

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is an aggressive disease. The prognosis is poor; less than 5% of those diagnosed are still alive five years after diagnosis, and complete remission is still rare. Tobacco smoking is a major risk factor of pancreatic cancer. However, the mechanism(s) through which it causes the disease remains unknown. Accumulating evidence indicates that carcinogenic compounds in cigarette smoke stimulate pancreatic cancer progression through induction of inflammation and fibrosis which act in concert with genetic factors leading to the inhibition of cell death and stimulation of proliferation resulting in the promotion of the PDAC.
I. Pancreatic ductal adenocarcinoma: Epidemiology and risk factors
I.1. Introduction
In the United States, over 40,000 individuals are diagnosed with pancreatic ductal adenocarcinoma (PDAC) and 37,000 die of the disease each year. The prognosis is poor with a 5 year survival of less than 5% (1). Even with combinations of treatment (surgery, chemotherapy, radiotherapy) the outcome for patients with this disease remains dismal. The economic impact of the disease is also of great concern as the total costs of the disease in the US are estimated to be $4.9 billion annually (2).
Over the past 5 years, there have been 10 negative Phase III trials investigating the effects of systemic treatment of advanced pancreatic cancer with chemotherapeutic agents (3,4). The failure of these trials likely reflects our limited knowledge of pancreatic cancer biology, particularly with regard to the initiation and progression of the disease. Indeed, very little is known about how PDAC risk factors promote carcinogenesis.
Among the important risk factors for PDAC are chronic pancreatitis and diabetes mellitus. Chronic pancreatitis increases the risk of pancreatic cancer by up to 13 times (5,6). Diabetes mellitus increases the risk by at least 2 times and the more recent the onset of diabetes the stronger the correlation with pancreatic cancer (7,8). Aging, heavy alcohol drinking, family history of the disease, male gender and African American ethnicity are other risk factors for pancreatic cancer (9).
The major environmental and the strongest avoidable risk factor for pancreatic cancer is tobacco smoking (vide infra for detailed discussion) (10-11). However, the pro-carcinogenic effects of smoking on the pancreas are inadequately studied. The goal of this review is to summarize information available on the pro-carcinogenic effects of smoking and underlying mechanisms; and to outline a research strategy designed to reveal the mechanisms through which smoking predisposes to PDAC.
I.2. PDAC characteristics: Role of genetic alterations, inflammation and fibrosis
PDAC presents all classical hallmarks of cancer including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis and activating invasion and metastasis resulting from genetic alterations developed during the progression of the disease from pre-malignant lesions to the PDAC stage (12). The somatic Kras mutation remains the major genetic mutation present in 90% of patients and perhaps the earliest genetic alteration associated with pancreatic cancer (13). Kras mutation leads to the activation of downstream proliferative signaling such as the BRAF/MEK/Erk-mediated proliferation and survival pathway. The pro-oncogenic effects of Kras are enhanced by inactivating somatic mutations of multiple tumor suppressor genes including p53, MADH4, P16, and BRCA2 which are present in the human tumors. These multiple genetic mutations are involved in proliferation, resistance to cell death, and metastasis.
While the role of genetic mutations in carcinogenesis has been a widely accepted concept for several decades, it is only in the recent past that the key influence of the micro-environment on tumor growth and spread has been recognized and acknowledged. (14-16). Thus, inflammation and fibrosis, two major features of the stroma of pancreatic cancer are receiving increasing attention with regard to their effects on tumour behavior. It has been shown that acute necroinflammation of the pancreas speeds up and promotes PDAC formation in mice containing pancreas-specific transgenes expressing Kras (17). Progression to the PDAC stage in Kras mice subjected to pancreatitis is mediated at least, in part, by pro-survival transcription factor STAT3 which induces matrix metalloproteinase 7 (MMP7) expression (18). STAT3 binding activity is increased by inflammatory cytokines known to be up-regulated during acute pancreatitis. Matrix metalloproteinases (MMPs) are important players in local invasion of cancer as well as angiogenesis and metastasis.
As noted earlier, PDAC is characterized by a prominent, dense desmoplastic reaction that surrounds the often scarce cancer cell clusters. One report showed that more extensive fibroblastic cell proliferation in PDAC correlated with poorer disease outcome (19). The key producer of the cancer desmoplasia is the pancreatic stellate cell (PSC), a cell type now established as the central player in pancreatic fibrogenesis. In normal pancreas, PSCs are in an inactive or quiescent state. During pancreatic injury such as chronic pancreatitis and pancreatic cancer, PSCs are activated to a myofibroblastic state (20-21) resulting in synthesis of excessive amounts of extracellular matrix (ECM) proteins causing pathological fibrosis. In vitro and in vivo studies showed that PSCs play a major role in facilitating local growth and distant spread of pancreatic cancer (22-24). The cells interact closely with cancer cells, inhibiting cancer cell apoptosis but promoting cancer cell proliferation, migration, invasion, and anchorage independent growth (25). In turn, cancer cells induce PSC proliferation, activation and migration. PSCs also interact with endothelial cells to promote angiogenesis (24). Interestingly, PSCs from the primary tumour have also been shown to travel to distant metastatic sites where they probably facilitate the seeding and growth of circulating cancer cells (24). Recent studies have also implicated PSCs in both chemoresistance (26) and resistance to radiotherapy (27) (both well-known features of pancreatic cancer).
The factors mediating the interactions between PSCs and cancer cells and PSCs and endothelial cells are being increasingly identified. One mechanism through which activated stellate cells support cancer cells is through secretion of ECM proteins (collagen, fibronectin and laminin), MMPs and growth factors such as transforming growth factor beta (TGF-β) and platelet-derived growth factor(PDGF). Both ECM proteins and growth factors promote survival of the cancer cells through activation of intracellular reactive oxygen species (ROS) generating systems in the pancreatic cancer cells such as nicotinamide adenine dinucleotide phosphate (NADPH) oxidase enzymes (28-31). Activation of NADPH oxidase by growth factors also occurs in PSCs and their hepatic counterparts, hepatic stellate cells during the process of stellate cell activation (32-33). Thus, oxidant stress is an important factor in desmoplasia and tumour growth in pancreatic cancer (25,34).
Figure 1 summarizes and illustrates the three major contributors to pancreatic carcinogenesis: genetic mutations, inflammation and fibrosis. They act synergistically. Both cancer and stellate cells stimulate each other through secretion of TGF-β and PDGF by the cancer cells; or by secretion of extracellular matrix proteins such as collagen, fibronectin and laminin, growth factors, and MMPs by stellate cells. ECM proteins stimulate NADPH oxidase activity and ROS production, proliferation and resistance to death. MMPs facilitate invasion and metastasis. Cytokines and chemokines produced by the inflammatory cells promote Kras-induced proliferation and stimulate activation of stellate cells resulting in fibrosis. Both cancer and stellate cells interact with endothelial cells to promote new blood vessel formation, thereby facilitating metastasis.
Figure 1.
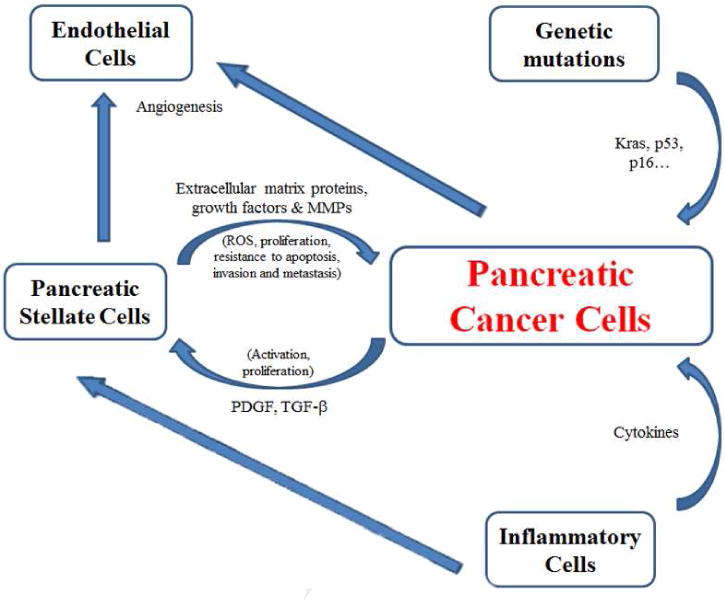
Major contributors to pancreatic carcinogenesis.
I.3. Animal models of PDAC
The development of the genetic mouse models of PDAC has significantly advanced our understanding of the possible mechanisms mediating the initiation of this disease. These models are based for the most part on pancreatic acinar cell-specific activation of Kras. Most of them develop pre-invasive pancreatic neoplastic lesions called pancreatic intraepithelial neoplasms (PanINs) similar to ones found in the human disease (35). However, most models of PanINs do not progress spontaneously to the PDAC stage of invasive and metastatic adenocarcinoma. Only three Kras models: Pdx1-Cre;LSL-Kras model (36), p48Cre;LSL-Kras (36), and EL-CreERT cLGL-Kras, fully replicate advanced PDAC phenotype including invasion, metastasis and fibrosis. Furthermore, a recent study showed that the extent of Kras activation determines PDAC induction, i.e., Kras mutation expression at a low level is not sufficient to trigger PDAC, whereas, higher levels of Kras expression result in advanced PDAC (37). On the other hand, combinations of Kras activating mutation with other genetic alterations characteristic for PDAC, such as p53 inactivating mutations speed up and promote the PDAC development in mice. These data strongly indicate that Kras activation is necessary but not sufficient to develop PDAC; and suggest contribution of other cancer promoting factors acting in concert with Kras.
II. Tobacco smoking and pancreatic cancer
II.1. Epidemiology
More than 400,000 people die each year in the United States alone as a result of past or current cigarette smoking; adult smokers lose an average 13 to 15 years of life-expectancy because of their smoking (38,39). In addition to regular cigarettes and cigars, other forms of tobacco include smokeless tobacco (also called chewing tobacco, snuff and snus), pipes and hookahs (water-pipes). Although most research has focused on the harms of cigarette smoking, all forms of tobacco are harmful. Importantly, both smokeless tobacco and smoking tobacco are known to cause cancer in humans. Lung cancer is by far the first and most studied tobacco-related cancer. In addition to PDAC, other tobacco-related cancers include, tongue, larynx, stomach and brain cancers (12,13). 80% to 90% of lung cancer patients in the United States involve smoking (40). In general, for people who have already developed cancer, quitting smoking reduces the risk of developing a second cancer (41-43).
As noted earlier, smoking is a major established risk factor for pancreatic cancer (10,11). Nearly one fourth of all pancreatic cancer deaths are linked to tobacco use (9). A study of 490 patients in Los Angeles County, California, revealed that smoking a pack or more a day was associated with a five-fold to six-fold increased risk of developing pancreatic cancer (44). The risk of developing pancreatic cancer from smoking cigarettes depends on the duration and the intensity of smoking as well as the age at which the smoker began smoking (45). The median age of diagnosis of pancreatic cancer is 15 years earlier in the tobacco smokers (56 years) compared to non-smokers (71 years) (46).
The incidence of pancreatic cancer correlates directly with smoking prevalence. Temporal trends in US cigarette smoking prevalence rates from 1920 to 1978 correlate with temporal trends in US pancreatic cancer mortality in both sexes. In males, increases and decreases in smoking prevalence are associated with increases and decreases in pancreatic cancer rates. A similar association was observed in females; the prevalence of smoking in women was observed to increase at a later time point than in men (1930 compared to 1920) and this was accompanied by an increase in pancreatic cancer incidence. A recent slight decline in smoking prevalence in women has been associated with a slowing of the rise in pancreatic cancer incidence (47).
In a study published in 1983 data were collected from 50,000 male former college students; the records of 126 men who died of pancreatic cancer in a 16-50 yr follow-up period were compared with those of 504 surviving classmates with respect to physical and social characteristics. Strong positive associations were found for cigarette smoking. Smoking 10 or more cigarettes per day during college corresponded to a relative risk of 2.6, and an otherwise positive smoking history yielded a relative risk of 2.4 (48). The meta-analysis by Iodice et al, of 82 published studies containing epidemiologic information about smoking and pancreatic cancer showed a slightly lower overall risk of 1.74 in current smokers (49).
II.2. Compounds implicated in smoking-induced cancer
Of the more than 7,000 chemicals in tobacco smoke, at least 250 are known to be harmful. Among them, at least 60 are carcinogenic (50,51). These cancer-causing chemicals include arsenic, benzene, ethylene oxide and nickel among others; and importantly, a family of nitrosamines such as: 4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone (NNK), N′-nitrosonornicotine (NNN), 1-(N-methyl-N-nitrosamino)-1-(3-pyridinyl)-4-butanal) (NNAA), 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol (NNAL) (51-53). Nicotine is a major component of tobacco smoke. Although by itself nicotine is not carcinogenic; its metabolites including NNK, NNN, and NNAL are highly carcinogenic. NNK is one of the most abundant carcinogens in tobacco smoke (53). It is a component of the smoke of cigarettes and is further produced in the body as a metabolite of nicotine. Cigarettes, cigars, and other tobacco products vary widely in their content of nicotine and carcinogenic nicotine metabolites. For example, in a cigar (which can contain as many as 20 grams of tobacco), the nicotine content can vary between 5.9 and 335.2 milligrams per gram of tobacco (54).
During smoking, nicotine reaches the lungs and is quickly absorbed into the bloodstream (55). Longer exposure to nicotine leads to a higher nicotine retention in esophagus, spleen, cecum, pancreas, testes, and heart (56). This is important given that nicotine is metabolized into carcinogenic compounds such as NNK and NNAL. Both nicotine and nitrosamine components of cigarette smoke have been shown to reach the pancreas and were found in the pancreatic juice of smokers (57). Nicotine levels in pancreatic juice are 7 times higher in smokers than in non-smokers (57).
II.3. Lack of animal models as a major obstacle for studying the mechanisms underlying smoking -induced PDAC
For over 30 years, multiple laboratories have attempted to develop animal models of pancreatic cancer induced by exposure to tobacco smoke. The first model was developed in hamsters in 1979 (58). In this model, a smoking condensate was delivered in utero by intra-peritoneal (i.p.) injections in pregnant female hamsters. The progeny were observed for 15-25 months and found to develop benign and malignant neoplasms in various organs, including pancreas, sex organs and liver (58).
In another study, rats which consumed drinking water containing NNK and NNAL for a lifetime developed lung and pancreatic cancer. Pancreatic cancers were observed in 9% of rats treated with 1.0 ppm NNK, and in 27% of rats treated with 5.0 ppm NNAL for two years, thus demonstrating a role of these compounds in triggering pancreatic carcinogenesis. The incidence of lung tumors was twice that of pancreatic tumors in these animals (59).
The significant disadvantage of the models described above is that cancer develops only in a small proportion of the animals and that even in these animals, several years of exposure to smoking compounds is required to produce pancreatic cancer. In a more recent model, mice were exposed for 45 days to the combination of carcinogenic chemical 7,12-dimethylbenzanthracene (DMBA) and nicotine. Although this model resulted in more rapid cancer development, the carcinogenic effect of DMBA was not limited to the pancreas. Furthermore, if cigarette smoke extract (CSE) is applied instead of nicotine, animals failed to develop pancreatic cancer (60).
Our Approach
Considering the role of Kras in PDAC development discussed above we proposed the hypothesis that smoking acts in cooperation with activating Kras to promote PDAC. To test this hypothesis we applied i.p. injections of NNK (100mg/kg) once per week for 4 weeks to mice with pancreas-specific Kras transgene expressed using the elastase promoter (EL-Kras) which results in Kras expression only in acinar cells of the pancreas. In these mice, treatment with NNK resulted in advanced PanIN lesions that did not develop in control treated mice. These results indicate that NNK acts synergistically with Kras to promote pancreatic carcinogenesis. Of note, the advanced lesions contained stimulated fibrosis, activated stellate cells and inflammation similar to human pancreatic cancer (61).
We now plan to develop a smoking-induced pancreatic cancer model using the PDX-Cre;LSL-Kras mice. These mice develop PanIN lesions which progress to the PDAC stage. Therefore, they are a good tool for studying whether exposure to cigarette smoke accelerates the progression of PanINs to overt cancer. We believe that using these models will advance our understanding of the mechanisms underlying smoking-induced progression of pancreatic cancer.
II.4. Signaling pathways activated by tobacco smoking leading to cancer
Pro-tumorigenic signaling pathways activated by smoking have been best studied in lung. A major established smoking-related cause of lung cancer is formation of DNA adducts (i.e. with NNK) resulting in Kras mutation. In lung cancer, smoking-induced Kras mutation is an established cause of the disease.
In addition to inducing DNA mutations, NNK interacts with cells through receptors. Indeed, NNK binding to β-adrenergic receptors triggers increases in cellular cyclic AMP (cAMP) and subsequent activation of protein kinase A which, in turn, activates phospholipase A2 releasing arachidonic acid from cell membrane phospholipids. Arachidonic acid metabolites are known to increase DNA synthesis and cell proliferation to promote lung cancer (62). Another pro-survival pathway activated by NNK in lung cancer cells is trans-activation of the EGF receptor (EGFR) and its downstream Raf1/MEK/ERK pathway (63).
Differently from lung cancer, smoking does not cause genetic alterations of the genes known to be mutated in pancreatic cancer (such as Kras and p53) in the pancreas suggesting that smoking enhances the risk for pancreatic cancer through mechanisms other than genetic mutation (64,65). However, it is worth noting that mutations present at lower frequency in pancreatic cancer (such as TTN gene mutation) were increased in the pancreas of smokers compared to non-smokers (65). Yet, and similarly to lung, NNK stimulates pancreatic cancer cell growth through β-adrenergic receptor-mediated activation of Cox2 (66). Furthermore, NNK, through interacting with β-adrenergic receptors 1 and 2, trans-activates EGFR, increases intracellular cAMP accumulation and stimulates Erk phosphorylation in pancreatic ductal cells (67).
In addition to the adrenergic receptors, smoking compounds such as nicotine and NNK interact with pancreatic cells through nicotinic acethylcholine receptors (nAChR), especially through α7-AChR (68,69).
II.5. Role of stroma and inflammatory cells in the oncogenic effects of smoking
Exposure to smoking causes direct activation of epithelial and immune cells in the oral and conducting airways, inducing the secretion of pro-inflammatory factors that promote the recruitment and survival of other immune cells, including neutrophils, macrophages, T-cells, and dendritic cells. Simultaneously, cigarette smoking impairs innate host defense mechanisms, subdues innate responses to pathogens, and alters adaptive immune responses to inhaled antigens. The net result of these effects is a state of chronic injury and inflammation in the airway (70).
As stated above, inflammation and fibrosis are characteristics of chronic pancreatitis, a known risk factor for pancreatic cancer. However, in contrast to the lung, little is known about the effects of smoking on inflammation and fibrosis in the pancreas, although one recent study has implicated cigarette smoking in the acceleration of chronic pancreatitis (71). Importantly, nicotine has been shown to increase the secretion and accumulation of digestive enzymes in rat pancreas, which likely facilitates premature intracellular activation, thereby leading to pancreatic damage (72-74). The damaging effect of nicotine in the exocrine pancreas can also be attributed to one of its pro-carcinogen metabolites.
The animal model of smoking-induced pancreatic cancer which we described above provides an ideal tool to study the role of fibrosis and inflammation in the pro-carcinogenic effects of smoking. We found that NNK administration increases pancreatic stellate cell numbers and activation, and stimulates fibrosis in neoplastic lesions of our model (61). These findings concur with those of Dr. S. Batra and colleagues who reported that exposure of rats to cigarette smoke stimulated fibrosis and inflammation in the pancreas (75). A recent study has shown that the hepatic counterparts of PSCs, i.e. hepatic stellate cells, express nicotinic acetylcholine receptors and respond to nicotine exposure by increased proliferation and ECM synthesis (76). However, little is known about the direct effects of nicotine and its metabolites on PSCs and this area is very worthy of study. Because activation of PSCs leads to inhibition of cell death pathways, especially apoptosis, and stimulation of proliferation in pancreatic cancer cells, it is likely that PSC activation by smoking compounds and the subsequent interaction of PSCs with cancer cells play an important role in the pro-carcinogenic effect of smoking on the pancreas.
In terms of smoking and inflammation, a recent study has reported a significant increase in the infiltration of macrophages into neoplastic lesions in mice treated with NNK. In addition, infiltration of immune cells accompanied by the expression of the inflammatory mediators such as macrophage inflammatory protein 1 alpha (MIP-1α), interleukin 1 beta (IL-1β), and TGF-β, has been demonstrated in the pancreas of rats exposed to cigarette smoke (61,75). Notably, macrophage infiltration is also observed in human pancreatic cancer (77).
Figure 2 shows the proposed mediators of the carcinogenic effect of tobacco smoke in the pancreas. Smoking carcinogens act directly on cancer cells and their precursors to stimulate proliferation and inhibit apoptosis. At the same time smoking compounds affect the micro-environment of the tumor cells by activating major components of the desmoplasia, namely stellate cells and immune cells, (leading to secretion of extracellular matrix proteins, growth factors, cytokines and MMPs). In turn, PSCs and immune cells interact with cancer cells to promote disease progression.
Figure 2.
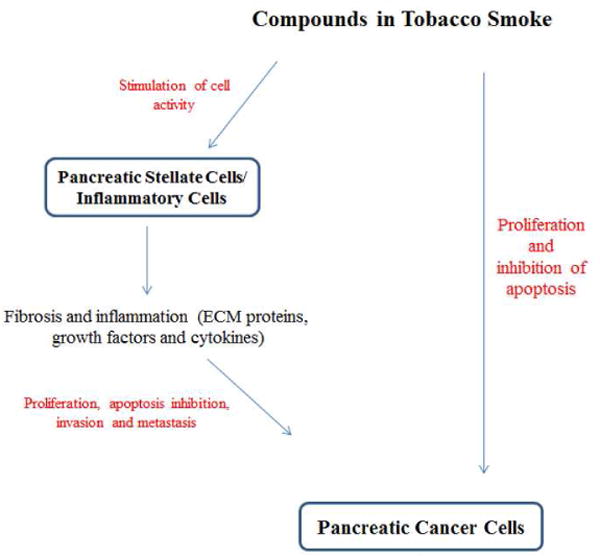
Proposed pathways mediating pancreatic cancer progression by tobacco smoking.
III. Conclusions and future directions
Our results strongly suggest that smoking is a factor that promotes pancreatic cancer rather than initiates it. Thus, understanding how smoking stimulates the progression of pancreatic cancer should be a key goal for the field. The use of animal-exposure smoking systems and genetically altered mice such as the EL-Kras or PDX-Kras mice are probably the most useful tools currently available to study the mechanisms of smoking-induced pancreatic cancer progression. Determining the mechanisms underlying the effect of smoking compounds on fibrosis and inflammation may provide important insights into the pathogenesis of pancreatic cancer. The dense desmoplastic reaction of pancreatic cancer and the recent data on the key role of stellate cells for providing a pro-carcinogenic environment justify this area of investigation. Thus, a detailed investigation of the effect of smoking compounds on stellate cell activation (ECM, growth factors) and then on the key mediators of the interaction with cancer cells will likely provide novel therapeutic targets.
Acknowledgments
Funding: Supported by NIH grant to M.E. (K01AA019996) and Department of Veterans Affairs.
Abbreviations
- cAMP
Cyclic AMP
- EGFR
Epidermal growth factor receptor
- ECM
Extracellular matrix protein
- IL-1β
interleukin 1 beta
- MIP-1α
Macrophage inflammatory protein 1 alpha
- MMPs
Matrix metalloproteinases
- nAChRs
Nicotinic acetylcholine receptors
- NADPH oxidase
nicotinamide adenine dinucleotide phosphate oxidase
- NNAA
1-(N-methyl-N-nitrosamino)-1-(3-pyridinyl)-4-butanal)
- NNAL
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanol
- NNK
4-(methylnitrosamine)-1-(3-pyridyl)-1-butanone
- NNN
N′-nitrosonornicotine
- PDAC
Pancreatic ductal adenocarcinoma
- PDGF
Platelet-derived growth factor
- TGF-β
Transforming growth factor beta
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Wilson LS, Lightwood JM. Pancreatic Cancer: Total Costs and Utilization of Health Services. J Surg Oncol. 1999;71:171–181. doi: 10.1002/(sici)1096-9098(199907)71:3<171::aid-jso7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 3.Philip PA. Targeted therapies for pancreatic cancer. Gastrointest Cancer Res. 2008;2(4 Suppl):S16–9. [PMC free article] [PubMed] [Google Scholar]
- 4.Bayraktar S, Bayraktar UD, Rocha-Lima CM. Recent developments in palliative chemotherapy for locally advanced and metastatic pancreas cancer. World J Gastroenterol. 2010;16(6):673–82. doi: 10.3748/wjg.v16.i6.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lowenfels AB, Maisonneuve P, Cavallini G, Ammann RW, Lankisch PG, Andersen JR, Dimagno EP, Andrén-Sandberg A, Domellöf L. Pancreatitis and the risk of pancreatic cancer. International Pancreatitis Study Group. N Engl J Med. 1993;328:1433–7. doi: 10.1056/NEJM199305203282001. [DOI] [PubMed] [Google Scholar]
- 6.Raimondi S, Lowenfels AB, Morselli-Labate AM, Maisonneuve P, Pezzilli R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract Res Clin Gastroenterol. 2010;24(3):349–58. doi: 10.1016/j.bpg.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Chari ST, Leibson CL, Rabe KG, Timmons LJ, Ransom J, de Andrade M, Petersen GM. Pancreatic cancer-associated diabetes mellitus: prevalence and temporal association with diagnosis of cancer. Gastroenterology. 2008;134:95–101. doi: 10.1053/j.gastro.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gupta S, Vittinghoff E, Bertenthal D, Corley D, Shen H, Walter LC, McQuaid K. New-onset diabetes and pancreatic cancer. Clin Gastroenterol Hepatol. 2006;4:1366–72. doi: 10.1016/j.cgh.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 9.Maisonneuve P, Lowenfels AB. Epidemiology of pancreatic cancer: an update. Dig Dis. 2010;28(4-5):645–56. doi: 10.1159/000320068. [DOI] [PubMed] [Google Scholar]
- 10.Lowenfels AB, Maisonneuve P. Environmental Factors and Risk of Pancreatic Cancer. Pancreatology. 2003;3:1–8. doi: 10.1159/000069140. [DOI] [PubMed] [Google Scholar]
- 11.Raimondi S, Maisonneuve P, Löhr JM, Lowenfels AB. Early Onset Pancreatic Cancer: Evidence of a Major Role for Smoking and Genetic Factors. Canc Epidem Biomark& Prev. 2007;16:1894. doi: 10.1158/1055-9965.EPI-07-0341. 1055-9965. [DOI] [PubMed] [Google Scholar]
- 12.Hanahan D, Weinberg RA. Hallmarks of Cancer: The Next Generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988;53:549–554. doi: 10.1016/0092-8674(88)90571-5. [DOI] [PubMed] [Google Scholar]
- 14.Fidler IJ. The organ microenvironment and cancer metastasis. Differentiation. 2002;70:498–505. doi: 10.1046/j.1432-0436.2002.700904.x. [DOI] [PubMed] [Google Scholar]
- 15.Liotta LA, Kohn EC. The microenvironment of the tumor-host interface. Nature. 2001;411:375–379. doi: 10.1038/35077241. [DOI] [PubMed] [Google Scholar]
- 16.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell Mar. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Carrière C, Young AL, Gunn JR, Longnecker DS, Korc M. Acute Pancreatitis Accelerates Initiation and Progression to Pancreatic Cancer in Mice Expressing Oncogenic Kras in the Nestin Cell Lineage. PLoS One. 2011;6(11):e27725. doi: 10.1371/journal.pone.0027725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fukuda A, Wang SC, Morris JP, 4th, Folias AE, Liou A, Kim GE, Akira S, Boucher KM, Firpo MA, Mulvihill SJ, Hebrok M. Stat3 and MMP7 Contribute to Pancreatic Ductal Adenocarcinoma Initiation and Progression. Cancer Cell. 2011;19(4):441–55. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watanabe I, Hasebe T, Sasaki S, Konishi M, Inoue K, Nakagohri T, Oda T, Mukai K, Kinoshita T. Advanced pancreatic ductal cancer: fibrotic focus and beta-catenin expression correlate with outcome. Pancreas. 2003;26:326–333. doi: 10.1097/00006676-200305000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Apte MV, Haber PS, Applegate TL, Norton ID, McCaughan GW, Korsten MA, Pirola RC, Wilson JS. Periacinar stellate shaped cells in rat pancreas: identification, isolation, and culture. Gut. 1998;43(1):128–33. doi: 10.1136/gut.43.1.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haber PS, Keogh GW, Apte MV, Moran CS, Stewart NL, Crawford DH, Pirola RC, McCaughan GW, Ramm GA, Wilson JS. Activation of pancreatic stellate cells in human and experimental pancreatic fibrosis. Am J Pathol. 1999;155(4):1087–95. doi: 10.1016/S0002-9440(10)65211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68(3):918–26. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vonlaufen A, Joshi S, Qu C, Phillips PA, Xu Z, Parker NR, Toi CS, Pirola RC, Wilson JS, Goldstein D, Apte MV. Pancreatic stellate cells: partners in crime with pancreatic cancer cells. Cancer Res. 2008;68(7):2085–93. doi: 10.1158/0008-5472.CAN-07-2477. 2008. [DOI] [PubMed] [Google Scholar]
- 24.Xu Z, Vonlaufen A, Phillips PA, Fiala-Beer E, Zhang X, Yang L, Biankin AV, Goldstein D, Pirola RC, Wilson JS, Apte MV. Role of pancreatic stellate cells in pancreatic cancer metastasis. Am J Pathol. 2010;177(5):2585–96. doi: 10.2353/ajpath.2010.090899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang RF, Moore T, Arumugam T, Ramachandran V, Amos KD, Rivera A, Ji B, Evans DB, Logsdon CD. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erkan M, Kleeff J, Gorbachevski A, Reiser C, Mitkus T, Esposito I, Giese T, Büchler MW, Giese NA, Friess H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007;132(4):1447–64. doi: 10.1053/j.gastro.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 27.Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through beta1-integrin signaling. Cancer Res. 2011;71(10):3453–8. doi: 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaquero EC, Edderkaoui M, Nam KJ, Gukovsky I, Pandol SJ, Gukovskaya AS. Extracellular matrix proteins protect pancreatic cancer cells from death via mitochondrial and nonmitochondrial pathways. Gastroenterology. 2003;125(4):1188–202. doi: 10.1016/s0016-5085(03)01203-4. [DOI] [PubMed] [Google Scholar]
- 29.Edderkaoui M, Hong P, Vaquero EC, Lee JK, Fischer L, Friess H, Buchler MW, Lerch MM, Pandol SJ, Gukovskaya AS. Extracellular matrix stimulates reactive oxygen species production and increases pancreatic cancer cell survival through 5-lipoxygenase and NADPH oxidase. Am J Physiol Gastrointest Liver Physiol. 2005;289(6):G1137. doi: 10.1152/ajpgi.00197.2005. [DOI] [PubMed] [Google Scholar]
- 30.Vaquero EC, Edderkaoui M, Pandol SJ, Gukovsky I, Gukovskaya AS. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J Biol Chem. 2004;279(33):34643–54. doi: 10.1074/jbc.M400078200. [DOI] [PubMed] [Google Scholar]
- 31.Lee JK, Edderkaoui M, Truong P, Ohno I, Jang KT, Berti A, Pandol SJ, Gukovskaya AS. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology. 2007;133(5):1637–48. doi: 10.1053/j.gastro.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 32.Edderkaoui M, Odinokova I, Ohno I, Gukovsky I, Go VL, Pandol SJ, Gukovskaya AS. Ellagic acid induces apoptosis through inhibition of nuclear factor kappa B in pancreatic cancer cells. World J Gastroenterol. 2008;14(23):3672–80. doi: 10.3748/wjg.14.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu R, Wang YL, Edderkaoui M, Lugea A, Apte MV, Pandol SJ. Ethanol augments PDGF-induced NADPH oxidase activity and proliferation in rat pancreatic stellate cells. Pancreatology. 2007;7:332–40. doi: 10.1159/000105499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandol S, Edderkaoui M, Gukovsky I, Lugea A, Gukovskaya A. Desmoplasia of pancreatic ductal adenocarcinoma. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S44–7. doi: 10.1016/j.cgh.2009.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grippo PJ, Nowlin PS, Demeure MJ, Longnecker DS, Sandgren EP. Preinvasive pancreatic neoplasia of ductal phenotype induced by acinar cell targeting of mutant Kras in transgenic mice. Cancer Res. 2003;63(9):2016–9. [PubMed] [Google Scholar]
- 36.Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4(6):437–50. doi: 10.1016/s1535-6108(03)00309-x. [DOI] [PubMed] [Google Scholar]
- 37.Ji B, Tsou L, Wang H, Gaiser S, Chang DZ, Daniluk J, Bi Y, Grote T, Longnecker DS, Logsdon CD. Ras activity levels control the development of pancreatic diseases. Gastroenterology. 2009;137(3):1072–82. doi: 10.1053/j.gastro.2009.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morris PD. Lifetime excess risk of death from lung cancer for a U.S. female never-smoker exposed to environmental tobacco smoke. Environ Res. 1995;68:3–9. doi: 10.1006/enrs.1995.1002. [DOI] [PubMed] [Google Scholar]
- 39.Heidrich J, Wellmann J, Heuschmann PU, Kraywinkel K, Keil U. Mortality and morbidity from coronary heart disease attributable to passive smoking. Eur Heart J. 2007;28:2498–2502. doi: 10.1093/eurheartj/ehm151. [DOI] [PubMed] [Google Scholar]
- 40.Alberg AJ, Ford JG, Samet JM American College of Chest Physicians. Epidemiology of lungcancer. ACCP Evidence-Based Clinical Practice Guidelines (2nd edition) [J] Chest. 2007;132(3 Suppl):29S–55S. doi: 10.1378/chest.07-1347. [DOI] [PubMed] [Google Scholar]
- 41.McBride CM, Ostroff JS. Teachable moments for promoting smoking cessation: The context of cancer care and survivorship. Cancer Control. 2003;10(4):325–333. doi: 10.1177/107327480301000407. [DOI] [PubMed] [Google Scholar]
- 42.Travis LB, Rabkin CS, Brown LM, Allan JM, Alter BP, Ambrosone CB, Begg CB, Caporaso N, Chanock S, DeMichele A, Figg WD, Gospodarowicz MK, Hall EJ, Hisada M, Inskip P, Kleinerman R, Little JB, Malkin D, Ng AK, Offit K, Pui CH, Robison LL, Rothman N, Shields PG, Strong L, Taniguchi T, Tucker MA, Greene MH. Cancer survivorship—genetic susceptibility and second primary cancers: Research strategies and recommendations. J Nat Cancer Inst. 2006;98(1):15–25. doi: 10.1093/jnci/djj001. [DOI] [PubMed] [Google Scholar]
- 43.Parsons A, Daley A, Begh R, Aveyard P. Influence of smoking cessation after diagnosis of early stage lung cancer on prognosis: Systematic review of observational studies with meta-analysis. Br Med J. 2010;340:b5569. doi: 10.1136/bmj.b5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mack TM, Yu MC, Hanisch R, Henderson BE. Pancreas cancer and smoking, beverage consumption, and past medical history. J Natl Cancer Inst. 1986;76(1):49–60. [PubMed] [Google Scholar]
- 45.Lynch SM, Vrieling A, Lubin JH, Kraft P, Mendelsohn JB, Hartge P, Canzian F, Steplowski E, Arslan AA, Gross M, Helzlsouer K, Jacobs EJ, LaCroix A, Petersen G, Zheng W, Albanes D, Amundadottir L, Bingham SA, Boffetta P, Boutron-Ruault MC, Chanock SJ, Clipp S, Hoover RN, Jacobs K, Johnson KC, Kooperberg C, Luo J, Messina C, Palli D, Patel AV, Riboli E, Shu XO, Rodriguez Suarez L, Thomas G, Tjønneland A, Tobias GS, Tong E, Trichopoulos D, Virtamo J, Ye W, Yu K, Zeleniuch-Jacquette A, Bueno-de-Mesquita HB, Stolzenberg-Solomon RZ. Cigarette smoking and pancreatic cancer: a pooled analysis from the pancreatic cancer cohort consortium. Am J Epidemiol. 2009;170(4):403–13. doi: 10.1093/aje/kwp134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Howes N, Lerch MM, Greenhalf W, Stocken DD, Ellis I, Simon P, Truninger K, Ammann R, Cavallini G, Charnley RM, Uomo G, Delhaye M, Spicak J, Drumm B, Jansen J, Mountford R, Whitcomb DC, Neoptolemos JP, European Registry of Hereditary Pancreatitis and Pancreatic Cancer (EUROPAC) Clinical and genetic characteristics of hereditary pancreatitis in Europe. Clin Gastro Hep. 2004;2(3):252–261. doi: 10.1016/s1542-3565(04)00013-8. [DOI] [PubMed] [Google Scholar]
- 47.Weiss W, Benarde MA. The temporal relation between cigarette smoking and pancreatic cancer. Am J Public Health. 1983;73(12):1403–4. doi: 10.2105/ajph.73.12.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Whittemore AS, Paffenbarger RS, Jr, Anderson K, Halpern J. Early precursors of pancreatic cancer in college men. J Chronic Dis. 1983;36(3):251–6. doi: 10.1016/0021-9681(83)90059-0. [DOI] [PubMed] [Google Scholar]
- 49.Iodice S, Gandini S, Maisonneuve P, Lowenfels AB. Tobacco and the risk of pancreatic cancer: a review and meta-analysis. Langenbecks Arch Surg. 2008;393(4):535–45. doi: 10.1007/s00423-007-0266-2. [DOI] [PubMed] [Google Scholar]
- 50.U.S. Department of Health and Human Services. How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2010. [Google Scholar]
- 51.Hecht SS. Cigarette smoking, cancer risks, carcinogens and mechanisms. Langenbecks Arch Surg. 2006;391:603–613. doi: 10.1007/s00423-006-0111-z. [DOI] [PubMed] [Google Scholar]
- 52.Wen J, Fu JH, Zhang W, Guo M. Lung carcinoma signaling pathways activated by smoking. Chin J Cancer. 2011;30(8):551–558. doi: 10.5732/cjc.011.10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoffmann D, Hoffmann I, El-Bayoumy K. The less harmful cigarette: a controversial issue. a tribute to Ernst L. Wynder. Chem Res Toxicol. 2001;14(7):767–90. doi: 10.1021/tx000260u. [DOI] [PubMed] [Google Scholar]
- 54.Henningfield JE, Fant RV, Radzius A, Frost S. Nicotine concentration, smoke pH and whole tobacco aqueous pH of some cigar brands and types popular in the United States. Nic Tobac Res. 1999;1(2):163–168. doi: 10.1080/14622299050011271. [DOI] [PubMed] [Google Scholar]
- 55.Le Houezec J. Role of nicotine pharmacokinetics in nicotine addiction and nicotine replacement therapy: a review. Int J Tuberc Lung Dis. 2003;7(9):811–9. [PubMed] [Google Scholar]
- 56.Chowdhury P, Doi R, Chang LW, Rayford PL. Tissue distribution of [3H]-nicotine in rats. Biomed Environ Sci. 1993;6(1):59–64. [PubMed] [Google Scholar]
- 57.Prokopczyk B, Hoffmann D, Bologna M, Cunningham AJ, Trushin N, Akerkar S, Boyiri T, Amin S, Desai D, Colosimo S, Pittman B, Leder G, Ramadani M, Henne-Bruns D, Beger HG, El-Bayoumy K. Identification of tobacco-derived compounds in human pancreatic juice. Chem Res Toxicol. 2002;15(5):677–85. doi: 10.1021/tx0101088. [DOI] [PubMed] [Google Scholar]
- 58.Nicolov IG, Chernozemsky IN. Tumors and hyperplastic lesions in Syrian hamsters following transplacental and neonatal treatment with cigarette smoke condensate. J Cancer Res Clin Oncol. 1979;94(3):249–56. doi: 10.1007/BF00419284. [DOI] [PubMed] [Google Scholar]
- 59.Rivenson A, Hoffmann D, Prokopczyk B, Amin S, Hecht SS. Induction of lung and exocrine pancreas tumors in F344 rats by tobacco-specific and Areca-derived N-nitrosamines. Cancer Res. 1988;48(23):6912–7. [PubMed] [Google Scholar]
- 60.Bersch VP, Osvaldt AB, Edelweiss MI, Schumacher Rde C, Wendt LR, Abreu LP, Blom CB, Abreu GP, Costa L, Piccinini P, Rohde L. Effect of nicotine and cigarette smoke on an experimental model of intraepithelial lesions and pancreatic adenocarcinoma induced by 7,12-dimethylbenzanthracene in mice. Pancreas. 2009;38(1):65–70. doi: 10.1097/MPA.0b013e318184d330. [DOI] [PubMed] [Google Scholar]
- 61.Edderkaoui M, Park C, Lee I, Nitsche C, Gerloff A, Grippo PJ, Pandol SJ, Gukovskaya AS. Novel model of pancreatic neoplastic lesions induced by smoking compound NNK. Pancreas. 2011;40(8):1320. Abstract. [Google Scholar]
- 62.Schuller HM, Tithof PK, Williams M, Plummer H., 3rd The tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone is a beta-adrenergic agonist and stimulates DNA synthesis in lung adenocarcinoma via beta-adrenergic receptor-mediated release of arachidonic acid. Cancer Res. 1999;59(18):4510–5. [PubMed] [Google Scholar]
- 63.Schuller HM, Cekanova M. NNK-induced hamster lung adenocarcinomas over-express beta2-adrenergic and EGFR signaling pathways. Lung Cancer. 2005;49(1):35–45. doi: 10.1016/j.lungcan.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 64.Porta M, Crous-Bou M, Wark PA, Vineis P, Real FX, Malats N, Kampman E. Cigarette smoking and K-ras mutations in pancreas, lung and colorectal adenocarcinomas: etiopathogenic similarities, differences and paradoxes. Mutat Res. 2009;682(2-3):83–93. doi: 10.1016/j.mrrev.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 65.Blackford A, Parmigiani G, Kensler TW, Wolfgang C, Jones S, Zhang X, et al. Genetic mutations associated with cigarette smoking in pancreatic cancer. Cancer Res. 2009;69(8):3681–8. doi: 10.1158/0008-5472.CAN-09-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Weddle DL, Tithoff P, Williams M, Schuller HM. Beta-adrenergic growth regulation of human cancer cell lines derived from pancreatic ductal carcinomas. Carcinogenesis. 2001;22(3):473–9. doi: 10.1093/carcin/22.3.473. [DOI] [PubMed] [Google Scholar]
- 67.Askari MD, Tsao MS, Schuller HM. The tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone stimulates proliferation of immortalized human pancreatic duct epithelia through beta-adrenergic transactivation of EGF receptors. J Cancer Res Clin Oncol. 2005;131(10):639–48. doi: 10.1007/s00432-005-0002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Askari MD, Tsao MS, Cekanova M, Schuller HM. Ethanol and the tobacco-specific carcinogen, NNK, contribute to signaling in immortalized human pancreatic duct epithelial cells. Pancreas. 2006;33:53–62. doi: 10.1097/01.mpa.0000226883.55828.e9. [DOI] [PubMed] [Google Scholar]
- 69.Yoshikawa H, Hellström-Lindahl E, Grill V. Evidence for functional nicotinic receptors on pancreatic beta cells. Metabolism. 2005;54:247–54. doi: 10.1016/j.metabol.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 70.Lee J, Taneja V, Vassallo R. Cigarette Smoking and Inflammation: Cellular and Molecular Mechanisms. J Dent Res. 2012;91(2):142–9. doi: 10.1177/0022034511421200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alexandre M, Pandol SJ, Gorelick FS, Thrower EC. The Emerging Role of Smoking in the Development of Pancreatitis. Pancreatology. 2011;11(5):469–474. doi: 10.1159/000332196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chowdhury P, Doi R, Tangoku A, Rayford PL. Structural and functional changes of rat exocrine pancreas exposed to nicotine. Int J Pancreatol. 1995;18(3):257–64. doi: 10.1007/BF02784950. [DOI] [PubMed] [Google Scholar]
- 73.Dubick MA, Palmer R, Lau PP, Morrill PR, Geokas MC. Altered exocrine pancreatic function in rats treated with nicotine. Toxicol Appl Pharmacol. 1988;96(1):132–9. doi: 10.1016/0041-008x(88)90255-4. [DOI] [PubMed] [Google Scholar]
- 74.Lau PP, Dubick MA, Yu GS, Morrill PR, Geokas MC. Dynamic changes of pancreatic structure and function in rats treated chronically with nicotine. Toxicol Appl Pharmacol. 1990;104(3):457–65. doi: 10.1016/0041-008x(90)90167-s. [DOI] [PubMed] [Google Scholar]
- 75.Wittel UA, Pandey KK, Andrianifahanana M, Johansson SL, Cullen DM, Akhter MP, Brand RE, Prokopczyk B, Batra SK. Chronic pancreatic inflammation induced by environmental tobacco smoke inhalation in rats. Am J Gastroenterol. 2006;101(1):148–59. doi: 10.1111/j.1572-0241.2006.00405.x. [DOI] [PubMed] [Google Scholar]
- 76.Soeda J, Morgan M, McKee C, Mouralidarane A, Lin C, Roskams T, Oben JA. Nicotine induces fibrogenic changes in human liver via nicotinic acetylcholine receptors expressed on hepatic stellate cells. Biochem Biophys Res Commun. 2012;417(1):17–22. doi: 10.1016/j.bbrc.2011.10.151. [DOI] [PubMed] [Google Scholar]
- 77.Esposito I, Menicagli M, Funel N, Bergmann F, Boggi U, Mosca F, Bevilacqua G, Campani D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J Clin Pathol. 2004;57:630–636. doi: 10.1136/jcp.2003.014498. [DOI] [PMC free article] [PubMed] [Google Scholar]