

Abstract
Early clearance (EC) is the successful eradication of inhaled Mycobacterium tuberculosis before an adaptive immune response develops. Evidence for EC comes from case contact studies that consistently show that a proportion of heavily exposed individuals do not develop M. tuberculosis infection. Further support for the existence of this phenotype comes from genetic loci associated with tuberculin reactivity. In this review we discuss aspects of the innate response that may underpin EC and hypotheses that can be tested through field laboratory link studies in M. tuberculosis case contacts. Specifically, we consider mechanisms whereby alveolar macrophages recognize and kill intracellular M. tuberculosis, and how other cell types, such as neutrophils, natural killer T cells, mucosa-associated invariant T cells and γδ T cells may assist. How EC may be impaired by HIV infection or vitamin D deficiency is also explored. As EC is a form of protective immunity, further study may advance the development of vaccines and immunotherapies to prevent M. tuberculosis infection.
Keywords: early clearance, innate immunity, Mycobacterium tuberculosis
Introduction
Mycobacterium tuberculosis is the second most common infectious killer worldwide: in 2013, 9 million people will have been diagnosed with tuberculosis (TB) and 1.2–1.4 million will die from the disease.1 A lack of feasible preventive strategies means that the public health response to M. tuberculosis focuses on case finding and management and has a modest impact on transmission. Better preventive strategies are desperately needed, yet efforts to develop a vaccine against infectious pulmonary TB have not been successful.2
Early clearance (EC) of M. tuberculosis infection can be defined as the eradication of infecting M. tuberculosis before an adaptive response develops. This phenotype has long been speculated to exist; however, its immunological correlates are yet to be described.3 Identification of the immunological mechanisms behind EC could present new opportunities to prevent M. tuberculosis infection. First, EC may be influenced by modifiable host factors that could be therapeutic targets. Second, EC is a model of protective immunity to M. tuberculosis and its study may yield sought after correlates of protection for vaccine development.4 For these reasons EC should be prioritized as a focus of M. tuberculosis research. In this review, we discuss putative immunological mechanisms of EC that can serve as hypotheses to be tested in field–laboratory link studies.
Evidence for early clearance
Presently, both epidemiological and genetic data support the existence of EC of M. tuberculosis. Case contact studies repeatedly find the exposure to M. tuberculosis does not always lead to a positive interferon-γ release assay (IGRA) or tuberculin skin test (TST). In 1941, Israel et al. remarked on heavily exposed nursing students: ‘the persistence of a negative reaction in these students, after the majority of other students had long been infected, is a striking phenomenon.’5 Case contact studies in TB show that approximately half of exposed persons are TST negative, even in high endemicity settings.6 Exposure to M. tuberculosis is a function of duration, proximity and grade of sputum positivity, so EC can be best appreciated when these dimensions of exposure are maximal (Table 1).7 Outbreaks in closed environments, such as a US naval ship where 66 sailors shared a cabin with seven others with pulmonary TB, and 13 remained TST negative after 6 months, illustrate this best.8 In such settings it is highly likely that M. tuberculosis was inhaled, contained and cleared before an adaptive response developed.
Table 1.
Selected highly exposed populations and proportion tuberculin skin test (TST) negative
| Highly exposed population | Duration of exposure | Proportion TST negative (%) | References |
|---|---|---|---|
| American sailors shared cabin with seven pulmonary tuberculosis (TB) cases | 6 months | 19.7 | 8 |
| Nurses, in 1930s New York hospital | 3 years | 7* | 96 |
| Household contacts in Jinan, China with average exposure 9 hr/day and proximity 30–40 cm | Not stated | 31 | 97 |
| Household case contacts in The Gambia sharing same bed | 43 | 98 | |
| Gambian adults sharing room with patient | 35 | 99 | |
| Contact at least alternate days with a patient with laryngeal TB | 6 months | 31 | 100 |
Boosting may occur in healthcare workers undergoing repeated TSTs.
Lurie demonstrated hereditable resistance to Mycobacterium bovis infection in rabbits in the 1940s.9 Today, genetic loci associated with TST reactivity have been identified in two populations, suggesting that host factors play an important role in failure to establish an infection after exposure. Recently, Cobat et al. used a genome-wide linkage study to show that TST status is highly hereditable: a single locus accounted for 65% of TST variability in a Columbian population,10 and two loci determined TST status in South Africa.11 The first (TST1) is associated with a lack of TST responsiveness and the second (TST2) is associated with the degree of response. Additionally, two candidate gene studies compared TST-positive and TST-negative participants and found associations with cytokine genes.12,13 The best explanation for these observations is EC. Alternative explanations can only be discounted by studies that address exposure level as a confounder and reduce the likelihood that a negative TST reflects anergy through post-exposure follow up.
Early versus delayed clearance
Early clearance is one of several phenotypes in the progression to TB disease after exposure (Fig. 1). By definition, EC occurs before the development of a positive IGRA or TST, i.e. before the development of an adaptive immune response. Clearance could also occur later in the course of infection. However, at present clearance in a person with a positive IGRA or TST could only be distinguished from latent tuberculosis infection (LTBI) by necropsy, so it is difficult to study delayed clearance. If EC fails, either primary progressive TB or LTBI develops. These phenotypes are influenced by both innate and adaptive immune responses and are beyond the scope of this review.
Figure 1.
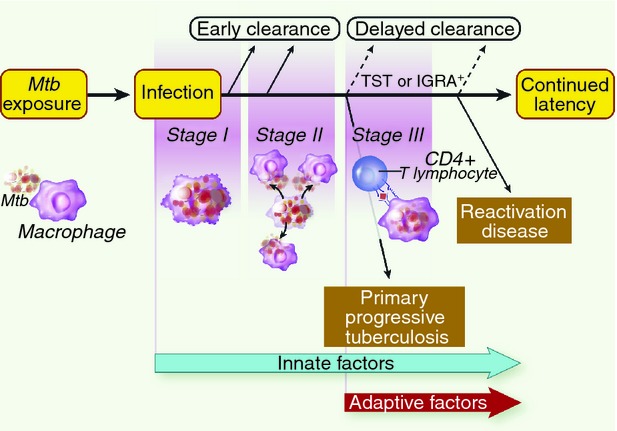
Phenotypes in the progression to tuberculosis (TB) disease after exposure. Mycobacterium tuberculosis (Mtb) exposure leads to infection. Early clearance (EC) occurs before the development of an adaptive immune response and is likely to be due to innate factors, during Stage I or II. If the pathogen evades EC mechanisms, an adaptive response develops (Stage III), measurable through a positive tuberculin skin test (TST) or interferon-γ release assay (IGRA). Early evasion of both innate and adaptive responses results in primary progressive TB. However, in the majority of infected individuals M. tuberculosis is contained as latent TB infection with only 5% later reactivating disease. Others speculate that clearance could also occur at the time of or after TST/IGRA conversion, we term this delayed clearance.
Early clearance of M. tuberculosis is most likely to be mediated by an effective innate immune response
If individuals exposed to M. tuberculosis clear the infection, what could be the possible mechanism? The most plausible hypothesis is that EC is an innate immune response that occurs before adaptive immunity can develop. Inter-individual variability in quantitative assessment of innate immune responses, such as mycobacterial whole blood stimulation assays14 and inhibition of mycobacterial growth in vitro, support the notion of variability in ability to clear M. tuberculosis between individuals. The Lübeck disaster in which 252 newborns were accidentally inoculated with virulent mycobacteria illustrates this variability most dramatically: approximately one-third died, but another third remained well and the remainder were only slightly ill.15
Others have argued that waning M. tuberculosis specific T-cell responses following point source exposure suggest a role for T cells in clearance of an acute infection.16 Similar claims have been made about IGRA ‘reversions’ in serially tested healthcare workers.17 Both observations could result from bias, due to random variation in the IGRA result.18 IGRA reversion was no more likely in those treated for LTBI than in controls in a clinical trial, so cannot be assumed to reflect EC.19
How does early clearance occur?
The alveolar macrophage is the primary target cell of early M. tuberculosis infection. Recognition by alveolar macrophages, and possibly epithelial cells, leads to pro-inflammatory cytokine production, chemokine secretion and antimicrobial peptides.20 This may influence the activation state of other target cells. The seminal rabbit studies by Lurie et al. identified infection of alveolar macrophages as the first stage of pulmonary TB, and point at which pathological findings in rabbits bred to resist infection diverged from those found in susceptible rabbits.21 However, if M. tuberculosis escapes the alveolar macrophage, a period of logarithmic growth (second stage) ensues with infection of newly recruited monocytes. More recently, a zebra fish Mycobacterium marinum model has indicated that granuloma formation occurs at this stage and facilitates further infection of new macrophages.22 The arrival of the CD4+ T cells secreting interferon-γ (IFN-γ) to activate recruited macrophages marks the third stage and the start of an adaptive response. Early clearance is more likely when intracellular mycobacterial killing in alveolar macrophages is successful in the first stage, and least likely once mycobacterial outgrowth and infection of recruited macrophages favour pathogen survival.
The role of alveolar macrophages in early clearance
As the dominant phagocyte in the healthy lung, alveolar macrophages probably play a key role in EC, by phagocytosis and immune recognition of M. tuberculosis, and the subsequent inflammatory response and M. tuberculosis killing. Mathematical models differ in their estimates of EC, because of differing assumptions about the killing capacity of alveolar macrophages.23,24 Some view alveolar macrophages as alternatively activated or promoting anergy.24,25 However, Flynn et al. argue that they may not fit either category, particularly in the early stages of infection, before cytokine activation.26 Notwithstanding these controversies, a model of M. tuberculosis infection of alveolar macrophages can be advanced drawing on murine studies and ex vivo human studies. Figure 2 describes how the outcome of infection can be influenced by different events including pathogen recognition, cytokine production, intracellular killing and the mode of macrophage death.
Figure 2.
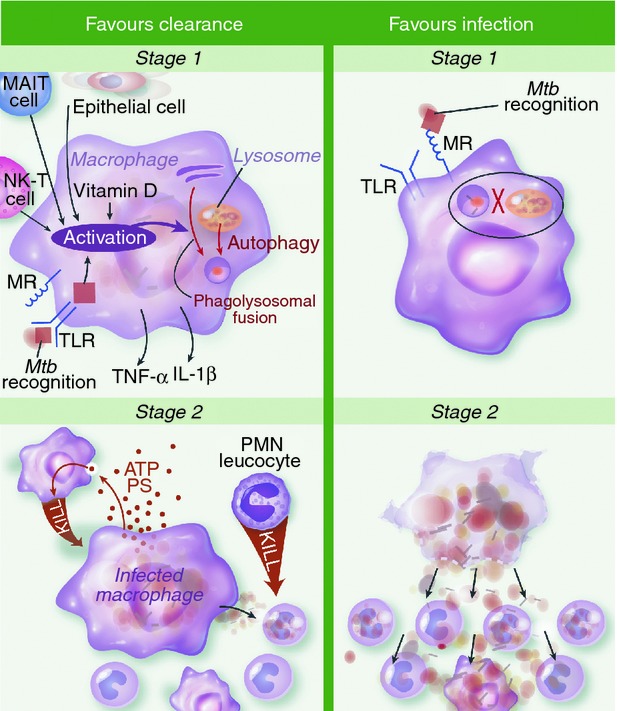
Hypothesized mechanisms of clearance versus infection during first and second stages of Mycobacterium tuberculosis (Mtb) infection. In stage one infection is primarily of alveolar macrophages. Activation of macrophages may be important for early clearance (EC), through recognition of M. tuberculosis via surface, cytosolic or phagosomal pattern recognition receptors (PRRs) and/or tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) secreted by other cells. This promotes killing of intracellular M. tuberculosis by phagosomal acidification, hydrolytic enzymes and generation of reactive nitrogen intermediates. Also, antimicrobial peptides and autophagy promote mycobacterial killing and are both induced by vitamin D. Alternatively, infection is favoured if the macrophage's initial interaction with M. tuberculosis is via ligation of the mannose receptor (MR). This promotes uptake without recognition and inhibition of phagolysosomal fusion. In stage two, infected macrophages undergo apoptosis and express adenosine triphosphate and phosphotidyl serine. This attracts monocytes and neutrophils that engulf the infected cell and deploy oxidative killing mechanisms to achieve clearance. Neutrophils activated in this fashion secrete antimicrobial peptides cathelicidin, human neutrophil peptides and Lipocalin 2 to kill infected monocytes. Sustained infection is most likely when infected macrophages undergo necrotic cell death. The disruption of the macrophage membrane facilitates mycobacterial outgrowth so newly recruited monocytes are infected and logarithmic growth ensues. IL-1β, interleukin-1β; MAIT, mucosa-associated invariant T; NK-T, natural killer T; PMN, polymorphonuclear cells; PS, phosphatidyl serine; TLR, Toll-like receptor
Phagocyosis, pattern recognition of M. tuberculosis and induction of protective cytokines
Uptake of M. tuberculosis by phagocytes occurs via specific receptors that can influence subsquent events within the cell. The mannose receptor (MR) is heavily expressed by alveolar macrophages and the primary avenue for uptake of non-opsonized M. tuberculosis in the alveolar space where complement is rare. Ligation of the MR by mannose-capped lipoarabinomannan induces phagocytosis.
When certain M. tuberculosis cell wall components bind to pattern recognition receptors (PRRs) on the surface or in the cytosol of host innate cells, this shapes the subsequent immune response. Toll-like receptors (TLRs), nucleotide-binding oligomerization domain (NOD)-like receptors and C-type lectins recognize M. tuberculosis and drive cytokine signals that influence mycobacterial clearance.27 TLR2 binds lipoarabinomannan, and as a dimer with TLR6 binds a 19 000 molecular weight M. tuberculosis lipoprotein. These signals are transduced through MyD88 and transcription of nuclear transcription factor-κB (NF-κB) and so promote inflammatory cytokine secretion, particularly tumour necrosis factor-α (TNF-α).28 TLR4 recognizes heat-labile cell-associated factor and also activates MyD88 and NF-κB, yet induces interleukin-1β (IL-1β) production.29 TLR8 and TLR9 are endosomally located and so are able to detect the nucleic acids of intraphagosomal M. tuberculosis. Stimulation results in the expression of NF-κB-dependent cytokines as well as type 1 interferons.30,31 TLR8 expression is increased in patients with active TB and in ex vivo cell stimulation experiments, and genetic variants have been associated with susceptibility to active TB in two populations.32
Similarly, ligation of NOD2, another PRR expressed by human alveolar macrophages, limits intracellular M. tuberculosis growth through up-regulation of autophagy and cathelicidin production.33 Polymorphisms in NOD2 have been associated with TB in some populations,34 albeit not in others.35,36 NOD-like receptor or IFN-inducible protein 2 receptor ligation also activates the inflammasome; the multiprotein complex containing caspase-1 that activates IL-1β.37 Polymorphisms in the inflammasome pathway, specifically caspase recruitment domain-containing protein 8, are associated with risk of TB in HIV-infected individuals.38
Besides MR, two other C-type lectins may recognize M. tuberculosis: dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin (DC-SIGN) and macrophage C type lectin. The M. tuberculosis induces expression of DC-SIGN in alveolar macrophages, and binding of DC-SIGN to M. tuberculosis mannose-capped lipoarabinomannan leads to an anti-inflammatory signal.39 Macrophage C type lectin (also called Clec4d) has recently been identified as a functional receptor for M. tuberculosis in experimental models,40 but genetic association studies in humans are needed.
Mycobacterial killing
Macrophages have a number of mechanisms to kill intracellular mycobacteria. After phagocytosis the M. tuberculosis-containing phagosome matures through interactions with the endocytic pathway. This includes acidification of the phagosome through expression of H+-ATPase as well as the hydrolytic enzyme cathepsin D.41 Mycobacteria are subjected to further oxidative stress through up-regulation of nicotinamide adenine dinucleotide phosphate oxidase and nitric oxide synthase to produce reactive nitrogen intermediates and possibly reactive oxygen intermediates. In mice this process is up-regulated by IFN-γ acting in synergy with TNF-α,42,43 whereas in humans 1,25-(OH)2 vitamin D3 is also required.44 The importance of this defence for killing of intraphagosomal M. tuberculosis has driven both pathogen evasion strategies and host counter strategies to influence phagolysosomal fusion. Mannose-capped lipoarabinomannan, through interaction with the MR, inhibits phagolysosomal fusion and, via peroxisome proliferator-activated receptor, down-regulates transcription of NF-κB, activating protein-1 and signal transducer and activator of transcription, favouring an anti-inflammatory cytokine profile and blunted oxidative response.45,46 Cell wall constituents from more virulent mycobacterial strains achieve greater inhibition of phagolysosomal fusion.47 Macrophages may counter this inhibition through deploying autophagosomes that engulf phagosomes and force lysosomal fusion.48,49 Autophagy is promoted by IFN-γ, vitamin D and TLR4 via Toll–IL-1 receptor domain-containing adaptor-inducing IFN-β signalling.48,50 Finally, M. tuberculosis-infected macrophages produce hepcidin and cathelicidin, which have direct activity against M. tuberculosis. 51,52
Macrophage death and survival of M. tuberculosis
Macrophages may die through apoptosis or necrosis, and experiments in macrophage cell cultures show that apoptosis inhibits M. tuberculosis replication. The apoptotic macrophage expresses ATP and phosphatidyl serine to promote its efferocytosis by other phagocytes that kill the bacterium.53 Virulent strains of M. tuberculosis may evade apoptosis and induce necrosis through production of lipoxin A4, which blocks prostaglandin E2 synthesis and prevents repair of the plasma membrane.54 This necrotic death favours membrane disruption and mycobacterial outgrowth. A zebra fish model in which the balance of pro-inflammatory and anti-inflammatory eicosanoid lipoxins was associated with macrophage necrosis and M. tuberculosis outgrowth.55
Neutrophils
To date, neutrophil responses are the only aspect of EC studied in a prospective cohort of case contacts. Martineau et al. found absolute neutrophil count associated with a negative IGRA response and greater killing capacity in an ex vivo bacille Calmette–Guérin lux whole blood stimulation assay. Killing was also associated with serum levels of the neutrophil antimicrobial peptides, cathelicidin, human neutrophil peptides 1 and 3 and Lipocalin 2.56 Lowe et al. argue for an influential role of neutrophils in EC,57 noting that early depletion or recruitment of neutrophils impacts the outcome of M. tuberculosis infection in animal models.58–60 Kisich et al. observed that neutrophils from some donors killed mycobacteria spontaneously whereas others required TNF-α activation; variability that may reflect varying capacities for EC.61 Neutrophils phagocytose M. tuberculosis and are in turn phagocytosed by monocyte/macrophages recruited in the second stage of infection. At this point the neutrophil can influence whether the macrophage undergoes apoptotic or necrotic cell death, thereby influencing the likelihood of clearance.57
Natural killer and T-cell subsets
Even less is known about the role of other immune cells in EC. Natural killer (NK), mucosa-associated invariant T (MAIT) cells and γδ T cells express germ-line encoded PRRs that give them innate type functions. The early appearance of NK cells in M. tuberculosis suggests a possible role in EC. The germ line encoded receptor NKp46 on NK cells recognizes infected monocytes.62 Natural killer cells produce IFN-γ and IL-22, which promote M. tuberculosis killing and macrophage apoptosis.63,64 The NK cells in experimentally infected animals have a variable impact on the course of infection.65,66
Mucosa-associated invariant T cells are a recently described T-cell population found in mucosal tissue including the lung. As they do not depend on clonal expansion, MAIT cells respond rapidly to danger signals and provide an early innate source of IFN-γ to drive macrophage activation. MAIT-cell-deficient mice challenged with aerosolized M. bovis have a significantly higher burden of infection at day 10.67 MAIT cell populations expanded early in infection in lungs and were required for timely recruitment of CD4 and CD8 cells in a Francisella tularensis vaccine – mouse model of acute infection.68
γδ T cells may also play a role in the early response to M. tuberculosis as they are present in alveoli, and recognize mycobacterial phosphoantigens expressed on the surface of infected macrophages.69 Their activation results in killing of infected macrophages through cytotoxic granules, IFN-γ and TNF-α signalling, and cell-contact-dependent help. As with other T-cell sub-sets, the role of γδ T cells in the response to early M. tuberculosis infection needs to be defined.
Acquired defects of early clearance
A variety of clinical conditions could impair clearance, including diabetes mellitus, smoking end-stage renal disease, corticosteroid use and anti-TNF-α agents among others. Conditions where laboratory and clinical observations support impaired clearance are HIV infection and vitamin D deficiency.
HIV
HIV is an established risk factor for developing active TB, and is probably also a risk for M. tuberculosis infection. HIV impairs cell-mediated immunity through depletion of CD4 T cells.70 Additionally, HIV impairs innate immunity in a manner that may impair EC: macrophage turnover is increased in HIV infection, and apoptosis is impaired;71,72 dendritic cell numbers are diminished and their autophagic and antigen-presenting functions are suppressed;73,74 NK cells have low levels of glutathione and permit higher rates of mycobacterial growth in co-cultured monocytes.75 Molecular epidemiological studies of M. tuberculosis suggest that susceptibility to infection is a greater contributor to risk of TB in HIV-positive persons than accelerated disease progression. HIV was strongly associated with re-infection in a study of previously treated South African miners.76 Additionally, a meta-analysis of molecular epidemiological studies shows high rates of clustering of M. tuberculosis strains, supporting susceptibility to infection rather than reactivation of previously circulating strains.77 A low baseline monocyte to lymphocyte ratio was associated with a higher risk of subsequent TB in HIV-positive persons, and may reflect impaired clearance.78
Vitamin D
Vitamin D deficiency (VDD) may be a modifiable risk factor for M. tuberculosis infection. Vitamin D is a potent modulator of innate immunity and was a treatment for TB in the pre-antibiotic era. Vitamin D promotes mycobacterial killing through modulation of the innate immune response including the production of antimicrobial peptides52,79 and up-regulation of autophagy in macrophages.79,80 The effect occurs at physiological concentrations: a single oral dose of vitamin D increased the killing of bacille Calmette–Guérin observed in whole blood assays performed on TB case contacts.81 Vitamin D receptor polymorphisms and VDD interact to cause susceptibility to TB infection among Guajarati Indians living in the UK.82
Whereas clinical studies of vitamin D supplementation in active TB have been disappointing,83 very few studies have considered whether VDD influences the development of M. tuberculosis infection. In a small Spanish study following TST-negative TB case contacts, all 12 who underwent TST conversion had VDD compared with 57 of 82 (69.5%) non-converters.84 In a double blind placebo-controlled randomized controlled trial among 120 Mongolian child case contacts, vitamin D supplementation seemed to protect against TST-conversion (relative risk 0.41; 95% confidence interval 0.16–1.09; P = 0.06).85 Therefore, there is some evidence that vitamin D influences EC, and more field studies in TB case contacts are needed.
Early clearance and vaccine development
Early clearance is a genuine example of protective immunity to M. tuberculosis and could offer new avenues for vaccine development. Although IFN-γ and M. tuberculosis-specific CD4+ T cells are clearly involved in the host response to M. tuberculosis, there is increasing awareness that these factors are not particularly robust correlates of protection.86 Furthermore, the possibility of concurrent infection with two M. tuberculosis strains, and exogenous re-infection following M. tuberculosis treatment suggest that the adaptive response to M. tuberculosis is not protective.87,88 The incidence of TB in previously treated South Africans was estimated to be four times higher than in the general population, implying a deficit in innate immunity rather than a protective acquired response.89 Early clearance provides a model of protective natural immunity to M. tuberculosis that is likely to represent a more fruitful avenue for vaccine development than adaptive responses.
Further research and conclusions
Early clearance is a priority for M. tuberculosis research as such studies have the potential to discover new immunotherapies and vaccines to prevent infection. Case contact studies allow EC to be observed in humans, through the integration of immunological phenotyping with a well established field study design.90 This approach is analogous to the study of highly exposed persistently HIV-negative sex workers to identify broadly neutralizing antibodies to HIV, now recognized as a new avenue for HIV vaccine development. Case contact studies allow investigators to quantify and adjust for environmental and pathogen factors, so host factors can be studied without confounding. Specifically, duration of index case infectivity, proximity and frequency of contact, sputum bacillary burden and strain type. Long-term follow up of putative ‘early clearers’, potentially over decades, can strengthen confidence that persistent TST negativity is in fact clearance, not anergy.
Identifying and validating biomarkers is a necessary first step for EC research. This will provide supporting evidence for EC as a distinct phenotype, assist in easier identification of those who will show EC in future studies, and suggest possible mechanisms for EC. Biomarkers may also enable study of EC in HIV, which is presently limited by the poor sensitivity of TST and IGRA. Other M. tuberculosis phenotypes have been studied in this way, for example, the cytokine and cell populations that distinguish LTBI progressors from non-progressors.91 Additionally, the effect of ex vivo M. tuberculosis stimulation on cytokine production or gene transcription can be studied for its correlation with phenotype.92 A challenge in EC research will be to assess the events that occur in the lungs of healthy case contacts. Some biomarkers may be detectable in blood, whereas others might only be found on bronchoalveolar lavage. The latter has been proposed but its invasiveness and complexity limit its use in large studies.93
In addition, more work is needed to validate the two published studies examining genetic risk factors for M. tuberculosis infection. In this respect, household contact studies have particular advantages.94 Poor selection of cases and controls may bias many studies of M. tuberculosis susceptibility.95 Comparison of TB cases to poorly defined controls naturally yields a variety of different associations that could reflect susceptibility to infection or disease progression. A narrower definition of phenotype, that focuses just on the shorter chain of events between exposure and infection (or clearance) is more likely to yield loci associated with EC.
This review has suggested a mechanism for EC based on animal and ex vivo human experiments, as well as genetic studies. Whether these pathways are in fact associated with EC must be tested in human studies. Understanding the mechanism of EC could enable its manipulation through drugs or vaccines, and open a new field in TB prevention.
Disclosures
The authors declare that they have no competing financial interests..
Glossary
- DC-SIGN
dendritic cell specific intracellular adhesion molecule-3-grabbing non-integrin
- EC
early clearance
- IGRA
interferon-γ release assay
- IFN-γ
interferon-γ
- LTBI
latent tuberculosis infection
- MAIT
mucosa-associated invariant T
- MR
mannose receptor
- NF-κB
nuclear transcription factor-κB
- NK
natural killer
- NOD
nucleotide-binding oligomerization domain
- PRR
pattern recognition receptors
- TB
tuberculosis (disease)
- TLR
Toll-like receptors
- TNF-α
tumour necrosis factor-α
- TST
tuberculin skin test
- VDD
vitamin D deficiency
References
- 1.WHO. Geneva: World Health Organization; http://www.who.int/mediacentre/factsheets/fs104/en/ [accessed on 12 March 2013]Tuberculosis [Internet]WHO Available from: [Google Scholar]
- 2.Tameris MD, Hatherill M, Landry BS, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet. 2013;381:1021–8. doi: 10.1016/S0140-6736(13)60177-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dheda K, Schwander SK, Zhu B-D, van Zyl-Smit RN, Zhang Y. The immunology of tuberculosis: from bench to bedside. Respirology. 2010;15:433–50. doi: 10.1111/j.1440-1843.2010.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brennan MJ, Thole J. Tuberculosis vaccines: a strategic blueprint for the next decade. Tuberculosis (Edinb) 2012;92(Suppl. 1):S6–13. doi: 10.1016/S1472-9792(12)70005-7. [DOI] [PubMed] [Google Scholar]
- 5.Israel HL, Hetherington HW, Ord JG. A study of tuberculosis among students of nursing. JAMA. 1941;117:839–44. [Google Scholar]
- 6.Morrison J, Pai M, Hopewell PC. Tuberculosis and latent tuberculosis infection in close contacts of people with pulmonary tuberculosis in low-income and middle-income countries: a systematic review and meta-analysis. Lancet Infect Dis. 2008;8:359–68. doi: 10.1016/S1473-3099(08)70071-9. [DOI] [PubMed] [Google Scholar]
- 7.Rieder HL. Epidemiologic basis of tuberculosis control. 1st edn. Paris: International Union Against Tuberculosis and Lung Disease (IUATLD); 1999. [Google Scholar]
- 8.Houk VN, Baker JH, Sorensen K, Kent DC. The epidemiology of tuberculosis infection in a closed environment. Arch Environ Health. 1968;16:26–35. doi: 10.1080/00039896.1968.10665011. [DOI] [PubMed] [Google Scholar]
- 9.Lurie MB. Experimental epidemiology of tuberculosis. J Exp Med. 1944;79:573–89. doi: 10.1084/jem.79.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cobat A, Barrera LF, Henao H, Arbelaez P, Abel L, García F, Schurr E, Alcaïs A. Tuberculin skin test reactivity is dependent on host genetic background in Colombian tuberculosis household contacts. Clin Infect Dis. 2012;54:968–71. doi: 10.1093/cid/cir972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cobat A, Gallant CJ, Simkin L, et al. Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med. 2009;206:2583–91. doi: 10.1084/jem.20090892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thye T, Browne EN, Chinbuah MA, et al. IL10 haplotype associated with tuberculin skin test response but not with pulmonary TB. PLoS ONE. 2009;4:e5420. doi: 10.1371/journal.pone.0005420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zembrzuski VM, Basta PC, Callegari-Jacques SM, Santos RV, Coimbra CE, Salzano FM, Hutz MH. Cytokine genes are associated with tuberculin skin test response in a native Brazilian population. Tuberculosis (Edinb) 2010;90:44–9. doi: 10.1016/j.tube.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 14.van Crevel R, van der Ven-Jongekrijg J, Netea MG, de Lange W, Kullberg BJ, van der Meer JW. Disease-specific ex vivo stimulation of whole blood for cytokine production: applications in the study of tuberculosis. J Immunol Methods. 1999;222:145–53. doi: 10.1016/s0022-1759(98)00192-6. [DOI] [PubMed] [Google Scholar]
- 15.The Lübeck catastrophy: a general review. BMJ. 1931;1:986. [PMC free article] [PubMed] [Google Scholar]
- 16.Ewer K, Millington KA, Deeks JJ, Alvarez L, Bryant G, Lalvani A. Dynamic antigen-specific T-cell responses after point-source exposure to Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2006;174:831–9. doi: 10.1164/rccm.200511-1783OC. [DOI] [PubMed] [Google Scholar]
- 17.Zanetti C, Peracchi M, Zorzi D, Fiorio S, Fallico L, Palu G. Outbreak of transient conversions of the QuantiFERON-TB gold in-tube test in laboratory health care worker screenings. Clin Vaccine Immunol. 2012;19:954–60. doi: 10.1128/CVI.05718-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nardell EA, Wallis RS. Here today—gone tomorrow: the case for transient acute tuberculosis infection. Am J Respir Crit Care Med. 2006;174:734–5. doi: 10.1164/rccm.200607-923ED. [DOI] [PubMed] [Google Scholar]
- 19.Adetifa IM, Ota MOC, Jeffries DJ, et al. Interferon γ ELISPOT as a biomarker of treatment efficacy in latent tuberculosis infection: a clinical trial. Am J Respir Crit Care Med. 2013;187:439–45. doi: 10.1164/rccm.201208-1352OC. [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Wang Y, Liu X. The role of airway epithelial cells in response to mycobacteria infection. Clin Dev Immunol. 2012;2012:791392. doi: 10.1155/2012/791392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lurie MB, Zappasodi P, Dannenberg AM, Weiss GH. On the mechanism of genetic resistance to tuberculosis and its mode of inheritance. Am J Hum Genet. 1952;4:302–14. [PMC free article] [PubMed] [Google Scholar]
- 22.Davis JM, Ramakrishnan L. The role of the granuloma in expansion and dissemination of early tuberculous infection. Cell. 2009;136:37–49. doi: 10.1016/j.cell.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gammack D, Doering CR, Kirschner DE. Macrophage response to Mycobacterium tuberculosis infection. J Math Biol. 2004;48:218–42. doi: 10.1007/s00285-003-0232-8. [DOI] [PubMed] [Google Scholar]
- 24.Day J, Friedman A, Schlesinger LS. Modeling the immune rheostat of macrophages in the lung in response to infection. Proc Natl Acad Sci U S A. 2009;106:11246–51. doi: 10.1073/pnas.0904846106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blumenthal RL, Campbell DE, Hwang P, DeKruyff RH, Frankel LR, Umetsu DT. Human alveolar macrophages induce functional inactivation in antigen-specific CD4 T cells. J Allergy Clin Immunol. 2001;107:258–64. doi: 10.1067/mai.2001.112845. [DOI] [PubMed] [Google Scholar]
- 26.Flynn JL, Chan J, Lin PL. Macrophages and control of granulomatous inflammation in tuberculosis. Mucosal Immunol. 2011;4:271–8. doi: 10.1038/mi.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thoma-Uszynski S, Stenger S, Takeuchi O, et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291:1544–7. doi: 10.1126/science.291.5508.1544. [DOI] [PubMed] [Google Scholar]
- 28.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96:14459–63. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones BW, Means TK, Heldwein KA, Keen MA, Hill PJ, Belisle JT, Fenton MJ. Different Toll-like receptor agonists induce distinct macrophage responses. J Leukoc Biol. 2001;69:1036–44. [PubMed] [Google Scholar]
- 30.Cervantes JL, Weinerman B, Basole C, Salazar JC. TLR8: the forgotten relative revindicated. Cell Mol Immunol. 2012;9:434–8. doi: 10.1038/cmi.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gantier MP, Irving AT, Kaparakis-Liaskos M, et al. Genetic modulation of TLR8 response following bacterial phagocytosis. Hum Mutat. 2010;31:1069–79. doi: 10.1002/humu.21321. [DOI] [PubMed] [Google Scholar]
- 32.Davila S, Hibberd ML, Hari Dass R, et al. Genetic association and expression studies indicate a role of toll-like receptor 8 in pulmonary tuberculosis. PLoS Genet. 2008;4:e1000218. doi: 10.1371/journal.pgen.1000218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juárez E, Carranza C, Hernández-Sánchez F, León-Contreras JC, Hernández-Pando R, Escobedo D, Torres M, Sada E. NOD2 enhances the innate response of alveolar macrophages to Mycobacterium tuberculosis in humans. Eur J Immunol. 2012;42:880–9. doi: 10.1002/eji.201142105. [DOI] [PubMed] [Google Scholar]
- 34.Austin CM, Ma X, Graviss EA. Common nonsynonymous polymorphisms in the NOD2 gene are associated with resistance or susceptibility to tuberculosis disease in African Americans. J Infect Dis. 2008;197:1713–6. doi: 10.1086/588384. [DOI] [PubMed] [Google Scholar]
- 35.Möller M, Nebel A, Kwiatkowski R, Van Helden PD, Hoal EG, Schreiber S. Host susceptibility to tuberculosis: CARD15 polymorphisms in a South African population. Mol Cell Probes. 2007;21:148–51. doi: 10.1016/j.mcp.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Stockton JC, Howson JMM, Awomoyi AA, McAdam KP, Blackwell JM, Newport MJ. Polymorphism in NOD2, Crohn's disease, and susceptibility to pulmonary tuberculosis. FEMS Immunol Med Microbiol. 2004;41:157–60. doi: 10.1016/j.femsim.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 37.Dorhoi A, Nouailles G, Jörg S, et al. Activation of the NLRP3 inflammasome by Mycobacterium tuberculosis is uncoupled from susceptibility to active tuberculosis. Eur J Immunol. 2011;42:374–84. doi: 10.1002/eji.201141548. [DOI] [PubMed] [Google Scholar]
- 38.Pontillo A, Carvalho MS, Kamada AJ, Moura R, Schindler HC, Duarte AJ, Crovella S. Susceptibility to Mycobacterium tuberculosis infection in HIV-positive patients is associated with CARD8 genetic variant. J Acquir Immune Defic Syndr. 2013;63:147–51. doi: 10.1097/QAI.0b013e31828f93bb. [DOI] [PubMed] [Google Scholar]
- 39.Tailleux L, Pham-Thi N, Bergeron-Lafaurie A, et al. DC-SIGN induction in alveolar macrophages defines privileged target host cells for mycobacteria in patients with tuberculosis. PLoS Med. 2005;2:e381. doi: 10.1371/journal.pmed.0020381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miyake Y, Toyonaga K, Mori D, et al. C-type lectin MCL is an FcRg-coupled receptor that mediates the adjuvanticity of mycobacterial cord factor. Immunity. 2013;38:1050–62. doi: 10.1016/j.immuni.2013.03.010. [DOI] [PubMed] [Google Scholar]
- 41.Welin A, Raffetseder J, Eklund D, Stendahl O, Lerm M. Importance of phagosomal functionality for growth restriction of Mycobacterium tuberculosis in primary human macrophages. J Innate Immun. 2011;3:508–18. doi: 10.1159/000325297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flesch IE, Kaufmann SH. Activation of tuberculostatic macrophage functions by γ interferon, interleukin-4, and tumor necrosis factor. Infect Immun. 1990;58:2675–7. doi: 10.1128/iai.58.8.2675-2677.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ding AH, Nathan CF, Stuehr DJ. Release of reactive nitrogen intermediates and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of activating cytokines and evidence for independent production. J Immunol. 1988;141:2407–12. [PubMed] [Google Scholar]
- 44.Denis M. Killing of Mycobacterium tuberculosis within human monocytes: activation by cytokines and calcitriol. Clin Exp Immunol. 1991;84:200–6. doi: 10.1111/j.1365-2249.1991.tb08149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang PB, Azad AK, Torrelles JB, Kaufman TM, Beharka A, Tibesar E, DesJardin LE, Schlesinger LS. The human macrophage mannose receptor directs Mycobacterium tuberculosis lipoarabinomannan-mediated phagosome biogenesis. J Exp Med. 2005;202:987–99. doi: 10.1084/jem.20051239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajaram MVS, Brooks MN, Morris JD, Torrelles JB, Azad AK, Schlesinger LS. Mycobacterium tuberculosis activates human macrophage peroxisome proliferator-activated receptor γ linking mannose receptor recognition to regulation of immune responses. J Immunol. 2010;185:929–42. doi: 10.4049/jimmunol.1000866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torrelles JB, Azad AK, Schlesinger LS. Fine discrimination in the recognition of individual species of phosphatidyl-myo-inositol mannosides from Mycobacterium tuberculosis by C-type lectin pattern recognition receptors. J Immunol. 2006;177:1805–16. doi: 10.4049/jimmunol.177.3.1805. [DOI] [PubMed] [Google Scholar]
- 48.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 49.Songane M, Kleinnijenhuis J, Alisjahbana B, et al. Carvalho LH, editor. Polymorphisms in autophagy genes and susceptibility to tuberculosis. PLoS ONE. 2012;7:e41618. doi: 10.1371/journal.pone.0041618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa NT. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–44. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol. 2007;82:934–45. doi: 10.1189/jlb.0407216. [DOI] [PubMed] [Google Scholar]
- 52.Martineau AR, Wilkinson KA, Newton SM, et al. IFN-γ-and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol. 2007;178:7190–8. doi: 10.4049/jimmunol.178.11.7190. [DOI] [PubMed] [Google Scholar]
- 53.Martin CJ, Booty MG, Rosebrock TR, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe. 2012;12:289–300. doi: 10.1016/j.chom.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Divangahi M, Chen M, Gan H, et al. Mycobacterium tuberculosis evades macrophage defenses by inhibiting plasma membrane repair. Nat Immunol. 2009;10:899–906. doi: 10.1038/ni.1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tobin DM, Roca FJ, Oh SF, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148:434–46. doi: 10.1016/j.cell.2011.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martineau AR, Newton SM, Wilkinson KA, et al. Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest. 2007;117:1988–94. doi: 10.1172/JCI31097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lowe DM, Redford PS, Wilkinson RJ, O'Garra A, Martineau AR. Neutrophils in tuberculosis: friend or foe? Trends Immunol. 2012;33:14–25. doi: 10.1016/j.it.2011.10.003. [DOI] [PubMed] [Google Scholar]
- 58.Sugawara I, Udagawa T, Yamada H. Rat neutrophils prevent the development of tuberculosis. Infect Immun. 2004;72:1804–6. doi: 10.1128/IAI.72.3.1804-1806.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pedrosa J, Saunders BM, Appelberg R, Orme IM, Silva MT, Cooper AM. Neutrophils play a protective nonphagocytic role in systemic Mycobacterium tuberculosis infection of mice. Infect Immun. 2000;68:577–83. doi: 10.1128/iai.68.2.577-583.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petrofsky M, Bermudez LE. Neutrophils from Mycobacterium avium-infected mice produce TNF-α, IL-12, and IL-1βand have a putative role in early host response. Clin Immunol. 1999;91:354–8. doi: 10.1006/clim.1999.4709. [DOI] [PubMed] [Google Scholar]
- 61.Kisich KO, Higgins M, Diamond G, Heifets L. Tumor necrosis factor α stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun. 2002;70:4591–9. doi: 10.1128/IAI.70.8.4591-4599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vankayalapati R, Wizel B, Weis SE, et al. The NKp46 receptor contributes to NK cell lysis of mononuclear phagocytes infected with an intracellular bacterium. J Immunol. 2002;168:3451–7. doi: 10.4049/jimmunol.168.7.3451. [DOI] [PubMed] [Google Scholar]
- 63.Brill KJ, Li Q, Larkin R, Canaday DH, Kaplan DR, Boom WH, Silver RF. Human natural killer cells mediate killing of intracellular Mycobacterium tuberculosis H37Rv via granule-independent mechanisms. Infect Immun. 2001;69:1755–65. doi: 10.1128/IAI.69.3.1755-1765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dhiman R, Indramohan M, Barnes PF, Nayak RC, Paidipally P, Rao LV, Vankayalapati R. IL-22 produced by human NK cells inhibits growth of Mycobacterium tuberculosis by enhancing phagolysosomal fusion. J Immunol. 2009;183:6639–45. doi: 10.4049/jimmunol.0902587. [DOI] [PubMed] [Google Scholar]
- 65.Junqueira-Kipnis AP, Kipnis A, Jamieson A, Juarrero MG, Diefenbach A, Raulet DH, Turner J, Orme IM. NK cells respond to pulmonary infection with Mycobacterium tuberculosis, but play a minimal role in protection. J Immunol. 2003;171:6039–45. doi: 10.4049/jimmunol.171.11.6039. [DOI] [PubMed] [Google Scholar]
- 66.Sugawara I, Yamada H, Mizuno S, Li CY, Nakayama T, Taniguchi M. Mycobacterial infection in natural killer T cell knockout mice. Tuberculosis (Edinb) 2002;82:97–104. doi: 10.1054/tube.2002.0331. [DOI] [PubMed] [Google Scholar]
- 67.Chua WJ, Truscott SM, Eickhoff CS, Blazevic A, Hoft DF, Hansen TH. Polyclonal mucosa-associated invariant T cells have unique innate functions in bacterial infection. Infect Immun. 2012;80:3256–67. doi: 10.1128/IAI.00279-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Meierovics A, Yankelevich W-JC, Cowley SC. MAIT cells are critical for optimal mucosal immune responses during in vivo pulmonary bacterial infection. Proc Natl Acad Sci U S A. 2013;110:E3119–28. doi: 10.1073/pnas.1302799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Meraviglia S, El Daker S, Dieli F, Martini F, Martino A. γδ T cells cross-link innate and adaptive immunity in Mycobacterium tuberculosis infection. Clin Dev Immunol. 2011;2011 doi: 10.1155/2011/587315. Article ID 587315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lawn SD, Butera ST, Shinnick TM. Tuberculosis unleashed: the impact of human immunodeficiency virus infection on the host granulomatous response to Mycobacterium tuberculosis. Microbes Infect. 2002;4:635–46. doi: 10.1016/s1286-4579(02)01582-4. [DOI] [PubMed] [Google Scholar]
- 71.Patel NR, Zhu J, Tachado SD, Zhang J, Wan Z, Saukkonen J, Koziel H. HIV impairs TNF-α mediated macrophage apoptotic response to Mycobacterium tuberculosis. J Immunol. 2007;179:6973–80. doi: 10.4049/jimmunol.179.10.6973. [DOI] [PubMed] [Google Scholar]
- 72.Hasegawa A, Liu H, Ling B, et al. The level of monocyte turnover predicts disease progression in the macaque model of AIDS. Blood. 2009;114:2917–25. doi: 10.1182/blood-2009-02-204263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Blanchet FP, Moris A, Nikolic DS, et al. Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010;32:654–69. doi: 10.1016/j.immuni.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cameron PU, Handley AJ, Baylis DC, Solomon AE, Bernard N, Purcell DF, Lewin SR. Preferential infection of dendritic cells during human immunodeficiency virus type 1 infection of blood leukocytes. J Virol. 2007;81:2297–306. doi: 10.1128/JVI.01795-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Guerra C, Johal K, Morris D, et al. Control of Mycobacterium tuberculosis growth by activated natural killer cells. Clin Exp Immunol. 2012;168:142–52. doi: 10.1111/j.1365-2249.2011.04552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sonnenberg P, Murray J, Glynn JR, Shearer S, Kambashi B, Godfrey-Faussett P. HIV-1 and recurrence, relapse, and reinfection of tuberculosis after cure: a cohort study in South African mineworkers. Lancet. 2001;358:1687–93. doi: 10.1016/S0140-6736(01)06712-5. [DOI] [PubMed] [Google Scholar]
- 77.Houben RMGJ, Crampin AC, Ndhlovu R, et al. Human immunodeficiency virus associated tuberculosis more often due to recent infection than reactivation of latent infection. Int J Tuberc Lung Dis. 2011;15:24–31. [PubMed] [Google Scholar]
- 78.Naranbhai V, Hill AV, Abdool Karim SS, Naidoo K, Abdool Karim Q, Warimwe GM, McShane H, Fletcher H. Blood monocyte–lymphocyte ratios identify adults at risk of incident tuberculosis amongst patients initiating antiretroviral therapy. J Infect Dis. 2013 doi: 10.1093/infdis/jit494. doi: 10.1093/infdis/jit494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311:1770–3. doi: 10.1126/science.1123933. [DOI] [PubMed] [Google Scholar]
- 80.Campbell GR, Spector SA. Vitamin D inhibits human immunodeficiency virus type 1 and Mycobacterium tuberculosis infection in macrophages through the induction of autophagy. PLoS Pathog. 2012;8:e1002689. doi: 10.1371/journal.ppat.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Martineau AR, Wilkinson RJ, Wilkinson KA, et al. A single dose of vitamin D enhances immunity to mycobacteria. Am J Respir Crit Care Med. 2007;176:208–13. doi: 10.1164/rccm.200701-007OC. [DOI] [PubMed] [Google Scholar]
- 82.Wilkinson RJ, Llewelyn M, Toossi Z, et al. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case–control study. Lancet. 2000;355:618–21. doi: 10.1016/S0140-6736(99)02301-6. [DOI] [PubMed] [Google Scholar]
- 83.Ralph AP, Lucas RM, Norval M. Vitamin D and solar ultraviolet radiation in the risk and treatment of tuberculosis. Lancet Infect Dis. 2013;13:77–88. doi: 10.1016/S1473-3099(12)70275-X. [DOI] [PubMed] [Google Scholar]
- 84.Arnedo-Pena A, Juan-Cerdán JV, Romeu-Garcia A, et al. Latent tuberculosis infection, tuberculin skin test and vitamin D status in contacts of tuberculosis patients: a cross-sectional and case–control study. BMC Infect Dis. 2011;11:349. doi: 10.1186/1471-2334-11-349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ganmaa D, Giovannucci E, Bloom BR, et al. Vitamin D, tuberculin skin test conversion, and latent tuberculosis in Mongolian school-age children: a randomized, double-blind, placebo-controlled feasibility trial. Am J Clin Nutr. 2012;96:391–6. doi: 10.3945/ajcn.112.034967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kagina BM, Abel B, Scriba TJ, et al. Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette–Guérin vaccination of newborns. Am J Respir Crit Care Med. 2010;182:1073–9. doi: 10.1164/rccm.201003-0334OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Braden CR, Morlock GP, Woodley CL, et al. Simultaneous infection with multiple strains of Mycobacterium tuberculosis. Clin Infect Dis. 2001;33:e42–7. doi: 10.1086/322635. [DOI] [PubMed] [Google Scholar]
- 88.van Rie A, Warren R, Richardson M, Victor TC, Gie RP, Enarson DA, Beyers N, van Helden PD. Exogenous reinfection as a cause of recurrent tuberculosis after curative treatment. N Engl J Med. 1999;341:1174–9. doi: 10.1056/NEJM199910143411602. [DOI] [PubMed] [Google Scholar]
- 89.Verver S, Warren RM, Beyers N, et al. Rate of reinfection tuberculosis after successful treatment is higher than rate of new tuberculosis. Am J Respir Crit Care Med. 2005;171:1430–5. doi: 10.1164/rccm.200409-1200OC. [DOI] [PubMed] [Google Scholar]
- 90.Hill PC, Ota MOC. Tuberculosis case-contact research in endemic tropical settings: design, conduct, and relevance to other infectious diseases. Lancet Infect Dis. 2010;10:723–32. doi: 10.1016/S1473-3099(10)70164-X. [DOI] [PubMed] [Google Scholar]
- 91.Sutherland JS, Hill PC, Adetifa IM, et al. Identification of probable early-onset biomarkers for tuberculosis disease progression. PLoS ONE. 2011;6:e25230. doi: 10.1371/journal.pone.0025230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Thuong NT, Dunstan SJ, Chau TT, et al. Identification of tuberculosis susceptibility genes with human macrophage gene expression profiles. PLoS Pathog. 2008;4:e1000229. doi: 10.1371/journal.ppat.1000229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schwander S, Dheda K. Human lung immunity against Mycobacterium tuberculosis. Am J Respir Crit Care Med. 2011;183:696–707. doi: 10.1164/rccm.201006-0963PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Stein CM. Genetic epidemiology of tuberculosis susceptibility: impact of study design. PLoS Pathog. 2011;7:e1001189. doi: 10.1371/journal.ppat.1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wilkinson RJ. Human genetic susceptibility to tuberculosis: time for a bottom-up approach? J Infect Dis. 2012;205:525–7. doi: 10.1093/infdis/jir792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Amberson JB, Riggins H. Tuberculosis among student nurses: a five year study at Bellevue hospital. Ann Intern Med. 1936:156–165. [Google Scholar]
- 97.Lutong L, Bei Z. Association of prevalence of tuberculin reactions with closeness of contact among household contacts of new smear-positive pulmonary tuberculosis patients. Int J Tuberc Lung Dis. 2000;4:275–7. [PubMed] [Google Scholar]
- 98.Lienhardt C, Fielding K, Sillah J, et al. Risk factors for tuberculosis infection in Sub-Saharan Africa. Am J Respir Crit Care Med. 2003;168:448–55. doi: 10.1164/rccm.200212-1483OC. [DOI] [PubMed] [Google Scholar]
- 99.Hill PC, Brookes RH, Fox A, et al. Large-scale evaluation of enzyme-linked immunospot assay and skin test for diagnosis of Mycobacterium tuberculosis infection against a gradient of exposure in The Gambia. Clin Infect Dis. 2004;38:966–73. doi: 10.1086/382362. [DOI] [PubMed] [Google Scholar]
- 100.Muecke C, Isler M, Menzies D, Allard R, Tannenbaum TN, Brassard P. The use of environmental factors as adjuncts to traditional tuberculosis contact investigation. Int J Tuberc Lung Dis. 2006;10:530–5. [PubMed] [Google Scholar]