

Normal cells and cancer cells rely on mitochondria for a range of essential functions including production of ATP, biosynthetic intermediates, heme and iron-sulfur clusters, and reactive oxygen species (ROS) for signaling. These functions are essential for cell survival and proliferation. Most cancer cells have functional mitochondria to fulfill these essential requirements; however, there is a subset of cancers which derive from a loss of function in tricarboxylic acid (TCA) cycle enzymes, including fumarate hydratase (FH).1 Cancer cells with loss of FH function are respiratory-deficient and accumulate high levels of fumarate. This raises an intriguing question: how do cancer cells with dysfunctional mitochondria fulfill their requirements for cell proliferation?
The isolation of FH-deficient renal cancer cells (UOK262) has allowed examination of how loss of FH impacts mitochondrial metabolism and cell proliferation.2 Loss of FH causes mitochondrial dysfunction; however, the essential cellular processes that typically occur in the mitochondria are maintained. ATP is maintained by increasing glycolysis. The mitochondrial TCA cycle generates intermediates that are required for biosynthesis reactions, such as citrate for fatty acid synthesis. Normal cells produce citrate by oxidative condensation of oxaloacetate and glycolytic acetyl-CoA; however, UOK262 cells do not produce acetyl-CoA from glycolysis. In UOK262 cells, glutamine-derived mitochondrial 2-oxoglutarate is metabolized in the reverse direction of the canonical TCA cycle to produce citrate by reductive carboxylation.3 Thus, cells are capable of generating the TCA cycle intermediates required for proliferation, whether it is by oxidative mitochondrial metabolism in normal cells or reductive mitochondrial metabolism in FH mutant cells.
A byproduct of oxidative metabolism is the generation of ROS by the electron transport chain. Increased ROS at low nM levels in cancer cells promotes signaling pathways to activate HIFs for metabolic adaptation, NFκB for cell survival, and MAP kinases for cell proliferation.4 However, ROS at higher levels can oxidize lipids, proteins, or DNA to promote damage or mutagenesis. To exploit ROS signaling, cancer cells both increase ROS to promote tumorigenic signaling and also increase antioxidant proteins and metabolites to keep ROS at levels compatible with growth. Antioxidants such as glutathione are oxidized by ROS and restored by consumption of NADPH by antioxidant enzymes. ROS levels can be increased by either increased generation of ROS or by decreased supply of antioxidants. How FH-null cells that engage in reductive metabolism increase ROS to promote pro-tumorigenic pathways is not fully understood.
Recently we reported the production and accumulation of fumarate in FH-deficient cells is required to increase ROS levels.5 Accumulated fumarate undergoes a covalent reaction with intracellular glutathione (GSH) to produce a novel metabolite, succinated glutathione (GSF). Surprisingly, GSF serves as a substrate for glutathione reductase. Normally, glutathione reductase reduces oxidized glutathione (GSSG) to GSH by coupling it to NADPH oxidation to NADP+. An in vitro mixture of glutathione reductase, NADPH, and GSF showed rapid depletion of NADPH and production of glutathione. Thus, glutathione reductase uses NADPH to split GSF into glutathione and, likely, succinate. Decreased levels of NADPH will result in less antioxidant activity and increased ROS levels. Indeed, FH-null cells show decreased NADPH and increased ROS compared with FH reconstituted cells. Also, treatment with GSF caused depletion of NADPH and enhanced endogenous ROS. At first glance, it would seem the accumulation of fumarate binding to glutathione would deplete glutathione levels, raising ROS to excessively high levels, resulting in cell death or suppression of proliferation. However, previous reports indicated that fumarate can cause the post-translational modification succination on KEAP1, the negative regulator of the master antioxidant transcription factor NRF2.6,7 Succination of KEAP1 allows NRF2 to be active and stimulate antioxidant genes. Thus, NRF2 is activated to mitigate increased ROS of FH-null cells. Indeed, we observed that RNAi of NRF2 in FH-deficient tumor cells caused excessively high levels of ROS, resulting in suppression of proliferation.
A consequence of this increased ROS production in FH-deficient cancer cells was the activation of HIF-1 and hyper-methylation of histones, regulated by inhibition of the 2-oxoglutarate dioxygenases prolyl hydroxylases and Jumonji-domain-containing histone demethylases, respectively.8 Fumarate is known to be a competitive inhibitor with 2-oxoglutrate for these dioxygenases. However, our data suggest, in addition to fumarate, these dioxygenases might be susceptible to inhibition through an unknown ROS-dependent mechanism. Based on our observations and previous research on ROS, we propose that loss of FH is advantageous for tumor cells, because it allows the accumulation of fumarate, which ultimately triggers an increase in ROS levels. The accumulation of fumarate and ROS can impinge on variety of pathways that control tumorigenesis (Fig. 1).
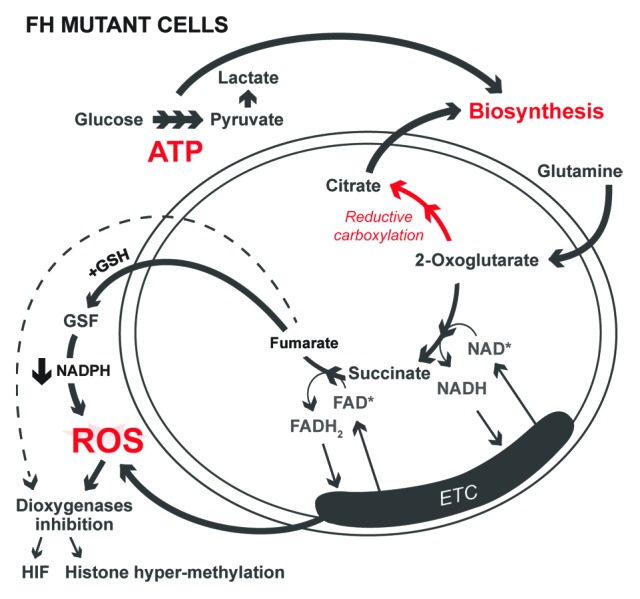
Figure 1. FH mutant cancer cells maintain proliferation requirements supplied by the mitochondria. Normal cells utilize oxidative metabolism to supply metabolites for biosynthesis, ATP for energy, and ROS for signaling. FH-null cells supply these intermediates despite having impaired mitochondrial function. Biosynthetic citrate is produced by reductive carboxylation, ATP levels are maintained by glycolysis, and ROS levels are increased by fumarate accumulation.
The role of the mitochondria in cancer cells has been contentious for decades. Otto Warburg’s initial hypothesis was that mitochondrial impairment was a prerequisite for transformation. Thus, many investigators have assumed that most cancer cells have impaired or dampened mitochondrial function. However, cancer cells can exhibit increased, decreased, or unchanged mitochondrial metabolism compared with normal cells. Our data indicate that mitochondria provide essential functions for cell proliferation even if they harbor mutations that impair their oxidative metabolism. It will be of interest in future studies to determine whether mitochondrial metabolism is an attractive target for cancer therapy.
Sullivan LB, et al. Mol Cell. 2013;51:236–48. doi: 10.1016/j.molcel.2013.05.003.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27513
References
- 1.Wallace DC. Nat Rev Cancer. 2012;12:685–98. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y, et al. Cancer Genet Cytogenet. 2010;196:45–55. doi: 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mullen AR, et al. Nature. 2012;481:385–8. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sena LA, et al. Mol Cell. 2012;48:158–67. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sullivan LB, et al. Mol Cell. 2013;51:236–48. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ooi A, et al. Cancer Cell. 2011;20:511–23. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 7.Adam J, et al. Cancer Cell. 2011;20:524–37. doi: 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loenarz C, et al. Trends Biochem Sci. 2011;36:7–18. doi: 10.1016/j.tibs.2010.07.002. [DOI] [PubMed] [Google Scholar]