

Abstract
Objectives
Highly active antiretroviral therapy (HAART) is the mainstay of treatment for HIV-1 infection. While current HAART regimens have been extremely effective, issues of associated toxicity, cost and resistance remain and there is a need for novel antiretroviral compounds to complement the existing therapy. We sought to develop a novel high-throughput method for identifying compounds that block later steps in the life cycle not targeted by current therapy.
Methods
We designed a high-throughput screen to identify inhibitors of post-integration steps in the HIV-1 life cycle. The screening method was applied to a library of compounds that included numerous FDA-approved small molecules.
Results
Among the small molecules that inhibited late stages in HIV-1 replication were members of the cardiac glycoside family. We demonstrate that cardiac glycosides potently inhibit HIV-1 gene expression, thereby reducing the production of infectious HIV-1. We demonstrate that this inhibition is dependent upon the human Na+/K+-ATPase, but independent of cardiac glycoside-induced increases in intracellular Ca2+.
Conclusions
We have validated a novel high-throughput screen to identify small molecule inhibitors of HIV-1 gene expression, virion assembly and budding. Using this screen, we have demonstrated that a number of FDA-approved compounds developed for other purposes potently inhibit HIV-1 replication, including the cardiac glycosides. Our work indicates that the entire cardiac glycoside family of drugs shows potential for antiretroviral drug development.
Keywords: HAART, digoxin, antiretroviral
Introduction
Current antiretroviral therapy effectively reduces the plasma level of HIV-1 in most infected individuals to below the limit of detection of clinical assays.1–3 However, the ongoing problems of undesirable off-target effects, high cost and drug-resistant viral strains indicate a need for additional classes of antiretroviral drugs.4,5 Viral assembly and budding have both been previously suggested as potential targets for such new drug classes. Late pre-assembly stages of the replication cycle include viral transcription, translation and transport of viral proteins and RNA to the plasma membrane. Although these stages are primarily mediated by host proteins, they could be targeted by novel antiretroviral drugs. With regard to virus assembly, previous studies have identified potentially targetable interactions mediated by both the C-terminal and N-terminal domains of the capsid, as well as interactions between zinc and the zinc finger of the nucleocapsid protein.6 In the case of budding, the interaction between the cellular protein Tsg101, a member of the ESCRT-1 family, and the PTAP sequence in the p6 domain of the viral protein Gag has been identified as a target.7,8 Despite several potential drug targets, there are currently no clinically approved drugs that inhibit stages of the HIV-1 replication cycle following integration and preceding maturation, leaving a significant portion of the HIV-1 replication cycle unopposed by antiretrovirals.
The aim of this study was to develop a high-throughput method to screen for compounds with late-stage inhibitory potential against HIV-1. To do so, we transfected 293T cells with plasmids containing HIV-1 proviral DNA and then measured virus production using a qPCR assay of nucleic acid extracted from the cellular supernatant.9 The reverse transcription step of this assay made use of an oligo-dT primer to prevent the detection of both HIV-1 DNA and unprocessed RNA transcripts. Additionally, the qPCR step used a primer specific for polyadenylated HIV-1 RNA, again preventing the detection of unprocessed HIV-1 RNA transcripts and HIV-1 DNA, including the plasmid DNA used in transfection. To test this approach, we screened the Spectrum 2000 drug library. Of the compounds that inhibited late stages of the HIV-1 replication cycle, there was a striking overrepresentation of drugs from the cardiac glycoside class. We identified compounds belonging to both subclasses of cardiac glycosides: the cardenolides and the bufadienolides. Although both are C(23) steroids, they differ in that cardenolides contain a butenolide five-membered lactone ring at C-17, whereas bufadienolides contain a six-membered lactone ring.10 Members of both classes of cardiac glycosides inhibited late stages of HIV-1 production and changes in structure resulted in changes in inhibition.
Methods
293T cell screen for inhibitors of post-integration stages of the HIV-1 replication cycle
Lipofectamine 2000 (Invitrogen, USA) was used to co-transfect 293T cells with a plasmid expressing a CXCR4-tropic HIV-1 envelope protein (pX4) as well as a plasmid containing a variant of the reference HIV-1 isolate NL4-3 in which the env gene has been replaced with green fluorescent protein (GFP) (pNL4-3ΔEnvGFP). After 6 h, the cells were treated with 0.05% Trypsin-EDTA (Gibco, USA), pelleted and plated in flat-bottomed 96-well plates at 180 000 cells/well in Dulbecco's modified Eagle's medium + 10% fetal bovine serum. Each test compound was added at a final concentration of 10 μM. Compounds were taken from the Spectrum 2000 compound library (Microsource, USA). The compounds were dissolved in DMSO and an equivalent concentration of DMSO (0.1% v/v) was present in each well, including the control wells. The proteasome inhibitor epoxomicin (Sigma–Aldrich) was added at a concentration of 1.8 μM as a positive control. The cells were incubated for 18 h at 37°C in 5% CO2. The supernatant was then removed and the released HIV-1 RNA was extracted and quantified.
HIV-1 RNA extraction and RT-qPCR
HIV-1 RNA was extracted from 60 μL of treated 293T cell supernatant using the ZR-96 Viral RNA Kit (Zymo Research, USA). Eluted RNA was reverse transcribed to cDNA using the Superscript III First-Strand Synthesis System (Invitrogen, USA). The reverse transcription reaction was primed with oligo-dT, as described in the manufacturer's protocol. The HIV-1 cDNA was detected using primers and probes specific for sequences corresponding to polyadenylated HIV-1 RNA.9 Inhibition was calculated relative to the untreated control.
Flow cytometry
Flow cytometry was performed using a BD FACS Canto II (BD Biosciences, USA). Analysis was performed using FlowJo software (Tree Star, USA). Evaluation of the toxicity of the compounds screened was performed by fixing the cells with 2.5% paraformaldehyde and examining the resulting forward/side scatter plots. Any compounds that caused significant toxicity were not included in the list of hits and were not used in subsequent experiments.
Measurement of intracellular Ca2+ levels with Fura Red AM dye
Increases in intracellular Ca2+ were visualized using Fura Red AM dye (Invitrogen, USA). After treatment, cells were incubated with Fura Red AM dye (10 μM) for 30 min and then washed with PBS. Cells were analysed for Fura Red AM by flow cytometry using the AmCyan channel. Decreases in the AmCyan signal corresponded to quenching of the Fura Red AM fluorescence as a result of increases in intracellular Ca2+. The inhibitor KB-R7943 was obtained from Sigma–Aldrich (USA).
Plasmids for the expression of the α-1 subunit of the Na+/K+-ATPase
The plasmid containing the murine α-1 subunit of the Na+/K+-ATPase was obtained from Open Biosystems (plasmid MMM1013-7512721). The plasmid containing the human α-1 subunit of the Na+/K+-ATPase was also obtained from Open Biosystems (plasmid MHS1010-9205172).
Results
Development of a cell-based screen for post-integration inhibitors of the HIV-1 replication cycle
We designed a cell-based system to screen for compounds that inhibit the release of packaged HIV-1 genomic RNA. Briefly, 293T cells were co-transfected with plasmids containing a CXCR4-tropic HIV-1 envelope (pX4) and an NL4-3 genome in which the env gene was deleted and replaced with GFP (pNL4-3ΔEnvGFP). The transfected 293T cells were transferred to 96-well plates and assay compounds were added at the screening concentration of 10 μM. After 18 h, cell-free supernatants were collected and viral RNA isolated. The eluted viral RNA was reverse transcribed to cDNA and then analysed by qPCR using a probe and primers specific to the DNA sequence corresponding to the polyadenylated HIV-1 RNA.9
To verify that this screen was capable of detecting compounds that inhibited the late stages of the HIV-1 life cycle, we utilized epoxomicin, a potent inhibitor of the 26S proteasome, as a positive control. Treatment of HIV-1-producing cells with epoxomicin has been shown to reduce the release of virus particles, presumably through the reduction in the levels of free ubiquitin required for HIV-1 gag processing.11 Treatment with epoxomicin reduced the amount of HIV-1 genomic RNA released into the supernatant (Figure 1), indicating that this system can detect known post-integration inhibitors of HIV-1 replication.
Figure 1.
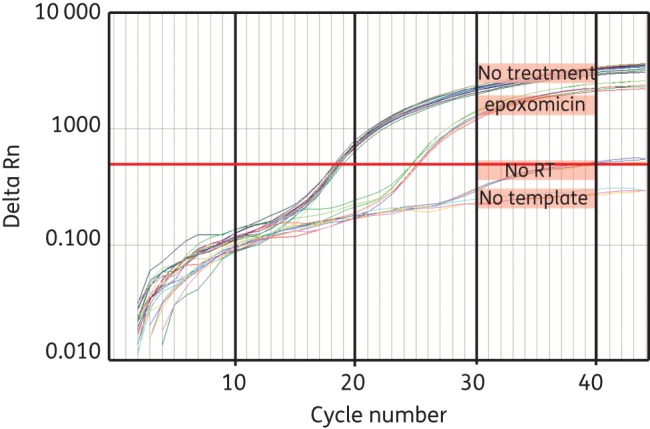
Screening for compounds that inhibit late stages of HIV-1 replication. 293T cells were transiently transfected with pNL4-3ΔEnvGFP and pX4 and then some wells were treated with epoxomicin (1.8 μM). Eighteen hours after treatment, RNA was extracted from culture supernatant, cDNA was synthesized and HIV-1 RNA-specific qPCR was performed. Supernatant from untreated cells was used as a control. No-template controls and no-reverse-transcription (RT) controls were also included. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Identification of the cardiac glycosides as potent inhibitors of late stages of the HIV-1 replication cycle
With the goal of identifying novel late-stage inhibitors of the HIV-1 replication cycle, we utilized the 293T cell-based system described above to screen the Spectrum 2000 compound library (Microsource Discovery), a 2000 compound collection of approved drugs, natural products and other bioactive molecules. Our screen yielded a number of compounds that inhibited HIV-1 genomic RNA release to varying degrees, with some compounds causing >5 log inhibition relative to the negative controls (data not shown).
Among the compounds identified in our screen were the transcription inhibitor actinomycin D and the translation inhibitor cycloheximide. Actinomycin D has been shown to inhibit the replication of several other viruses.12–14 The identification of these general inhibitors served to validate the screening approach. Furthermore, other previously described inhibitors of HIV-1 were identified, including celastrol and digoxin.15,16 Recently, digoxin has been shown to strongly inhibit HIV-1 structural protein synthesis via alteration of RNA processing.16 Whether other cardiac glycosides also inhibit HIV-1 was previously unknown. Here, we observed that numerous other cardiac glycosides inhibited HIV-1 production (Table 1). Of these, the majority of the screened cardiac glycosides were cardenolides; the single bufadienolide that was screened also inhibited HIV-1 production.
Table 1.
Inhibition of HIV-1 RNA release from 293T cells by cardiac glycosides
| Cardiac glycoside | Log inhibition | FDA approved? | Structural class |
|---|---|---|---|
| Convallatoxin | 5.35 | no | cardenolide |
| Periplocymarin | 4.45 | no | cardenolide |
| Digitoxin | 2.41 | yes | cardenolide |
| Digoxin | 2.36 | yes | cardenolide |
| Strophanthidin | 2.25 | no | cardenolide |
| Gitoxigenin diacetate | 2.02 | no | cardenolide |
| Digoxigenin | 2.01 | no | cardenolide |
| Cymarin | 1.94 | no | cardenolide |
| Sarmentogenin | 1.76 | no | cardenolide |
| Gitoxin | 1.18 | no | cardenolide |
| Gitoxigenin | 0.84 | no | cardenolide |
| Strophanthidinic acid lactone acetate | 0 | no | cardenolide |
| Strophanthidin semicarbazide | 0 | no | cardenolide |
Because of the striking overrepresentation of cardiac glycosides among the inhibitors identified in the screen, we chose to focus specifically on these compounds. To determine whether the reduction in supernatant HIV-1 RNA by the cardiac glycosides resulted in a functional reduction in the infection of primary CD4+ T cells by those virus-containing supernatants, we spinoculated primary CD4+ lymphoblasts from healthy donors with the supernatant from 293T cells that had been transfected with pX4 and pNL4-3ΔEnvGFP and then treated with cardiac glycosides. Because we observed that the cardiac glycosides inhibited HIV-1 production in our 293T cell screen, we expected that the supernatant from treated transfected cells would contain fewer viruses than supernatant from transfected cells that were not treated with cardiac glycosides. The resulting level of infection in CD4+ lymphoblasts therefore reflects how much virus was produced in the presence of the cardiac glycosides. Infection was measured 3 days after spinoculation using flow cytometry to detect GFP expression. The percentage inhibition was calculated relative to the maximum infection observed following infection with the supernatants from untreated, transfected 293T cells and IC50 values were calculated. We observed dose-dependent reductions in the infection of CD4+ lymphoblasts by supernatants from cardiac glycoside-treated cells, with IC50 values ranging from 349.5 nM down to 4.3 nM for the compounds tested (Table 2). This confirms that the reductions in supernatant HIV-1 RNA caused by cardiac glycoside treatment resulted in reductions of infectious virus in the supernatant.
Table 2.
IC50 values for inhibition of infection of primary CD4+ lymphoblasts with supernatant from transfected 293T cells treated with cardiac glycosides
| Compound | IC50 (nM) | FDA approved? | Structural class |
|---|---|---|---|
| Bufalin | 4.3 | no | bufadienolide |
| Strophanthidin | 218.6 | no | cardenolide |
| Digoxin | 17.0 | yes | cardenolide |
| Gitoxigenin | 100.6 | no | cardenolide |
| Gitoxigenin diacetate | 349.5 | no | cardenolide |
| Digitoxin | 99.2 | yes | cardenolide |
| Ouabain octahydrate | 13.0 | no | cardenolide |
We sought to further verify that the cardiac glycosides inhibit production of HIV-1 structural proteins. Therefore, we directly examined the effects of cardiac glycosides on the expression of GFP from a proviral construct with GFP in the env ORF. We observed dose-dependent reductions in HIV-1 gene expression by the cardiac glycosides in 293T cells transfected with pNL4-3ΔEnvGFP and pX4, as measured by flow cytometry analysis of gene expression (data not shown). Expression of GFP is expected to require a single splicing reaction of the type used to generate env mRNAs. Our finding is consistent with a previous report indicating that digoxin interferes with the Rev-dependent export of singly spliced HIV-1 RNA.16 Our results extend these findings by demonstrating that the cardiac glycosides as a class inhibit HIV-1 gene expression.
Cardiac glycoside inhibition of HIV-1 gene expression is dependent upon the Na+/K+-ATPase
Cardiac glycosides inhibit the Na+/K+-ATPase by binding to the α-1 subunit, blocking Na+ ion extrusion from treated cells.17,18 However, the murine α-1 subunit is resistant to inhibition by cardiac glycosides.19 To determine whether the inhibition of HIV-1 gene expression by the cardiac glycosides was dependent upon the inhibition of the human Na+/K+-ATPase, 293T cells were transiently co-transfected with the murine or human α-1 subunit in addition to pNL4-3ΔEnvGFP and pX4. Overexpression of the murine α-1 subunit in 293T cells abrogated the HIV-1 inhibition by digoxin, as measured by GFP expression (Figure 2a versus b). Thus, expression of the digoxin-resistant murine α-1 subunit of the Na+/K+-ATPase is sufficient to overcome digoxin inhibition. When the human α-1 subunit was overexpressed in 293T cells, HIV-1 inhibition by digoxin was still observed (Figure 2c versus d), indicating that overexpression of an α-1 subunit of the Na+/K+-ATPase is not sufficient to overcome digoxin inhibition. Taken together, these data suggest that digoxin inhibits HIV-1 gene expression via inhibition of the α-1 subunit of the Na+/K+-ATPase.
Figure 2.

Overexpression of the murine α-1 subunit of the Na+/K+-ATPase abrogates HIV-1 inhibition by digoxin. (a) Flow cytometric analysis of GFP expression in 293T cells transfected with the murine α-1 subunit of the Na+/K+-ATPase, pNL4-3ΔEnv and pX4. (b) Flow cytometric analysis of GFP expression in 293T cells transfected with the murine α-1 subunit of the Na+/K+-ATPase, pNL4-3ΔEnv and pX4 and then treated with digoxin (10 μM) for 18 h. (c) Flow cytometric analysis of GFP expression in 293T cells transfected with the human α-1 subunit of the Na+/K+-ATPase, pNL4-3ΔEnv and pX4. (d) Flow cytometric analysis of GFP expression in 293T cells transfected with the human α-1 subunit of the Na+/K+-ATPase, pNL4-3ΔEnv and pX4 and then treated with digoxin (10 μM) for 18 h. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Cardiac glycoside inhibition of HIV-1 gene expression is independent of increases in intracellular Ca2+ concentration
Cardiac glycoside inhibition of the Na+/K+-ATPase has been shown to induce increases in intracellular Ca2+ concentrations.17,18 Therefore, cardiac glycoside inhibition of HIV-1 via interaction with the α-1 subunit of the Na+/K+-ATPase may also depend upon increases in intracellular Ca2+. Previously, the small molecule KB-R7943 was shown to inhibit digoxin-induced increases in intracellular Ca2+ via inhibition of a sodium–calcium exchanger.20,21 We first verified that coadministration of digoxin and KB-R7943 prevents digoxin-induced increases in intracellular Ca2+(Figure 3a). Next, we tested whether HIV-1 inhibition by digoxin depended on digoxin-induced increases in intracellular Ca2+. Thus, we coadministered digoxin and KB-R7943 to 293T cells transfected with pNL4-3ΔEnvGFP and pX4. Co-administration of digoxin with KB-R7943 did not relieve the inhibitory effect of digoxin on HIV-1, as measured by GFP expression using flow cytometry (Figure 3b). This result demonstrates that HIV-1 inhibition by cardiac glycosides is independent of increases in intracellular Ca2+.
Figure 3.
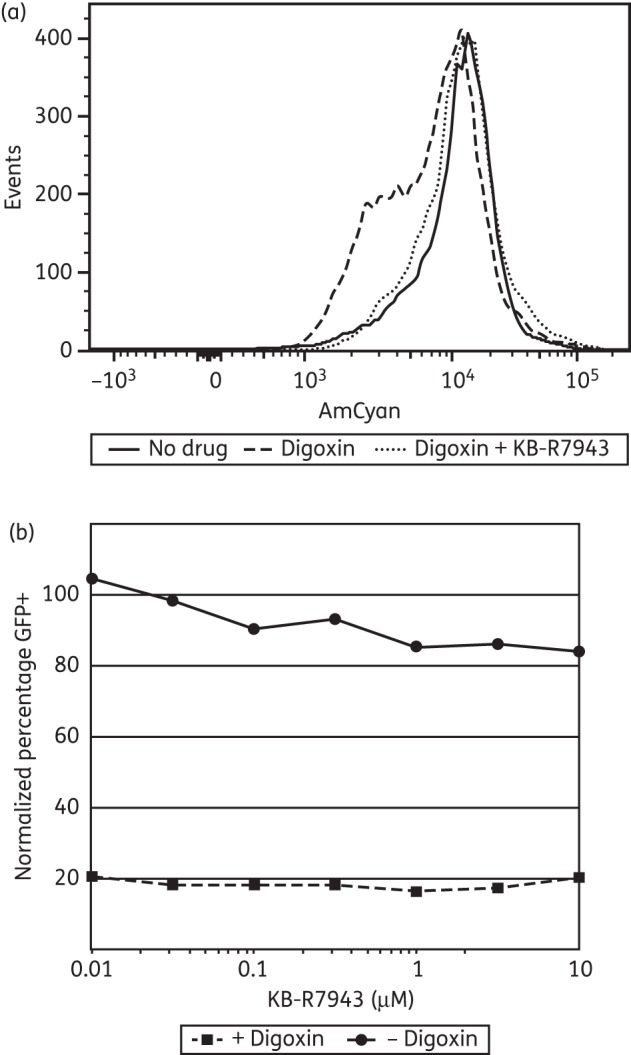
Digoxin inhibition of HIV-1 is independent of digoxin-induced increases in intracellular Ca2+. (a) Flow cytometric analysis of 293T cells treated with DMSO alone, digoxin (1 μM) or digoxin (1 μM) plus KB-R7943 (10 μM) for 18 h and then incubated with Fura Red AM dye (10 μM) for 30 min. Reduced AmCyan fluorescence indicates increased intracellular Ca2+. (b) Flow cytometric analysis of GFP expression in 293T cells transiently transfected with pNL4-3ΔEnvGFP and pX4 and then treated with the indicated concentrations of KB-R7943 alone or in combination with digoxin (1 μM) for 18 h. The percentage of cells expressing GFP is normalized to GFP expression in transfected DMSO-only control 293T cells.
Discussion
The principal components of current highly active antiretroviral therapy (HAART) regimens target only select stages of the HIV-1 life cycle: entry, reverse transcription, integration and maturation. While there is no doubt that HAART has successfully reduced the morbidity and mortality associated with HIV-1 infection, the problems of drug resistance, associated toxicities and cost remain. As a result, the development of novel classes of inhibitors remains a priority. Few inhibitors of HIV-1 gene expression and budding have been identified to date. Therefore, we sought to identify compounds that inhibit HIV-1 at these stages, with a preference for compounds that have been tested or approved for use in humans. To accomplish this, we developed a cell-based screen that could be scaled for rapid testing of large compound libraries.
Our screening uncovered a number of compounds that inhibit HIV-1 virion production by cells transfected with proviral DNA constructs, including numerous compounds from the cardiac glycoside family. The cardiac glycosides were initially described as inhibitors of the Na+/K+-ATPase and have enjoyed wide use in the treatment of heart failure and arrhythmia. Recent studies have suggested that this family of compounds may also have potent anticancer,22 immunomodulatory23 and antihypertensive properties24 as well as activity against HIV-1 replication.16
With respect to the antiretroviral effects of the cardiac glycosides, a recent study has shown that a single drug in the cardiac glycoside class, digoxin, inhibits HIV-1 replication by altering viral RNA splice site use, resulting in the decreased production of the Rev protein.16 This effect is mediated through the activity of a subset of the SR family of cellular splicing factors. However, overexpression of the Rev protein alone did not counter the inhibitory effect of digoxin, suggesting that other mechanisms of inhibition may be involved. Wong et al.16 indicate that digitoxin, a compound that is closely related to digoxin, has a similar effect on certain SR proteins25 and suggest that other cardiac glycosides may also be effective at preventing HIV-1 replication. Here, we demonstrate that this is indeed the case.
Our studies demonstrate that not only digoxin but also the entire cardiac glycoside family of compounds exhibit antiretroviral effects. Cardiac glycosides are known Na+/K+-ATPase inhibitors; this inhibition results in the inhibition and then reversal of the sodium–calcium exchanger, leading to the build-up of calcium in the cell.17,18,20 Although Wong et al.16 demonstrated that the inhibition of HIV-1 replication was related to certain SR splicing proteins, we questioned whether the action of the cardiac glycosides depended on both the Na+/K+-ATPase and the cardiac glycoside-induced increase in intracellular Ca2+. Our study demonstrates that although HIV-1 inhibition is dependent on the Na+/K+-ATPase, it was independent of the induced increase in intracellular Ca2+. Previous studies have shown that the Na+/K+-ATPase plays a role in several signal transduction pathways.26–32 Additionally, some previous studies have also shown that the induction of signalling is independent of the intracellular ion concentrations.33 It is possible that the inhibition of this ATPase triggers a signalling mechanism leading to modulation of the action of the SR splicing proteins, another question that warrants further investigation. A detailed analysis of the mechanism of inhibition by each member of the cardiac glycoside family along with structure–activity relationship studies are extremely important to the potential development of the cardiac glycosides as antiretrovirals. Of note, the dose-limiting toxicities observed with cardiac glycosides in humans are related to toxic increases in cardiac contractility, driven by increases in intracellular Ca2+. As the mechanism of cardiac glycoside inhibition of HIV-1 appears to be independent of such Ca2+ increases, it is possible that structural modification of the cardiac glycosides or development of alternative inhibitors could avoid the cardiac toxicity while maintaining HIV-1 inhibition.
A preponderance of evidence in the literature suggests that current HAART regimens are capable of fully suppressing ongoing HIV-1 replication in treated individuals. However, treatment intensification with raltegravir has been shown to lead to a rapid increase in episomal 2-LTR circles in a proportion of treated individuals, even though no effect on residual low-level viraemia in these individuals was observed.34 Such reductions in low-level viraemia upon intensification would further suggest that ongoing replication is occurring. As the cardiac glycosides inhibit HIV-1 at a post-integration stage of the viral life cycle, treatment intensification studies employing the cardiac glycosides may offer some new insights into the source of low-level viraemia. Our present study demonstrates that the cardiac glycosides prevent the production and release of HIV-1 RNA-containing virions. Therefore, we predict that intensification with cardiac glycosides would lead to a decline in low-level plasma HIV-1 RNA. The kinetics of this decline could provide evidence for the source of this low-level plasma HIV-1 RNA, be it ongoing replication in activated CD4+ T cells, release of virus from cognate antigen-reactivated cells of the latent reservoir or another source entirely.
Funding
This work was supported by the Howard Hughes Medical Institute, an ARCHE grant from the Foundation for AIDS Research, and by the National Institutes of Health (AI081600).
Transparency declarations
None to declare.
Acknowledgements
We would like to acknowledge the High Throughput Biology Center at The Johns Hopkins University Institute for Basic Biomedical Sciences for assistance with compound library preparation.
References
- 1.Hammer SM, Squires KE, Hughes MD, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337:725–33. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, Mellors JW, Havlir D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337:734–9. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 3.Perelson AS, Essunger P, Cao Y, et al. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature. 1997;387:188–91. doi: 10.1038/387188a0. [DOI] [PubMed] [Google Scholar]
- 4.Little SJ, Holte S, Routy JP, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N Engl J Med. 2002;347:385–94. doi: 10.1056/NEJMoa013552. [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Picado J, DePasquale MP, Kartsonis N, et al. Antiretroviral resistance during successful therapy of HIV type 1 infection. Proc Natl Acad Sci USA. 2000;97:10948–53. doi: 10.1073/pnas.97.20.10948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiang Y, Liu X, De Clercq E. New therapeutic approaches targeted at the late stages of the HIV-1 replication cycle. Curr Med Chem. 2011;18:16–28. doi: 10.2174/092986711793979751. [DOI] [PubMed] [Google Scholar]
- 7.Goila-Gaur R, Demirov DG, Orenstein JM, et al. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77:6507–19. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen H, Liu X, Li Z, et al. TSG101: a novel anti-HIV-1 drug target. Curr Med Chem. 2010;17:750–8. doi: 10.2174/092986710790514444. [DOI] [PubMed] [Google Scholar]
- 9.Shan L, Rabi SA, Laird GM, et al. A novel PCR assay for quantification of HIV-1 RNA. J Virol. 2013;87:6521–5. doi: 10.1128/JVI.00006-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melero CP, Medarde M, San Feliciano A. A short review on cardiotonic steroids and their aminoguanidine analogues. Molecules. 2000;5:51–81. [Google Scholar]
- 11.Schubert U, Ott DE, Chertova EN, et al. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci USA. 2000;97:13057–62. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duesberg PH, Robinson WS. Inhibition of mouse leukemia virus (MLV) replication by actinomycin D. Virology. 1967;31:742–6. doi: 10.1016/0042-6822(67)90211-5. [DOI] [PubMed] [Google Scholar]
- 13.Bader JP. The role of deoxyribonucleic acid in the synthesis of Rous sarcoma virus. Virology. 1964;22:462–8. doi: 10.1016/0042-6822(64)90067-4. [DOI] [PubMed] [Google Scholar]
- 14.Bases RE, King AS. Inhibition of Rauscher murine leukemia virus growth in vitro by actinomycin D. Virology. 1967;32:175–83. doi: 10.1016/0042-6822(67)90268-1. [DOI] [PubMed] [Google Scholar]
- 15.Narayan V, Ravindra KC, Chiaro C, et al. Celastrol inhibits Tat-mediated human immunodeficiency virus (HIV) transcription and replication. J Mol Biol. 2011;410:972–83. doi: 10.1016/j.jmb.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wong RW, Balachandran A, Ostrowski MA, et al. Digoxin suppresses HIV-1 replication by altering viral RNA processing. PLoS Pathog. 2013;9:e1003241. doi: 10.1371/journal.ppat.1003241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hauptman PJ, Kelly RA. Digitalis. Circulation. 1999;99:1265–70. doi: 10.1161/01.cir.99.9.1265. [DOI] [PubMed] [Google Scholar]
- 18.Iwamoto T, Kita S. Hypertension, Na+/Ca2+ exchanger, and Na+, K+-ATPase. Kidney Int. 2006;69:2148–54. doi: 10.1038/sj.ki.5000421. [DOI] [PubMed] [Google Scholar]
- 19.Lin Y, Dubinsky WP, Ho DH, et al. Determinants of human and mouse melanoma cell sensitivities to oleandrin. J Exp Ther Oncol. 2008;7:195–205. [PubMed] [Google Scholar]
- 20.Weiss M, Baek M, Kang W. Systems analysis of digoxin kinetics and inotropic response in the rat heart: effects of calcium and KB-R7943. Am J Physiol Heart Circ Physiol. 2004;287:H1857–67. doi: 10.1152/ajpheart.01121.2003. [DOI] [PubMed] [Google Scholar]
- 21.Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–7. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- 22.Prassas I, Diamandis EP. Novel therapeutic applications of cardiac glycosides. Nat Rev Drug Discov. 2008;7:926–35. doi: 10.1038/nrd2682. [DOI] [PubMed] [Google Scholar]
- 23.Huh JR, Leung MW, Huang P, et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORgammat activity. Nature. 2011;472:486–90. doi: 10.1038/nature09978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Qian DZ, Tan YS, et al. Digoxin and other cardiac glycosides inhibit HIF-1α synthesis and block tumor growth. Proc Natl Acad Sci USA. 2008;105:19579–86. doi: 10.1073/pnas.0809763105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson ES, Lin CH, Xiao X, et al. The cardiotonic steroid digitoxin regulates alternative splicing through depletion of the splicing factors SRSF3 and TRA2B. RNA. 2012;18:1041–9. doi: 10.1261/rna.032912.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kometiani P, Li J, Gnudi L, et al. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J Biol Chem. 1998;273:15249–56. doi: 10.1074/jbc.273.24.15249. [DOI] [PubMed] [Google Scholar]
- 27.Xie Z, Kometiani P, Liu J, et al. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J Biol Chem. 1999;274:19323–8. doi: 10.1074/jbc.274.27.19323. [DOI] [PubMed] [Google Scholar]
- 28.Haas M, Askari A, Xie Z. Involvement of Src and epidermal growth factor receptor in the signal-transducing function of Na+/K+-ATPase. J Biol Chem. 2000;275:27832–7. doi: 10.1074/jbc.M002951200. [DOI] [PubMed] [Google Scholar]
- 29.Haas M, Wang H, Tian J, et al. Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J Biol Chem. 2002;277:18694–702. doi: 10.1074/jbc.M111357200. [DOI] [PubMed] [Google Scholar]
- 30.Aizman O, Uhlen P, Lal M, et al. Ouabain, a steroid hormone that signals with slow calcium oscillations. Proc Natl Acad Sci USA. 2001;98:13420–4. doi: 10.1073/pnas.221315298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aydemir-Koksoy A, Abramowitz J, Allen JC. Ouabain-induced signaling and vascular smooth muscle cell proliferation. J Biol Chem. 2001;276:46605–11. doi: 10.1074/jbc.M106178200. [DOI] [PubMed] [Google Scholar]
- 32.Miyakawa-Naito A, Uhlen P, Lal M, et al. Cell signaling microdomain with Na,K-ATPase and inositol 1,4,5-trisphosphate receptor generates calcium oscillations. J Biol Chem. 2003;278:50355–61. doi: 10.1074/jbc.M305378200. [DOI] [PubMed] [Google Scholar]
- 33.Liu J, Tian J, Haas M, et al. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J Biol Chem. 2000;275:27838–44. doi: 10.1074/jbc.M002950200. [DOI] [PubMed] [Google Scholar]
- 34.Buzon MJ, Massanella M, Llibre JM, et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat Med. 2010;16:460–5. doi: 10.1038/nm.2111. [DOI] [PubMed] [Google Scholar]