

Significance
Cellular misfolded proteins are transported to the cell surface by MHC class II molecules via association with the peptide-binding groove without processing to peptides. We found that IgG heavy chain is transported to the cell surface by MHC class II molecules. Furthermore, IgG heavy chain associated with MHC class II molecules is recognized by autoantibodies in rheumatoid arthritis patients. Autoantibody binding to IgG heavy chain complexed with different MHC class II alleles was strongly associated with rheumatoid arthritis susceptibility conferred by certain MHC class II alleles. These findings suggest that misfolded proteins complexed with MHC class II molecules could be targets for autoantibodies in autoimmune diseases, which might be involved in autoimmune disease susceptibility.
Keywords: autoimmune disease, major histocompatibility complex
Abstract
Specific HLA class II alleles are strongly associated with susceptibility to rheumatoid arthritis (RA); however, how HLA class II regulates susceptibility to RA has remained unclear. Recently, we found a unique function of HLA class II molecules: their ability to aberrantly transport cellular misfolded proteins to the cell surface without processing to peptides. Rheumatoid factor (RF) is an autoantibody that binds to denatured IgG or Fc fragments of IgG and is detected in 70–80% of RA patients but also in patients with other diseases. Here, we report that intact IgG heavy chain (IgGH) is transported to the cell surface by HLA class II via association with the peptide-binding groove and that IgGH/HLA class II complexes are specifically recognized by autoantibodies in RF-positive sera from RA patients. In contrast, autoantibodies in RF-positive sera from non-RA individuals did not bind to IgGH/HLA class II complexes. Of note, a strong correlation between autoantibody binding to IgG complexed with certain HLA-DR alleles and the odds ratio for that allele’s association with RA was observed (r = 0.81; P = 4.6 × 10−5). Our findings suggest that IgGH complexed with certain HLA class II alleles is a target for autoantibodies in RA, which might explain why these HLA class II alleles confer susceptibility to RA.
Autoantibodies are produced in most autoimmune diseases and are frequently used in their diagnosis. Rheumatoid factor (RF) is an IgM autoantibody that binds to denatured IgG or Fc fragments of IgG and is detected in about 80% of RA patients but also in 5–10% of healthy individuals, as well as in other autoimmune diseases (1). However, the natural antigens that are recognized by RF are unknown, partly because such denatured IgG does not exist in physiological situations. Specific HLA class II alleles are strongly associated with susceptibility to many autoimmune diseases (2). Because peptide repertoires presented on different HLA class II alleles differ (3, 4), it has been proposed that specific peptide–HLA class II combinations affect T-cell development and/or tolerance, which may confer susceptibility or resistance to autoimmune diseases (5). A recent report of a large-scale genetic study indicates that several amino acid residues within the HLA-DR peptide-binding groove determine susceptibility to rheumatoid arthritis (RA) (6). However, it has remained enigmatic for decades how specific HLA class II molecules control the immune response in autoimmune diseases (7).
We recently found that misfolded HLA class I molecules are present on the cell surface of HLA class II-expressing cells. Unexpectedly, we discovered that these misfolded HLA class I molecules were transported to the cell surface by association with the peptide-binding groove of HLA class II molecules (8). Indeed, direct association between HLA class II molecules and misfolded HLA class I molecules was detected in HLA class II-expressing B-cell lines. Furthermore, the intact misfolded proteins transported to the cell surface by HLA class II molecules efficiently stimulated antigen-specific B cells (8). Accordingly, we hypothesized that misfolded proteins aberrantly transported to the cell surface by HLA class II molecules might be targets for autoantibodies detected in autoimmune diseases.
Results
IgG Heavy Chain Alone Is Expressed on the Cell Surface in Association with MHC Class II Molecules.
Because denatured IgG is a target for RF, we addressed whether intracellular misfolded IgG heavy chains (IgGHs) are transported to the cell surface by the RA-susceptible HLA-DR4 (HLA-DRA*01:01/DRB1*04:04) molecule. Five different secreted forms of IgG1 heavy chains cloned from human peripheral blood mononuclear cells (PBMCs) transfected into 293T cells were not detected on the cell surface (Fig. 1A) but were detected intracellularly in similar amounts (Fig. 1B). In contrast, the secreted form of IgGH were detectable on the cell surface in the presence of HLA-DR molecules, with the amount of IgG detected being different for each of the five different heavy chains (Fig. 1A). This suggests that the variable (V) region of IgG influences the association with HLA-DR molecules. Because the antibody light chain was not required for expression, the structure of the IgGH complexed with the HLA-DR molecule seems to be different from that of IgGH in normal antibody that is associated with light chain.
Fig. 1.

IgGH is transported to the cell surface by HLA-DR. (A) The secreted forms of IgGHs cloned from human PBMCs that have different V regions were cotransfected with HLA-DRα, DRB1*04:04 (HLA-DR4), and GFP. Cell surface IgG on GFP-expressing cells was detected with anti-human IgG Fc-specific Ab (red lines). Cells transfected with IgGH alone were stained as control (shaded histograms). (B) The secreted form of IgGH clones was cotransfected with GFP, and intracellular IgGH was detected by intracellular staining with anti-human IgG Fc-specific Ab (red lines). Cells transfected with GFP alone were stained as control (shaded histograms). MFIs of IgG complexed with HLA-DR or intracellular IgG are shown in the figure (B). Data are representative of three independent experiments. (C) HLA-DR4 (red lines), Cw3-pep-HLA-DR4 (blue lines), or control plasmids (black lines) were cotransfected with IgGH clone 1 and GFP. Expression of IgG and HLA-DR on GFP-positive cells is shown. (D) HLA-DR4, DR3, Cw3-pep-DR4, and IgGH clone 1 (IgGH) were transfected, HLA-DR was immunoprecipitated from cell lysates, and IgGH and HLA-DR in precipitates and whole-cell lysates were immunoblotted. (E) N-terminal Flag-tagged or C-terminal His-tagged IgGH was transfected with GFP in the presence (red lines) or absence (shaded histograms) of HLA-DR4. Cell surface IgG on GFP-positive cells was detected by anti-Flag or anti-His mAb, respectively. Data are representative of at least three independent experiments.
Next, we analyzed involvement of the peptide-binding groove of HLA class II molecules in the transport of IgGH to the cell surface. We generated HLA-DR4 covalently attached with an HLA-Cw3 peptide, one of the most common naturally bound peptides in the peptide-binding groove of HLA-DR4 (9). Wild-type HLA-DR4 transported IgGH to the cell surface but HLA-DR4 covalently attached with HLA-Cw3 peptide failed to induce cell surface expression of IgGH (Fig. 1C). Furthermore, full-length IgGH (50 kDa) was coprecipitated with HLA-DR4 but not with HLA-DR4 containing a covalently attached peptide (Fig. 1D). When IgGH with a His tag at the C terminus or a Flag tag at the N terminus was transfected with HLA-DR, cell surface IgGH was detected using anti-Flag or anti-His mAb (Fig. 1E). These data indicate that full-length IgGH, rather than degraded IgGH, is expressed on the cell surface in association with the peptide-binding groove of HLA-DR and that the V region of IgG influences the association with HLA-DR. To determine where HLA-DR associates with IgGH, we analyzed the association using pulse–chase experiments (Fig. 2). Endoglycosidase H (Endo H)-sensitive HLA-DR was coimmunoprecipitated with IgGH just after pulsing cells for 15 min with [35S]methionine and cysteine (0 h). Association of IgGH with HLA-DR was also detected after 2 and 4 h. These data indicate that IgGH associates with nascent HLA-DR molecules in the endoplasmic reticulum (ER).
Fig. 2.

Association of IgGH with HLA-DR in the ER. IgGH and HLA-DR were transfected into 293T cells and were labeled with [35S]methionine and cysteine for 15 min and chased for the indicated times in the absence of radioactivity. IgGH was precipitated with protein A (Upper) and both IgGH and HLA-DR were precipitated with protein A plus anti–HLA-DR mAb (Lower). The precipitates were boiled, treated with Endo H or mock, and resolved by SDS/PAGE. HLA-DRαβ and IgGH are indicated. Data are representative of three independent experiments.
Autoantibodies from RA Patients Bind to IgGH Complexed with MHC Class II Molecules.
We analyzed binding of IgM autoantibodies in a RF standard serum to IgGH bound to HLA-DR4 molecules (Fig. 3A). Autoantibodies in the RF standard serum bound strongly to the secreted form of IgGH complexed with HLA-DR molecules but only weakly to the membrane-bound form of IgG expressed on the cell surface by cotransfection with light chain, Igα, and Igβ. The level of expression of the membrane-bound form of IgG compared with the secreted form of IgGH bound to HLA-DR was almost the same. The presence of light chain did not affect IgGH cell surface transportation by HLA-DR and autoantibody binding to IgGH complexed with HLA-DR. Autoantibodies in the RF standard serum did not bind to the membrane-bound form of IgG even in the presence of HLA-DR molecules. To confirm that autoantibodies bind to the IgGH/HLA class II complexes, we used a human monoclonal RF antibody derived from an RA patient, RF61, which shows similar specificity to RF (10, 11). RF61 bound to the secreted form of IgGH complexed with HLA-DR molecules but not to the membrane-bound form of IgG. This indicated that IgGH bound to HLA-DR is a target for autoantibodies in RA patients.
Fig. 3.

IgGH complexed with HLA-DR is specifically recognized by autoantibodies in RA patients. (A) The IgGH, light chain (L), and GFP were cotransfected with (red) or without (blue) HLA-DR4. The membrane form of IgGH containing the same V region as IgGH clone 1 (mIgGH) was transfected with the light chain, Igα, Igβ (Igαβ), and GFP with (red) or without (blue) HLA-DR. Cell surface expression of HLA-DR or IgG and binding of autoantibodies in the RF standard serum and RF61 mAb by GFP-positive cells are shown. (B) IgGH, HLA-DR4, and GFP were cotransfected. Binding of IgM autoantibody in sera from RA patients and healthy controls to GFP-positive cells expressing IgGH/HLA-DR complexes is shown (red lines). The same cells were stained by APC-conjugated anti-human IgM Ab only as control (shaded histograms). RF activities (units per milliliter) of serum samples are shown in the figure. (C) RF titers of sera from 151 RA patients, 20 SLE patients, 117 APS patients, and 128 healthy controls were plotted against IgM autoantibody titers of each serum to IgGH complexed with HLA-DR4 (anti-IgGH/HLA-DR Ab titer). Samples with autoantibody titers under 400 were plotted in red and those between 400 and 2,000 were plotted in blue. Linear regression line (dashed line), the correlation coefficient (r), and P value of the regression line are shown in the plots of RA patients.
We analyzed serum samples from RA patients and healthy donors for binding to IgGH/ HLA class II complexes. RF-positive sera (defined as sera that bind to IgG Fc fragments) from RA patients contained IgM autoantibodies against IgGH complexed with HLA-DR molecules (Fig. 3B). On the other hand, sera from healthy donors, including some RF-positive sera from healthy donors, did not contain autoantibodies against IgGH/HLA-DR complexes. When sera from 151 RA patients were analyzed, autoantibody titers against IgGH complexed with HLA-DR molecules were well correlated with their RF titers as determined by binding to human IgG Fc fragments (r = 0.85, P = 5.9 × 10−44) (Fig. 3C). On the other hand, autoantibody titers against IgGH complexed with HLA-DR molecules were low in sera from systemic lupus erythematosus (SLE) patients, anti-phospholipid syndrome (APS) patients, and healthy donors, including individuals with high RF titers. These data suggest that RA patients possess autoantibodies to both Fc fragments of IgG and IgGH/HLA-DR complexes. IgGH/HLA-DR complexes, but not Fc fragments of IgG, seem to represent a specific target for autoantibodies in RA patients.
A Strong Correlation Between Autoantibody Binding to IgG Complexed with Each HLA-DR Allele and the Odds Ratio for That Allele’s Association with RA.
We compared the efficiency of IgGH transport by different HLA-DR molecules. Although RA-susceptible HLA-DR4 efficiently induced cell surface IgGH expression, cell surface IgGH transport by RA-resistant HLA-DR3 (HLA-DRA*01:01/HLA-DRB1*03:01) was quite inefficient, although there was no difference in the expression of these HLA-DR molecules on the cell surface (Fig. 4A). Moreover, IgGH was coprecipitated with HLA-DR4 but not HLA-DR3 (Fig. 1D). Invariant chain (Ii), which blocks the peptide-binding groove of nascent MHC class II molecules and transports MHC class II to the endosomal components, partially inhibited transport of IgGH to the cell surface by these HLA-DR molecules. However, HLA-DR4, not HLA-DR3, still efficiently transported IgGH to the cell surface (Fig. 4A). Autoantibodies from an RF-positive RA patient bound to the IgGH complexed with HLA-DR4 but less efficiently with IgGH complexed with HLA-DR3. A more pronounced difference was observed in the autoantibody binding to IgGH between HLA-DR4 and HLA-DR3 in the presence of Ii. A cysteine-mutant misfolded Hen egg lysozyme (HEL) protein (8) was transported to the cell surface by HLA-DR3 better than HLA-DR4, indicating that the failure of HLA-DR3 to transport IgGH did not reflect a general inability to associate with all misfolded proteins (Fig. 4B). In addition, Ii blocked mutated HEL (mutHEL) transport by HLA-DR4 more efficiently than HLA-DR3, suggesting that the ability of Ii to block the transport of misfolded proteins by HLA-DR is allele-specific (Fig. 4B). On the other hand, HLA-DM destabilizes the association of Ii-derived peptides with HLA class II molecules in endosomal compartments, which promotes HLA class II molecules to associate with peptides derived from endosomes (12). Therefore, there is a possibility that HLA-DM also destabilizes the association of IgGH with HLA-DR. However, HLA-DM did not affect the IgGH transported by HLA-DR4, although HLA-DM blocked cell surface expression of IgGH transported by HLA-DR3 (Fig. 4C). This suggests that HLA-DM affects the association of IgGH with the RA-susceptible HLA-DR protein in the ER less than Ii.
Fig. 4.

Autoantibody binding to IgGH complexed with HLA-DR molecules is different between RA-susceptible and RA-resistant alleles. (A and C) The IgGH and GFP were cotransfected together with HLA-DR into 293T cells in the presence (red lines) or absence (blue lines) of Ii (A) or HLA-DM (C), and surface expression of HLA-DR and IgG and binding of autoantibodies in the RF standard serum on GFP-positive cells were analyzed. Cells transfected with the IgGH alone were stained as a control (shaded histograms). Expression of Ii (A) or HLA-DM (C) was analyzed by intracellular staining. (B) Flag-tagged cysteine mutant HEL and HLA-DR were cotransfected with GFP in the presence (red lines) or absence (blue lines) of Ii. The transfectants were stained with anti-Flag and anti–HLA-DR mAbs, and expression on GFP-positive cells was determined. Cells transfected with mutHEL alone were stained as a control (shaded histograms). Data are representative of at least three independent experiments.
We further tested whether there is a correlation between binding of IgM autoantibodies from RA patients to IgGH complexed with different HLA-DRB1 alleles and the RA susceptibility conferred by certain HLA-DRB1 alleles in the presence of Ii and light chain to represent physiological situations (Fig. 5A). Strikingly, a strong correlation was observed between the binding of autoantibodies from RA patients to IgGH complexed with each HLA-DR allele and the odds ratio for that allele’s association with RA (r = 0.81; P = 4.6 × 10−5). There was no correlation between misfolded HEL transport and the odds ratio for each of these HLA-DR alleles (Fig. 5B). These data suggest that autoantibody binding to IgGH complexed with certain HLA-DR molecules is involved in susceptibility to RA.
Fig. 5.

A strong correlation between autoantibody binding to IgG complexed with each HLA-DR allele and the odds ratio for that allele’s association with RA. (A) The IgGH was cotransfected with Ig light chain (L), Ii, and HLA-DRα in combination with each HLA-DRB1 allele indicated, and the MFIs of binding of autoantibodies in the RF standard serum to IgG complexed with HLA-DR were plotted against the odds ratios for RA susceptibility for each HLA-DRB1 allele. The odds ratios for the association between different HLA-DRB1 alleles and rheumatoid arthritis, which were obtained from a recently reported large-scale genetic study (6), were log-transformed to normalize the distribution. Linear regression line (dashed line), the correlation coefficient (r), and P value of the regression line are shown. (B) Cysteine mutant HEL was cotransfected with Ii and HLA-DRα in combination with each HLA-DRB1 allele, and the MFIs of HEL staining of the transfectants were plotted against the odds ratios for RA susceptibility for each HLA-DRB1 allele. Data are representative of four independent experiments.
In Situ Association of IgGH with HLA-DR in Synovial Membrane from RA Patients.
The presence of specific autoantibodies against IgGH complexed with HLA-DR suggests that HLA-DR/IgGH complexes exist in tissues of RA patients as a target antigen for the autoantibodies. To test this possibility, we analyzed the association of IgGH and HLA-DR using a proximity ligation assay (PLA) that detects protein–protein interactions closer than 40 nm (13). PLA signals between HLA-DR and IgGH were observed in synovial membrane from RA patients but not from osteoarthritis patients (Fig. 6). These data suggested that IgGH and HLA-DR complexes are present in synovial membrane in RA patients as a target for autoantibodies.
Fig. 6.
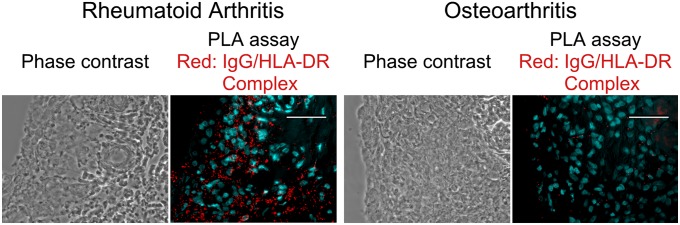
In situ association of IgGH with HLA-DR in synovial membranes of RA patients. In situ association of IgGH with HLA-DR in tissue sections from synovial membranes of RA patients (Left) and osteoarthritis (Right) patients were analyzed by PLA. PLA signals and nuclei are shown as red and light blue, respectively. (Scale bars, 50 μm.) Representative data of tissue sections from three different patients are shown.
Discussion
Ii binds newly synthesized HLA class II molecules and prevents associations with other proteins (3, 14). However, the affinity of Ii-derived peptide (CLIP peptide) binding to HLA class II proteins is not always higher than other peptide antigens (15). Therefore, linear epitopes exposed in IgGH might associate with HLA class II molecules in the ER in place of Ii if they have a high affinity or high abundance. Indeed, in our study, the efficiency of the blockade of the binding of IgGH to HLA class II molecules by Ii varied according to the allele examined. Ii efficiently blocked transportation of IgGH by HLA-DR3, whereas a significant amount of IgGH was still transported to the cell surface by RA-susceptible HLA-DR4 and was recognized by autoantibodies from RA patients even in the presence of Ii and light chain. These are compatible with a prior report that an Ii-derived CLIP peptide has a higher affinity for HLA-DR3 than HLA-DR4 (16). However, Ii blocked the transport of misfolded HEL by HLA-DR4 more efficiently than that by HLA-DR3. Because several epitopes in Ii are involved in the association with MHC class II molecules (4), affinity of a single CLIP peptide to MHC class II molecules alone seems not to be correlated with the efficiency of Ii to block the misfolded protein binding to MHC class II molecules. On the other hand, the HLA class II molecules that associate with IgGH will not be transported to the endosomal compartments because, unlike invariant chain, IgGH does not have an endosomal-sorting signal. Therefore, IgGH associated with HLA class II molecules might be directly transported to the cell surface in an intact form by HLA class II molecules.
Binding of IgGH to the peptide-binding groove of HLA-DR suggested that linear epitopes revealed on unfolded IgGH are involved in the association with HLA-DR. Indeed, peptide fragments of IgGH, including V regions, are often eluted from HLA-DR (4, 17, 18). On the other hand, when extracellular protein antigens are endocytosed and unfolded in endosomes, these proteins form a large molecular complex with MHC class II molecules via the peptide-binding groove, followed by cleavage and/or trimming of the bound protein by endosomal proteinases (19, 20). Therefore, it is possible that misfolded IgGH transported to the cell surface by MHC class II molecules are also cleaved and/or trimmed to peptide fragments after endocytosis of IgGH/MHC class II complex. The MHC class II molecules that appear on the cell surface with the IgGH-derived peptides after endosomal processing might elicit antigen-specific T-cell responses to enhance autoantibody production. Indeed, IgG RF is produced in some RA patients, although most RF is IgM (1).
RF is frequently used in the diagnosis of RA, and a recent large-scale prospective cohort study indicated that healthy individuals with high RF have a 26-fold greater long-term risk of RA than RF-negative individuals (21). However, RF is not thought to be directly involved in pathogenicity of RA, partly because RF is detected in other diseases that lack symptoms of arthritis (1). Most RFs from RA patients and non-RA individuals bind to the Fc portions of IgG1, IgG2, and IgG4. In contrast, RFs from non-RA individuals rarely bind to the Fc portion of IgG3 (less than 5%), whereas RFs from some RA patients bind to IgG3 (about 30%) (22–24). These observations suggested that epitopes recognized by RFs from RA patients are different from RFs of non-RA individuals (22–24). In our study, we found that autoantibodies against IgG1 complexed with HLA-DR are specifically detected in most RA patients but not in non-RA individuals. Therefore, RF epitopes on IgG1 complexed with HLA-DR seem to be different from those on the Fc portion of IgGs not complexed with HLA-DR.
At present, it is unclear whether autoantibodies against IgGH/HLA-DR complexes are directly involved in pathogenesis of RA. However, a strong correlation between autoantibody binding to IgGH/HLA-DR complex and RA susceptibility was observed. In addition, it is well recognized that autoantibodies against ubiquitously expressed self-antigens (for example, glucose-6-phosphate isomerase or type II collagen) can induce arthritis in mouse model systems (25, 26). Considering these observations, autoantibodies to IgGH/HLA-DR complexes might also play a role in the pathogenesis of RA.
IgG-positive B cells in PBMCs, including those from RA patients, are not recognized by autoantibodies from RA patients, suggesting that normal peripheral IgG-positive B cells expressing HLA class II molecules do not display IgGH/HLA-DR complexes. We have shown that IgGH/HLA-DR complexes are detectable in synovial membrane from RA patients by using a PLA assay. Furthermore, an unfolded protein response is detected in plasma cells in the synovial membrane of RA patients (27). These observations suggested that abnormal B cells producing unusual IgG/HLA-DR complexes specifically differentiate in the synovial membrane of RA patients. The diffuse distribution of PLA signals in synovial membranes from RA patients suggests that abnormal B cells producing these IgG/HLA-DR complexes might have secreted these complexes. MHC class II molecules can be secreted from B cells or antigen-presenting cells as exosomes (28). Therefore, abnormal B cells might have secreted these IgGH/HLA-DR complexes in synovial membranes as exosomes, which could be a target for autoantibodies in RA patients. Autoantibodies against citrullinated proteins are also detected in RA patients (29, 30). Because citrullination is known to cause protein misfolding (31), such proteins might be effectively transported to the cell surface by HLA class II molecules and might induce autoantibody production. Therefore, it is important to characterize abnormal B cells generating IgGH/HLA-DR complexes and to investigate in vivo situations in which misfolded or structurally altered proteins are complexed with HLA class II molecules. Further analyses of misfolded or structurally altered proteins complexed with HLA class II molecules might help us to better understand HLA class II-related autoimmune diseases.
Materials and Methods
Human Samples.
The collection and use of human sera and synovial tissues were approved by the institutional reviewer board (IRB) of Hokkaido University, Dohgo Spa Hospital, and Osaka University. Written informed consent was obtained from all participants according to the relevant guidelines of the IRB. Diagnoses of RA and SLE were based on the 1987 American College of Rheumatology classification revised criteria for RA (32) and the 1982 American College of Rheumatology revised criteria for classification of SLE (33), respectively. The diagnosis of APS was based on the preliminary classification criteria for definite APS (34). Some sera were purchased from George King Bio-Medical. RF standard serum (1,060 U/mL) was purchased from GenWay Biotech.
Plasmids.
cDNA for different HLA class II alleles, HLA-DMα (DMA*01:02), HLA-DMβ (DMB*01:01), Ii (accession no. NM_004355.2) were cloned from cDNA prepared from pooled human PBMCs (3H Biomedical) or human cell lines. Igα (accession No. NM_007655.3) and Igβ (accession No. NM_008339.2) were cloned from cDNA prepared from mouse spleen. Some HLA-DR alleles were generated using QuikChange multimutagenesis kits (Agilent Technologies) from HLA genes with similar sequences. All cDNA sequences for HLA were based on information contained in the Immunogenetics/HLA Database (www.ebi.ac.uk/imgt/hla/index.html). HLA-Cw3-pep-HLA-DRB1*04:04 containing a covalently attached peptide was generated by adding the peptide sequence (Cw3: GSHSMRYFYTAVSRPGR) and linker (GSGSGS) between the signal sequence and extracellular domain of these MHC class II cDNA as previously described (35). A cDNA for the mutHEL in which two cysteine residues at positions 30 and 64 were replaced with alanine was generated as described (8). V regions of RF61 RF mAb were synthesized according to the published sequence (accession numbers: for heavy chain, X54437; λ chain, X54438). These V regions were cloned into pME18S expression vectors containing mouse secreted form of IgG1 constant region (accession no. L27437.1) or human λ constant region (accession no. X06876). Plasmids containing heavy and light chain genes for the Abs were cotransfected into 293T cells. The culture supernatants were collected 3 d later and were used for staining. Nucleotide sequences of all of the constructs were confirmed by DNA sequencing (ABI3130xl). IgGHs were amplified from human PBMCs (3H Biomedical) using sense primers for V regions (5′-GTCTTGTCCCAGGTCACCTTGAAGGAG-3′ for clone 1 and 3, 5′-GCCCACTCCCAGGTGCAGCTGGTGCAG-3′ for clone 2, 5′-GTGCAGCTGGTGCAGTCTGGAGCAGAG-3′ for clone 4, 5′-GTCCAGTGTGAAGTGCAGCTGGTGGAG-3′ for clone 5), and antisense primer for constant region of secreted IgG (5′-TCATTTACCCGGAGACAGGGAG-3′) and cloned into pME18S expression vector containing a CD150 signal sequence. The accession numbers for each IgG clone are as follows: clone 1, JQ917464; clone 2, JX292761; clone 3, JX292762; clone 4, JX292763; and clone 5, JX292764. Membrane-bound forms of IgGH, C terminus His-tagged IgGH, and N terminus Flag-tagged IgGH were constructed from secreted form of IgGH (clone 1) by using PCR.
Antibodies.
HL-40 (EXBIO) mAb was used for detection of HLA-DR by flow cytometry, and L243 (ATCC) mAb was used for immunoprecipitation of HLA-DR proteins. Anti-Flag mAb (clone M2, Sigma), anti-DYKDDDDK tag (Flag) mAb (BioLegend), anti-His mAb (Wako), anti-human CD74 (Ii) mAb (LN2, BD Biosciences), and anti–HLA-DM mAb (Map.DM1; Santa Cruz Biotechnology) were used for flow cytometry. Rabbit anti–HLA-DRα Ab (FL-254; Santa Cruz Biotechnology) and peroxidase-conjugated donkey anti-human IgG (H+L) Ab (Jackson ImmunoResearch) were used for Western blotting.
Transfection.
Expression plasmids containing each cDNA were transiently transfected into human embryonic kidney-derived 293T cells, which lack Fc receptors, using Polyethylenimine Max (Polyscience).
Flow Cytometry.
Cells were incubated with primary mouse mAbs, followed by allophycocyanin (APC)-conjugated anti-mouse IgG Ab (all from Jackson ImmunoResearch). For intracellular staining, cells were fixed and permeabilized with Fixation/Permeabilization solution (BD Bioscience). Intracellular Ii or HLA-DM was detected with anti-human Ii (CD74) mAb (BD Bioscience) or anti–HLA-DM mAb (Santa Cruz Biotechnology), followed by APC-conjugated anti-mouse IgG Ab. Intracellular IgGH was detected with APC-conjugated anti-mouse IgG Fc Ab. Stained cells were analyzed on a FACSCalibur (Becton Dickinson). Normal human IgG was reduced by 2-mercaptoethanol (37 °C for 30 min) and was used as denatured IgG after gel filtration.
Immunoprecipitation and Immunoblotting.
Cells were lysed in lysis buffer (10 mM Tris, 150 mM NaCl, pH 7.5) containing 0.5% Nonidet P-40 (Sigma). HLA-DR and IgGH were precipitated with biotin–anti–HLA-DR mAb (L243) plus streptavidin–Sepharose (GE Healthcare) and protein A–Sepharose (GE Healthcare), respectively. The immunoprecipitates were eluted by boiling with SDS/PAGE sample buffer, separated on 5–20% (wt/vol) polyacrylamide gels (Atto), and transferred onto PVDF membranes (Millipore). The membranes were incubated with anti–HLA-DRα Ab (Santa Cruz Biotechnology), followed by anti-rabbit IgG (Thermo Fisher Scientific) or anti-human IgG (Jackson ImmunoResearch) Ab. Peroxidase activity was detected with the SuperSignal reagent (Thermo Fisher Scientific).
Metabolic Labeling.
Transfectants (2.5 × 106) were labeled with 0.1 mCi of [35S]methionine and cysteine (PerkinElmer) in 1 mL of methionine and cysteine-free DMEM (Sigma) for 15 min at 37 °C and then were incubated for various times in complete culture medium containing a 100-fold excess of unlabeled methionine and cysteine. Cells were lysed at 4 °C in a solution containing 10 mM Tris (pH 7.5), 150 mM NaCl, and 0.5% Nonidet P-40. IgGH was precipitated with protein A. As a control, both HLA-DR and IgGH were precipitated with anti–HLA-DR mAb L243 mixed with protein A. For determination of Endo H resistance, the immunoprecipitates were eluted from the beads in 20 μL of Endo H digestion buffer (40 mM DTT, 0.5% SDS) by heating at 100 °C for 10 min, and the eluted proteins were then incubated for 1 h at 37 °C in the absence or the presence of 250 U of Endo H (NEB). The reaction mixtures were then analyzed by SDS/PAGE on a separated on 5–20% polyacrylamide gels and autoradiography was analyzed by using FLA-7000 (Fujifilm).
Autoantibody-Binding Analysis to IgG/HLA-DR Complex.
Human secreted IgG1 (clone 1) or membrane IgG1 containing the same V region as clone 1 was cotransfected together with HLA-DRαβ, Ig κ chain, Igα, Igβ, and GFP. The transfectants were mixed with 1/300-diluted RF standard serum (1,060 U/mL) or sera from RF-positive and -negative patients, followed by biotin-conjugated anti-human IgM Ab (Jackson ImmunoResearch) and then APC-labeled streptavidin (eBioscience). RF61 mAb was premixed with APC-labeled anti-mouse IgG Fc Ab and used directly for the staining. Cell surface IgG was detected by APC-labeled anti-human IgG Fc Ab (Jackson ImmunoResearch). IgG bound on the GFP-expressing cells were detected by APC-labeled anti-mouse IgG Fc Ab. Mean fluorescence intensities (MFIs) of autoantibody binding to HLA-DR– and IgGH-transfected cells were calculated by subtracting MFI of autoantibody binding to GFP-negative transfectants. Anti-IgGH/HLA-DR complex Ab titers were calculated based on IgM autoantibody binding to IgGH bound to HLA-DR4 by a standard RF serum of which the RF titer is known (1,060 U/mL).
Measuring RF Titers.
RF titers were measured by a sandwich ELISA. Human IgG Fc fragment (Jackson ImmunoResearch) was coated onto 96-microwell plates (Costar), and IgM bound to the plates was detected by peroxidase-conjugated rabbit anti-human IgM Ab (Jackson ImmunoResearch). Peroxidase activities were detected using OptEIA (BD Bioscience). RF titers were calculated by using the standard RF serum of which the RF titer is known (1,060 U/mL; GenWay Biotech).
Proximity Ligation Assay.
A Duolink in situ PLA kit was used according to the manufacturer’s instructions (Olink Bioscience) for in situ proximity-ligation assays. Paraffin-embedded tissue sections from RA patients and osteoarthritis patients were treated with target retrieval solution (S1700; Dako) according to the manufacturer’s instructions to retrieve epitopes for Abs and were incubated with goat anti-human IgG Fc-specific Ab (Jackson ImmunoResearch) together with anti–HLA-DR mAb (TAL.1B5; Dako), and Cy3 PLA signals were developed using anti-mouse MINUS and anti-goat PLUS PLA probes. Nuclei were stained with DAPI fluorescence dye. The assayed tissue sections were analyzed by Axioplan 2 fluorescence microscopy.
Statistics.
To assess the significance of the correlation, Pearson’s product–moment correlation coefficient was used, and the correlation coefficient (r) and P value of the linear regression line were calculated. Odds ratios for the association between different HLA-DRB1 alleles and RA, which were obtained from a recently reported large-scale genetic study (6), were log-transformed to normalize the distribution. P values of <0.05 were regarded as statistically significant.
Acknowledgments
We thank S. Sakaguchi for critical reading of the manuscript; H. Kikutani, M. Murakami, N. Sakaguchi, and J. Wang for helpful comments; M. Matsumoto and K. Shida for technical assistance; and members of the Department of Dermatology, Osaka University Graduate School of Medicine, for collection of clinical samples. This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas (HLA Disease and Evolution) from the Ministry of Education, Culture, Sports, Science and Technology Japan (to T. Sasazuki and H.A.); Grants-in-Aid for Scientific Research (B) (to H.A.), (C) (to M.K., T. Suenaga), and Young Scientists (B) (to K.H.) from the Japan Society for the Promotion of Science; and National Center for Global Health and Medicine Grant 21A-106 (to H.A. and N. T.-S.). L.L.L. is an American Cancer Society Professor and is supported by National Institutes of Health Grant AI068129.
Footnotes
The authors declare no conflict of interest.
References
- 1.Dörner T, Egerer K, Feist E, Burmester GR. Rheumatoid factor revisited. Curr Opin Rheumatol. 2004;16(3):246–253. doi: 10.1097/00002281-200405000-00013. [DOI] [PubMed] [Google Scholar]
- 2.Jones EY, Fugger L, Strominger JL, Siebold C. MHC class II proteins and disease: A structural perspective. Nat Rev Immunol. 2006;6(4):271–282. doi: 10.1038/nri1805. [DOI] [PubMed] [Google Scholar]
- 3.Neefjes J, Jongsma ML, Paul P, Bakke O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat Rev Immunol. 2011;11(12):823–836. doi: 10.1038/nri3084. [DOI] [PubMed] [Google Scholar]
- 4.Chicz RM, et al. Specificity and promiscuity among naturally processed peptides bound to HLA-DR alleles. J Exp Med. 1993;178(1):27–47. doi: 10.1084/jem.178.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nicholson MJ, Hahn M, Wucherpfennig KW. Unusual features of self-peptide/MHC binding by autoimmune T cell receptors. Immunity. 2005;23(4):351–360. doi: 10.1016/j.immuni.2005.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Raychaudhuri S, et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat Genet. 2012;44(3):291–296. doi: 10.1038/ng.1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton JL, Harney SM, Wordsworth BP, Brown MA. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004;5(3):151–157. doi: 10.1038/sj.gene.6364045. [DOI] [PubMed] [Google Scholar]
- 8.Jiang Y, et al. Transport of misfolded endoplasmic reticulum proteins to the cell surface by MHC class II molecules. Int Immunol. 2013;25(4):235–246. doi: 10.1093/intimm/dxs155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayden JB, McCormack AL, Yates JR, 3rd, Davey MP. Analysis of naturally processed peptides eluted from HLA DRB1*0402 and *0404. J Neurosci Res. 1996;45(6):795–802. doi: 10.1002/(SICI)1097-4547(19960915)45:6<795::AID-JNR16>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 10.Harindranath N, et al. Complete sequence of the genes encoding the VH and VL regions of low- and high-affinity monoclonal IgM and IgA1 rheumatoid factors produced by CD5+ B cells from a rheumatoid arthritis patient. Int Immunol. 1991;3(9):865–875. doi: 10.1093/intimm/3.9.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burastero SE, Casali P, Wilder RL, Notkins AL. Monoreactive high affinity and polyreactive low affinity rheumatoid factors are produced by CD5+ B cells from patients with rheumatoid arthritis. J Exp Med. 1988;168(6):1979–1992. doi: 10.1084/jem.168.6.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schulze MS, Wucherpfennig KW. The mechanism of HLA-DM induced peptide exchange in the MHC class II antigen presentation pathway. Curr Opin Immunol. 2012;24(1):105–111. doi: 10.1016/j.coi.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Söderberg O, et al. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods. 2006;3(12):995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 14.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 15.Davenport MP, et al. Naturally processed peptides from two disease-resistance-associated HLA-DR13 alleles show related sequence motifs and the effects of the dimorphism at position 86 of the HLA-DR β chain. Proc Natl Acad Sci USA. 1995;92(14):6567–6571. doi: 10.1073/pnas.92.14.6567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Patil NS, et al. Rheumatoid arthritis (RA)-associated HLA-DR alleles form less stable complexes with class II-associated invariant chain peptide than non-RA-associated HLA-DR alleles. J Immunol. 2001;167(12):7157–7168. doi: 10.4049/jimmunol.167.12.7157. [DOI] [PubMed] [Google Scholar]
- 17.Muntasell A, et al. HLA-DR4 molecules in neuroendocrine epithelial cells associate to a heterogeneous repertoire of cytoplasmic and surface self peptides. J Immunol. 2002;169(9):5052–5060. doi: 10.4049/jimmunol.169.9.5052. [DOI] [PubMed] [Google Scholar]
- 18.Seward RJ, Drouin EE, Steere AC, Costello CE. Peptides presented by HLA-DR molecules in synovia of patients with rheumatoid arthritis or antibiotic-refractory Lyme arthritis. Mol Cell Proteomics. 2011;10(3):002477. doi: 10.1074/mcp.M110.002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castellino F, Zappacosta F, Coligan JE, Germain RN. Large protein fragments as substrates for endocytic antigen capture by MHC class II molecules. J Immunol. 1998;161(8):4048–4057. [PubMed] [Google Scholar]
- 20.Sercarz EE, Maverakis E. Mhc-guided processing: Binding of large antigen fragments. Nat Rev Immunol. 2003;3(8):621–629. doi: 10.1038/nri1149. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen SF, Bojesen SE, Schnohr P, Nordestgaard BG. Elevated rheumatoid factor and long term risk of rheumatoid arthritis: A prospective cohort study. BMJ. 2012;345:e5244. doi: 10.1136/bmj.e5244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonagura VR, et al. Mapping studies reveal unique epitopes on IgG recognized by rheumatoid arthritis-derived monoclonal rheumatoid factors. J Immunol. 1993;151(7):3840–3852. [PubMed] [Google Scholar]
- 23.Sasso EH, Barber CV, Nardella FA, Yount WJ, Mannik M. Antigenic specificities of human monoclonal and polyclonal IgM rheumatoid factors. The C γ 2-C γ 3 interface region contains the major determinants. J Immunol. 1988;140(9):3098–3107. [PubMed] [Google Scholar]
- 24.Milosević-Jovcić N, Cirić D, Hajduković-Dragojlović L, Mircetić V. Differences in the relationship of specificity to titre and functional affinity between circulating Ga- and pan-reactive IgM rheumatoid factors in rheumatoid arthritis. Rheumatology (Oxford) 2004;43(9):1190–1193. doi: 10.1093/rheumatology/keh287. [DOI] [PubMed] [Google Scholar]
- 25.Matsumoto I, et al. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3(4):360–365. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 26.Griffiths MM, Remmers EF. Genetic analysis of collagen-induced arthritis in rats: A polygenic model for rheumatoid arthritis predicts a common framework of cross-species inflammatory/autoimmune disease loci. Immunol Rev. 2001;184:172–183. doi: 10.1034/j.1600-065x.2001.1840116.x. [DOI] [PubMed] [Google Scholar]
- 27.Reimold AM, et al. Plasma cell differentiation requires the transcription factor XBP-1. Nature. 2001;412(6844):300–307. doi: 10.1038/35085509. [DOI] [PubMed] [Google Scholar]
- 28.Théry C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9(8):581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki A, et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet. 2003;34(4):395–402. doi: 10.1038/ng1206. [DOI] [PubMed] [Google Scholar]
- 30.Schellekens GA, et al. The diagnostic properties of rheumatoid arthritis antibodies recognizing a cyclic citrullinated peptide. Arthritis Rheum. 2000;43(1):155–163. doi: 10.1002/1529-0131(200001)43:1<155::AID-ANR20>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 31.Tarcsa E, et al. Protein unfolding by peptidylarginine deiminase. Substrate specificity and structural relationships of the natural substrates trichohyalin and filaggrin. J Biol Chem. 1996;271(48):30709–30716. doi: 10.1074/jbc.271.48.30709. [DOI] [PubMed] [Google Scholar]
- 32.Arnett FC, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31(3):315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- 33.Tan EM, et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25(11):1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 34.Miyakis S, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS) J Thromb Haemost. 2006;4(2):295–306. doi: 10.1111/j.1538-7836.2006.01753.x. [DOI] [PubMed] [Google Scholar]
- 35.Scott CA, Peterson PA, Teyton L, Wilson IA. Crystal structures of two I-Ad-peptide complexes reveal that high affinity can be achieved without large anchor residues. Immunity. 1998;8(3):319–329. doi: 10.1016/s1074-7613(00)80537-3. [DOI] [PubMed] [Google Scholar]