a local version on their server system or run Galaxy on the Amazon EC2 Cloud.
‘LONI’ [143] is a workflow processing application that can be used to wrap any executable for use in the LONI system. In order to access the tools, users need to connect to either public or private pipeline servers. Workflows can be divided into several modules and researchers can either use existing modules or create new ones from scratch. Several NGS analysis workflows have been published that are ready to be imported and used.
The ‘Taverna’ [144] workflow management system stores workflows in a format that is simple to share and manipulate outside the editor. Initially, it was not shipped with any prepackaged NGS analysis tools, and integrating tools required some programming experience. However, workflows for NGS have been made public that use a specific queuing system to distribute jobs to a computational cluster. These workflows can be downloaded and imported, and additionally edited.
In summary, the analytical pipelines provide out-of-the box solutions for analyzing whole-genome or whole-exome sequencing data. However, due to the fixed choice of included tools they do not provide as much flexibility as the workflow systems. In contrary, the tested workflow systems offer greater flexibility for integrating analytical tools but often require to manually install analysis tools and may not offer predefined solutions to analyze sequencing data.
DISCUSSION
In our report, we provide a comprehensive survey of tools available for the analysis of whole-genome/whole-exome data covering all analytical steps: quality assessment, alignment, variant identification, variant annotation and visualization. The information we provide represents a valuable guideline for both an expert in the field and a less-experienced user to select the appropriate tools for a specific application and assemble an optimal analytical pipeline. The analysis of NGS data is a fast moving field and recommendations which tools to use might quickly change. Nevertheless, we make the following general recommendations.
First, tools for quality assessment and alignment matured to a great extent and the choice is rather straightforward. Depending on the supporting platform, several tools are available that report properties and quality of obtained sequencing reads. Among the evaluated tools, FASTQC and NGSQC are capable of providing reports for FASTQ as well as the ABI SOLiD file format. Based on the obtained quality results, tools can be used to trim or remove reads that do not suffice the predefined quality standards. We recommend tools that are in active development and provide trimming as well as filtering functionality such as FASTX-Toolkit or PRINSEQ. Similarly, alignment software suites have been constantly improved over the past few years and several tools are widely used and supported by a large user community. For example, BWA, Bowtie and SOAP have matured greatly and are actively used in NGS data analysis projects. It is noteworthy that support for the ABI SOLiD platform has been dropped in recent versions of some of the alignment tools (e.g. BWA > 1.6.0, Bowtie2).
Second, for variant identification, we suggest a consensus approach, e.g. running CRISP, GATK and SAMtools on the same data set. It has been reported that no single approach comprehensively captures all genetic variations, and therefore, several variant identification tools should be applied. Variants will then only be considered if they fulfill certain criteria (e.g. identified by a minimum number of independent variant callers) [145]. However, applying such stringent criteria inevitably filters out true positives. If the candidate variant is missed, the search can be broadened and additional biological criteria can be applied. It is well known that the used library preparation method and the selected sequencing technology directly influence the reported outcome. It is therefore good practice to consider certain metrics (e.g. strand bias) and use heuristic approaches to separate true positives from false positives. Finally, additional aspects worth considering are the availability of up-to-date documentation and the existence of an active user community.
Third, the choice of an annotation tool is largely dependent on the desired selection of variant annotations. Four of the evaluated tools (ANNOVAR, SeattleSeq, NGS–SNP and VEP) include prediction scores, which are used to reflect the estimated deleterious impact of a particular variant. Several tools have been published that generate multiple variant annotations at once, which can be tremendously useful for shortening the analysis time (e.g. ANNOVAR, SeattleSeq or VARIANT). Furthermore, tools should be selected that regularly update the included information of publicly available databases. In addition, users should consider possible security issues when using web-based annotation tools.
Fourth, visualization tools are usually easy to install and it might therefore be a valid approach to test different software suites. In addition, some visualization programs such as IGV, Savant or the UCSC Genome Browser offer the possibility to obtain annotation tracks—such as reference genomes—which facilitates the usage for inexperienced users. Further consideration for the choice of the visualization tool is the possibility to handle the data format used in previous analysis methods for reporting variants and variant annotations. When choosing web-based systems, users should be aware of security and legal issues that might arise.
Prioritization of candidate variants
With the use of whole-exome and whole-genome sequencing, the challenge of the ‘next-generation genetics’ is one of narrowing down the list of candidate variants and interpreting remaining variants within a biological context [146]. A widely used approach to substantially reduce the candidate list is to exclude known variants which are present in public SNP databases, published studies or in-house databases as it is assumed that common variants represent harmless variations [147]. Another way to narrow down the genomic search space is the use of pedigree information, i.e. sequencing distantly related individuals with the phenotype of interest which might be sufficient to identify the causing mutation [148]. This approach is also helpful to identify the cause of common disorders that are genetically highly heterogeneous. However, since each generation introduces up to 4.5 deleterious mutations [149], it might be as well that a de novo mutation is causing the disease. Furthermore, without family information, it is often difficult to predict whether a disease follows a recessive or dominant inheritance [147].
In the case of cancer genomes, all potential variations need to be considered, including germline susceptibility loci, somatic SNPs and INDELs, CNVs and SVs [150]. A common method is to use pairwise comparison of tumor and normal tissues from the same individual to distinguish somatic coding mutations [10] and to identify driver mutations [18, 19]. In addition, tools have been published that can determine significant mutations in cancer by using groups of tumor/normal sample pairs, clinical data and identified variants from the cohort under investigation [151].
All prioritization methods have in common that researchers risk removing the pathogenic variant [147] which is reflected by the currently reported high rates of false-positive and false-negative predictions [152, 153]. Therefore, prediction tools should be used with caution since they may not be reliable enough to infer a definitive diagnosis [154]. The use of different prioritization approaches [147] and the combination of prediction results with phenotypic and pedigree data as well as data from databases might be the best approach to determine the potential cause of the disease under investigation [154].
Outlook
As sequencing costs continue to fall, we will experience a shift from sequencing human whole-exomes to whole-genomes, which will create the need for even more sophisticated methods to find the mutations causal for diseases [10]. We believe that future developments of workflow and pipeline systems will tremendously facilitate the analysis of NGS data, as they do not require complex installation routines and necessary data conversion steps from end-users.
SUPPLEMENTARY DATA
Supplementary data are available online at http://bib.oxfordjournals.org/.
Key points.
Next-generation sequencing technologies provide unprecedented opportunities to characterize individual genomic landscapes and identify mutations relevant for diagnosis and therapy for Mendelian disorders, complex diseases and cancers.
In this article, the variant analysis workflow for whole-genome and whole-exome sequencing is described, and software tools supporting each step are outlined.
The main focuses of this review article were the steps of variant identification, variant annotation and visualization.
We selected 32 tools and tested them using four different data sets.
Acknowledgements
We thank the reviewers for their numerous constructive suggestions, which helped us to considerably improve the article.
FUNDING
This work was supported by a grant from the Tiroler Standortagentur (Bioinformatics Tyrol), from the Austrian Science Fund (Projects Doktoratskolleg W11 Molecular Cell Biology and Oncology and SFB F21 Cell Proliferation and Cell Death in Tumors) and the FFG project Oncotyrol.
Biographies
Stephan Pabinger, PhD, is a research fellow at the Division for Bioinformatics at Innsbruck Medical University.
Andreas Dander is a research assistant at Oncotyrol and a PhD student at Innsbruck Medical University.
Maria Fischer, PhD, is a research fellow at the Division for Bioinformatics at Innsbruck Medical University.
Rene Snajder is a technical assistant at the Division for Bioinformatics at Innsbruck Medical University and Oncotyrol.
Michael Sperk is a PhD student at Innsbruck Medical University.
Mirjana Efremova is a PhD student at Innsbruck Medical University.
Birgit Krabichler, PhD, is a research fellow at the Division of Human Genetics, Innsbruck Medical University.
Michael Speicher, MD, is chair of the Institute of Human Genetics at the Medical University of Graz.
Johannes Zschocke, MD, is chair of the Division of Human Genetics at Innsbruck Medical University.
Zlatko Trajanoski, PhD, is chair of the Division for Bioinformatics at Innsbruck Medical University.
References
- 1.Gonzaga-Jauregui C, Lupski JR, Gibbs RA. Human genome sequencing in health and disease. Annu Rev Med. 2012;63:35–61. doi: 10.1146/annurev-med-051010-162644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ng SB, Turner EH, Robertson PD, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–6. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hodges E, Xuan Z, Balija V, et al. Genome-wide in situ exon capture for selective resequencing. Nat Genet. 2007;39:1522–7. doi: 10.1038/ng.2007.42. [DOI] [PubMed] [Google Scholar]
- 4.Rothberg JM, Hinz W, Rearick TM, et al. An integrated semiconductor device enabling non-optical genome sequencing. Nature. 2011;475:348–52. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- 5.Eisenstein M. Oxford Nanopore announcement sets sequencing sector abuzz. Nat Biotechnol. 2012;30:295–6. doi: 10.1038/nbt0412-295. [DOI] [PubMed] [Google Scholar]
- 6.Ng SB, Bigham AW, Buckingham KJ, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–3. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42:30–5. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Girard SL, Gauthier J, Noreau A, et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat Genet. 2011;43:860–3. doi: 10.1038/ng.886. [DOI] [PubMed] [Google Scholar]
- 9.O’Roak BJ, Deriziotis P, Lee C, et al. Exome sequencing in sporadic autism spectrum disorders identifies severe de novo mutations. Nat Genet. 2011;43:585–9. doi: 10.1038/ng.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shendure J. Next-generation human genetics. Genome Biol. 2011;12:408. doi: 10.1186/gb-2011-12-9-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106:19096–101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng PC, Levy S, Huang J, et al. Genetic variation in an individual human exome. PLoS Genet. 2008;4:e1000160. doi: 10.1371/journal.pgen.1000160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Robinson PN, Krawitz P, Mundlos S. Strategies for exome and genome sequence data analysis in disease-gene discovery projects. Clin Genet. 2011;80:127–32. doi: 10.1111/j.1399-0004.2011.01713.x. [DOI] [PubMed] [Google Scholar]
- 14.Bentley DR, Balasubramanian S, Swerdlow HP, et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bamshad MJ, Shendure JA, Valle D, et al. The centers for Mendelian genomics: a new large-scale initiative to identify the genes underlying rare Mendelian conditions. Am J Med Genet A. 2012;158 A:1523–5. doi: 10.1002/ajmg.a.35470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amberger J, Bocchini CA, Scott AF, et al. McKusick’s Online Mendelian Inheritance in Man (OMIM) Nucleic Acids Res. 2009;37:D793–6. doi: 10.1093/nar/gkn665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stitziel NO, Kiezun A, Sunyaev S. Computational and statistical approaches to analyzing variants identified by exome sequencing. Genome Biol. 2011;12:227. doi: 10.1186/gb-2011-12-9-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varela I, Tarpey P, Raine K, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–42. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wei X, Walia V, Lin JC, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mardis ER, Wilson RK. Cancer genome sequencing: a review. Hum Mol Genet. 2009;18:R163–8. doi: 10.1093/hmg/ddp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castle JC, Kreiter S, Diekmann J, et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 2012;72:1081–91. doi: 10.1158/0008-5472.CAN-11-3722. [DOI] [PubMed] [Google Scholar]
- 23.Schadt EE, Linderman MD, Sorenson J, et al. Computational solutions to large-scale data management and analysis. Nat Rev Genet. 2010;11:647–57. doi: 10.1038/nrg2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bao S, Jiang R, Kwan W, et al. Evaluation of next-generation sequencing software in mapping and assembly. J Hum Genet. 2011;56:406–14. doi: 10.1038/jhg.2011.43. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen R, Paul JS, Albrechtsen A, et al. Genotype and SNP calling from next-generation sequencing data. Nat Rev Genet. 2011;12:443–51. doi: 10.1038/nrg2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li H, Homer N. A survey of sequence alignment algorithms for next-generation sequencing. Brief Bioinformatics. 2010;11:473–83. doi: 10.1093/bib/bbq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koboldt DC, Larson DE, Chen K, et al. Massively parallel sequencing approaches for characterization of structural variation. Methods Mol Biol. 2012;838:369–84. doi: 10.1007/978-1-61779-507-7_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Datta S, Datta S, Kim S, et al. Statistical analyses of next generation sequence data: a partial overview. J Proteomics Bioinform. 2010;3:183–90. doi: 10.4172/jpb.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Botstein D, Risch N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003;33(Suppl):228–37. doi: 10.1038/ng1090. [DOI] [PubMed] [Google Scholar]
- 30.Ku C-S, Naidoo N, Pawitan Y. Revisiting Mendelian disorders through exome sequencing. Hum Genet. 2011;129:351–70. doi: 10.1007/s00439-011-0964-2. [DOI] [PubMed] [Google Scholar]
- 31.Lalonde E, Albrecht S, Ha KCH, et al. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum Mutat. 2010;31:918–23. doi: 10.1002/humu.21293. [DOI] [PubMed] [Google Scholar]
- 32.Parla JS, Iossifov I, Grabill I, et al. A comparative analysis of exome capture. Genome Biol. 2011;12:R97. doi: 10.1186/gb-2011-12-9-r97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lettice LA, Hill AE, Devenney PS, et al. Point mutations in a distant sonic hedgehog cis-regulator generate a variable regulatory output responsible for preaxial polydactyly. Hum Mol Genet. 2008;17:978–85. doi: 10.1093/hmg/ddm370. [DOI] [PubMed] [Google Scholar]
- 34.Marian AJ. Molecular genetic studies of complex phenotypes. Transl Res. 2012;159:64–79. doi: 10.1016/j.trsl.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Visscher PM, Brown MA, McCarthy MI, et al. Five years of GWAS discovery. Am J Hum Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–97. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 37.Kathiresan S, Srivastava D. Genetics of human cardiovascular disease. Cell. 2012;148:1242–57. doi: 10.1016/j.cell.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Day-Williams AG, Zeggini E. The effect of next-generation sequencing technology on complex trait research. Eur J Clin Invest. 2011;41:561–7. doi: 10.1111/j.1365-2362.2010.02437.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boyden LM, Choi M, Choate KA, et al. Mutations in kelch-like 3 and cullin 3 cause hypertension and electrolyte abnormalities. Nature. 2012;482:98–102. doi: 10.1038/nature10814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Norton N, Li D, Rieder MJ, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–82. doi: 10.1016/j.ajhg.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nejentsev S, Walker N, Riches D, et al. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science. 2009;324:387–9. doi: 10.1126/science.1167728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med. 2008;359:2143–53. doi: 10.1056/NEJMra0802968. [DOI] [PubMed] [Google Scholar]
- 43.Speicher MR, Geigl JB, Tomlinson IP. Effect of genome-wide association studies, direct-to-consumer genetic testing, and high-speed sequencing technologies on predictive genetic counselling for cancer risk. Lancet Oncol. 2010;11:890–8. doi: 10.1016/S1470-2045(09)70359-6. [DOI] [PubMed] [Google Scholar]
- 44.Chung CC, Chanock SJ. Current status of genome-wide association studies in cancer. Hum Genet. 2011;130:59–78. doi: 10.1007/s00439-011-1030-9. [DOI] [PubMed] [Google Scholar]
- 45.Ghoussaini M, Fletcher O, Michailidou K, et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nat Genet. 2012;44:312–8. doi: 10.1038/ng.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walsh T, King M-C. Ten genes for inherited breast cancer. Cancer Cell. 2007;11:103–5. doi: 10.1016/j.ccr.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 47.Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet. 2010;42:410–4. doi: 10.1038/ng.569. [DOI] [PubMed] [Google Scholar]
- 48.Jabbour E, Fava C, Kantarjian H. Advances in the biology and therapy of patients with chronic myeloid leukaemia. Best Pract Res Clin Haematol. 2009;22:395–407. doi: 10.1016/j.beha.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 49.Walther A, Johnstone E, Swanton C, et al. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–99. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 50.Mardis ER. Next-generation DNA sequencing methods. Annu Rev Genomics Hum Genet. 2008;9:387–402. doi: 10.1146/annurev.genom.9.081307.164359. [DOI] [PubMed] [Google Scholar]
- 51.Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 52.Margulies M, Egholm M, Altman WE, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437:376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Medvedev P, Stanciu M, Brudno M. Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009;6:S13–20. doi: 10.1038/nmeth.1374. [DOI] [PubMed] [Google Scholar]
- 54.Dai M, Thompson RC, Maher C, et al. NGSQC: cross-platform quality analysis pipeline for deep sequencing data. BMC Genomics. 2010;11(Suppl 4):S7. doi: 10.1186/1471-2164-11-S4-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cox MP, Peterson DA, Biggs PJ. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010;11:485. doi: 10.1186/1471-2105-11-485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dohm JC, Lottaz C, Borodina T, et al. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 2008;36:e105. doi: 10.1093/nar/gkn425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmieder R, Edwards R. Quality control and preprocessing of metagenomic datasets. Bioinformatics. 2011;27:863–4. doi: 10.1093/bioinformatics/btr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cibulskis K, McKenna A, Fennell T, et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics. 2011;27:2601–2. doi: 10.1093/bioinformatics/btr446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Blankenberg D, Gordon A, Von Kuster G, et al. Manipulation of FASTQ data with Galaxy. Bioinformatics. 2010;26:1783–5. doi: 10.1093/bioinformatics/btq281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Planet E, Attolini CS-O, Reina O, et al. htSeqTools: high-throughput sequencing quality control, processing and visualization in R. Bioinformatics. 2012;28:589–90. doi: 10.1093/bioinformatics/btr700. [DOI] [PubMed] [Google Scholar]
- 61.Martínez-Alcántara A, Ballesteros E, Feng C, et al. PIQA: pipeline for Illumina G1 genome analyzer data quality assessment. Bioinformatics. 2009;25:2438–9. doi: 10.1093/bioinformatics/btp429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dolan PC, Denver DR. TileQC: a system for tile-based quality control of Solexa data. BMC Bioinformatics. 2008;9:250. doi: 10.1186/1471-2105-9-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schmieder R, Lim YW, Rohwer F, et al. TagCleaner: Identification and removal of tag sequences from genomic and metagenomic datasets. BMC Bioinformatics. 2010;11:341. doi: 10.1186/1471-2105-11-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Raney BJ, Cline MS, Rosenbloom KR, et al. ENCODE whole-genome data in the UCSC genome browser (2011 update) Nucleic Acids Res. 2011;39:D871–5. doi: 10.1093/nar/gkq1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.The Genome Reference Consortium. http://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc. [Google Scholar]
- 66.Genome Bioinformatics Group (UCSC). Comparison of UCSC and NCBI human assemblies. http://genome.ucsc.edu/FAQ/FAQreleases.html#release4. [Google Scholar]
- 67.Yu X, Guda K, Willis J, et al. How do alignment programs perform on sequencing data with varying qualities and from repetitive regions? BioData Mining. 2012;5:6. doi: 10.1186/1756-0381-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8. doi: 10.1101/gr.078212.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alkan C, Kidd JM, Marques-Bonet T, et al. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat Genet. 2009;41:1061–7. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li R, Yu C, Li Y, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–7. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 75.Ning Z, Cox AJ, Mullikin JC. SSAHA: a fast search method for large DNA databases. Genome Res. 2001;11:1725–9. doi: 10.1101/gr.194201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–9. doi: 10.1101/gr.111120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Galinsky VL. YOABS: yet other aligner of biological sequences—an efficient linearly scaling nucleotide aligner. Bioinformatics. 2012;28:1070–7. doi: 10.1093/bioinformatics/bts102. [DOI] [PubMed] [Google Scholar]
- 78.Ruffalo M, LaFramboise T, Koyutürk M. Comparative analysis of algorithms for next-generation sequencing read alignment. Bioinformatics. 2011;27:2790–6. doi: 10.1093/bioinformatics/btr477. [DOI] [PubMed] [Google Scholar]
- 79.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burrows M, Wheeler DJ. A block sorting lossless data compression algorithm. Technical Report 124, Digital Equipment Corporation. 1994 [Google Scholar]
- 81.Lee H, Schatz MC. Genomic Dark Matter: The reliability of short read mapping illustrated by the Genome Mappability Score. Bioinformatics. 2012;28:2097–105. doi: 10.1093/bioinformatics/bts330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim SY, Li Y, Guo Y, et al. Design of association studies with pooled or un-pooled next-generation sequencing data. Genet Epidemiol. 2010;34:479–91. doi: 10.1002/gepi.20501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Neuman JA, Isakov O, Shomron N. Analysis of insertion-deletion from deep-sequencing data: software evaluation for optimal detection. Brief Bioinformatics. 2013;14:46–55. doi: 10.1093/bib/bbs013. [DOI] [PubMed] [Google Scholar]
- 84.Sathirapongsasuti JF, Lee H, Horst BAJ, et al. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–54. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nielsen CB, Cantor M, Dubchak I, et al. Visualizing genomes: techniques and challenges. Nat Methods. 2010;7:S5–15. doi: 10.1038/nmeth.1422. [DOI] [PubMed] [Google Scholar]
- 86.Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Darzentas N. Circoletto: visualizing sequence similarity with Circos. Bioinformatics. 2010;26:2620–1. doi: 10.1093/bioinformatics/btq484. [DOI] [PubMed] [Google Scholar]
- 88.O’Brien TM, Ritz AM, Raphael BJ, et al. Gremlin: an interactive visualization model for analyzing genomic rearrangements. IEEE Trans Vis Comput Graph. 2010;16:918–26. doi: 10.1109/TVCG.2010.163. [DOI] [PubMed] [Google Scholar]
- 89.Wang J, Kong L, Gao G, et al. A brief introduction to web-based genome browsers. Brief Bioinformatics. 2013;14:131–43. doi: 10.1093/bib/bbs029. [DOI] [PubMed] [Google Scholar]
- 90.Cline MS, Kent WJ. Understanding genome browsing. Nat Biotechnol. 2009;27:153–5. doi: 10.1038/nbt0209-153. [DOI] [PubMed] [Google Scholar]
- 91.Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Schossig A, Wolf NI, Fischer C, et al. Mutations in ROGDI cause Kohlschütter-Tönz syndrome. Am J Hum Genet. 2012;90:701–7. doi: 10.1016/j.ajhg.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xi R, Hadjipanayis AG, Luquette LJ, et al. Copy number variation detection in whole-genome sequencing data using the Bayesian information criterion. Proc Natl Acad Sci USA. 2011;108:E1128–36. doi: 10.1073/pnas.1110574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spector MS, Iossifov I, Kritharis A, et al. Mast-cell leukemia exome sequencing reveals a mutation in the IgE mast-cell receptor β chain and KIT V654A. Leukemia. 2012;26:1422–5. doi: 10.1038/leu.2011.354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ju YS, Lee W-C, Shin J-Y, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–45. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wu TD, Nacu S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics. 2010;26:873–81. doi: 10.1093/bioinformatics/btq057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299. [Google Scholar]
- 98.Bansal V. A statistical method for the detection of variants from next-generation resequencing of DNA pools. Bioinformatics. 2010;26:i318–24. doi: 10.1093/bioinformatics/btq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wei Z, Wang W, Hu P, et al. SNVer: a statistical tool for variant calling in analysis of pooled or individual next-generation sequencing data. Nucleic Acids Res. 2011;39:e132. doi: 10.1093/nar/gkr599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang W, Hu W, Hou F, et al. SNVerGUI: a desktop tool for variant analysis of next-generation sequencing data. J Med Genet. 2012 doi: 10.1136/jmedgenet-2012-101001. [DOI] [PubMed] [Google Scholar]
- 102.Koboldt DC, Zhang Q, Larson DE, et al. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–76. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Larson DE, Harris CC, Chen K, et al. SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics. 2012;28:311–7. doi: 10.1093/bioinformatics/btr665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Abyzov A, Urban AE, Snyder M, et al. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84. doi: 10.1101/gr.114876.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li J, Lupat R, Amarasinghe KC, et al. CONTRA: copy number analysis for targeted resequencing. Bioinformatics. 2012;28:1307–13. doi: 10.1093/bioinformatics/bts146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yoon S, Xuan Z, Makarov V, et al. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–92. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen K, Wallis JW, McLellan MD, et al. BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009;6:677–81. doi: 10.1038/nmeth.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sun R, Love MI, Zemojtel T, et al. Breakpointer: using local mapping artifacts to support sequence breakpoint discovery from single-end reads. Bioinformatics. 2012;28:1024–5. doi: 10.1093/bioinformatics/bts064. [DOI] [PubMed] [Google Scholar]
- 109.Marschall T, Costa I, Canzar S, et al. CLEVER: clique-enumerating variant finder. Bioinformatics. 2012;28(22):2875–288. doi: 10.1093/bioinformatics/bts566. [DOI] [PubMed] [Google Scholar]
- 110.Sindi SS, Onal S, Peng L, et al. An integrative probabilistic model for identification of structural variation in sequencing data. Genome Biol. 2012;13:R22. doi: 10.1186/gb-2012-13-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sindi S, Helman E, Bashir A, et al. A geometric approach for classification and comparison of structural variants. Bioinformatics. 2009;25:i222–30. doi: 10.1093/bioinformatics/btp208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong K, Keane TM, Stalker J, et al. Enhanced structural variant and breakpoint detection using SVMerge by integration of multiple detection methods and local assembly. Genome Biol. 2010;11:R128. doi: 10.1186/gb-2010-11-12-r128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kalender Atak Z, De Keersmaecker K, Gianfelici V, et al. High accuracy mutation detection in leukemia on a selected panel of cancer genes. PLoS ONE. 2012;7:e38463. doi: 10.1371/journal.pone.0038463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Makarov V, O’Grady T, Cai G, et al. AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics. 2012;28:724–5. doi: 10.1093/bioinformatics/bts032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grant JR, Arantes AS, Liao X, et al. In-depth annotation of SNPs arising from resequencing projects using NGS-SNP. Bioinformatics. 2011;27:2300–1. doi: 10.1093/bioinformatics/btr372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ge D, Ruzzo EK, Shianna KV, et al. SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27:1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cingolani P, Patel VM, Coon M, et al. Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front Genet. 2012;3:35. doi: 10.3389/fgene.2012.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Medina I, De Maria A, Bleda M, et al. VARIANT: command line, web service and web interface for fast and accurate functional characterization of variants found by next-generation sequencing. Nucleic Acids Res. 2012;40:W54–8. doi: 10.1093/nar/gks572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP effect predictor. Bioinformatics. 2010;26:2069–70. doi: 10.1093/bioinformatics/btq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Davydov EV, Goode DL, Sirota M, et al. Identifying a high fraction of the human genome to be under selective constraint using GERP++ PLoS Comput Biol. 2010;6:e1001025. doi: 10.1371/journal.pcbi.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chun S, Fay JC. Identification of deleterious mutations within three human genomes. Genome Res. 2009;19:1553–61. doi: 10.1101/gr.092619.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schwarz JM, Rödelsperger C, Schuelke M, et al. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 124.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Pollard KS, Hubisz MJ, Rosenbloom KR, et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110–21. doi: 10.1101/gr.097857.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 127.Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862–4. doi: 10.1126/science.185.4154.862. [DOI] [PubMed] [Google Scholar]
- 129.Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Loraine AE, Helt GA. Visualizing the genome: techniques for presenting human genome data and annotations. BMC Bioinformatics. 2002;3:19. doi: 10.1186/1471-2105-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Spudich GM, Fernández-Suárez XM. Touring Ensembl: a practical guide to genome browsing. BMC Genomics. 2010;11:295. doi: 10.1186/1471-2164-11-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Dreszer TR, Karolchik D, Zweig AS, et al. The UCSC Genome Browser database: extensions and updates 2011. Nucleic Acids Res. 2012;40:D918–23. doi: 10.1093/nar/gkr1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Loveland J. VEGA, the genome browser with a difference. Brief Bioinformatics. 2005;6:189–93. doi: 10.1093/bib/6.2.189. [DOI] [PubMed] [Google Scholar]
- 135.Carver T, Harris SR, Berriman M, et al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–9. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Carver T, Harris SR, Otto TD, et al. BamView: visualizing and interpretation of next-generation sequencing read alignments. Brief Bioinformatics. 2013;14:203–12. doi: 10.1093/bib/bbr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinformatics. 2013;14:178–92. doi: 10.1093/bib/bbs017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fiume M, Williams V, Brook A, et al. Savant: genome browser for high-throughput sequencing data. Bioinformatics. 2010;26:1938–44. doi: 10.1093/bioinformatics/btq332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lam HYK, Pan C, Clark MJ, et al. Detecting and annotating genetic variations using the HugeSeq pipeline. Nat Biotechnol. 2012;30:226–9. doi: 10.1038/nbt.2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fischer M, Snajder R, Pabinger S, et al. SIMPLEX: cloud-enabled pipeline for the comprehensive analysis of exome sequencing data. PLoS ONE. 2012;7:e41948. doi: 10.1371/journal.pone.0041948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Asmann YW, Middha S, Hossain A, et al. TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. 2012;28:277–8. doi: 10.1093/bioinformatics/btr612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goecks J, Nekrutenko A, Taylor J. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Rex DE, Ma JQ, Toga AW. The LONI pipeline processing environment. Neuroimage. 2003;19:1033–48. doi: 10.1016/s1053-8119(03)00185-x. [DOI] [PubMed] [Google Scholar]
- 144.Hull D, Wolstencroft K, Stevens R, et al. Taverna: a tool for building and running workflows of services. Nucleic Acids Res. 2006;34:W729–32. doi: 10.1093/nar/gkl320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mills RE, Walter K, Stewart C, et al. Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Cooper GM, Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat Rev Genet. 2011;12:628–40. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 147.Gilissen C, Hoischen A, Brunner HG, et al. Disease gene identification strategies for exome sequencing. Eur J Hum Genet. 2012;20:490–7. doi: 10.1038/ejhg.2011.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 149.Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci USA. 2010;107:961–8. doi: 10.1073/pnas.0912629107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mardis ER. Genome sequencing and cancer. Curr Opin Genet Dev. 2012;22:245–50. doi: 10.1016/j.gde.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hindorff LA, Gillanders EM, Manolio TA. Genetic architecture of cancer and other complex diseases: lessons learned and future directions. Carcinogenesis. 2011;32:945–54. doi: 10.1093/carcin/bgr056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Mathe E, Olivier M, Kato S, et al. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Res. 2006;34:1317–25. doi: 10.1093/nar/gkj518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wei Q, Wang L, Wang Q, et al. Testing computational prediction of missense mutation phenotypes: functional characterization of 204 mutations of human cystathionine beta synthase. Proteins. 2010;78:2058–74. doi: 10.1002/prot.22722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lindblom A, Robinson PN. Bioinformatics for human genetics: promises and challenges. Hum Mutat. 2011;32:495–500. doi: 10.1002/humu.21468. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.