

Significance
Mitochondria play a vital role in cellular life. Understanding how mitochondria are regulated may uncover new ways to restore mitochondrial function in disease states. Here we use an unbiased, pooled ORFeome library screening strategy to identify 94 candidate mitochondrial regulator genes. We show that one of the candidate genes, glioma tumor-suppressor candidate region gene 2 (GLTSCR2), controls mitochondrial function in cultured cells and in Caenorhabditis elegans. The transcription factor Myc is a key downstream mediator of GLTSCR2 signaling. Furthermore, GLTSCR2 is itself induced by mitochondrial stress, pointing to a novel mitochondrial signaling pathway.
Abstract
Mitochondrial defects underlie a multitude of human diseases. Genetic manipulation of mitochondrial regulatory pathways represents a potential therapeutic approach. We have carried out a high-throughput overexpression screen for genes that affect mitochondrial abundance or activity using flow-cytometry–based enrichment of a cell population expressing a high-complexity, concentration-normalized pool of human ORFs. The screen identified 94 candidate mitochondrial regulators including the nuclear protein GLTSCR2, also known as PICT1. GLTSCR2 enhances mitochondrial function and is required for the maintenance of oxygen consumption, consistent with a pivotal role in the control of cellular respiration. RNAi inactivation of the Caenorhabditis elegans ortholog of GLTSCR2 reduces respiration in worms, indicating functional conservation across species. GLTSCR2 controls cellular proliferation and metabolism via the transcription factor Myc, and is induced by mitochondrial stress, suggesting it may constitute a significant component of the mitochondrial signaling pathway.
In the eukaryotic cell, mitochondria generate energy to support cellular life and regulate diverse processes such as apoptosis and calcium signaling. Mitochondrial insufficiency can carry deleterious consequences, including impaired oxidative phosphorylation (OXPHOS) and reduced ATP synthesis, which can culminate in human disease (1, 2). For example, respiratory chain disorders can be caused by inherited or spontaneous mutations in mitochondrial DNA or nuclear genes that encode respiratory chain subunits. Defects in oxidative phosphorylation may also occur as a secondary effect of mutations in genes encoding mitochondrial proteins involved in other aspects of mitochondrial physiology. Mitochondrial disorders commonly exhibit tissue selectivity and clinical heterogeneity, which may reflect varying bioenergetics thresholds of different cell types, intrinsic complexities of mitochondrial genetics and biochemistry, and environmental influences that introduce further variability. In addition to the primary mitochondrial disorders, mitochondrial dysfunction is implicated in a broad spectrum of age-related diseases such as neurodegeneration, metabolic syndrome, and cancer (1, 2). That mitochondrial defects feature so prominently in a wide range of disease processes points to the potential utility of targeting this organelle for therapeutic purposes.
One possible approach to compensate for inherited or acquired mitochondrial respiratory defects may be to actively induce mitochondrial OXPHOS capacity. Several recent studies with mouse models of defective cytochrome c-oxidase activity have reported beneficial effects of genetic or pharmacological manipulations that enhance OXPHOS activity (3, 4). Such manipulations help preserve ATP levels in mutant mouse tissues and in cultured cells from human patients, and have been observed to improve motor function in mice deficient in cytochrome c oxidase genes. Endurance exercise, which has been shown to counteract the accelerated aging phenotype in the PolG mitochondrial mutator mice, restores mitochondrial abundance and cytochrome c-oxidase activity (5). These results suggest that partial restoration of mitochondrial function in some disease states may be achievable by harnessing endogenous regulatory pathways controlling mitochondrial biogenesis or activity. It thus becomes worthwhile to ascertain which specific cellular pathways may be experimentally exploited for this purpose.
To address this issue, we carried out an unbiased, large-scale gain-of-function genetic screen to identify genes whose overproduction can enhance mitochondrial abundance or activity. Because traditional cDNA libraries commonly suffer from incomplete clones and skewed gene representation, we used the human ORFeome (hORFeome v5.1), a normalized collection of 15,483 human ORFs in the Gateway cloning system (6, 7). To enable a high-throughput screening platform, we adapted the ORFeome library for use in a pooled format, allowing simultaneous evaluation of all ORFs, and deployed fluorescent mitochondria-selective probes as reporters of mitochondrial activity or abundance in live cells. We have identified 76 candidate genes that increase the mitochondrial reporter signals upon overexpression and 18 genes that have the opposite effect. These genes encode secreted factors, transcription factors, as well as predicted polypeptides of unknown function, and represent a resource that will facilitate further efforts to understand and manipulate mitochondrial regulatory mechanisms.
Results
A Genome-Scale Gain-of-Function Screen for Mitochondrial Regulators.
Our strategy was to perform a high-throughput flow cytometry-based mitochondrial screen with an expression library containing the human ORFeome collection (Fig. 1). Whereas large-scale RNAi screens have become commonplace in recent years, aided in part by the ease of transfecting siRNA oligonucleotides into most cell types, genome-scale gain-of-function screens have been hampered by the relative difficulty of introducing expression plasmids into mammalian cells at a high efficiency. We transferred the entire human ORFeome collection as a pool into a lentiviral expression vector (pHAGE T-Rex) to ensure efficient gene delivery and stable expression. The use of pooled ORF clones for lentiviral packaging and transduction was compatible with rapid, phenotype-based selection of cell subpopulations that we designed to be a central feature of the screen, as opposed to testing individual ORF clones arrayed in a microwell plate format. The human ORFeome offers superior gene representation relative to traditional genomic cDNA libraries and the viral titers are largely preserved across different ORF sizes ranging from 75 bp to 10.5 kb, with a median ORF size of 1 kb. We selected the viral transduction conditions that would provide an approximately 1,000-fold coverage of each ORF in the cell population being screened. The frequency of multiple integration events was reduced by using a relatively low multiplicity of infection (MOI = 1) at the time of lentiviral transduction and is mitigated by the representation of 1,000 for each given viral construct.
Fig. 1.

Schematic diagram of the genome-scale overexpression screen. The human ORFeome collection was cloned into the lentiviral vector pHAGE T-Rex by Gateway cloning and packaged into viruses. C2C12 mouse myoblast cells were transduced with the viruses, selected, and stained with NAO and MitoTracker deep red dyes for FACS sorting. Microarray hybridization was used to compare the genomic DNA samples collected from high NAO, high MT-DR cells (shown as grayed out areas in the FACS profiles) vs. the unsorted cells. Similarly, the low NAO, low MT-DR cells and the unsorted cells were compared.
We performed the screen in C2C12 myoblast cells, which are a well-characterized, nontransformed cellular system that has been extensively used for studies of mitochondrial regulation in cultured cells. Following lentiviral transduction, antibiotic selection, and propagation for 3 days, we carried out flow-cytometry–based cell sorting in conjunction with mitochondria-selective fluorescent reporters to enrich for genes that increase or reduce mitochondrial abundance or activity. Live cells were stained with two different mitochondria-selective probes, nonyl acridine orange (NAO) and MitoTracker deep red (MT-DR), and were then sorted to isolate subpopulations exhibiting concordant changes in the signal intensities of both reporters, thus increasing specificity. These two dyes have previously been found to be sensitive to both mitochondrial abundance and membrane potential (8–10). We confirmed that C2C12 cells expressing the mitochondrial biogenesis regulator peroxisome proliferator-activated receptor γ coactivator-1α displayed higher signal intensities of the two reporter dyes (Fig. S1).
We subsequently harvested genomic DNA from the high signal (overlap between the top 5% based on NAO fluorescence and the top 5% based on MT-DR fluorescence) and low signal (bottom 5% in NAO fluorescence and in MT-DR fluorescence) fractions collected by cell sorting and compared each of these fractions to the unsorted fraction by PCR amplifying and labeling the DNA samples with Cy3 and Cy5, and hybridizing competitively to custom microarrays. Following deconvolution of the microarray data, the ratio of the abundance of each ORF in the high signal fraction versus the unsorted fraction was used to identify a set of genes that were consistently enriched by more than 2.5-fold in two independent experiments. These genes represented potential positive regulators of mitochondrial abundance or activity. Likewise, we compared the enrichment in the low signal fraction versus the unsorted fraction to identify potential negative regulators. The candidate genes were then tested individually by lentivirally mediated stable expression in C2C12 cells, which were assayed directly for NAO and MT-DR signals. We scored candidate genes as confirmed hits if there was more than a 20% increase in either NAO or MT-DR signal relative to cells transduced with the empty control vector. Based on these criteria, we obtained 76 positive mitochondrial regulators and 18 negative regulators (Tables S1 and S2).
These confirmed genes are a functionally diverse group. Based on the PANTHER gene ontology system, the majority of these genes were classified as being involved in metabolic processes, cellular processes, and cellular communication. Represented protein classes included hydrolases, nucleic acid binding proteins, transferases, and transcription factors. Of interest were genes linked to positive regulation of cell communication (HTR2B, PTGS2, BCL10, ZDHHC13, GPC3, and ALS2), calcium ion transport and signaling (CACNA2D1, CASQ1, PLCG2, and PTGS2), and cellular stress response (NFE2L2 and EIF2AK4), each of which may have significant connections to mitochondrial physiology. The composition of the validated gene subset is consistent with the idea that mitochondrial abundance and activity are subject to modulation by a myriad of external effector signals and intracellular signaling cascades converging upon metabolic pathways.
Measurements of Mitochondrial Membrane Potential and Assessment of Loss-of-Function Phenotypes.
To further examine the effects of these 94 reconfirmed candidates on mitochondrial physiology, we measured gene overexpression-induced changes in mitochondrial membrane potential using the potentiometric dye tetramethylrhoadamine methyl ester (TMRM), which distributes within polarized mitochondria in a Nernstian fashion. The majority of the genes we tested produced only small alterations in mitochondrial membrane potential relative to the control, with less than a 20% change in magnitude (Fig. 2 A and B). We suspect that one reason for this low percentage may be that genes causing much larger changes in mitochondrial membrane potential may have reduced cell growth and viability. Dissipation of mitochondrial membrane potential interferes with ATP production and is often seen in early apoptosis, whereas high mitochondrial membrane potential may excessively increase reactive oxygen species production. In contrast, the NAO and MT-DR signals showed much wider variations (Fig. 2 C and D). Whereas nearly all of the confirmed genes showed simultaneous increases or decreases in both NAO and MT-DR signal intensities, as expected from the constraints imposed at the time of cell sorting, the correlation between the two reporters is not strong (r = 0.10), suggesting significant differences in the properties of the two reporters (Fig. 2E).
Fig. 2.

Further analysis of the confirmed hits from the screen using secondary assays. (A) Distribution of TMRM signals among the 94 confirmed hits expressed in C2C12 cells. The genes that produce less than a 20% change in the TMRM signal relative to the control cells are colored in a lighter shade. (B) Distribution of the NAO vs. TMRM signals among the 94 confirmed hits. (C) Distribution of NAO signals among the 94 confirmed hits. The genes whose overexpression produce less than a 20% change in the NAO signal relative to the control cells are colored in a lighter shade. (D) Distribution of MitoTracker deep red signals among the 94 confirmed hits. The genes that produce less than a 20% change in the MT-DR signal relative to the control cells are colored in a lighter shade. (E) Distribution of the NAO versus MitoTracker deep red signals among the 94 confirmed hits. (F) NAO signal values for the 11 genes that scored in the mini-RNAi screen in IMR90 cells with at least two of three independent siRNAs. (G) MitoTracker deep red signal values for the 11 genes that scored in the mini-RNAi screen in IMR90 cells with at least two of three independent siRNAs indicated by different colored bars.
To identify genes that may potentially serve as a control point in mitochondrial regulation, we also assessed loss-of-function phenotypes by small interfering RNA (siRNA)-based gene depletion in conjunction with flow cytometry. We reasoned that a given gene may be a key regulator of mitochondrial function in vivo if its depletion had the opposite effect of its overproduction. A mini-RNAi screen targeting the 94 confirmed hits was performed in IMR90 human primary fibroblast cells using siRNAs targeting the corresponding human orthologs, thus also evaluating functional conservation across distinct cell types and species. We considered a gene to score in the mini-RNAi screen if at least two of three independent siRNA oligonucleotides targeting the same gene produced a significant change in the NAO or MT-DR signal, compared with the control siRNA. Eight such genes produced a statistically significant (P < 0.05) loss-of-function phenotype that was the opposite of the gain-of-function phenotype noted in the ORFeome screen, and three came close (0.05 ≤ P < 0.1) (Fig. 2 F and G). Seven of these 11 genes (CASQ1, GLTSCR2, GPC3, MAT1A, SLC44A5, TGFBRAP1, and USP33) were predicted to be positive mitochondrial regulators, and the other four (CCDC33, CPNE2, TERF1, and WDR33) negative regulators. This subset of 11 genes included genes involved in calcium-mediated signaling, cell adhesion, transcription, proteolysis, and other cellular processes.
GLTSCR2 Regulates Mitochondrial Respiration.
We focused on glioma tumor-suppressor candidate region gene 2 (GLTSCR2, also known as PICT1), because it displayed consistency between the loss- and gain-of-function phenotypes and has previously been implicated in cancer (11). GLTSCR2 significantly increased both NAO and MT-DR signals when overexpressed in C2C12 cells in the ORFeome screen (Fig. 3A). siRNAs targeting GLTSCR2 decreased the NAO signals in the mini-RNAi screen we subsequently performed in IMR90 cells (Fig. 2F). These results suggested conservation of function across multiple cell types.
Fig. 3.

GLTSCR2 regulates mitochondrial respiration in primary human fibroblasts and in the nematode C. elegans. *P < 0.05 by unpaired t test; **P < 0.01. (A) Stable expression of GLTSCR2 cDNA in C2C12 cells increases both NAO and MitoTracker deep red signals. (B) Stable expression of GLTSCR2 cDNA in IMR90 cells increases oxygen consumption. (C) Depletion of GLTSCR2 in IMR90 cells by siRNA transfection reduces oxygen consumption. Cells were examined 4 d after transfection with the Seahorse XF24 flux analyzer. (D) Depletion of GLTSCR2 in IMR90 cells by stable shRNA expression reduces oxygen consumption, which can be rescued by expressing an RNAi-resistant GLTSCR2 cDNA. (E) Stable expression of GLTSCR2 cDNA in IMR90 cells increases cellular ATP content. (F) Depletion of GLTSCR2 in IMR90 cells by stable shRNA expression reduces cellular ATP content. (G) RNAi inactivation of the GLTSCR2 ortholog Y39B6A.33 in C. elegans reduces oxygen consumption.
For a more direct assessment of mitochondrial OXPHOS activity, we examined oxygen consumption rates in intact cells. Measurements of cellular oxygen consumption in IMR90 fibroblasts stably expressing GLTSCR2 showed a 25% enhanced respiration relative to the control (Fig. 3B). Depletion of GLTSCR2 in IMR90 fibroblasts by siRNA reduced oxygen consumption by 30% (Fig. 3C), and similar results were obtained with shRNA-based depletion (Fig. 3D). This phenotype could be rescued by expressing an shRNA-resistant cDNA (Fig. 3D and Fig. S2). The cellular ATP levels appeared to reflect the changes in respiration in IMR90 cells occurring upon stable overexpression of the GLTSCR2 cDNA or depletion by shRNA (Fig. 3 E and F).
These data indicate that GLTSCR2 acts to regulate respiration in mammalian cells but whether this function is evolutionarily conserved is unknown. To evaluate a potentially conserved role for GLTSCR2 across species, we assayed oxygen consumption rates of Caenorhabditis elegans worms undergoing RNAi inactivation of Y39B6.33, the C. elegans ortholog of GLTSCR2, and found lower oxygen consumption rates relative to the control RNAi worms (Fig. 3G). RNAi inactivation of another mitochondrial gene, T06D8.6, did not produce a change of similar magnitude, consistent with previous results (12). Together, these findings suggest that GLTSCR2 has a physiological role in regulating mitochondrial respiration that is functionally conserved across different species in an in vivo setting.
GLTSCR2 Controls Cellular Proliferation and Respiration via the Transcription Factor Myc.
The role of GLTSCR2 in cancer appears to be complex and may depend upon cellular context. GLTSCR2 was initially proposed to be a potential tumor suppressor based on its location on chromosome 19q13.32, which is frequently deleted in human tumors, particularly in gliomas, and its interaction with phosphatase and tensin homolog (PTEN) (13, 14). Overexpression of GLTSCR2 in glioblastoma cell lines was reported to induce apoptotic cell death (15). However, it was recently suggested that GLTSCR2 promotes oncogenesis based on the observation that the GLTSCR2+/− mice were more resistant to chemically induced skin cancers (11). GLTSCR2 null ES cells accumulated p53 and underwent apoptosis, and shRNA-mediated depletion of GLTSCR2 in cancer cell lines induced p53 as well (11). This group also noted that lower expression of the GLTSCR2 mRNA transcript correlated with improved survival in some colorectal and esophageal cancers with intact p53.
To further investigate a possible role for GLTSCR2 in cancer, we examined the effect of overexpressing or depleting GLTSCR2 in IMR90 primary fibroblasts. Unexpectedly, we found that siRNA-mediated depletion of GLTSCR2 significantly reduced p53 levels in IMR90 primary cells, as did stable expression of an shRNA targeting a different region of the GLTSCR2 gene (Fig. 4A). Stable overexpression of GLTSCR2 induced p53 (Fig. 4B). Despite elevated basal levels of p53, the GLTSCR2-overexpressing IMR90 cells proliferated substantially faster than the control cells, implying a potential involvement of a growth-promoting effector protein (Fig. 4C). We thus considered possible downstream target proteins that may impact upon cell proliferation and mitochondrial respiration.
Fig. 4.
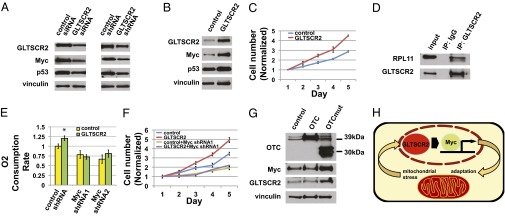
GLTSCR2 responds to mitochondrial stress and controls cell proliferation and respiration via Myc. (A) Depletion of GLTSCR2 in IMR90 cells by siRNA transfection (Left) or by stable shRNA expression (Right) induces Myc and p53 levels. Protein samples were isolated 72 h after siRNA transfection. (B) Stable expression of GLTSCR2 in IMR90 cells induces Myc and p53 levels. (C) GLTSCR2 increases cell proliferation in IMR90 cells. Cells were counted in triplicates every day for 5 consecutive days. (D) GLTSCR2 interacts with RPL11. The 293T-Rex cells stably carrying HA-GLTSCR2 under a tetracycline-inducible promoter were treated with doxycycline for 24 h and the lysates were immunoprecipitated with anti-HA antibody or IgG. (E) Depletion of Myc in IMR90 cells by stable shRNA expression eliminates GLTSCR2-mediated increases in oxygen consumption. (F) Depletion of Myc in IMR90 cells by stable shRNA expression abolishes GLTSCR2-mediated increases in cell proliferation. Cells were counted in triplicates every day for 5 consecutive days. (G) Expression of mutant OTC in IMR90 cells induces GLTSCR2 and Myc expression. The wild-type OTC protein runs at 39 kDa, whereas the deletion mutant lacking amino acids 30–114 runs at 30 kDa. (H) Model of mitochondrial stress signaling involving GLTSCR2 and Myc.
GLTSCR2 was previously shown to physically interact with RPL11 and sequester it in the nucleolus (11). Others have reported that RPL11 inhibits Myc activity by competing with Myc coactivators at the target gene promoters and also reduces Myc levels by binding and destabilizing the Myc mRNA (16–18). This suggested that GLTSCR2 may regulate Myc. We observed that overexpression of GLTSCR2 in IMR90 cells induces Myc protein levels, whereas depletion of GLTSCR2 by RNAi reduced Myc levels in these cells (Fig. 4 A and B). We also confirmed the interaction between GLTSCR2 and RPL11 (Fig. 4D).
Furthermore, depletion of Myc by RNAi abrogated GLTSCR2-mediated effects on cell proliferation and oxygen consumption in IMR90 cells (Fig. 4 E and F and Fig. S3). Myc activation is known to induce p53 in normal human fibroblasts via p14 alternate reading frame (p14ARF)-independent mechanisms, which may explain the concomitant increase in p53 levels in GLTSCR2-expressing IMR90 cells (19). These results argue that the induction of Myc is a key downstream event in GLTSCR2 signaling and that GLTSCR2 may act to promote proliferation and possibly oncogenesis through Myc.
GLTSCR2 Is Regulated by Mitochondrial Stress.
GLTSCR2 is primarily localized to the nucleolus, where the production of ribosomal subunits must be carefully coordinated with changing cellular needs and external signals (20). The nucleolus has increasingly become recognized as a sensor and integrator for several forms of cellular stress (21). We were therefore interested to know if GLTSCR2 could be subject to regulation by stresses emanating from mitochondria. Impaired mitochondrial function is known to activate stress response pathways that signal to the nucleus and set off nuclear changes (22). An important example of mitochondrial stress signaling is triggered by misfolded mitochondrial proteins, analogous to the unfolded protein response activated in the endoplasmic reticulum in response to proteotoxic stress in that organelle. In mammalian cells, the mitochondrial unfolded protein response has been primarily studied using overexpression of a deletion mutant form of the mitochondrial matrix protein ornithine transcarbamylase (OTC) (23). We found that GLTSCR2 is induced by overexpression of the mutant OTC with a concomitant induction of Myc (Fig. 4G). This suggests the possibility that GLTSCR2 and Myc may be part of the mitochondrial unfolded protein response.
Together, our data are consistent with the model that mitochondrial stress signaling pathways activate the GLTSCR2–Myc axis by mechanisms yet to be determined, resulting in adaptive responses (Fig. 4H). In this context, it is noteworthy that Myc is known to enhance mitochondrial respiration, and importantly, also to stimulate glycolysis, which can provide an alternative source of energy for cellular needs even when oxidative phosphorylation remains impaired (24).
Discussion
Mitochondrial function is central to cellular physiology and human health. Understanding how mitochondria are regulated is therefore a major research goal in cell biology. Here we have performed a gain-of-function genetic screen for proteins that alter mitochondrial abundance or function. Using the human ORFeome as a pool of high complexity allowed unbiased, simultaneous screening of tens of millions of lentiviral particles carrying some 15,483 human ORFs in the target cell population. The lentiviral expression system provided highly efficient transduction and long-term gene expression. The strategies for enrichment, based on the fluorescent intensities of mitochondria-selective reporters, and the microarray-based strategies for recovery of the ORF identities produced a set of 94 candidate proteins involved in mitochondrial regulation.
Among the candidate proteins identified in the screen, we focused on the nuclear protein GLTSCR2 and have demonstrated that it controls mitochondrial respiration in primary human cells. Depletion of GLTSCR2 reduces mitochondrial respiration, whereas increasing its levels enhances respiration. The role of GLTSCR2 in regulating oxygen consumption is conserved across evolution as reduction of the GLTSCR2 ortholog in C. elegans also lowers oxygen consumption. Thus, GLTSCR2 possesses several of the hallmarks of a physiological regulator of mitochondrial respiration in mammals.
How GLTSCR2 executes its regulation of mitochondria is a key question. Our observations point to Myc as a main link between GLTSCR2 and its effects on cellular respiration and proliferation. Furthermore, the GLTSCR2 and Myc protein levels are regulated by mitochondrial proteotoxic stress. Consistent with our observations, Myc has previously been reported to be up-regulated in response to mitochondrial dysfunction associated with depletion of mitochondrial DNA in mammalian cell lines (25, 26). Other studies have suggested that interfering with Myc’s activity may contribute to mitochondrial dysfunction in aging. Myc controls mitochondrially encoded OXPHOS genes by directly activating mitochondrial transcription factor A, and a pseudohypoxic state induced during aging may cause a decline in mitochondrial function via disruption of Myc’s activity (27).
The induction of Myc in conditions of mitochondrial stress may serve an adaptive function of stimulating glucose uptake and glycolysis to compensate for the impairment of oxidative phosphorylation, while also attempting to restore mitochondrial activity. Previous studies have suggested that GLTSCR2 is induced by genotoxic stressors, such as ionizing radiation and UV radiation, and low-dose actinomycin D, which is thought to cause ribosomal stress by inhibiting rRNA synthesis (28, 29). It is possible that some of these agents may also impinge upon mitochondrial function and activate pathways leading to the induction of GLTSCR2.
It remains unknown how mitochondrial stress regulates the GLTSCR2 levels. The mitochondrial unfolded protein response has been primarily associated with misfolded mitochondrial proteins, but it is now known that this pathway can be activated by multiple types of mitochondrial stress that perturb the balance of mitochondrial proteins, such as loss of mitochondrial DNA, deficiencies in certain electron transport proteins, or disruption of the mitochondrial import machinery (30). In the nematode C. elegans, the mitochondrial unfolded protein signaling is apparently required for the lifespan-extending effect of electron transport chain defects in that organism (31). The mitochondrial unfolded protein response in mammalian cells remains poorly understood at a mechanistic level, representing a wealth of opportunities to obtain new insights into this crucial signaling pathway. Overall, much remains to be learned about the signaling pathways to and from mitochondria, and as the present study illustrates, systematic screens for the identification of regulatory proteins in the mitochondrial signaling networks provide a potentially useful approach.
Materials and Methods
Constructs and Reagents.
Antibodies were purchased from commercial vendors including GLTSCR2 (Abnova), p53 (EMD), Myc (Cell Signaling), RPL11 (Abcam), OTC (Novus), and vinculin (Sigma).
Cell Culture.
C2C12 cells (American Type Culture Collection) were maintained in DMEM containing 10% (vol/vol) FCS (Invitrogen). IMR90 cells were maintained in DMEM with 10% FCS in a low-oxygen (3%) incubator.
Lentiviral Library Screening.
The human ORFeome collection version 5.1 (hORFeome v5.1) was used for this study. The ORFs were divided into 10 separate subpools and transferred to the pHAGE T-Rex lentiviral expression vector via the LR recombinase reaction. The pHAGE T-Rex vector combines the promoter and the DEST cassette regions of pT-Rex DEST 30 (Life Technologies; 12301-016) and the backbone of the pHAGE-TRE-HA-Puro-DEST vector. The expression vectors containing the ORFs were transfected into 293T cells along with packaging plasmids to produce lentiviral supernatants. The viruses were collected 48 h after transfection, filtered with a 0.45-mm filter, and stored at −80 °C. The titers were determined for each subpool. Fifteen million C2C12 cells were transduced with pooled viruses in the presence of 5 μg/mL polybrene (Sigma) with an average representation of 1,000 cells per ORF at a MOI of 1. The viruses were removed after an overnight incubation. The transduced cell populations were selected with 2 μg/mL of puromycin and propagated until they reached a number of ∼100 million cells. Cells were then stained with NAO and MitoTracker deep red (Invitrogen) by incubation at 37 °C for 30 min, collected by trypsinization, and sorted by flow cytometry (BD FACSAria II) based on the NAO and MitoTracker signal intensities. The high signal (top 5% in both NAO and MT-DR) and the low signal (bottom 5% in both NAO and MT-DR) fractions were collected and genomic DNA was harvested from these samples as well as the unsorted cells.
Microarray Hybridization.
The ORF inserts were amplified from genomic DNA by PCR using Ex Taq HS (Takara), using the forward primer GATCCCTACCGGTGATATCC and the reverse primer TAATACGACTCACTATAGGGAGAC. The PCR products were used as templates for in vitro transcription with a MEGAscript kit (Ambion). The RNA probes were then labeled with Cy3 and Cy5 with a ULS labeling kit (Kreatech), column purified, fragmented at 60 °C, and hybridized to custom microarrays synthesized by Agilent. Each ORF was represented by an average of three different probes on the microarray and nearly all ORFs (>95%) were detected. Scanning and feature extraction were performed with an Agilent DNA microarray scanner (G2505C).
Validation Screen.
The individual ORFs were shuttled into the pHAGE T-Rex vector using LR Clonase. Viral supernatants were produced as described above and cells were stained with NAO and MitoTracker deep red, and analyzed with a BD LSRII flow cytometer. The NAO signal was collected using the FITC channel at 530 nm, whereas the MitoTracker deep red signal was collected using the APC channel.
Mini siRNA Screen.
siRNAs targeting the 94 confirmed hits were cherry picked from the Ambion Silencer Select Human Genome siRNA Library and were reformatted into 96-wells at the Harvard Medical School Institute for Chemistry and Chemical Biology. At least three distinct siRNAs targeted each gene. The siRNAs were transfected into IMR90 cells with Lipofectamine RNAiMax (Invitrogen) at a 15-nM final concentration. After 5 d, cells were stained with NAO and MitoTracker deep red, trypsinized, and analyzed with a BD LSRII flow cytometer.
Western Analysis.
Whole cell extracts were prepared by cell lysis, and equal amounts of lysates were resolved on SDS/PAGE, transferred to Immobilon-P membrane (Millipore), and probed with the appropriate antibodies. The proteins were visualized by ECL chemiluminscence (Pierce).
Oxygen Consumption Measurements in Cells and in C. elegans.
Real-time measurements of oxygen tension and pH in cultured cells were obtained in 24-well microplates using the XF24 flux analyzer (Seahorse Bioscience). The oxygen consumption rate in each well was calculated from these measurements and normalized to protein content from cell lysates. For the C. elegans studies, synchronized 1-d-old adult worms were fed bacteria expressing dsRNA targeting Y39B6A.33 or T06D8.6, or an RNAi control for 3 d. Culture plates contained 5-fluorodeoxyuridine to prevent egg hatching. Worms were washed off the plates with M9 buffer and pipetted into 24-microplates for oxygen consumption rate measurements with the XF24 flux analyzer. Each well contained 100 worms, with five replicate wells per condition. The oxygen consumption rate was normalized to protein content from worm lysates. Each experiment was performed twice.
ATP Measurements.
Cellular ATP content was determined using ATP Bioluminescence Assay kit HS (Roche) using quadruplicate samples.
Immunoprecipitation of Protein Complexes.
293T-Rex cells stably carrying HA-GLTSCR2 under a tetracycline-inducible promoter were treated for 24 h with 2 μg/mL doxycycline, washed with PBS, and lysed in 1× lysis buffer (20 mM Tris⋅HCl at pH 8, 137 mM NaCl, 10% glycerol, 1% Nonidet P-40, 2 mM EDTA). Immunoprecipitation was performed with anti-HA antibody or mouse IgG conjugated to agarose beads (Sigma).
Gene Overexpression and Depletion.
For stable lentiviral expression of cDNAs or shRNAs, viral supernants were prepared by transfecting 293E cells with packaging plasmids as described above. The shRNA hairpin inserts included a nontargeting control sequence (CCTAAGGTTAAGTCGCCCTCG), GLTSCR2 shRNA (AAGTCCAGAAGAAGTCACTGC), Myc shRNA1 (TTGAGGCAGTTTACATTATGG), and Myc shRNA2 (TTTAAGGATAACTACCTTGGG). For transient gene depletion, nontargeting control siRNA (d-001210-02-05) and GLTSCR2 siRNA were purchased from Dharmacon and transfected into cells with Lipofectamine RNAiMax at a 15-nM concentration. The protein and gene expression were analyzed 3 d following siRNA transfection.
Cell Growth Measurement.
IMR90 cells were seeded at 75,000 cells per 60-mm dish and counted in triplicates each day for 5 consecutive days using a Z2 Coulter cell counter.
Supplementary Material
Acknowledgments
We thank the Institute of Chemistry and Cell Biology for assistance with the mini-RNAi screening. This work was supported by K08DK081612 (to J.C.Y.), R01GM094398 (to T.K.B.), and National Institutes of Health research grants (to D.A.S. and S.J.E.). D.A.S. is also supported by the Paul F. Glenn Foundation, the United Mitochondrial Disease Foundation, the Juvenile Diabetes Foundation, and a gift from the Schulak family. S.J.E. is an investigator with the Howard Hughes Medical Institute.
Footnotes
D.A.S. is a scientific consultant for GlaxoSmithKline, Cohbar, Ovascience, and Metrobiotech.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400705111/-/DCSupplemental.
References
- 1.Schapira AH. Mitochondrial diseases. Lancet. 2012;379(9828):1825–1834. doi: 10.1016/S0140-6736(11)61305-6. [DOI] [PubMed] [Google Scholar]
- 2.Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491(7424):374–383. doi: 10.1038/nature11707. [DOI] [PubMed] [Google Scholar]
- 3.Wenz T, Diaz F, Spiegelman BM, Moraes CT. Activation of the PPAR/PGC-1alpha pathway prevents a bioenergetic deficit and effectively improves a mitochondrial myopathy phenotype. Cell Metab. 2008;8(3):249–256. doi: 10.1016/j.cmet.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4.Viscomi C, et al. In vivo correction of COX deficiency by activation of the AMPK/PGC-1α axis. Cell Metab. 2011;14(1):80–90. doi: 10.1016/j.cmet.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Safdar A, et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci USA. 2011;108(10):4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamesch P, et al. hORFeome v3.1: A resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89(3):307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang X, et al. A public genome-scale lentiviral expression library of human ORFs. Nat Methods. 2011;8(8):659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacobson J, Duchen MR, Heales SJ. Intracellular distribution of the fluorescent dye nonyl acridine orange responds to the mitochondrial membrane potential: Implications for assays of cardiolipin and mitochondrial mass. J Neurochem. 2002;82(2):224–233. doi: 10.1046/j.1471-4159.2002.00945.x. [DOI] [PubMed] [Google Scholar]
- 9.Lugli E, et al. Characterization of cells with different mitochondrial membrane potential during apoptosis. Cytometry A. 2005;68(1):28–35. doi: 10.1002/cyto.a.20188. [DOI] [PubMed] [Google Scholar]
- 10.Hallap T, Nagy S, Jaakma U, Johannisson A, Rodriguez-Martinez H. Mitochondrial activity of frozen-thawed spermatozoa assessed by MitoTracker Deep Red 633. Theriogenology. 2005;63(8):2311–2322. doi: 10.1016/j.theriogenology.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Sasaki M, et al. Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat Med. 2011;17(8):944–951. doi: 10.1038/nm.2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee SS, et al. A systematic RNAi screen identifies a critical role for mitochondria in C. elegans longevity. Nat Genet. 2003;33(1):40–48. doi: 10.1038/ng1056. [DOI] [PubMed] [Google Scholar]
- 13.Smith JS, et al. A transcript map of the chromosome 19q-arm glioma tumor suppressor region. Genomics. 2000;64(1):44–50. doi: 10.1006/geno.1999.6101. [DOI] [PubMed] [Google Scholar]
- 14.Okahara F, Ikawa H, Kanaho Y, Maehama T. Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J Biol Chem. 2004;279(44):45300–45303. doi: 10.1074/jbc.C400377200. [DOI] [PubMed] [Google Scholar]
- 15.Yim JH, et al. The putative tumor suppressor gene GLTSCR2 induces PTEN-modulated cell death. Cell Death Differ. 2007;14(11):1872–1879. doi: 10.1038/sj.cdd.4402204. [DOI] [PubMed] [Google Scholar]
- 16.Challagundla KB, et al. Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol Cell Biol. 2011;31(19):4007–4021. doi: 10.1128/MCB.05810-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai MS, Arnold H, Sun XX, Sears R, Lu H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007;26(14):3332–3345. doi: 10.1038/sj.emboj.7601776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai MS, Sun XX, Lu H. Ribosomal protein L11 associates with c-Myc at 5 S rRNA and tRNA genes and regulates their expression. J Biol Chem. 2010;285(17):12587–12594. doi: 10.1074/jbc.M109.056259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindström MS, Wiman KG. Myc and E2F1 induce p53 through p14ARF-independent mechanisms in human fibroblasts. Oncogene. 2003;22(32):4993–5005. doi: 10.1038/sj.onc.1206659. [DOI] [PubMed] [Google Scholar]
- 20.Kalt I, Levy A, Borodianskiy-Shteinberg T, Sarid R. Nucleolar localization of GLTSCR2/PICT-1 is mediated by multiple unique nucleolar localization sequences. PLoS ONE. 2012;7(1):e30825. doi: 10.1371/journal.pone.0030825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010;40(2):216–227. doi: 10.1016/j.molcel.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ryan MT, Hoogenraad NJ. Mitochondrial-nuclear communications. Annu Rev Biochem. 2007;76:701–722. doi: 10.1146/annurev.biochem.76.052305.091720. [DOI] [PubMed] [Google Scholar]
- 23.Zhao Q, et al. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21(17):4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med. 2013;3(8) doi: 10.1101/cshperspect.a014217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Morais R. Up-regulation of nuclear genes in response to inhibition of mitochondrial DNA expression in chicken cells. Biochim Biophys Acta. 1997;1352(3):325–334. doi: 10.1016/s0167-4781(97)00035-3. [DOI] [PubMed] [Google Scholar]
- 26.Biswas G, Guha M, Avadhani NG. Mitochondria-to-nucleus stress signaling in mammalian cells: Nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene. 2005;354:132–139. doi: 10.1016/j.gene.2005.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomes AP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155(7):1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JY, et al. Involvement of GLTSCR2 in the DNA damage response. Am J Pathol. 2011;179(3):1257–1264. doi: 10.1016/j.ajpath.2011.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, et al. Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 2012;19(10):1613–1622. doi: 10.1038/cdd.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haynes CM, Fiorese CJ, Lin YF. Evaluating and responding to mitochondrial dysfunction: The mitochondrial unfolded-protein response and beyond. Trends Cell Biol. 2013;23(7):311–318. doi: 10.1016/j.tcb.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.