

Significance
Cytokine storm plays an essential and commanding role in the clinical outcome and pathogenesis of influenza virus infection. We previously documented that a small molecule that activates sphingosine-1-phosphate-1 receptor (S1P1R) signaling is primarily responsible for blunting cytokine storm to protect the infected host from the consequences of influenza infection. In the present study, we map host innate signaling pathways of cytokine storm and chart where along those pathways the drug is effective. We find that the efficacy of S1P1R agonist in blunting cytokine storm is through global inhibition downstream of myeloid differentiation primary response gene 88 and IFN-β promoter stimulator-1 signaling.
Keywords: pathology, pulmonary
Abstract
During pathogenic influenza virus infection, robust cytokine production (cytokine storm), excessive inflammatory infiltrates, and virus-induced tissue destruction all contribute to morbidity and mortality. Earlier we reported that modulation of sphingosine-1-phosphate-1 receptor (S1P1R) signaling provided a chemically tractable approach for the effective blunting of cytokine storm, leading to the improvement of clinical and survival outcomes. Here, we show that S1P1R agonist treatment suppresses global cytokine amplification. Importantly, S1P1R agonist treatment was able to blunt cytokine/chemokine production and innate immune cell recruitment in the lung independently of endosomal and cytosolic innate sensing pathways. S1P1R signaling suppression of cytokine amplification was independent of multiple innate signaling adaptor pathways for myeloid differentiation primary response gene 88 (MyD88) and IFN-β promoter stimulator-1 signaling, indicating a common pathway inhibition of cytokine storm. We identify the MyD88 adaptor molecule as responsible for the majority of cytokine amplification observed following influenza virus challenge.
Overabundant innate immune responses correlate with increased morbidity and mortality during multiple pathogenic respiratory viral infections (1–5). When studying human pandemic H1N1/2009 influenza virus in mice and ferrets, we found direct evidence that cytokine storm was chemically tractable using a sphingosine-1-phosphate receptor-1 (S1P1R) selective agonists. S1P1R agonist therapy suppressed innate immune cell recruitment, cytokine-chemokine production, and improved survival without altering viral clearance, indicating that cytokine storm was causative to disease pathogenesis and that S1P therapy could suppress detrimental innate immune responses without hindering virus control (6, 7). The identification that S1P1R agonists suppress detrimental innate immune responses without hindering virus control indicates that S1P1R probes may serve as both viable drug leads to curb influenza virus morbidity and mortality, and as research tools to identify additional cellular signaling pathways that can be targeted to improve clinical outcomes during respiratory viral infection.
To generate a molecular understanding how S1P1R agonist therapy effectively blunts pathological innate inflammatory responses, we systematically assessed the role various innate signaling pathways play in S1P1R-mediated suppression of inflammation following influenza virus infection. Using an S1P1R selective agonist synergistically with genetic and biochemical tools, we reveal that S1P1R signaling effectively suppressed global cytokine amplification at a point that converged downstream of multiple innate signaling pathways. We reveal that S1P1R signaling can suppress innate cellular recruitment and cytokine amplification downstream of both endosome and cytosolic innate sensing pathways. Moreover, we identify myeloid differentiation primary response gene 88 (MyD88) as the predominant signaling molecule required for innate immune cell recruitment and cytokine/chemokine production following influenza virus infection. This study indicates that S1P1R agonist efficacy during influenza virus infections occurs through its capacity for common pathway inhibition downstream of multiple innate pathogen-sensing molecules of cytokine amplification.
Results
S1P1R Agonism Blunts Innate Cellular and Cytokine Responses Independently of Toll-Like Receptor 3 or -7 Signaling.
Multiple pattern recognition receptors have been documented to sense influenza virus infection and elicit innate immune responses, including Toll-like receptors (TLR3 and -7), cytosolic receptors (retinoic acid-inducible gene, RIG-I) and nucleotide binding domain and leucine-rich repeat containing proteins (NLRs, NLRP3) (8). We asked whether recognition of influenza virus generated double-stranded (ds)RNA (TLR3) or single-strand (ss)RNA (TLR7) was required for S1P1R-mediated inhibition of cytokine and innate cellular responses. To address this question, we infected TLR3- or TLR7-deficient mice with the H1N1 WSN strain of influenza virus and treated infected mice with either vehicle or the S1P1R agonist, CYM5442. Influenza virus infection in TLR3-deficient mice resulted in slightly elevated production of IFN-α, IL-6, chemokine (C-C) ligand (CCL)2, and CCL5, along with a significant increase in the production of IFN-γ compared with wild-type controls (Fig. 1A). Moreover, treatment of TLR3-deficient mice with S1P1R agonist following influenza virus infection resulted in significant reduction of IFN-α as well as all cytokines and chemokines analyzed (Fig. 1A). In contrast to TLR3-deficient mice, influenza virus infection of TLR7-deficient mice resulted in significantly reduced levels of IFN-α in the bronchoalveolar lavage fluid (BALF) 48 h postinfection compared with wild-type controls (Fig. 1B). Interestingly, similar levels of IL-6, CCL2, CCL5, and IFN-γ were detected in the BALF of TLR7-deficient mice compared with wild-type controls (Fig. 1B). Furthermore, treatment of influenza virus-infected mice with S1P1R agonist resulted in a significant >twofold suppression of IFN-α compared with vehicle-treated controls (Fig. 1B, Inset) as well as significant suppression of all cytokine and chemokines measured (Fig. 1B), collectively demonstrating that S1P1R agonism can suppress innate cytokine and chemokine production independently of TLR3 or -7 signaling.
Fig. 1.
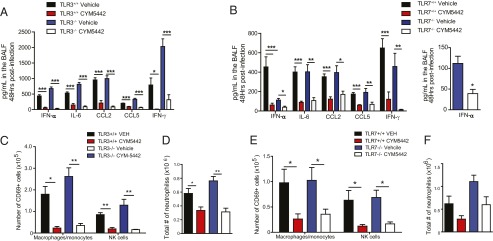
S1P1R agonism blunts innate cellular and cytokine responses independently of TLR3 or -7 signaling. TLR3−/− (A) or TLR7−/− (B) mice were infected with 1 × 104 PFU WSN influenza virus and either vehicle (water) or CYM5442 (2 mg/kg) were administered intratracheally to mice. Proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. Total numbers of activate macrophages/monocytes and NK cells (C and E) were quantified from collagenase-digested lungs at 48 h postinfluenza virus infection. (D and F) Total numbers of neutrophils isolated from collagenase-digested lungs. *P < 0.05, **P < 0.01, ***P < 0.005. Results are representative of two to three independent experiments and five mice per group.
In addition to suppressing cytokine and chemokine production, we demonstrated that S1P1R agonist therapy also inhibits innate immune cell activation and recruitment into the lung following influenza virus infection (6, 7, 9). Thus, we asked what role, if any, TLR3 and -7 signaling played in innate immune cell activation and recruitment into the lung. We observed similar numbers of activated macrophages/monocytes and natural killer cells (NK cells) in the lung 48 h postinfection in both TLR3 and TLR7-deficient mice compared with wild-type controls (Fig. 1 C and E). Moreover, S1P1R agonist therapy reduced numbers of activated macrophages/monocytes and NK cells in the lung 48 h postinfection in both TLR3- and TLR7-deficient mice (Fig. 1 C and E). We also observed reduced expression of the CD69 activation marker on macrophages/monocytes and NK cells following S1P1R agonist treatment in both TLR3- and TLR7-deficient mice following influenza virus infection (Fig. S1). S1P1R agonist treatment following influenza virus infection reduced numbers of neutrophils in the lung in both TLR3- and TLR7-deficient mice (Fig. 1 D and F). These data indicate that S1P1R agonist therapy is efficacious in reducing innate cytokine/chemokine production and immune cell recruitment independently of either TLR3 or TLR7 signaling.
Endosome and Cytosolic Innate Recognition Pathways Are Not Required for S1P1R Agonist Inhibition of Influenza Virus-Induced Innate Cellular and Cytokine Responses.
Two major intracellular signaling pathways activated by influenza virus infection are the endosome (TLR3 and -7) and cytosolic (RIG-I) pathways. To address if either endosome or cytosolic sensing pathways were targets of S1P1R agonism, we used mice defective in endosome [Unc93b1 (3d mouse)] and cytosolic (IFN-β promoter stimulator-1, IPS-1−/−) signaling. The 3d mutant mice respond to endosome TLR ligands (TLR3, -7, and -9) (10). Conversely, mitochondrial IPS-1 is the central adaptor protein for RIG-I signaling and IPS-1−/− mice are unable to signal through the RIG-I or melanoma differentiation-associated-5 sensors (11). We infected either 3d or IPS-1−/− mice with influenza virus, treated infected mice with either vehicle or S1P1R agonist, and measured innate immune responses following infection. Similar to TLR7-deficient mice, infection of 3d mice resulted in significant reduction in the levels of IFN-α in the BALF 48 h following infection compared with wild-type controls (Fig. 2A). Reductions in IL-6 and IFN-γ levels were also observed in 3d mice compared with controls (Fig. 2A). Despite reduced cytokine/chemokine production in 3d mice following influenza virus infection, significant levels of IFN-α, IL-6, CCL2, CCL5, and IFN-γ were produced (Fig. 2A), suggesting that additional innate signaling pathways (likely RIG-I and NLRP3) are able to elicit cytokine/chemokine responses in the absence of endosome TLR signaling. Importantly, S1P1R agonist therapy further suppressed IFN-α production (Fig. 2A, Inset) as well as all other cytokines/chemokines analyzed in 3d mice (Fig. 2A), indicating that S1P1R agonism blunts cytokine/chemokine production independently of endosomal TLR signaling. Alternatively, influenza virus infection in IPS-1−/− mice resulted in reduced IFN-α and IFN-γ levels in BALF despite similar levels of IL-6, CCL2, and CCL5 compared with wild-type controls (Fig. 2B). Similar to 3d mice, IPS-1−/− mice produced significant levels of all cytokines and chemokines analyzed 48 h postinfection (Fig. 2B), suggesting that endosomal TLR and NLRP3 signaling can compensate for the absence of IPS-1. Despite significant production of cytokines and chemokines in IPS-1−/− mice following influenza virus infection, S1P1R agonist treatment suppressed all cytokines and chemokines produced (Fig. 2B).
Fig. 2.
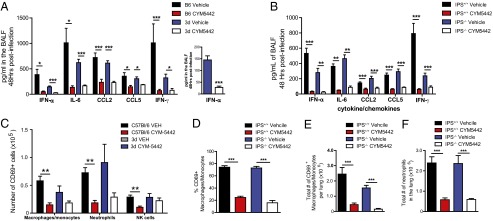
S1P1R agonist inhibition of influenza virus induced cytokine amplification is independent of endosomal and cytosolic innate sensing pathways. The 3d (A) or IPS-1−/− (B) mice were infected with 1 × 104 PFU WSN influenza virus and either vehicle (water) or CYM5442 (2 mg/kg) were administered intratracheally to mice. Proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. (C) Total numbers of activated macrophages/monocytes, NK cells, and neutrophils were quantified from collagenase-digested lungs at 48 h postinfluenza virus infection in 3d mice. Percentage (D) and total numbers (E) of CD69 expressing macrophages in the lung of vehicle or CYM-5442 treated IPS-1+/+ or IPS-1−/− mice 48 h postinfluenza virus infection. (F) Total numbers of neutrophils in the lungs of IPS-1+/+ or IPS-1−/− mice 48 h postinfluenza virus infection. *P < 0.05, **P < 0.01, ***P < 0.005. Results are representative of two to three independent experiments and five mice per group.
We also analyzed innate cellular recruitment in 3d and IPS−/− mice following influenza virus infection and treatment with S1P1R agonist. Despite reductions in IFN-α and multiple cytokines/chemokines in 3d mice, no significant differences in recruitment of innate immune cells, including macrophages/monocytes, neutrophils, and NK cells, occurred compared with wild-type controls (Fig. 2C). Moreover, S1P1R agonist treatment reduced recruitment of macrophages/monocytes and neutrophils into the lung in 3d mice following influenza virus infection (Fig. 2C). We observed reduced expression of the early activation marker CD69 on macrophages/monocytes and NK cells in 3d mice compared with WT controls (Fig. S2). Similar to 3d mice, we observed no difference in activation or recruitment of activated macrophages or neutrophils to the infected lung comparing wild-type and IPS-1−/− mice following influenza virus infection (Fig. 2 D and E). S1P1R agonist treatment inhibited the total numbers of macrophages/monocytes (Fig. 2E) and neutrophils (Fig. 2F) in the lungs of IPS-1−/− mice. The cytokine and cellular data above demonstrate that S1P1R agonist therapy inhibits early influenza virus innate inflammatory responses independent of endosomal or cytosolic signaling, suggesting that S1P1R agonist acts downstream of multiple innate signaling pathways.
IL-1R Signaling Is Required for Cytokine Amplification and Innate Cellular Recruitment Following Influenza Virus Infection.
In addition to endosome (TLR3, -7) and cytosolic (RIG-I) sensors, influenza virus infected cells can activate the NLRP3 inflammasome (12, 13). The end result of inflammasome activation is the cleavage of pro–IL-1 and pro–IL-18 to active proteins. In turn, secreted IL-1 can signal through the IL-1R to induce inflammatory gene production through MyD88 signaling. Signaling of IL-1 through the IL-1R contributes to both host protection and immune pathology following influenza virus infection. We asked what role IL-1–IL-1R signaling played in early cytokine amplification during influenza virus infection. IL-1R–deficient mice made significantly less IFN-α, as well as IL-6, CCL5, CXCL10, and IFN-γ compared with IL-1R–sufficient mice (Fig. 3A). We observed significant reduction in the numbers of activated macrophages/monocytes and NK cells (Fig. S3A), as well as expression levels of CD69 on these cellular populations in the lung in IL-1R–deficient mice (Fig. S3B). Furthermore, we detected significant reductions in neutrophils in IL-1R–deficient compared with IL-1R–sufficient lungs (Fig. S3C), which correlates with previous studies demonstrating reduced neutrophil recruitment in IL-1R–deficient mice following influenza virus infection (14). Collectively, our results demonstrate that IL-1R signaling contributes to early cytokine amplification following influenza virus challenge and may be a mechanism by which S1P1R signaling blunts cytokine storm.
Fig. 3.
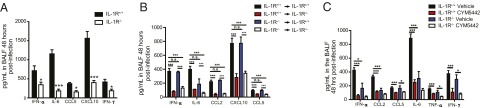
IL-1R signaling is required for cytokine amplification following influenza virus infection. (A) IL-1R−/− or IL-1R+/+ control mice were infected with 1 × 104 PFU WSN influenza virus and proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. (B) Bone marrow chimeras between IL-1R+/+ and IL-1R−/− mice by injection of either IL-1R+/+ bone marrow cells into lethally irradiated IL-1R−/− mice or vice versa. IL-1R+/+ bone marrow cells injected into irradiated IL-1R+/+ mice and IL-1R−/− bone marrow cells into irradiated IL-1R−/− mice as controls. Chimeric mice were infected with 1 × 104 PFU WSN influenza virus and proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. (C) IL-1R−/− or IL-1R+/+ control mice were infected with 1 × 104 PFU WSN influenza virus and either vehicle (water) or CYM5442 (2 mg/kg) were administered intratracheally to mice. Proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. *P < 0.05, **P < 0.01, ***P < 0.005. Results are representative of two to three independent experiments and five mice per group.
Endothelial cells (EC) are activated by IL-1α and -β and TNF-α to induce chemokine production and adhesion molecule expression on the endothelium of inflamed tissues facilitating recruitment of inflammatory immune cells. We asked whether IL-1R signaling was required on hematopoietic or nonhematopoeitic cells in the lung. To address this question, we made bone-marrow chimeras between IL-1R+/+ and IL-1R−/− mice by injection of either IL-1R+/+ bone marrow cells into lethally irradiated IL-1R−/− mice or vice versa. In addition, IL-1R+/+ bone marrow cells were injected into irradiated IL-1R+/+ mice and IL-1R−/− bone marrow cells into irradiated IL-1R−/− mice as controls. Mice were infected with influenza virus and cytokines and chemokines in the BALF assessed 48 h postinfection. Importantly, IL-1R expression was required on nonhematopoietic cells for cytokine amplification because IL-1R−/− mice receiving IL-1R+/+ bone marrow cells displayed reduced levels of IFN-α and cytokines/chemokines in the BALF 48 h postinfection compared with IL-R+/+ mice receiving either IL-1R+/+ or IL-1R−/− bone marrow cells (Fig. 3B). Recruitment of activated macrophages and NK cells, as well as expression levels of the activation marker CD69, was also reduced in mice deficient in IL-1R on nonhematopoeitic cells (Fig. S3 D and E). These results suggest that IL-1R signaling (likely through MyD88) is essential on nonhematopietic cells for cytokine amplification and recruitment of activate leukocytes following influenza virus infection. Moreover, pulmonary ECs express S1P1R and S1P1R signaling may suppress chemokine production from pulmonary ECs following IL-1 signaling, which may explain the S1P1R-mediated suppression of innate immune cell recruitment following influenza virus infection.
Finally, we asked whether S1P1R agonist treatment could further suppress cytokine/chemokine production in IL-1R–deficient mice. In agreement with our previous results, IL-1R–deficient mice produced significantly less IFN-α, CCL2, and IL-6 compared with IL-1R–sufficient mice following influenza virus infection (Fig. 3C), whereas only modest reductions in TNF-α, CCL5, and IFN-γ were observed when comparing IL-1R–sufficient to IL-1R–deficient mice (Fig. 3C). We observed significant suppression of CCL5, IFN-γ, and TNF-α in IL-1R–deficient mice upon treatment with S1P1R agonist following influenza virus infection (Fig. 3C), as well as reduction of IFN-α, CCL2, and IL-6, although the reduction did not reach statistical significance (Fig. 3C). Whereas the frequency and total numbers of activated macrophages were reduced in the lung of IL-1R–deficient mice, S1P1R agonism was able to further suppress the recruitment of activated macrophages (Fig. S3 F and G). However, although we observed reduced numbers of neutrophils in IL-1R–deficient mice, numbers of neutrophils in the lung were not reduced by S1P1R agonist treatment in IL-1R–deficient mice (Fig. S3H), suggesting that neutrophil recruitment into the lung requires IL-1R signaling.
Influenza Virus Induced Cytokine Amplification Requires MyD88 Signaling.
We next asked how ablating global TLR and IL-1R signaling would affect early innate cytokine production and innate immune cell recruitment following influenza virus challenge. We infected TIR-domain-containing adapter-inducing interferon-β (TRIF) signaling-deficient (Lps2) × MyD88−/− mice (referred to as dKO hereafter) with influenza virus and treated them with either vehicle of S1P1R agonist. The absence of MyD88 and TRIF signaling resulted in significant reduction of IFN-α and multiple cytokines and chemokines following influenza virus infection (Fig. 4A). Despite a modest inhibition of IFN-α production, treatment of dKO mice with S1P1R agonist following influenza virus infection did not result in further significant inhibition of any cytokine or chemokine measured (Fig. 4A), indicating that MyD88 and TRIF signaling is important for early cytokine amplification and that S1P1R agonist suppression of cytokine amplification likely occurs through suppressing one or both of these pathways. S1P1R agonist treatment of dKO mice following influenza virus infection resulted in significant inhibition of activated macrophages/monocytes and NK cells in the lung (Fig. 4 B–D).
Fig. 4.
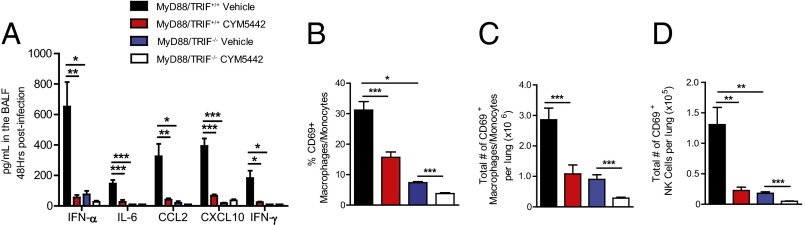
MyD88/TRIF signaling is required for cytokine amplification following influenza virus infection. Wild-type or MyD88−/−/TRIF−/− double-knockout mice were infected with 1 × 104 PFU WSN influenza virus and either vehicle (water) or CYM5442 (2 mg/kg) were administered intratracheally to mice. (A) Proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. Percentage (B) and total numbers (C) of activated macrophages/monocytes or (D) NK cells were quantified from collagenase-digested lungs at 48 h postinfluenza virus infection in MyD88−/−/TRIF−/− mice. *P < 0.05, **P < 0.01, ***P < 0.005. Results are representative of two independent experiments and five mice per group.
To determine whether MyD88, TRIF, or both pathways were necessary for cytokine amplification following influenza virus infection, we infected MyD88−/− or LPS2 mice separately with influenza virus and measured the production of IFN-α and multiple cytokine and chemokines. We observed significant reductions in IFN-α as well as multiple cytokines and chemokines in MyD88−/− mice compared with wild-type controls (Fig. 5A). In contrast to MyD88−/− mice, we observed no measureable changes in IFN-α or multiple cytokines and chemokines in the BALF of LPS2 compared with wild-type mice following influenza virus infection (Fig. 5A), indicating that MyD88 signaling, not TRIF, is essential for early cytokine amplification following influenza virus infection.
Fig. 5.

MyD88 is the predominant signaling pathway required for cytokine amplification following influenza virus infection. Wild-type, MyD88−/− mice (A and B) were infected with 1 × 104 PFU WSN influenza virus and either treated with vehicle or CYM-5442 (B) and proinflammatory cytokines and chemokines were measured 48 h postinfection in BALF by ELISA. (C) Total numbers of macrophages/monocytes and neutrophils were quantified from collagenase-digested lungs at 48 h postinfluenza virus infection in MyD88−/− mice. (D) Percentage of CD69 expressing macrophages in the lung of MyD88+/+ or MyD88−/− (vehicle or CYM-5442 treated) mice 48 h postinfluenza virus infection. *P < 0.05, **P < 0.01, ***P < 0.005. Results are representative of two independent experiments and five mice per group.
We next tested whether S1P1R agonist therapy suppressed cytokine amplification in MyD88−/− mice following influenza virus infection. MyD88−/− mice produced significantly less IFN-α and cytokines and chemokines following influenza challenge compared with MyD88+/+ controls (Fig. 5B). Remarkably, S1P1R agonist treatment significantly suppressed IFN-α production in MyD88−/− mice (Fig. 5B, Inset). We also observed modest but significant reduction in CXCL10 production in S1P1R agonist treated compared with vehicle treated MyD88−/− mice (Fig. 5B). Despite the low levels of cytokines in vehicle-treated mice, we did not observe significant reductions in IL-6, CCL2, or IFN-γ levels following CYM-5442 treatment in influenza virus-infected MyD88−/− mice (Fig. 5B). The numbers of macrophages and neutrophils in the lung following influenza virus infection were significantly reduced in MyD88−/− compared with MyD88+/+ mice (Fig. 5C). Moreover, we observed significant reductions in the frequencies and total numbers of activated macrophages/monocytes in MyD88−/− mice (Fig. 5 D and E). S1P1R agonist treatment inhibited the frequency and numbers of activated macrophages in the lung of MyD88−/− mice, however, did not reduce neutrophil numbers (Fig. 5 D and E). Taken together, our results suggest that MyD88 signaling is essential for the early amplification of cytokine production in the lung following influenza virus infection.
Discussion
Influenza virus infection of humans is associated with dysregulated and overabundant cytokine/chemokine production and innate inflammatory infiltrates, termed cytokine storm. Earlier we reported, using experimental animal models, that the cytokine storm accompanying H1N1 human pathogenic influenza virus infection was an essential component of the morbidity and mortality observed (6, 7). We observed that S1P1R agonists could blunt influenza virus induced cytokine storm and produce a significantly better clinical outcome than that of an antiviral neuraminidase inhibitor (7). This report extends these observations by uncovering several unique findings. First, S1P1R agonism decreases cytokine storm independently of TLR3 and -7, as well as endosomal or cytosolic signaling pathways. Second, IL-1R signaling is necessary for cytokine amplification and innate immune cell recruitment and activation. Moreover, we identify that IL-1R signaling on nonhematopoietic cells is essential for cytokine amplification. Third, MyD88/TRIF signaling is essential for cytokine amplification and innate immune cell recruitment and activation, with MyD88 being the predominant signaling adaptor with little involvement of TRIF. Moreover, although S1P1R agonist treatment likely suppresses the majority of cytokine/chemokine production following influenza infection via targeting MyD88 signaling, S1P1R agonist likely also suppresses additional pathways (at or downstream of the IPS-1 adaptor), through as of yet unidentified mechanisms (Fig. S4). Identification of the mechanism of S1P1R agonist-mediated suppression with respect to signaling pathways targeted and the connection with S1P1R signaling will be important areas of future investigation.
Poor outcomes following infection with influenza and other respiratory viruses in humans and experimental animals correlate with early dysregulated innate cytokine/chemokine production and immune cell recruitment, collectively called cytokine storm (1, 2, 4, 5, 15, 16). Thus, modulating this detrimental immune response via S1P1R agonist serves two purposes. First, it is a potential therapeutic strategy to curb morbidity and mortality, and second it is a signaling platform to explore additional checkpoints to further our understanding and designing of novel therapeutic targets to blunt cytokine storm. Because of the redundant signaling pathways that generate innate immune responses, we anticipate effective therapy will require blunting of multiple signaling pathways. Prior studies showed that neutralization of any one inflammatory mediator linked to morbidity and mortality provided little protection to pathogenic influenza virus infection in animal models (14, 17, 18). The ability of S1P1R-agonist treatment to prevent morbidity and mortality in murine and ferret models underscores the utility of S1P1R agonists as tools to illuminate signaling pathways that mediate pathology following human pathogenic influenza virus infection and point the way toward therapeutic control of influenza virus-induced immune pathology. Our study emphasizes the redundant sensing of influenza virus by multiple innate signaling pathways. We reveal that S1P1R agonist treatment suppresses innate immune cell recruitment and cytokine production independently of multiple innate sensing pathways; thus, our data—together with published reports—strongly suggest that successful modulation of immune pathology during influenza and other respiratory viral infections may require global blunting of innate immune signaling. The fact that MyD88 signaling was responsible for the majority of innate immune cell recruitment and cytokine/chemokine production suggests that therapies that focus to target different components of MyD88 signaling will likely prove successful for therapeutic intervention of influenza virus infection and dissection of signaling pathways necessary for cytokine storm.
Our previous work documented a critical role for pulmonary endothelial cells in promoting cytokine amplification and innate immune cell recruitment (6). The ability of S1P1R signaling to globally blunt innate immune responses suggests that S1P1R signaling likely targets other cell types in addition to endothelial cells. The identification of MyD88 as an essential signaling pathway required for cytokine amplification is likely to aid in identification of additional pulmonary cell types targeted by S1P1R agonist during influenza virus infection. Recent evidence has pointed toward a role for innate immune cell recruitment into the lung as causal to influenza virus induced morbidity and mortality (19, 20). Importantly, S1P1R agonist therapy inhibits both cytokine/chemokine production as well as innate immune cell recruitment.
Aberrant proinflammatory innate immune responses have been implicated in the pathogenesis of multiple viral infections (2, 21), bacterial infections (22), and autoimmune conditions (23, 24). A more detailed understanding of the cellular populations and signaling pathways targeted by S1P1R-signaling should provide insight into how to temper immune pathology and uncover novel signaling pathways that can be targeted to blunt cytokine storm. This information should point the way toward curbing mortality during respiratory viral infections.
Materials and Methods
Mice, Virus, and Compounds.
Six- to 8-wk-old C57BL/6 male mice were bred and maintained in a closed breeding facility at The Scripps Research Institute. All mouse experiments were approved by the Scripps Research Institute Institutional Animal Care and Use Committee. IPS-1−/− mice were a kind gift from Michael Gale (University of Washington, Seattle). Unc93b1 mutant mice (3d) were obtained from Bruce Beutler (The Scripps Research Institute, San Diego). Influenza A/WSN/33 (WSN; H1N1) virus was amplified and plaqued on Madin-Darby Canine Kidney cells. Mice were infected intratracheally with 1 × 104 PFU of influenza A/WSN/33 virus under isoflurane anesthesia. One hour postinfection, mice were anesthetized by isoflurane inhalation for intratracheal delivery of vehicle (100 μL of water) or CYM-5442 (2 mg/kg dissolved in water) were administered 1,13, 25, and 37 h postinfection.
Bone Marrow Chimeras.
Femurs were removed from Ly5.2+ IL-1R+/+ and IL-1R−/− mice and the bone marrow was extracted, disrupted through a 100-μm mesh screen, and red blood cells were lysed. Next, 1 × 107 Ly5.2+ IL-1R+/+ and IL-1R−/− isolated bone marrow cells were adoptively transferred into lethally irradiated (1,000 rads) Ly5.1+ IL-1R+/+ mice. As controls, IL-1R−/− bone marrow was transferred into irradiated IL-1R−/− mice. Bone marrow was allowed to reconstitute for 2 mo, during which the mice were maintained on water supplemented with antibiotics.
Cytokine and Chemokine Analysis.
The trachea of killed mice was exposed, transected, and intubated with a blunt 18-gauge needle. One milliliter of PBS supplemented with Complete Mini, EDTA-free Protease Inhibitor Mixture (Roche) was infused and recovered four times. The recovered BALF was spun at 3,000 × g for 3 min at 4 °C and stored at −800 C until use. Multiplex ELISA was performed on supernatant by Quansys Biosciences to detect TNF-α, MIP-1α, MCP-1, GM-CSF, IFN-γ, and RANTES. ELISAs were also performed using CCL2 (MCP-1), CCL5 (RANTES), CXCL10 (IP-10), IL-6, TNF-α, and IFN-γ Duoset kits (R&D Systems), as well as the VeriKineTM Mouse IFN-Alpha ELISA Kits (R&D Systems).
Cellular Analysis by Flow Cytometry.
Lungs were harvested from PBS-perfused mice and mechanically diced into small tissue pieces using surgical scissors. Diced lungs were suspended in 4 mL of CDTI buffer [0.5 mg/mL collagenase from Clostridium histolyticum Type IV (Sigma), 0.1 mg/mL Dnase I from bovine pancreas grade II (Roche), 1 mg/mL Trypsin inhibitor Type Ii-s (Sigma) in DMEM] for 1 h at 37 °C. Lung was then disrupted mechanically through a 100-nm filter, and red blood cells were lysed using red blood cell lysis buffer (0.02 Tris⋅HCL and 0.14 NH4Cl). Inflammatory cells were purified by centrifugation in 35% (vol/vol) PBS-buffered Percoll (GE Healthcare Life Sciences) at 500 × g for 15 min. Cell pellets were resuspended in staining buffer and Fc receptors were blocked using 25 µg/mL anti-mouse CD16/32 (BD Biosciences). Cells were stained with the following anti-mouse antibodies: Pacific blue-conjugated CD45.2 (BioLegend; clone 104), PerCP-Cy5.5-conjugated NK1.1 (BD Biosciences; clone PK136), Pacific blue-conjugated B220 (BD Biosciences; clone RA3-6B2), PE-Cy7-conjugated CD11b (eBiosciences; clone M1/70), PerCP-Cy5.5-conjugated CD11c (eBiosciences; clone N418), APC-conjugated Gr-1 (BD Biosciences; clone RB6-8C5), Pacific blue- and PE-conjugated Ly6G (BD Biosciences; clone IA8), APC-conjugated F480 (eBioscience; clone BM8), FITC-conjugated CD69 (BD Biosciences; clone H1.2F3). Flow cytometry acquisition was performed with BD FACSDiva-driven BD LSR II flow cytometer (Becton, Dickinson). Data were then analyzed with FlowJo software (Treestar).
Supplementary Material
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400593111/-/DCSupplemental.
References
- 1.Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445(7125):319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 2.Thiel V, Weber F. Interferon and cytokine responses to SARS-coronavirus infection. Cytokine Growth Factor Rev. 2008;19(2):121–132. doi: 10.1016/j.cytogfr.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacNeil A, Ksiazek TG, Rollin PE. Hantavirus pulmonary syndrome, United States, 1993–2009. Emerg Infect Dis. 2011;17(7):1195–1201. doi: 10.3201/eid1707.101306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macneil A, Nichol ST, Spiropoulou CF. Hantavirus pulmonary syndrome. Virus Res. 2011;162(1–2):138–147. doi: 10.1016/j.virusres.2011.09.017. [DOI] [PubMed] [Google Scholar]
- 5.de Jong MD, et al. Fatal outcome of human influenza A (H5N1) is associated with high viral load and hypercytokinemia. Nat Med. 2006;12(10):1203–1207. doi: 10.1038/nm1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teijaro JR, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walsh KB, et al. Suppression of cytokine storm with a sphingosine analog provides protection against pathogenic influenza virus. Proc Natl Acad Sci USA. 2011;108(29):12018–12023. doi: 10.1073/pnas.1107024108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ichinohe T, Iwasaki A, Hasegawa H. Innate sensors of influenza virus: Clues to developing better intranasal vaccines. Expert Rev Vaccines. 2008;7(9):1435–1445. doi: 10.1586/14760584.7.9.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsolais D, et al. A critical role for the sphingosine analog AAL-R in dampening the cytokine response during influenza virus infection. Proc Natl Acad Sci USA. 2009;106(5):1560–1565. doi: 10.1073/pnas.0812689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7(2):156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 11.Kumar H, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203(7):1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med. 2009;206(1):79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichinohe T, Pang IK, Iwasaki A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat Immunol. 2010;11(5):404–410. doi: 10.1038/ni.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrone LA, Szretter KJ, Katz JM, Mizgerd JP, Tumpey TM. Mice lacking both TNF and IL-1 receptors exhibit reduced lung inflammation and delay in onset of death following infection with a highly virulent H5N1 virus. J Infect Dis. 2010;202(8):1161–1170. doi: 10.1086/656365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cillóniz C, et al. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009;5(10):e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baskin CR, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc Natl Acad Sci USA. 2009;106(9):3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salomon R, Hoffmann E, Webster RG. Inhibition of the cytokine response does not protect against lethal H5N1 influenza infection. Proc Natl Acad Sci USA. 2007;104(30):12479–12481. doi: 10.1073/pnas.0705289104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Szretter KJ, et al. Role of host cytokine responses in the pathogenesis of avian H5N1 influenza viruses in mice. J Virol. 2007;81(6):2736–2744. doi: 10.1128/JVI.02336-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154(1):197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aldridge JR, Jr, et al. TNF/iNOS-producing dendritic cells are the necessary evil of lethal influenza virus infection. Proc Natl Acad Sci USA. 2009;106(13):5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Borges AA, et al. Hantavirus cardiopulmonary syndrome: Immune response and pathogenesis. Microbes Infect. 2006;8(8):2324–2330. doi: 10.1016/j.micinf.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 22.Bergeron Y, et al. Cytokine kinetics and other host factors in response to pneumococcal pulmonary infection in mice. Infect Immun. 1998;66(3):912–922. doi: 10.1128/iai.66.3.912-922.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kawane K, Tanaka H, Kitahara Y, Shimaoka S, Nagata S. Cytokine-dependent but acquired immunity-independent arthritis caused by DNA escaped from degradation. Proc Natl Acad Sci USA. 2010;107(45):19432–19437. doi: 10.1073/pnas.1010603107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Link H. The cytokine storm in multiple sclerosis. Mult Scler. 1998;4(1):12–15. doi: 10.1177/135245859800400104. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.