

Abstract
The mammalian/mechanistic target of rapamycin complex 1 (mTORC1) signaling pathway is hyperactivated in a variety of cancers and disorders, including lymphangioleiomyomatosis (LAM) and tuberous sclerosis complex (TSC), which are characterized by mutations in tumor suppressors TSC1 or TSC2. The concern with the use of mTORC1 inhibitors, such as rapamycin or its analogs (rapalogs), is that they cause upregulation of autophagy and suppress the negative feedback loop to Akt, which promotes cell survival, causing the therapy to be only partially effective, and relapse occurs upon cessation of treatment. In this study, we investigate the use of rapamycin in combination with resveratrol, a naturally occurring polyphenol, in TSC2-deficient cells. We tested whether such combination would prevent rapamycin-induced upregulation of autophagy and shift the cell fate toward apoptosis. We found that this combination treatment blocked rapamycin-induced upregulation of autophagy and restored inhibition of Akt. Interestingly, the combination of rapamycin and resveratrol selectively promoted apoptosis of TSC2-deficient cells. Thus, the addition of resveratrol to rapamycin treatment may be a promising option for selective and targeted therapy for diseases with TSC loss and mTORC1 hyperactivation.
Keywords: mTOR, rapamycin, resveratrol, TSC, LAM, autophagy, apoptosis
Introduction
The mammalian target of rapamycin (also known as the mechanistic target of rapamycin or mTOR) is a serine/threonine kinase that interacts with other proteins to form 2 distinct complexes: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). These complexes differ in the mode of their upstream activation and in the network of downstream proteins they phosphorylate and regulate. mTORC1 is composed of regulatory-associated protein of mTOR (Raptor), proline-rich AKT substrate 40kDa (PRAS40), mLST8 and DEPTOR. mTORC1 is activated in response to growth factors, amino acids, oxidative stress, energy and oxygen levels of the cell and regulates a plethora of biosynthetic processes in the cell, including protein, nucleotide and lipid synthesis, mitochondrial homeostasis, as well as control of cell size, proliferation, and autophagy.1
Most upstream signals are relayed to mTORC1 via the tuberous sclerosis complex (TSC) proteins hamartin (TSC1) and tuberin (TSC2). These tumor suppressors negatively regulate mTORC1 signaling.2,3 Mutations in TSC1/2 cause inappropriate activation of mTORC1 signaling and result in tumor formation. In this context, the tumors are generally benign, because mTORC1 hyperactivation causes inactivation of the oncogenic phosphatidylinositide-3-kinase (PI3K) pathway and Akt signaling via a negative feedback loop.4
Lymphangioleiomyomatosis (LAM) and TSC are 2 disorders that are caused by mutations in TSC1/2. TSC is an autosomal-dominant genetic disease, which occurs in about 1 in 6000 births and has a 95% penetrance.5,6 TSC manifestations include benign multi-organ tumors with life-threatening consequences. LAM is a rare sporadic lung disease that predominantly affects women of childbearing age, as well as some adult female patients with TSC,7 and is characterized by proliferation of smooth muscle-like cells in the lung, causing cystic lung destruction.8-10
Because TSC and LAM pathogenesis is closely linked to mTORC1 hyperactivation,11,12 use of rapamycin and its analogs (or rapalogs) to inhibit mTORC1 has proved to be a viable strategy in treatment of some of the disease manifestations. However, some major concerns with the use of rapalogs are that the therapy effect is partial, rapalogs are associated with well-described toxicity and adverse events, and the benefits are not maintained after therapy withdrawal.13
Data suggest that TSC-null cells are particularly dependent on autophagy for survival.14 Autophagy is a homeostatic process that is conserved in all eukaryotic cells, which enables the cell to degrade and recycle organelles and misfolded proteins. In contrast, during nutrient deprivation autophagy allows the cell to survive by reusing intracellular proteins until extracellular nutrients become available. Upon initiation of autophagy, cellular components that are targeted for degradation are sequestered in double-membrane autophagosomes, which are, in turn, fused with lysosomes and degraded.15,16 The mTORC1 signaling pathway is a major regulator of autophagy in response to nutrient levels. Under nutrient-rich conditions, mTORC1 promotes cell growth and blocks autophagy by inhibiting ULK1 complex formation.17-19 Consequently, rapalog-mediated mTORC1 inactivation is a potent inducer of autophagy, which provides a survival advantage to TSC-deficient cells.14 Concordantly, the combination of the autophagy inhibitor chloroquine with rapalogs was shown to be more effective in inhibiting the growth of TSC2-deficient tumors than either agent alone,20 and this combination caused cell death in melanoma cells.21
Another consequence of mTORC1 inactivation with rapalog treatment is reactivation of the oncogenic Akt signaling. The activity of mTORC1 and its downstream target the 40S ribosomal S6 kinase 1 (S6K1) negatively regulate the PI3K/Akt signaling pathway.22 One of the major concerns with the use of rapalogs is that negative feedback signaling from S6K1 to Akt is abrogated, which leads to reactivation of Akt. Akt can, in turn, lead to upregulation of mTORC1 signaling and tumor cell survival.23 Therefore, current efforts focus on identifying agents that can be used together with rapalogs to inhibit mTORC1 while suppressing Akt activity, impeding cell survival.
In this study, we describe the use of resveratrol in combination with rapamycin as a strategy for inhibition of growth of TSC-deficient cells. Resveratrol (Trans-3,5,4’-trihydroxystilbene) is a naturally occurring polyphenol that is found in red wine, grapes, and peanuts and possesses disease-protective and anti-aging properties.24 Resveratrol was shown to suppress proliferation of human cells in vitro and mediate anti-tumor effects through several signaling pathways by modulating cellular events associated with autophagy, cell growth, and proliferation.25,26 Evidence suggests that resveratrol activates Sirt1, a class II histone deacetylase.27 However, the effects on autophagy are proposed to be Sirt1-independent, specifically through modulation of the mTORC1/S6K1 activity by resveratrol.26,28-32 We report that resveratrol reduces rapamycin-induced autophagic activity, prevents rapamycin-induced Akt reactivation, and preferentially promotes apoptosis of TSC2-deficient cells. Therefore, the combination of rapamycin and resveratrol may be an effective strategy for treatment of diseases caused by TSC loss.
Results
Combination of rapamycin and resveratrol treatment restores inhibition of Akt, while suppressing mTORC1 signaling
We first investigated the effect of rapamycin and resveratrol, alone or in combination, on mTORC1 and Akt signaling pathways in wild-type (WT) and TSC2−/− mouse embryonic fibroblasts (MEFs) (Fig. 1A). As expected, basal levels of mTORC1 signaling were higher in TSC2−/− cells, while WT MEFs had higher basal levels of Akt phosphorylation compared with TSC2−/− MEFs. We observed that rapamycin effectively suppressed mTORC1 signaling in both cell types, as measured by markers of S6K1 activity, such as phosphorylation of S6K1 on Thr389, phosphorylation of S6K1 downstream targets eIF4B and S6, and stabilization of the protein levels of PDCD4. Due to inhibition of the negative feedback loop, rapamycin treatment increased Akt signaling, as measured by phosphorylation of Akt and its target PRAS40. Resveratrol treatment alone suppressed basal Akt signaling and slightly decreased S6K1 pathway activation. Interestingly, resveratrol was able to prevent rapamycin-induced reactivation of Akt signaling as well as phosphorylation of its downstream substrate, PRAS40, while simultaneously effectively suppressing mTORC1/S6K1 signaling. To confirm our observations, we examined ELT3 cells, TSC2-null Eker rat uterine leiomyoma-derived smooth muscle cells, and compared them to ELT3 cells overexpressing (o/e) TSC2. Cells were treated with rapamycin and resveratrol alone or in combination, and similarly, inhibition of Akt and mTORC1 signaling were observed (Fig. 1B). Additionally, the inhibition of Akt and mTORC1 signaling occurred after rapamycin and resveratrol combination treatment in human LAM-derived TSC2-null 621–101 cells (Fig. 1C). Because in LAM the TSC2-deficient cells metastasize into healthy lung tissue, we next examined the effects of the combination of rapamycin and resveratrol in normal human bronchial epithelial cells (NHBE). In contrast to 621–101 cells, NHBE cells show greater induction of Akt phosphorylation upon rapamycin treatment, probably owing to the presence of wild-type TSC2. However, similar to 621–101 cells, the combination of rapamycin and resveratrol treatment prevented rapamycin-induced activation of Akt signaling in NHBEs, while still maintaining inhibition of the mTORC1 signaling pathway (Fig. 1D). These data indicate that combination of rapamycin and resveratrol treatment is a powerful tool for inhibiting mTORC1 signaling while preventing Akt upregulation in normal and TSC2-deficient cells.
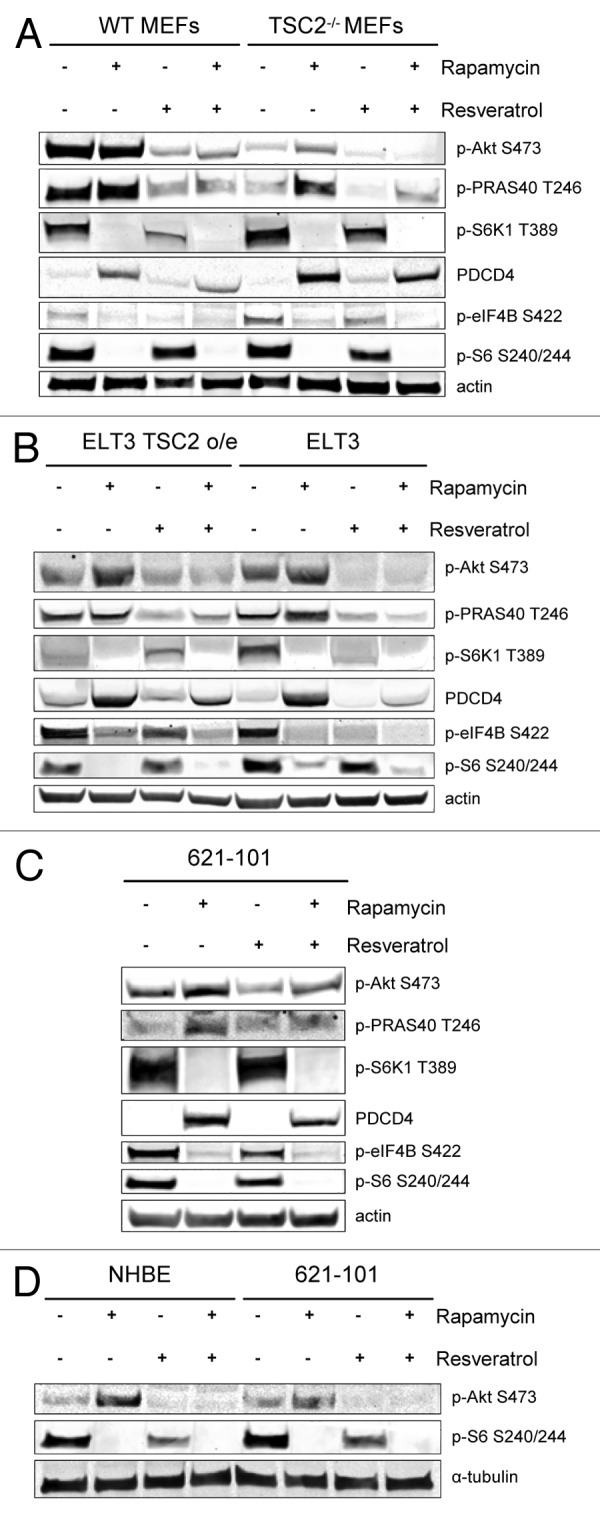
Figure 1. The combination of rapamycin and resveratrol inhibits PI3K/Akt and mTOR signaling pathways. (A) WT MEFs and TSC2−/− MEFs were treated with 20 nM rapamycin and/or 100 µM resveratrol for 24 h. Cells were lysed and the indicated proteins were detected by immunoblot. (B) ELT3 TSC2 o/e and ELT3 cells were treated as described in (A) and protein levels were detected by immunoblot. (C) 621–101 cells were treated as described in (A), and protein levels were detected by immunoblot. (D) 621–101 and NHBE cells were treated as described in (A), and protein levels were detected by immunoblot.
Combination of rapamycin and resveratrol downregulates autophagy in TSC2-null cells
Both rapamycin and resveratrol are known modulators of autophagy. Rapamycin potently induces autophagy,14 while resveratrol has pleiotropic effects on autophagy based on nutrient availability of the cell.26 We next tested the effect of the combination of rapamycin and resveratrol on autophagy induction in WT and TSC2−/− MEFs . Levels of p62/SQSTM1 protein inversely correlate with autophagy induction, because p62 is degraded in the lysosome upon rapamycin-induced increase in the autophagic flux. We observed that resveratrol alone did not have an effect on p62 levels. Interestingly, the combination treatment of rapamycin and resveratrol blocked autophagy induction in WT MEFs and TSC2−/− MEFs and restored p62 levels back to baseline (Fig. 2A).

Figure 2. Resveratrol prevents rapamycin-induced induction of autophagy. (A) WT MEFs and TSC2−/− MEFs were treated with 20 nM rapamycin and/or 100 µM resveratrol for 24 h. Cells were lysed, and protein levels of p62 and actin were analyzed by immunoblot. (B) TSC2−/− MEFs were treated with 20nM rapamycin and/or 100 µM resveratrol, with or without 4-h pretreatment with 100 nM BafA1. Cells were lysed and blotted for LC3 and actin. The histogram shows LC3-II levels (lower band) normalized to actin levels. LC3-II and actin levels were quantified using Image Studio 3.1 (LI-COR), and plotted using Excel. (C) ELT3 TSC2 o/e and ELT3 cells were treated with 20 nM rapamycin and/or 100 µM resveratrol, with or without 4-h pretreatment with 100 nM BafA1. Cells were lysed and blotted for LC3 and actin. The histogram shows LC3-II levels (lower band) normalized to actin levels. LC3-II and actin levels were quantified using Image Studio 3.1 (LI-COR), and plotted using Excel. (D) 621–101 cells were treated with 20 nM rapamycin and/or 100µM resveratrol for 24 h. Cells were lysed and probed for p62 and α-tubulin levels. (E) 621–101 and 621–101 TSC2 o/e cells were treated with 20 nM rapamycin and/or 100 µM resveratrol for 24 h Cells were lysed and probed for p-S6 (T240/244) and actin levels. (F) 621–101 and 621–101 TSC2 o/e cells were treated as described in (B). Histograms show LC3-II levels (lower band) normalized to either actin or LC3-I levels (upper band).
Another way of measuring autophagy levels is by examining LC3 expression. Upon autophagy induction, LC3-I is cleaved to LC3-II. The amount of LC3-II correlates with the number of autophagosomes; however, LC3-II is degraded by autophagy. Treatment of cells with Bafilomycin A1 (BafA1) blocks lysosomal degradation and causes LC3-II to accumulate, allowing detection of increased autophagic flux. We examined whether autophagy responses assayed by LC3-II levels are consistent with the observed p62 levels after rapamycin and resveratrol treatment (Fig. 2B). As expected, treatment of TSC2−/− MEFs with rapamycin in the presence of BafA1 upregulated autophagy induction, as evidenced by the increase in LC3-II expression compared with the untreated cells. Combination treatment of rapamycin and resveratrol in the presence of BafA1 blocked autophagy induction and decreased LC3-II levels, similar to the untreated controls. We next compared autophagy induction in ELT3 TSC2 o/e and ELT3 cells in response to combination of rapamycin and resveratrol. In ELT3 cells, resveratrol was able to prevent rapamycin-induced autophagy and accumulation of LC3-II, reducing its levels to baseline (Fig. 2C). Interestingly, rapamycin was not able to upregulate LC3II levels in ELT3 cells where TSC2 is overexpressed, indicating that this mechanism of autophagy induction is dependent on TSC2.
We next investigated the effects on autophagy in human TSC2-deficient 621–101 cells. We observed that while rapamycin caused reduction in the levels of the autophagy marker p62, the combination of rapamycin and resveratrol prevented p62 degradation, albeit to a lesser degree, possibly due to cell type-specific differences with regard to autophagy induction and markers of autophagic flux (Fig. 2D).
We also tested whether resveratrol was able to prevent rapamycin-induced upregulation of LC3-II in 621–101 cells and compared them to 621–101 cells in which TSC2 was overexpressed (o/e). In the 621–101 TSC2 o/e cells, mTORC1 signaling is reduced compared with the parental cell line, as measured by levels of phosphorylated S6 (Fig. 2E). 621–101 and 621–101 TSC2 o/e cells were treated with rapamycin and/or resveratrol in the presence of BafA1 and analyzed for LC3 levels (Fig. 2F). As expected, rapamycin treatment alone strongly increased LC3-II levels compared with the untreated controls. Interestingly, rapamycin treatment alone did not upregulate LC3-II levels in 621–101 TSC2 o/e cells, consistent with the fact that this mechanism of autophagy induction is dependent on the TSC2 status of 621–101 cells. Combination treatment of 621–101 cells with both rapamycin and resveratrol effectively blocked rapamycin-induced upregulation of LC3-II and reduced LC3-II levels to below those of untreated controls (Fig. 2F). Our results indicate that combination of rapamycin and resveratrol treatment is an effective tool in preventing autophagy induction in all 3 models of TSC2-deficeints cells examined, TSC2−/− MEFs, human 621–101 cells, and rat ELT3 cells.
Apoptosis is induced in TSC2-null cells, but not in control cells
Because TSC2-null cells are dependent on autophagy for survival, we next investigated whether blocking rapamycin-induced upregulation of autophagy in TSC2-deficient cells would cause resveratrol treatment to preferentially tilt the balance toward apoptosis. WT MEFs and TSC2−/− MEFs were treated with rapamycin and/or resveratrol for 24 h and assayed for apoptosis induction by examining cleavage of 2 markers, PARP and Caspase 3 (Fig. 3A). Strikingly, resveratrol treatment alone or in combination with rapamycin was able to strongly increase cleavage of PARP and Caspase 3 specifically in TSC2−/− MEFs, and not in WT MEFs. Similar effect was seen in ELT3 cells, where an increase in cleaved PARP and Caspase 3 fragments was observed specifically in TSC2-deficient ELT3 cells, and not in TSC2-overexpressing cells (Fig. 3B). Similarly, in 621–101 cells, resveratrol treatment in combination with rapamycin induced cleaved PARP and Caspase 3 fragments (Fig. 3C). We also compared apoptotic markers in 621–101 cells and wild-type NHBE cells (Fig. 3D). Consistent with our observations in MEFs, the combination of rapamycin and resveratrol treatment specifically upregulated cleavage of PARP and Caspase 3 in 621–101 cells, but not in NHBE cells. Our results indicate that rapamycin and resveratrol treatment preferentially induces apoptosis in TSC2-deficient cells, TSC2−/− MEFs, ELT3 cells, and 621–101 cells, but not in control WT MEFs, normal human NHBE cells and ELT3 TSC2 o/e cells.
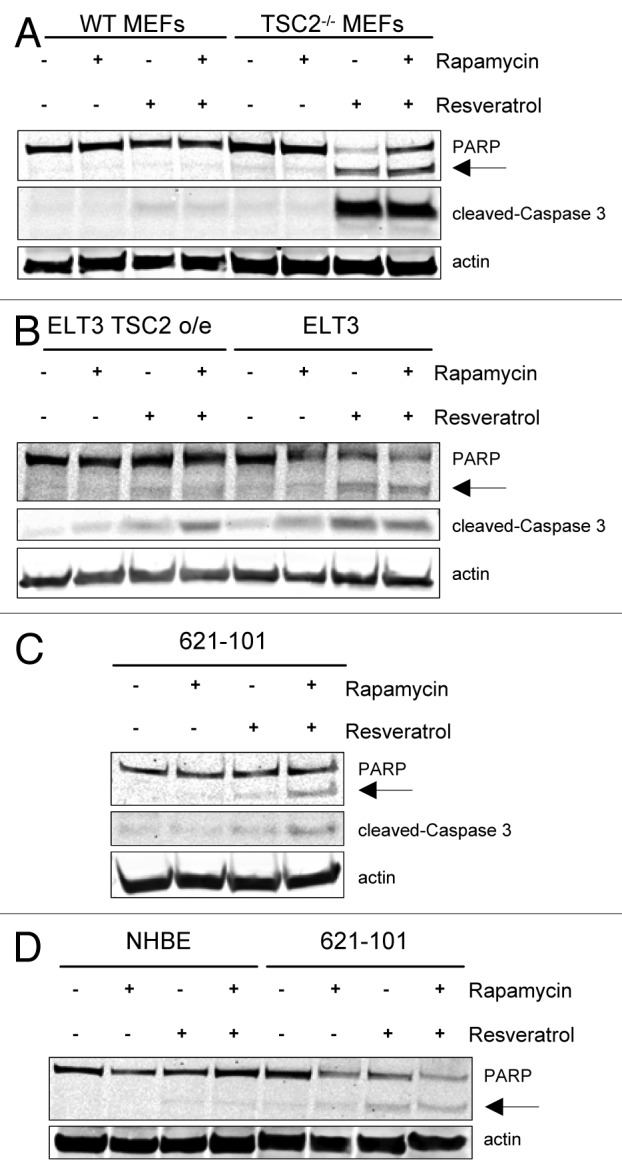
Figure 3. Resveratrol and rapamycin combination selectively induces markers of apoptosis in TSC2-null cells. (A) WT MEFs and TSC2−/− MEFs were treated with 20 nM rapamycin and/or 100 µM resveratrol for 24 h. Cells were lysed and probed for PARP, cleaved-Caspase 3, and actin. The arrow indicates cleaved PARP isoform. (B) ELT3 TSC2 o/e and ELT3 cells were treated as described in (A). (C) 621–101 cells were treated as described in (A). (D) NHBE and 621–101 cells were treated with 20 nM rapamycin and/or 100 µM resveratrol for 24 h. Cells were lysed and probed for PARP and actin.
We next examined whether TSC2 status correlates with cellular morphology and apoptotic responses during rapamycin and resveratrol treatment. WT and TSC2−/− MEFs were treated with rapamycin and/or resveratrol for 24 h. As depicted in Figure 4A, in response to the resveratrol, TSC2−/− MEFs demonstrate rounded off morphology and cell detachment that are more pronounced than in WT MEFs, and this phenotype is even more severe in cells treated with the combination of the 2 agents, where clusters of detached cells are visible. To quantify the levels of apoptosis on a single-cell level, we performed a cytometric Annexin V assay. TSC2−/− MEFs treated with rapamycin and resveratrol showed significant upregulation in the proportion of apoptotic cells compared with either untreated cells or rapamycin-treated cells (Fig. 4B). The fraction of apoptotic TSC2−/− MEFs treated with rapamycin and resveratrol were much higher than that of WT MEFs (Fig. 4B). We further quantified the amount of early apoptotic cells in WT MEFs as compared with TSC2−/− MEFs (Fig. 4C). Similar effect on induction of apoptosis using the combination treatment was observed when measuring early apoptotic cells in ELT3 cells as compared with ELT3 TSC2 o/e cells (Fig. 4D). These results provide additional support for our finding that TSC2−/− MEFs and ELT3 cells exhibit sensitivity to the combination of rapamycin and resveratrol, which preferentially induces apoptosis in TSC2-deficient cells, compared with either WT MEFs or ELT3 TSC2 o/e cells.

Figure 4. Analysis of apoptosis induction in WT and TSC2−/− MEFs. (A) WT MEFs and TSC2−/− MEFs were treated with 20 nM rapamycin and/or 50 µM resveratrol for 24 h. Cells were photographed using Zeiss light microscope under 20× magnification. (B) WT MEFs and TSC2−/− MEFs were treated as described in (A). Cells were pelleted and single cell suspension was incubated with the Guava Nexin Reagent for 20 min at room temperature, and analyzed for Annexin V staining by flow cytometry. (C) WT MEFs and TSC2−/− MEFs were treated as described in (A). Histogram represents quantification of early apoptotic cells from 3 experiments. (D) Histogram represents quantification of early apoptotic ELT3 TSC2 o/e and ELT3 cells as described in (C). Student t test was performed on treated samples relative to untreated controls. **P < 0.01.
We next investigated the cellular morphology of 621–101 cells treated with rapamycin and/or resveratrol. As depicted in Figure 5A, consistent with our observations in TSC2−/− MEFs, 621–101 cells treated with rapamycin and resveratrol were more rounded off, detaching in clusters. We examined the proliferation rates of 621–101 cells treated with rapamycin and/or resveratrol (Fig. 5B). We observed that while both rapamycin and resveratrol reduced cell numbers, the combination of the 2 agents was more effective than each drug alone. In contrast, TSC2-overexpressing 621–101 cells were less sensitive to the resveratrol’s inhibitory effects, alone or in combination with rapamycin, further supporting the role of TSC2 in mediating the balance of autophagy and apoptosis in these cells. To quantify the extent of apoptosis on a single-cell basis in 621–101 cells, Annexin V assay was performed after 24 h treatment with rapamycin and/or resveratrol (Fig. 5C). Our results indicate that the proportion of apoptotic 621–101 cells following treatment with the combination of rapamycin and resveratrol was higher compared with either untreated cells or cells treated with either rapamycin or resveratrol alone (Fig. 5C and D). While the increase in the fraction of apoptotic cells treated with both rapamycin and resveratrol was smaller in 621–101 cells compared with TSC2−/− MEFs, it was still significant and consistent with the presence of apoptotic markers detected by immunoblotting (Fig. 3).

Figure 5. 621–101 cells show induction of apoptosis and decrease in proliferation upon combination rapamycin and resveratrol treatment. (A) 621–101 cells were treated with either 20 nM rapamycin and/or 100 µM resveratrol for 24 h. Cells were photographed using Zeiss light microscope under 10× magnification. (B) 621–101 and 621–101 TSC2 o/e cells were treated with 20 nM rapamycin and/or 100 µM resveratrol for 48 h, proliferation assay was performed as described in “Materials and Methods”. (C) 621–101 cells were treated as described in (A). Cells were subsequently scraped, pelleted and incubated with the Guava Nexin Reagent for 20 min at room temperature, and analyzed for Annexin V staining by flow cytometry. (D) 621–101 cells were treated as described in (A). Histogram represents quantification of early apoptotic cells from 3 experiments. Student’s t test was performed on treated samples relative to untreated controls. *P < 0.05, **P < 0.01.
Finally, we examined the survival and metastatic capacity of TSC2-null ELT3 cells in vivo by testing whether the combination of rapamycin and resveratrol is effective in reducing lung metastases following tail vein injection in mice, using a previously established model33 (Fig. 6). Our results indicate that after 24 h treatment, mice treated with the combination therapy had significantly fewer lung metastases as assayed by photon flux compared with control mice or mice treated with either rapamycin or resveratrol alone. This provides further support for the clinical potential of the combination rapamycin and resveratrol therapy in treatment of lung manifestations in TSC and LAM.

Figure 6. Combination of rapamycin and resveratrol strongly reduces the survival of Tsc2-null cells in vivo. Mice were treated with vehicle, rapamycin, resveratrol, or rapamycin plus resveratrol. ELT3-luciferase-expressing cells were inoculated intravenously. (A) Representative bioluminescent images of cells after each drug treatment were shown. The total flux (photons/second) of cells was illustrated. (B) Representative bioluminescent images of lung colonization at 1, 6, and 24 h post-cell injection. Total photon flux/second present in the chest regions were quantified and compared among treatment groups. *P < 0.05, **P < 0.01, Student t test.
Discussion
Loss of TSC1/2 and the subsequent hyperactivation of mTORC1 signaling in TSC and LAM provide the basis for use of rapalogs in treatment of these diseases. The effectiveness of rapalogs in a monotherapy setting is limited by the fact that they cause reactivation of oncogenic PI3K/Akt signaling and potently induce autophagy, which allows for cell survival. Therefore, we set out to determine whether the addition of resveratrol, a compound with inhibitory effects on the PI3K/Akt/mTORC1 signaling would maintain inhibition of Akt and prevent autophagy induction in TSC2-deficient cells, causing these cells to undergo apoptosis. Because rapamycin and resveratrol have non-overlapping biological activities, the combination of the 2 agents has the potential to prevent the side effects associated with each agent alone and to provide additional beneficial effects.34
In wild-type cells, the TSC1/TSC2 complex moderates mTORC1 signaling, which limits the cell’s anabolic capacity, growth, and proliferation. In TSC2-null cells, unchecked mTORC1 and S6K1 signaling leads to uncontrolled cell growth. However, mTORC1/S6K1 hyperactivation inhibits PI3K/Akt signaling via a negative feedback loop, preventing neoplastic transformation of TSC2-null cells (Fig. 7A). When mTORC1 signaling is inhibited by rapamycin in TSC2-deficient cells, growth and proliferation are reduced, but several consequences emerge. Inhibition of mTORC1 removes the negative regulation of autophagy, increasing the cell’s ability to survive via this mechanism. Additionally, inhibition of mTORC1/S6K1 signaling removes the negative feedback loop to Akt, upregulating its activity, allowing it to further promote cell survival by inhibiting apoptosis (Fig. 7B). We found that resveratrol alone moderately inhibited mTORC1/S6K1 signaling, which is consistent with published data describing inhibitory activity toward mTORC1 either directly, or by positive regulation of AMPK.26,28,29 Data presented hereby demonstrate that the addition of resveratrol to rapamycin prevents reactivation of Akt. We hypothesize that in this setting Akt may be affected by inhibition of PI3K,35 and not by upregulation of expression of the inhibitory phosphatase PTEN,36 as we did not observe changes in PTEN expression (data not shown).

Figure 7. Attenuation of the PI3K/Akt/mTORC1 signaling pathway upon combination therapy. (A) Schematic representation of the PI3K/mTORC1 signaling pathway in WT and TSC2 null cells. Loss of TSC2 results in hyperactivation of mTORC1 signaling promoting cell growth and proliferation and causing feedback inhibition of PI3K/Akt. (B) Schematic representation of the PI3K/mTORC1 signaling pathway in TSC2-null cells upon rapamycin treatment. Inhibition of mTORC1 induces autophagy, Akt signaling and cell survival. (C) Schematic representation of the PI3K/mTORC1 signaling in TSC2-null cells upon combination of rapamycin and resveratrol treatment. Addition of resveratrol treatment prevents rapamycin-induced Akt activation and autophagy induction, promoting the progession to apoptosis.
Here we report the exciting finding that resveratrol in combination with rapamycin prevents induction of autophagy, and promotes the transition to apoptosis (Fig. 7C). Moreover, we found that TSC2-null cells are exquisitely sensitive to resveratrol-mediated induction of apoptosis, underscoring the potential of the combination of rapamycin and resveratrol as an effective and selective treatment strategy. Previous studies in breast cancer and neuroblastoma cell lines found that resveratrol caused cytotoxic effects resulting in upregulation of apoptosis.37,38 In the present study, we discover a novel finding that resveratrol treatment together with rapamycin specifically induces apoptosis in TSC2−/− cells, and not in wild-type cells.
It is thought that TSC2-deficient cells have lower basal levels of autophagy, and inhibition of mTORC1 by rapamycin treatment affects the induction of autophagy in TSC2-null LAM cells, promoting their survival.14 We observed that the combination treatment restores autophagy to basal levels in TSC2-null cells, as evidenced by p62 and LC3 markers. It is interesting to note that there were cell-specific differences in response to treatment among different cell lines. Resveratrol restored p62 levels upon combination treatment more potently in TSC2−/− MEFs than in 621–101 cells, although LC3-II levels were depressed more robustly upon combination treatment in 621–101 cells compared with TSC2−/− MEFs. It is also of note that rapamycin upregulated LC3-II levels in 621–101 and ELT3 cells that lack TSC2 but not in TSC2-overexpressing 621–101 and ELT3 cells, suggesting that this mechanism is dependent on TSC2. Recent evidence suggests that the mTORC1/S6K1 pathway is an important regulator of autophagy. mTORC1 regulates autophagy in at least 2 ways. First, mTORC1 is a negative regulator of the Atg1, a complex essential for autophagy induction, by direct phosphorylation of several component proteins.39 Second, mTORC1 is a central node in the signaling pathway that includes autophagy regulating factors, such as AMPK (positive regulator), Akt (negative regulator), and S6K1 (positive regulator in mammals).26,40,41 Data also show that mTORC1/S6K1 may positively regulate autophagy via negative feedback inhibition of Akt.23 Because of the role of autophagy in the pathogenesis of diseases with TSC inactivation, autophagy inhibitors such as chloroquine may have therapeutic potential, possibly in combination with rapalogs. The advantage of resveratrol is that in addition to modulating autophagy, it has the compounding benefit of preventing the potentially harmful rapamycin-induced Akt reactivation.
We were also excited to find that the combination of rapamycin and resveratrol treatment specifically upregulated apoptosis in TSC2-null cells, but not in WT cells. In TSC2−/− MEFs, 621–101 and ELT3 cells, resveratrol, as well as the combination treatment potently induced apoptotic markers, which is in stark contrast to WT MEFs, normal bronchial cells, and TSC2-overexpressing ELT3 cells, where very low levels of cleaved PARP and Caspase 3 were observed. The implication of such finding is tremendous as it indicates that combination of rapamycin and resveratrol selectively causes induction of apoptosis in TSC2-deficient cells.
The effect of rapamycin and resveratrol is also seen upon closer examination of cellular morphology. It is clear that TSC2-deficient cells are much more sensitive to resveratrol in combination with rapamycin than TSC2-expressing cells. In addition, flow cytometry-based analysis of early apoptosis confirmed that compared with wild-type cells, TSC2-deficient cells were preferentially undergoing early apoptosis after treatment with the combination of rapamycin and resveratrol. This supports immunoblot data indicating that such combination of agents specifically caused upregulation of apoptosis in cells lacking TSC2. Finally, we demonstrated the clinical potential of the combination of rapamycin and resveratrol in preventing survival and lung metastasis of TSC2-deficient ELT3 cells following injection into mice.
Taken together, our results show that the addition of resveratrol to rapamycin treatment in TSC2-null and LAM cells maintains inhibition of the mTORC1/S6K1 signaling, which reduces cell growth and proliferation and prevents induction of autophagy, while at the same time restores inhibition of Akt, which allows for progression to apoptosis. Importantly, upregulation of apoptosis is selective to TSC2-null cells and may serve as a basis for treatment of diseases with TSC2 inactivation, such as LAM. Because rapamycin is an FDA-approved drug and resveratrol is a widely available nutritional supplement, future steps may be taken to determine the efficacy of this combination in pre-clinical and clinical trials.
Materials and Methods
Cell culture
Cells were cultured in Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) in a humidified incubator with 5% CO2 at 37 °C. Lymphangioleiomyomatosis-derived cells (LAM 621–101 TSC2−/− cells) were obtained from Elizabeth P Henske (Harvard Medical School). 621–101 cells stably overexpressing TSC2 (621–101 TSC2 o/e) were obtained from Hayley McDaid (Albert Einstein College of Medicine). Wild-type mouse embryonic fibroblasts (WT MEFs) and TSC2−/− MEFs were obtained from David Kwiatkowski (Harvard Medical School). Normal human bronchial/tracheal epithelial (NHBE) cells were from Lonza and cultured in bronchial epithelial cell growth medium (BEGM).
Cell treatment
Cells were treated with 20 nM Rapamycin, 100 µM Resveratrol (Sigma-Aldrich), or combination of the 2 agents for 24 h. For detection of LC3-I/II, cells were treated with 100 nM Bafilomycin A1 (BafA1; Sigma-Aldrich) for 4 h, following treatment with 20 nM Rapamycin and/or 100 µM Resveratrol.
Immunoblots
Following treatment, cells were lysed in ice-cold lysis buffer (10 mM KPO4, 1 mM EDTA, 10 mM MgCl2, 50 mM β-glycerophosphate, 5 mM EGTA, 0.5% Nonidet P-40 [NP-40], 0.1% Brij 35, 1 mM sodium orthovanadate, 40 µg/ml phenylmethylsulfonyl fluoride, 10 µg/ml leupeptin, 5 µg/ml pepstatin, pH 7.28). Lysates were cleared of insoluble material by centrifugation at 15 000 g for 10 min at 4 °C.
Protein concentrations in cell extracts were measured by Bradford assay (BioRAD) according to the manufacturer’s protocol using Eppendorf BioPhotometer. Samples were equalized for protein concentration and denatured using 4× NuPAGE® LDS Sample buffer and 10× Reducing agent (Invitrogen) at 70 °C for 10 min. Samples were resolved using Bis-Tris Plus gels (Invitrogen) and transferred onto nitrocellulose membrane (GE Healthcare). Membranes were probed with the following primary antibodies: p-Akt (Ser473), p-S6K1 (Thr389), p-eIF4B (Ser422), p-S6 (240/244), p-PRAS40 (Thr246), p62, LC3, and Caspase 3; all from Cell Signaling Technologies; PDCD4 (Proteintech), actin (Santa Cruz Biotechnology), α-tubulin, and PARP (Abcam). Blots were incubated with IRDye-conjugated anti-rabbit, anti-mouse or anti-goat secondary antibodies (LI-COR), and imaged using Odyssey infrared detection instrument (LI-COR). All immunoblots were performed at least 3× to ensure reproducibility.
Apoptosis assay
Annexin V detection was performed using Guava Nexin Assay with the Guava EasyCyte Mini System flow cytometer (EMD-Millipore), according to the manufacturer’s protocol. 621–101 cells were treated with 20nM Rapamycin and/or 100 µM Resveratrol for 24 h. MEFs and ELT3 cells were treated with 20 nM Rapamycin and/or 50 µM Resveratrol for 24 h. Cells were collected by trypsinization and resuspended in growth media at approximate concentration of 2 × 104−1 × 105 cells/ml. 100 μL of cells was mixed with 100 μL of Guava Nexin Reagent and incubated for 20 min at room temperature in the dark. Quantification of the early apoptotic cells was performed using Guava Cell Cycle protocol according to the manufacturer’s instructions. The apoptosis assays were performed trice to ensure reproducibility and data was analyzed using WinMDI 2.9 software.
Microscopy
Cells were treated as indicated and visualized using Zeiss invertoscope under 10× or 20× magnification.
Proliferation assay
Cells were seeded at a concentration of 2500 cells/well in a 96-well plate, and allowed to attach. Cells were treated in quadruplicate with 20 nM Rapamycin and/or 100 µM Resveratrol for 48 h. To detect viable cells, cells were incubated with 100 μg/mL solution of neutral red dye in growth media for 30 min at 37 °C. Cells were washed and fixed in a 0.5% formalin-1% CaCl2 solution and permeabilized in 1% acetic acid-50% ethanol solution to release the incorporated neutral red. Absorbance was measured at 540 nm using a microtiter plate spectrophotometer, quantified and plotted using Excel.
Animal studies
All animal work was performed in accordance with protocols approved by the Institutional Animal Care and Use Committee, Children Hospital Boston. Female CB17 scid mice (Taconic) were supplemented with 17-β-estradiol (500 nM in drinking water) for 2 d, and then treated with rapamycin (6 mg/kg, p.o.), resveratrol (200 mg/kg, p.o.), rapamycin plus resveratrol (6 mg/kg, p.o. + 200 mg/kg, p.o.), or vehicle (100 μL PBS, p.o.) for 24 h before and 2 h after cell inoculation. Tsc2-null rat uterine leiomyoma-derived ELT3-luciferase cells were pre-treated with rapamycin (2 nM), resveratrol (20 μM), rapamycin plus resveratrol, or vehicle for 12 h. Cells were harvested, suspended in 100 μL PBS, and then injected intravenously into mice (n = 5/group). Lung colonization was monitored using bioluminescent live imaging at 1, 6, and 24 h post-cell injection. Ten minutes prior to imaging, animals were injected with D-luciferin (Perkin Elmer, 120 mg/kg, i.p.). Bioluminescent signals were recorded using the Xenogen IVIS System. Total photon flux of chest area was analyzed.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants to MKH from the NIH (CA151112), Atol Charitable Trust, LAM Foundation (098P0113), American Cancer Society (RSG-13-287-01-TBE), NIH grant to JJY (HL098216), fellowship support to AA from the National Cancer Center, and funding from Yeshiva University.
Glossary
Abbreviations:
- LAM
lymphangioleiomyomatosis
- MEFs
mouse embryonic fibroblasts
- mTORC
mammalian target of rapamycin complex
- PDCD4
programmed cell death protein 4
- PI3K
phosphatidylinositide-3-kinase
- PTEN
phosphatase and tensin homolog
- S6K1
40S ribosomal S6 Kinase 1
- TSC
tuberous sclerosis complex
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27355
References
- 1.Alayev A, Holz MK. mTOR signaling for biological control and cancer. J Cell Physiol. 2013;228:1658–64. doi: 10.1002/jcp.24351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893–904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–13. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 4.Zhang H, Cicchetti G, Onda H, Koon HB, Asrican K, Bajraszewski N, Vazquez F, Carpenter CL, Kwiatkowski DJ. Loss of Tsc1/Tsc2 activates mTOR and disrupts PI3K-Akt signaling through downregulation of PDGFR. J Clin Invest. 2003;112:1223–33. doi: 10.1172/JCI17222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Osborne JP, Jones AC, Burley MW, Jeganathan D, Young J, O’Callaghan FJ, Sampson JR, Povey S. Non-penetrance in tuberous sclerosis. Lancet. 2000;355:1698. doi: 10.1016/S0140-6736(00)02247-9. [DOI] [PubMed] [Google Scholar]
- 6.Wataya-Kaneda M, Tanaka M, Hamasaki T, Katayama I. Trends in the prevalence of tuberous sclerosis complex manifestations: an epidemiological study of 166 Japanese patients. PLoS One. 2013;8:e63910. doi: 10.1371/journal.pone.0063910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97:6085–90. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133:507–16. doi: 10.1378/chest.07-0898. [DOI] [PubMed] [Google Scholar]
- 9.Taveira-DaSilva AM, Steagall WK, Moss J. Lymphangioleiomyomatosis. Cancer Control. 2006;13:276–85. doi: 10.1177/107327480601300405. [DOI] [PubMed] [Google Scholar]
- 10.Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006;27:1056–65. doi: 10.1183/09031936.06.00113303. [DOI] [PubMed] [Google Scholar]
- 11.Goncharova EA, Goncharov DA, Spaits M, Noonan DJ, Talovskaya E, Eszterhas A, Krymskaya VP. Abnormal growth of smooth muscle-like cells in lymphangioleiomyomatosis: Role for tumor suppressor TSC2. Am J Respir Cell Mol Biol. 2006;34:561–72. doi: 10.1165/rcmb.2005-0300OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348–9. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 13.Kinder B, McCormack FX. Clinical trials for rare lung diseases: lessons from lymphangioleiomyomatosis. Lymphat Res Biol. 2010;8:71–9. doi: 10.1089/lrb.2009.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yu J, Parkhitko A, Henske EP. Autophagy: an ‘Achilles’ heel of tumorigenesis in TSC and LAM. Autophagy. 2011;7:1400–1. doi: 10.4161/auto.7.11.17652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505–10. doi: 10.1038/nrm1666. [DOI] [PubMed] [Google Scholar]
- 16.Rabinowitz JD, White E. Autophagy and metabolism. Science. 2010;330:1344–8. doi: 10.1126/science.1193497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ganley IG, Lam H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284:12297–305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parkhitko A, Myachina F, Morrison TA, Hindi KM, Auricchio N, Karbowniczek M, Wu JJ, Finkel T, Kwiatkowski DJ, Yu JJ, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108:12455–60. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie X, White EP, Mehnert JM. Coordinate autophagy and mTOR pathway inhibition enhances cell death in melanoma. PLoS One. 2013;8:e55096. doi: 10.1371/journal.pone.0055096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holz MK. The role of S6K1 in ER-positive breast cancer. Cell Cycle. 2012;11:3159–65. doi: 10.4161/cc.21194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng X, Kinsella TJ. Mammalian target of rapamycin and S6 kinase 1 positively regulate 6-thioguanine-induced autophagy. Cancer Res. 2008;68:2384–90. doi: 10.1158/0008-5472.CAN-07-6163. [DOI] [PubMed] [Google Scholar]
- 24.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–42. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bishayee A. Cancer prevention and treatment with resveratrol: from rodent studies to clinical trials. Cancer Prev Res (Phila) 2009;2:409–18. doi: 10.1158/1940-6207.CAPR-08-0160. [DOI] [PubMed] [Google Scholar]
- 26.Armour SM, Baur JA, Hsieh SN, Land-Bracha A, Thomas SM, Sinclair DA. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging (Albany NY) 2009;1:515–28. doi: 10.18632/aging.100056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–91. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dasgupta B, Milbrandt J. Resveratrol stimulates AMP kinase activity in neurons. Proc Natl Acad Sci U S A. 2007;104:7217–22. doi: 10.1073/pnas.0610068104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem. 2010;285:36387–94. doi: 10.1074/jbc.M110.169284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolinsky VW, Chan AY, Robillard Frayne I, Light PE, Des Rosiers C, Dyck JR. Resveratrol prevents the prohypertrophic effects of oxidative stress on LKB1. Circulation. 2009;119:1643–52. doi: 10.1161/CIRCULATIONAHA.108.787440. [DOI] [PubMed] [Google Scholar]
- 31.Demidenko ZN, Blagosklonny MV. At concentrations that inhibit mTOR, resveratrol suppresses cellular senescence. Cell Cycle. 2009;8:1901–4. doi: 10.4161/cc.8.12.8810. [DOI] [PubMed] [Google Scholar]
- 32.Ghosh HS, McBurney M, Robbins PD. SIRT1 negatively regulates the mammalian target of rapamycin. PLoS One. 2010;5:e9199. doi: 10.1371/journal.pone.0009199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu JJ, Robb VA, Morrison TA, Ariazi EA, Karbowniczek M, Astrinidis A, Wang C, Hernandez-Cuebas L, Seeholzer LF, Nicolas E, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. 2009;106:2635–40. doi: 10.1073/pnas.0810790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leontieva OV, Paszkiewicz G, Demidenko ZN, Blagosklonny MV. Resveratrol potentiates rapamycin to prevent hyperinsulinemia and obesity in male mice on high fat diet. Cell Death Dis. 2013;4:e472. doi: 10.1038/cddis.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matter WF, Brown RF, Vlahos CJ. The inhibition of phosphatidylinositol 3-kinase by quercetin and analogs. Biochem Biophys Res Commun. 1992;186:624–31. doi: 10.1016/0006-291X(92)90792-J. [DOI] [PubMed] [Google Scholar]
- 36.Waite KA, Sinden MR, Eng C. Phytoestrogen exposure elevates PTEN levels. Hum Mol Genet. 2005;14:1457–63. doi: 10.1093/hmg/ddi155. [DOI] [PubMed] [Google Scholar]
- 37.Chen Y, Tseng SH, Lai HS, Chen WJ. Resveratrol-induced cellular apoptosis and cell cycle arrest in neuroblastoma cells and antitumor effects on neuroblastoma in mice. Surgery. 2004;136:57–66. doi: 10.1016/j.surg.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Liu J, Liu X, Xing K, Wang Y, Li F, Yao L. Resveratrol-induced cell inhibition of growth and apoptosis in MCF7 human breast cancer cells are associated with modulation of phosphorylated Akt and caspase-9. Appl Biochem Biotechnol. 2006;135:181–92. doi: 10.1385/ABAB:135:3:181. [DOI] [PubMed] [Google Scholar]
- 39.Chang YY, Neufeld TP. Autophagy takes flight in Drosophila. FEBS Lett. 2010;584:1342–9. doi: 10.1016/j.febslet.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, et al. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007;9:218–24. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 41.Esclatine A, Chaumorcel M, Codogno P. Macroautophagy signaling and regulation. Curr Top Microbiol Immunol. 2009;335:33–70. doi: 10.1007/978-3-642-00302-8_2. [DOI] [PubMed] [Google Scholar]