

Abstract
We demonstrate the de novo fabrication of a biosensor, based upon virus-containing PEDOT nanowires, that detects prostate-specific membrane antigen (PSMA). This development process occurs in three phases: 1) isolation of a M13 virus with a displayed polypeptide receptor, from a library of ≈1011 phage-displayed peptides, which binds PSMA with high affinity and selectivity, 2) microfabrication of poly(3,4-ethylene-dioxythiophene) (PEDOT) nanowires that entrain these virus particles using the lithographically-patterned nanowire electrodeposition (LPNE) method, and, 3) electrical detection of the PSMA in high ionic strength (150 mM salt) media, including synthetic urine, using an array of virus-PEDOT nanowires using the electrical resistance of these nanowires for transduction. The electrical resistance of an array of these nanowires increases linearly with the PSMA concentration from 20 nM to 120 nM in high ionic strength PBF buffer, yielding a LOD for PSMA of 56 nM.
Keywords: biosensing, conducting polymers, LPNE, nanowires, PEDOT, phage display, PSMA
INTRODUCTION
Diagnostic tools resulting from the interface of nanoscopic materials and biological molecules have the potential to enable the early detection and monitoring of diseases.1 One specific example is the development of biosensors based upon nanowires of conductive polymers especially poly(pyrrole), PPy, and poly(aniline), PA.2–7 In 2005, Myung, Mulchandani and coworkers2 first demonstrated the feasibility of biosensing using single PPy nanowires in which a receptor was entrained during polymerization. In that work, PPy nanowires containing avidin-congugated QDs provided for the detection of 1 nM of this molecule with good signal-to-noise ratios.2 Subsequently, Mulchandani and coworkers3 showed that covalent attachment chemistries could be used to insure stable attachment of an antibody to the cancer marker CA-125 to a PPy nanowire. These biofunctionalized nanowires detected CA-125 as a decreased electrical conductivity, ΔS/S0 ≈ −0.5, for 1.0 unit/ml in human blood plasma. |ΔS/S0| increased linearly with CA-125 concentration to −0.8 at 1000 units/ml.3 Recently, Yun and coworkers6, 7 have further extended this strategy by demonstrating the introduction of both antibodies6 and DNA aptamers7 into PA nanowires. In the latter study,7 aptamer biofunctionalized PA nanowires provided for the detection of IfG down to 0.56 pg/mL, corresponding to ≈3 fM.7 Collectively, the results of Myung, Mulchandani, and Yun demonstrate tremendous potential for biofunctionalized nanowires for the label-free detection of proteins and peptides.
We have been studying the concept of using virus particles as receptors in biosensors for several years.8–13 Uniquely, M13 viruses can be engineered to recognize essentially any analyte through binding to polypeptides displayed on the viral surface.14, 15 Libraries of phage-displayed proteins and peptides can be created for engineering recognition by manipulating the virus-packaged DNA.10, 16, 17 The phage display approach also provides an efficient method for improving the affinity of a known peptide sequence, termed affinity maturation. Following selections from a naïve library of phage-displayed peptides, the apparent affinities of the first generation selectants can be improved through additional phage-based mutagenesis and selections. One proven affinity maturation method applies a library composed of wild-type and homologous amino acid substitutions in every position of a starting peptide sequence, termed homolog shotgun scanning.18, 19 In this study, we isolate a virus using a M13 phage homolog shotgun library that was screened for high affinity binding to the prostate cancer marker, prostate specific membrane antigen (PSMA).
Prostate cancer, the most frequently diagnosed malignancy in American men, kills over 250,000 men world-wide annually.20 Early detection of prostate cancer biomarkers assists in the diagnosis and successful treatment of the disease.21 Presently, the standard biomarker for detection of prostate cancer is prostate specific antigen (PSA), which is detected in elevated levels in blood when a malignancy is present. A 2004 study22 concluded, however, that prostate cancers, including high-grade cancers, occur at a rate of 15% for men 61 to 91 years who have a serum PSA level in the normal range (≤ 4 ng/mL); this finding underscores the need for a more sensitive biomarker for this disease. Prostate specific membrane antigen (PSMA), a candidate biomarker, is a 750-residue, 100 kDa glycoprotein that is overexpressed as a homodimer on the surface of prostate cancer cells.23, 24 PSMA has been well-characterized as a protein with diagnostic and prognostic capabilities for prostate cancer.25 For example, levels of PSMA in prostate tissue correlate with the aggressiveness of tumor growth.26 Serum PSMA is elevated to 0.60 μg/mL, or 6 nM, in patients with prostate cancer from baseline concentrations of 50 ng/mL (0.5 nM) in healthy patients.27 PSMA is also a biomarker of interest for prostate cancer screening involving urine and seminal fluid,28 but the concentrations of the protein in these body fluids for both healthy and diseased patients has not yet been reported due to the unavailability of a satisfactory assay. The absence of these data in the literature provides another motivation for the development of biosensor capable of measuring this potentially important cancer marker.
In this paper, we describe the development of a biosensor for the selective detection of PSMA in high ionic strength buffer (150 mM) and in synthetic urine. This biosensor, based upon a concept we have recently demonstrated at the proof-of-concept level8, consists of an array of nanowires composed of the conductive polymer poly(3,4-ethylenedioxythiophene) or PEDOT into which M13 virus particles have been entrained. These virus particles “display” a polypeptide that selectively recognizes and binds PSMA; the peptide is fused to the major coat protein, g8p or P8, for multicopy display per phage. PSMA is selectively captured by these entrained virus particles. Such binding causes the electrical resistance of the nanowire to increase, and permits the detection and quantitation of PSMA.
This study is the first to showcase all four phases of virus-biosensor development (Fig 1): 1) isolation of M13 virus particles displaying a non-natural polypeptide that recognizes and selectively binds PSMA (Fig S1), 2) affinity maturation of the displayed peptide for higher affinity binding, 3) incorporation of these viruses by electrodeposition into nanowires composed of the conductive polymer PEDOT, and, 3) detection of PSMA as a resistance increase for an array of these virus-PEDOT composite nanowires, culminating in a linear biosensor response down to [PSMA] = 20 nM. This limit-of-detection is not yet low enough to detect PSMA in blood serum and additional work will be required to reduce the LOD by an order of magnitude and hopefully more. However, this paper does demonstrate that virus particles that strongly bind PSMA, and that are also suitable for incorporation into biosensors, can be identified. We have further demonstrated that these viruses retain their binding affinity for PSMA after incorporation into PEDOT nanowires, including exposure to the harsh processing conditions associated with microelectronics fabrication. And finally, we have demonstrated that the resulting virus-nanowire biosensors can function in high ionic strength solutions including urine.
Figure 1.

Three phases of development of a virus-PEDOT nanowire biosensor for prostate specific membrane antigen (PSMA).
MATERIALS AND METHODS
Materials
All chemicals and reagents were purchased from Sigma-Aldrich and used as received unless otherwise noted. Anti-M13 antibody (p-Ab, GE Healthcare Life Sciences), KO7 helper phage, -M13-HRP monoclonal antibody (Amersham Biosciences), and anti-FLAG-M2 antibody (n-Ab, Sigma) were used as received. PSMA was a generous gift from Drs. William Ernst and Gary Fuji at Molecular Express, Inc. T4 Polynucleotide Kinase, T4 DNA Ligase, T7 DNA Polymerase, and all cell strains were obtained from New England Biolabs. All primers were synthesized by MWG Biotech. Milli-Q UV water was used as the solvent for all solutions. Phosphate-buffered fluoride (PBF, 4.2 mM Na2HPO4, 1.5 mM KH2PO4, and 140 mM NaF) was pH adjusted to 7.2 and sterile-filtered through a 0.22 mm filter (Corning).
Synthesis of PSMA-2 homolog library
The DNA degeneracies used in the homolog scanning oligonucleotide are represented in the IUB code (K = G/T, M = A/C, R = A/G, S = G/C, W = A/T, Y = C/T, N = A/C/G/T, B =C/G/T, V =A/C/G) in the following oligonucleotide. Dsb-D6 HomScn: 5′–GCGTTTAGCGCCAGCGCGTBGGASTGCGYCGA SGYCTWCMAVMAVKSGTGCGASTKGGGTGGCGGCAGCGGCAGCTCCAGCGGTGGA GGACCGGAGGAG–3′ The homolog scanning libraries were constructed using an optimized29 oligonucleotide-directed mutagenesis protocol30 with the oligonucleotide Dsb-D6 HomScn and phagemid pJ1156 as template. Phagemid pJ1156 was identical to a previously described phagemid designed for phage display of proteins (pM1165a)19 with the following exception. The codons encoding the Stu II signal sequence upstream of the displayed protein were changed to a Dsb-A signal sequence31 to increase display levels.
PSMA-binding enzyme-linked immunosorbent assay (ELISA)
A Nunc Maxisorp plate was coated with 10 μg/mL PSMA monomer or dimer protein for 1 h. The coating solution was removed, and the plate was blocked for 1 h with 0.2% BSA in PBS and washed 5 times with PT (300 μL/well). The wells were then incubated with phage displaying PSMA-binding peptides (100 μL/well) at the indicated concentrations for 1 h. After washing five times with PT, the plate was incubated with HRP-conjugated anti-M13 phage antibody (100 μL, 1:5000) in 0.1% PBT buffer for 30 min, and washed three times with PT, and twice with PBS. The plate was then developed using an o-phenylenediamine dihydrochloride/ H2O2 solution (100 μL, 1mg mL−1/ 0.02%) in citric acid buffer (50 mM citric acid, 50 mM Na2HPO4, pH 5.0), and the HRP activity was measured as absorbance at 450 nm.
Synthesis of PSMA-binding Nanowire Devices
Device fabrication applied lithographically patterned nanowire electrodeposition (LPNE)32 for the directed polymerization of EDOT mixed with phage (Scheme 1). Control over the nanowire assembly allowed facile synthesis of several hundred, parallel virus-PEDOT nanowires on a glass substrate. This process (Fig 2a) first required vapor-based deposition of a nickel film on the glass substrate (Fig 2a–i). After coating the nickel with a (+)-photoresist, the nickel was patterned using photolithography and the exposed nickel was removed using nitric acid (Fig 2a–ii). The acid etching also undercuts the photoresist to produce a nickel-edged nanotrench (Fig 2a–iii). Using the nickel edge as the working electrode in a three-electrode cell, virus-PEDOT nanowires were electropolymerized from aqueous 2.5 mM EDOT, 10 nM PSMA-binding viruses, and 12.5 mM LiClO4 (Fig 2a–iv); a primer coat of neat PEDOT, deposited by analogous protocols onto the nickel, helped nucleate formation of the virus-PEDOT nanowires (Fig 2b). The remaining photoresist was removed by acetone treatment, followed by further etching of the exposed nickel with nitric acid (0.8 M) (Fig 2a–v). Unfortunately, the cyclic voltammograms generated during nanowire growth do not contain information on the number of virus particles that are entrained in the electrodeposited PEDOT film. We are working now to develop a gravimetric method for quantitatively determining this important quantity.
Scheme 1.
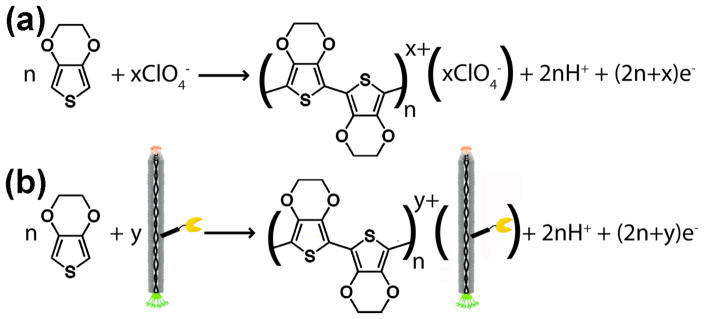
(a) Electrochemical oxidation of EDOT to form PEDOT in perchlorate-containing electrolyte. (b) Electrochemical oxidation of EDOT in the presence of negatively-charged M13 virus particles to form PEDOT. The yellow “pacman” symbolizes a displayed polypeptide epitope optimized to bind PSMA.
Figure 2.

Synthesis of virus-PEDOT nanowires: a) Process flow for the preparation, using the LPEN process, of virus-PEDOT composite nanowires for the detection of PSMA. b) Cyclic voltammograms for the electrodeposition of virus-PEDOT nanowires within the LPNE microfabricated template. Two primer scans were first carried out in aqueous 2.5 mM EDOT, 12.5 mM LiClO4 (black). Then five additional scans in 2.5 mM EDOT, 10 nM PSMA-binding viruses, and 12.5 mM LiClO4 were used to build up a virus-PEDOT composite nanowire 200–300 nm in total width. c, d) Optical micrographs of the resulting nanowire array on glass after the application of silver paste electrical contacts, e, f) Scanning electron micrographs of single nanowires of pure PEDOT (e), and the virus-PEDOT composite (f). The net-like structures observed in (f) are aggregates of filamentous M13 virus particles.
Each nanowire in the parallel array had a rectangular cross section, in which the duration of the electrodeposition and the thickness of the nickel layer dictated nanowire widths (≈200 nm) and heights (≈60 nm), respectively. As described below, the nanowires encapsulated PSMA-binding viruses, and retained electrical conductivity.
The electrical resistance of an array of virus-PEDOT nanowires was measured in order to transduce the binding of PSMA to these nanowires. To connect the virus-PEDOT nanowires to an electrical circuit, colloidal silver was manually painted onto the nanowire array to produce two contacts separated by ≈100 μm. After drying, these silver contacts were covered with an insulating lacquer to prevent the colloidal silver from leaching into the analyte solution (Fig 2c). The exposed region of the virus-PEDOT nanowires included hundreds of parallel nanowires of ≈200 to 300 μm in length (Fig 2c). The encapsulated viruses were visualized by scanning electron microscopy (Fig 2e, f).
In addition to incorporation of the M13 viruses, electrodeposition provides site-specific control over the polymerization. Only the nickel nanotrenches subjected to an oxidative potential become decorated with the virus-PEDOT nanowires, unlike less precise spotting techniques. During electrodeposition, perchlorate anions closely associate with the anodic, and thus positively charged, PEDOT nanowires. Here, a PSMA-binding peptide (amino acid sequence SECVEVFQNSCDW) is displayed on the protein coat of the negatively charged M13 viruses, which are then incorporated into the PEDOT nanowires. The displayed peptide did not interfere with the association or encapsulation of the viruses into the PEDOT nanowires. Furthermore, the viruses and the PSMA-binding scaffolds remained stable and functional, despite the harsh treatments required for device assembly, including drying, washing, rinsing with acetone, and soaking in nitric acid as described above.
RESULTS AND DISCUSSION
Affinity Maturation by Homolog Shotgun Scanning and Phage Display
The previously reported PSMA binding peptides, PSMA-1 (amino acid sequence, CALCEFLG) and PSMA-2 (amino acid sequence, LDCVEVFQNSCDW),13 were compared for affinity to the homodimeric and monomeric isoforms of PSMA. Though diagnostically irrelevant, the monomeric isoform of PSMA provides a control for the binding specificity. As shown by phage-based ELISA, PSMA-2 binds with high affinity and specificity to the PSMA dimer, but not the monomer (Fig 3a). PSMA-1 binds to both isoforms of PSMA. Chosen for its specificity to the PSMA dimer, the PSMA-2 sequence provided the template for an affinity maturation library targeting the PSMA dimer.
Figure 3.

Affinity maturation of phage-displayed, PSMA-binding ligands. (a) Phage-based ELISA with the first generation PSMA-1 and PSMA-2 ligands. Throughout this report, error bars in ELISAs indicate standard error (n=3). (b) The homolog-shotgun scanning library design used the PSMA-2 sequence as a template with the programmed mutations shown in red. The selectants incorporated the mutations highlighted in red. (c) Phage-based ELISA with the affinity-matured, PSMA-binding selectants. (d) Phage-based ELISA comparing PSMA-1,-2, and -3.
Homolog shotgun scanning of the PSMA-2 ligand applied combinatorial libraries of wild-type and closely related sidechains substituted for every amino acid of the peptide (Fig 3c). The two Cys residues, expected to enforce conformational rigidity, remained invariant in the library. The experimental diversity of the Homolog library (≈2 × 109 different variants of PSMA-2) vastly exceeded the theoretical diversity for the library (1.84 × 104); thus, the library likely included all possible combinations of the wild-type and the homologous substitutions. Enrichment for five PSMA-binding peptide sequences emerged after three rounds of library selections. Anti-selections removed members of the library capable of binding to the transferrin receptor (TfR), which is a homolog of PSMA (54% identity), and the blocking agent bovine serum albumin (BSA).
Next, the relative binding affinities of the five peptides selected from the homolog shotgun scanning library were compared. All selectants demonstrated equivalent or better affinity for the PSMA homodimer, than the parent peptide, PSMA-2. To provide a more sensitive test for evaluating binding affinity, the peptides were challenged to bind in half the time previously used for PSMA binding. Additionally, the target PSMA was coated on the microtiter plate with 10-fold lower concentration. This higher screening stringency can better distinguish high from moderate affinity binders. The more stringent ELISA demonstrates that peptides PSMA-3 and PSMA-4 possess a 100- to 1000-fold higher affinity towards PSMA, than the starting template, PSMA-2 (Figure 3b). As expected, the negative control phage, which lack a displayed peptide, fail to bind PSMA.
Analysis of the peptide sequences and their relative binding affinities could uncover structure-activity relationships required for high affinity binding to PSMA. The 11 C-terminal residues of PSMA-3 were largely conserved for all selectants from the homolog library (Figure 3d). Thus, this core sequence appears critical for the molecular recognition of PSMA. Two substitutions incorporated into the sequence of PSMA-3 (L1S and D2E) dramatically enhanced the apparent binding affinity for PSMA. The Ser substitution provides a more polar sidechain, which can potentially donate a hydrogen bond, improving the surface complementarity to PSMA. The selection of Glu in place of Asp extends the sidechain length by one methylene; this substitution maintains the contribution to binding by the carboxylate functionality, and the length extension could allow for new interactions. Increased avidity could also enhance the apparent binding affinity of the PSMA-3 ligand for PSMA, if the PSMA-3 peptide expresses more robustly on the phage surface than the PSMA-2 sequence. PSMA-3 shows specific binding for the PSMA dimer, but not the PSMA monomer, TfR, casein, or various other proteins (Figures S2, S3). In summary, PSMA-3 offers substantially higher binding affinity than PSMA-2 to the PSMA homodimer, and such improved binding can provide sensitive detection of PSMA in nanodevices.
Real-time Biosensing with PSMA-binding Nanowires in Buffer and Synthetic Urine
Following encapsulation of PSMA-3 viruses into PEDOT and nanowire device construction (Fig. 2), biosensing measurements monitored changes in the electrical resistance across the virus-PEDOT nanowires. Using an applied bias, Eapp= 100 mV across the nanowire array, the current, I, was measured, and converted into resistance, R = Eapp/I. The measurements were recorded in real-time during immersion in phosphate-buffered fluoride solution with various analytes. The analyte was pipetted directly onto the nanowire array (15 μl aliquots), and real-time changes in electrical resistance were observed. After each analyte injection, the wires were washed thoroughly with buffer.
In monitoring the change in resistance of the virus-PEDOT wires, nanowires with PSMA-3 viruses encapsulated showed a sensitive and specific increase in resistance upon addition of the prostate cancer marker PSMA (Fig 4a). The binding of PSMA to the nanowires results in a concentration dependent increase in the electrical resistance. A calibration plot for PSMA sensing by tailored virus-PEDOT nanowires demonstrates a linear correlation between the normalized change in resistance (ΔR / R0) and the concentration of the added PSMA (Fig 4b), where R0 is the initial electrical resistance measured in pure buffer. The slope of this calibration curve is 2.1 × 10−3 nM−1 and the intercept on the ΔR / R0 axis is 0.9 nM, which closely approximates the ΔR / R0 values obtained for the n-Ab control across a range of concentrations (Fig 4b). The determination of the limit-of-detection (LOD) for a linear calibration curve like that of Fig 4b has been considered by Lavagnin et al.33 who derived the following equation:
| (1) |
where n is the number of measurements in the calibration curve (n=10 for Fig 4b), tn−2 is the student’s t-parameter for n−2 degrees of freedom (1.86 for 90% and d.f. = 8), sy/x is the regression residual standard deviation (0.03), and b is the slope of the calibration curve. Eq. (1) affords an LOD estimate of 56 nM for PSMA. As reported here, no aspect of the device has been optimized and changes to the concentrations of encapsulated phage and displayed peptide, the device configuration and the nanowire dimensions could also improve device sensitivity.
Figure 4.

Real-time biosensing with PSMA-binding PEDOT nanowires. (a) A real-time trace of biosensing data, with the indicated injections of negative antibody (n-Ab), PSMA, or washes with PBF buffer (black arrows). (b) A compilation of all real-time biosensing data, depicted as a calibration curve with error bars indicating standard deviation (n=3). The change in resistance upon injection is plotted versus analyte concentration. (c) A plot demonstrating the change in resistance upon injection of analytes in a solution of synthetic urine (errors bars indicate standard deviation with n=3).
More work will be required to unambiguously establish the mechanism of signal transduction in these composite nanowires, and indeed, in all prior work involving biosensing with conductive polymer nanowires. Electrical conduction in PEDOT, a derivative of poly(thiophene), occurs as a consequence of the formation of positively charged bipolarons. These bipolarons function as vacancies, imparting mobility to the pi electrons present along the backbone of the polymer just as p-doping imparts mobility to electrons in the valance band of a semiconductor.34–36 The transport of electron vacancies through PEDOT, and between bipolaron cites, occurs in accordance with the variable range hopping model.34–36 Within this framework, how does transduction occur in our virus-doped PEDOT nanowires? Our hypothesis is that the PEDOT matrix of the composite nanowire functions as a “distributed gate” with an adjustable conductivity that depends upon the charge and quantity of the analyte molecules that are bound to the immobilized virus particles. This proposed mechanism is identical, in principle, to the function of ion-selective field effect transistors (ISFETs)37, 38 and biosensors based upon semiconductor nanowires.1, 39, 40 We have measured the pI of PSMA as 4.0 using zeta potential measurements conducted with dynamic light scattering, and this value implies that PSMA is negatively charged in the pH = 7.2 buffer used in the reported measurements. Consequently, in order for the binding of PSMA to increase the nanowire resistance, the PSMA must be gating electrons in the PEDOT, not holes as would be expected from the p-type thermopower measured for PEDOT.41 The mechanism of conduction in PPy mimics that seen in PEDOT, also involving charge localization in positively charged bipolarons. In the already-mentioned work involving the binding of the protein CA-1253 and biotin2 by PPy nanowires, the electrical resistance increased with increasing biotin and CA-125 concentrations, analogous to our reported observations, but the charge state of these two analyte molecules was not indicated in that work. The same trend was seen for the binding of His5-Syntaxin by Cu2+-functionalized PPy nanowires where again the charge state of the protein analyte was undefined, but, in this study, exposure of NTA-modified PPy nanowires to Cu2+ caused an increase in the nanowire conductivity, just as expected for an n-type semiconducting material and qualitatively in accord with our observations.42 In contradiction to this trend, Yun et al.6, 7 have shown that the binding of negatively charged IgE at single p-type PANI nanowires induces a decrease in the nanowire electrical resistance, an inversion of the response that we are reporting here. Collectively, these data for nanowires composed of three different but closely-related polymers – PEDOT, PPy, and PANI – do not provide a consistent picture of the transduction mechanism. More work will be required to elucidate the mechanism.
The robustness of virus-PEDOT nanowire biosensors and their potential applicability to clinical diagnostics were explored for the detection of PSMA added to artificial urine. Such measurements can be challenging, as synthetic urine includes a large number of components (including water, nitric acid, urea, sodium sulfate, and 14 other compounds), high salt and a pH of 5.8 as described previously.43 PSMA (### mg/mL solution in ***) was added to this artificial urine solution to provide the indicated final concentrations, and used for biosensing with PSMA-3-phage PEDOT nanowires; PEDOT nanowires, lacking incorporated viruses provide the negative control. Wires bearing the PSMA-binding recognition motif showed specific binding to PSMA in synthetic urine, with sensitivity measureable for PSMA concentrations above 66 nM (Fig 4c).
CONCLUSIONS
The de-novo fabrication of a biosensor for a particular biomarker using viruses as bioaffinity reagents has been demonstrated for the first time. The generalizability of M13 viruses and the robustness of virus-PEDOT nanowires makes this technique amenable to the molecular recognition of a wide range of analytes including disease markers, and thus offers broad applicability to clinical diagnostics. The fabrication process encompasses three main phases, starting with the isolation of a virus that displays a polypeptide epitope capable of selectively binding PSMA. Here we have demonstrated how affinity maturation by homolog shotgun scanning can be used to identify a PSMA binding peptide (PSMA-3) with 100- to 1000-fold greater PSMA binding affinity than the parent peptide PSMA-2. The second step is the fabrication by electrodeposition of PEDOT nanowires containing these viruses. We have demonstrated that our LPNE process is an expedient and general method for accomplishing this objective. And finally, the resulting PSMA-binding nanowire array must transduce the binding of PSMA in physiologically relevant media. Previously, we showed that the electrical resistivity of virus- composite nanowires increases when a 148 kDa antibody was selectively bound. The encapsulation of PSMA-3 viruses into PEDOT nanowires provides an effective route for electrical resistance-based sensing of the cancer marker in a buffer and synthetic urine matrix at physiologically relevant composition, pH, ionicity, and temperature. Specifically, we report here a higher sensitivity for the 100 kDa PSMA molecule (as compared with the 148 kDa antibody) with a limit-of-detection of 56 nM in high ionic strength buffer, and somewhat higher than this in synthetic urine.
This sensitivity is insufficient for the purpose of detecting PSMA in serum, and the general application of this approach will require an improvement in sensitivity and limit-of-detection of 2 to 3 orders of magnitude. To achieve this objective, the first priority must be the elucidation of the detailed mechanism of transduction.
Acknowledgments
R.M.P. acknowledges support from the National Science Foundation (CHE-0956524) and G.A.W. acknowledges support from the NAID (1 R43 AI074163) and the NCI (R01 CA33592-01) of the National Institutes of Health.
Footnotes
SUPPORTING INFORMATION
Experimental details and additional ELISA data are available. This material is available free of charge via the Internet at http://pubs.acs.org
Contributor Information
Reginald M. Penner, Email: rmpenner@uci.edu.
Gregory A. Weiss, Email: gweiss@uci.edu.
References
- 1.Patolsky F, Zheng G, Lieber CM. Anal Chem. 2006;78:4260–4269. doi: 10.1021/ac069419j. [DOI] [PubMed] [Google Scholar]
- 2.Ramanathan K, Bangar MA, Yun M, Chen W, Myung NV, Mulchandani A. Journal of the American Chemical Society. 2005;127:496–497. doi: 10.1021/ja044486l. [DOI] [PubMed] [Google Scholar]
- 3.Mulchandani A, Bangar MA, Shirale DJ, Chen W, Myung NV. Anal Chem. 2009;81:2168–2175. doi: 10.1021/ac802319f. [DOI] [PubMed] [Google Scholar]
- 4.Myung NV, Hangarter CM, Bangar M, Mulchandani A. Journal of Materials Chemistry. 2010;20:3131–3140. [Google Scholar]
- 5.Mulchandani A, Bangar MA, Shirale DJ, Purohit HJ, Chen W, Myung NV. Electroanalysis. 2011;23:371–379. [Google Scholar]
- 6.Yun M, Lee I, Luo XL, Cui XT. Biosens Bioelectron. 2011;26:3297–3302. doi: 10.1016/j.bios.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yun MH, Luo XL, Lee I, Huang JY, Cui XYT. Chem Commun. 2011;47:6368–6370. doi: 10.1039/c1cc11353d. [DOI] [PubMed] [Google Scholar]
- 8.Arter JA, Taggart DK, McIntire TM, Penner RM, Weiss GA. Nano Lett. 2010;10:4858–4862. doi: 10.1021/nl1025826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donavan KC, Arter JA, Pilolli R, Cioffi N, Weiss GA, Penner RM. Anal Chem. 2011;83:2420–2424. doi: 10.1021/ac2000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss GA, Penner RM. Anal Chem. 2008;80:3082–3089. doi: 10.1021/ac086009h. [DOI] [PubMed] [Google Scholar]
- 11.Weiss GA, Yang LMC, Diaz JE, McIntire TM, Penner RM. Anal Chem. 2008;80:5695–5705. doi: 10.1021/ac8008109. [DOI] [PubMed] [Google Scholar]
- 12.Weiss GA, Yang LMC, Diaz JE, McIntire TM, Penner RM. Anal Chem. 2008;80:933–943. doi: 10.1021/ac071470f. [DOI] [PubMed] [Google Scholar]
- 13.Yang LM, Tam PY, Murray BJ, McIntire TM, Overstreet CM, Weiss GA, Penner RM. Anal Chem. 2006;78:3265–3270. doi: 10.1021/ac052287u. [DOI] [PubMed] [Google Scholar]
- 14.Mao CB, Liu AH, Cao BR. Angewandte Chemie-International Edition. 2009;48:6790–6810. doi: 10.1002/anie.200900231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nanduri V, Sorokulova IB, Samoylov AM, Simonian AL, Petrenko VA, Vodyanoy V. Biosens Bioelectron. 2007;22:986–992. doi: 10.1016/j.bios.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Petrenko VA, Smith GP. Protein Eng. 2000;13:589–592. doi: 10.1093/protein/13.8.589. [DOI] [PubMed] [Google Scholar]
- 17.Scott JK, Smith GP. Science. 1990;249:386–390. doi: 10.1126/science.1696028. [DOI] [PubMed] [Google Scholar]
- 18.Levin AM, Murase K, Jackson PJ, Flinspach ML, Poulos TL, Weiss GA. Acs Chemical Biology. 2007;2:493–500. doi: 10.1021/cb700055t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murase K, Morrison KL, Tam PY, Stafford RL, Jurnak F, Weiss GA. Chem Biol. 2003;10:161–168. doi: 10.1016/s1074-5521(03)00025-5. [DOI] [PubMed] [Google Scholar]
- 20.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Ca-a Cancer Journal for Clinicians. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 21.Madu CO, Lu Y. J Cancer. 2010;1:150–177. doi: 10.7150/jca.1.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson IM, Pauler DK, Goodman PJ, Tangen CM, Lucia MS, Parnes HL, Minasian LM, Ford LG, Lippman SM, Crawford ED, Crowley JJ, Coltman CA. New England Journal of Medicine. 2004;350:2239–2246. doi: 10.1056/NEJMoa031918. [DOI] [PubMed] [Google Scholar]
- 23.Fair WR, Israeli RS, Heston WD. Prostate. 1997;32:140–148. doi: 10.1002/(sici)1097-0045(19970701)32:2<140::aid-pros9>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 24.Schulke N, Varlamova OA, Donovan GP, Ma DS, Gardner JP, Morrissey DM, Arrigale RR, Zhan CC, Chodera AJ, Surowitz KG, Maddon PJ, Heston WDW, Olson WC. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:12590–12595. doi: 10.1073/pnas.1735443100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Montironi R, Cheng L, Mazzucchelli R, Santinelli A, Bono A, Lopez-Beltran A. Urologia. 2008;75:149–155. [PubMed] [Google Scholar]
- 26.Ross JS, Sheehan CE, Fisher HA, Kaufman RP, Jr, Kaur P, Gray K, Webb I, Gray GS, Mosher R, Kallakury BV. Clin Cancer Res. 2003;9:6357–6362. [PubMed] [Google Scholar]
- 27.Wright GL, Xiao Z, Adam BL, Cazares LH, Clements MA, Davis JW, Schellhammer PF, Dalmasso EA. Cancer Res. 2001;61:6029–6033. [PubMed] [Google Scholar]
- 28.Sokoloff RL, Norton KC, Gasior CL, Marker KM, Grauer LS. Prostate. 2000;43:150–157. doi: 10.1002/(sici)1097-0045(20000501)43:2<150::aid-pros10>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 29.Sidhu SS, Lowman HB, Cunningham BC, Wells JA. Methods Enzymol. 2000;328:333–363. doi: 10.1016/s0076-6879(00)28406-1. [DOI] [PubMed] [Google Scholar]
- 30.Kunkel TA, Roberts JD, Zakour RA. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- 31.Steiner D, Forrer P, Stumpp MT, Pluckthun A. Nat Biotechnol. 2006;24:823–831. doi: 10.1038/nbt1218. [DOI] [PubMed] [Google Scholar]
- 32.Menke EJ, Thompson MA, Xiang C, Yang LC, Penner RM. Nat Mater. 2006;5:914–919. doi: 10.1038/nmat1759. [DOI] [PubMed] [Google Scholar]
- 33.Lavagnini I, Antiochia R, Magno F. Electroanal. 2007;19:1227–1230. [Google Scholar]
- 34.Kirchmeyer S, Reuter K. J Mater Chem. 2005;15:2077–2088. [Google Scholar]
- 35.Crispin X, Marciniak S, Osikowicz W, Zotti G, Van der Gon AWD, Louwet F, Fahlman M, Groenendaal L, De Schryver F, Salaneck WR. J Polym Sci Pol Phys. 2003;41:2561–2583. [Google Scholar]
- 36.Groenendaal BL, Jonas F, Freitag D, Pielartzik H, Reynolds JR. Adv Mater. 2000;12:481–494. [Google Scholar]
- 37.Vanderschoot BH, Bergveld P, Bos M, Bousse LJ. Sensor Actuator. 1983;4:267–272. [Google Scholar]
- 38.Bergveld P. Sensor Actuator. 1981;1:17–29. [Google Scholar]
- 39.Hahm J, Lieber CM. Nano Lett. 2004;4:51–54. [Google Scholar]
- 40.Cui Y, Wei QQ, Park HK, Lieber CM. Science. 2001;293:1289–1292. doi: 10.1126/science.1062711. [DOI] [PubMed] [Google Scholar]
- 41.Taggart DK, Yang YA, Kung SC, McIntire TM, Penner RM. Nano Lett. 2011;11:125–131. doi: 10.1021/nl103003d. [DOI] [PubMed] [Google Scholar]
- 42.Mulchandani A, Aravinda CL, Cosnier S, Chen W, Myung NV. Biosensors & Bioelectronics. 2009;24:1451–1455. doi: 10.1016/j.bios.2008.08.044. [DOI] [PubMed] [Google Scholar]
- 43.McCurdy D, Lin Z, Inn KGW, Bell R, Wagner S, Efurd DW, Steiner R, Duffy C, Hamilton TF, Brown TA, Marchetti AA. Journal of Radioanalytical and Nuclear Chemistry. 2005;263:447–455. [Google Scholar]