

Abstract
Cells with aberrations in chromosomal ploidy are normally removed by apoptosis. However, aneuploid neurons have been shown to remain functional and active both in the cortex and in the retina. Lim1 horizontal progenitor cells in the chicken retina have a heterogenic final cell cycle, producing some cells that enter S-phase without proceeding into M-phase. The cells become heteroploid but do not undergo developmental cell death. This prompted us to investigate if the final cell cycle of these cells is under the regulation of an active DNA damage response. Our results show that the DNA damage response pathway, including γ-H2AX and Rad51 foci, is not triggered during any phase of the different final cell cycles of horizontal progenitor cells. However, chemically inducing DNA adducts or double-strand breaks in Lim1 horizontal progenitor cells activated the DNA damage response pathway, showing that the cells are capable of a functional response to DNA damage. Moreover, manipulation of the DNA damage response pathway during the final cell cycle using inhibitors of ATM/ATR, Chk1/2, and p38MAPK, neither induced apoptosis nor mitosis in the Lim1 horizontal progenitor cells. We conclude that the DNA damage response pathway is functional in the Lim1 horizontal progenitor cells, but that it is not directly involved in the regulation of the final cell cycle that gives rise to the heteroploid horizontal cell population.
Keywords: ATM/ATR, apoptosis, cell cycle regulation, cisplatin, DNA damage response, H2AX, programmed cell death, Rad51, retina
Introduction
Aberrations in chromosomal ploidy have recently been demonstrated in cortical and retinal neurons.1-3 The occurrence of aneuploidy correlates with periods of programmed cell death (PCD), and inhibition of key regulators of PCD leads to an increase in aneuploid neurons.4 As the nervous system matures, these aneuploid neurons seem to remain functional and active.5 Our recent results showing heteroploidy in chicken Lim1 expressing (+) retinal horizontal cells (HCs) are consistent with the presence of aneuploid neurons in the vertebrate central nervous system. The transcription factor, Lim1, is expressed by the H1 HC subtype and is used as a specific marker for these cells.2 PCD is part of neural development, from early embryonic proliferative stages until adult stages.6 The retina is not an exception; 2 waves of PCD occur in the developing retina, and depending on the species, the timing and magnitude of retinal cell death varies.7 The early wave overlaps with the period of neurogenesis and differentiation, and in the chicken retina this occurs between embryonic day (E) 4 and 7, and the late wave overlaps with the period of neurotrophic interactions between post-mitotic neurons and their targets (E10 to 14).7 The early wave of PCD has been suggested to remove cells with DNA damage caused by erroneous regulation of cell cycle or differentiation.8,9
Although a majority of the retinal cell types are affected by PCD, the HCs do not undergo apoptosis during development of the chicken retina.10,11 The absence of apoptosis in HCs could be due to that the HCs are spared from developmental cell cycle or differentiation errors or that they have a high ability to repair the damage and thus recover. It has been reported that the HCs are able to sustain persistent DNA damage. In the conditional Rb1-inactivated mouse retina, rapid degeneration of most retinal cells, except HCs, occurs. The phosphorylated form of histone H2AX (γ-H2AX), a marker of DNA damage, was present in the HCs, while they were able to remain in the cell cycle for an extended period of time and consequently survive for months.12 These observations, together with the heterogenic final cell cycle seen in Lim1+ HCs, prompted us to investigate if the DNA damage response pathway in Lim1 horizontal progenitor cells (HPCs) is active and contributes to the regulation of the final cell cycle. We have previously shown that the presence of heteroploid HCs is a result of S-phase progression without subsequent mitosis and not a result of a mitotic catastrophe.2 The omitted mitosis may be an effect of disturbances during replication or DNA damage, leading to a block in S/G2-phase transition. Stalled replication forks and/or DNA damage will trigger cell cycle checkpoint responses that arrest the cell cycle in order to promote repair. Stalled replication forks will trigger the activation of ataxia telangiectasia Rad-3-related protein (ATR), while DNA damage triggers ataxia telangiectasia mutated (ATM) and ATR. Both ATM and ATR are members of the phosphatidylinositol-3 kinase-like family and phosphorylate H2AX.13,14 ATM/ATR will, in turn, activate checkpoint kinase 1 (Chk1), checkpoint kinase 2 (Chk2), and/or stress-induced p38 mitogen-activated protein kinase (p38MAPK),15 leading to S/G2-phase arrest.16 The repair mechanisms during the S/G2-phases include activation of DNA-dependent protein kinase (DNA-PK)17 and recruitment of the recombinase Rad51.18 Rad51 mediates assembly of DNA damage repair proteins and DNA strand invasion of an intact sister-chromatid during homologous recombination repair of DNA double-strand breaks.18,19
Our results show that the DNA damage response pathway, including γ-H2AX and Rad51 foci is not triggered during the heterogenic final cell cycle of Lim1+ HPCs. However, chemically inducing DNA adducts or double-strand breaks in Lim1+ HPCs activated the DNA damage response pathway, showing that these cells have the capacity to respond to DNA damage. Furthermore, manipulation of the DNA damage response pathway during the final cell cycle of these cells neither induced apoptosis nor mitosis, indicating that the DNA damage response pathway is not directly involved in the regulation of the final cell cycle that gives rise to the heteroploid HC population.
Results
Lim1+ HPCs do not express γ-H2AX or Rad51
In our previous studies, we did not find any Lim1+ HPCs labeled for activated caspase-3 (C-casp-3) during their final cell cycle2 or for fragmented DNA using TdT-mediated-dUTP-nick-end-labeling (TUNEL),20 indicating that these cells do not undergo PCD. During the final cell cycle, up to 40% of the Lim1+ cells arrest during or after the S-phase; this may indicate that the cell cycle is regulated by the DNA damage response pathway. We studied early markers for the DNA damage and repair pathway, γ-H2AX and Rad51, by analyzing γ-H2AX, Lim1 double- and Rad51, Lim1 double-immunolabeling in wild-type chicken retina during the final cell cycle of Lim1+ HPCs (stage [st] 25 and st27). These stages can be used to differentiate between the final cell cycle behaviors of the Lim1+ HPCs. At st25, HPCs have a final cell cycle with an S-phase that will not proceed into mitosis. At st27, the Lim1+ HPCs with basal mitoses can be observed in the very center of the retina, and they were used as an internal control for Lim1+ cells that proceeded into mitoses after S-phase.2 Co-labeling of Lim1 and γ-H2AX could not be detected in the inspected Lim1+ cells (274 Lim1+ cells at st25 and 566 Lim1+ cells at st27). None of the investigated Lim1+ cells (109 Lim1+ cells at st25 and 182 Lim1+ cells at st27) were Rad51-positive. Nuclei were also inspected by confocal microscopy but Lim1+ cells were neither found to be γ-H2AX nor Rad51+ (data not shown). To distinguish the Lim1+ HPCs that had entered S-phase, we labeled cells with the thymidine analog EdU at st25. EdU was injected into the yolk, and the retinas were analyzed after 3 h. Lim1, EdU double-positive cells, which represent the HPCs that give rise to the heteroploid cells,2 were neither γ-H2AX nor Rad51+ (Fig. 1A and B). Labeling of γ-H2AX was observed in Lim1-negative cells, confirming that the immunohistochemical analysis was working, and that retinal progenitor cells exhibit γ-H2AX immunoreactivity during development.
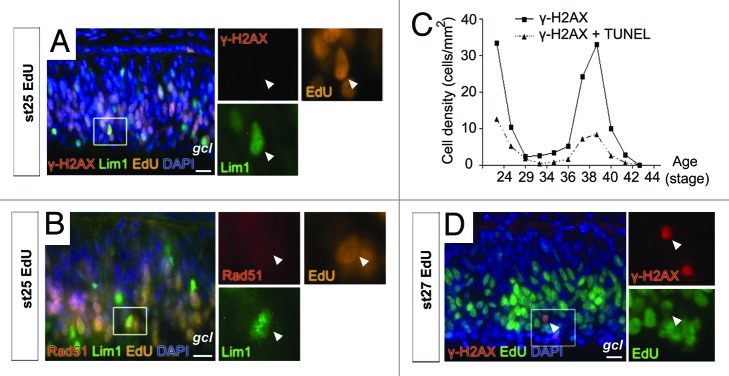
Figure 1. γ-H2AX and Rad51 in the developing horizontal progenitor cells. Fluorescence micrographs of phosphorylated histone H2AX (γ-H2AX), Lim1 and EdU (A), and Rad51, Lim1 and EdU (B) in st25 untreated retina. (C) Diagram summarizing the density of γ-H2AX+ and γ-H2AX, TUNEL double-positive cells in the central part of st22 to st44 retinas. (D) Fluorescence micrograph of γ-H2AX, EdU double-positive cells in the central part of st27 retina. Arrowhead, double-positive cell; gcl, ganglion cell layer; st, Hamburger and Hamilton stages. Scale bar is 10 µm.
H2AX is phosphorylated in the normal developing chicken retina
To compare the negative results observed for the Lim1+ population with the total retinal cell population, we analyzed the full series of st22 – 44 (E3.5 – 18) embryonic chicken retinas for γ-H2AX. The γ-H2AX labeling was compared with TUNEL, and the number of γ-H2AX+ and γ-H2AX, TUNEL double-positive cells in the central part of the retina were counted. A bimodal pattern was seen with peaks in the labeled cell density before st29 and at st38–40 (Fig. 1C). Overall, the number of γ-H2AX+ and γ-H2AX, TUNEL double-positive cells paralleled, and at every developmental time point, there were more γ-H2AX single-positive cells than γ-H2AX, TUNEL double-positive cells (Fig. 1C), suggesting that not all γ-H2AX+ cells develop DNA fragmentation and proceed into apoptosis. From st37, the majority of the γ-H2AX+ cells were seen in the inner nuclear layer and in the ganglion cell layer, consistent with the corresponding to PCD in post-mitotic retinal neurons.10,11 At st37 we noted γ-H2AX+ cells in the peripheral part of the retina, where proliferation still occurs at this stage (data not shown). The result was consistent with γ-H2AX being present in mitotic cells during the early phase (younger than st29, Fig. 1C).
To confirm that γ-H2AX was present in proliferating cells, the thymidine analog EdU was used to label cells in S-phase. Stage 25 and st27 embryos were injected with EdU and staining for EdU and γ-H2AX was performed after 3 h. γ-H2AX, EdU double-positive cells were seen at both st25 and 27 (Fig. 1D), and 14% of the γ-H2AX+ cells had incorporated EdU (102 γ-H2AX+ cells, n = 7). The presence of EdU, γ-H2AX double-positive cells indicated that the DNA damage response was active in cells during S-phase or in cells that had just left S-phase.
Lim1+ HPCs respond to DNA breaks with γ-H2AX, activated caspase-3, and Rad51 foci
The results suggested that Lim1+ and Lim1-negative cells may respond differently to signals that generate γ-H2AX. Whereas, some Lim1-negative cells have γ-H2AX during retinal development, the Lim1+ cells did not display γ-H2AX. We therefore tested if the Lim1+ HPCs had the capacity to respond with phosphorylation of H2AX and formation of Rad51 foci. We induced DNA damage by injecting the DNA damaging drugs neocarzinostatin or cisplatin. Neocarzinostatin is a radiomimetic drug that induces double-strand breaks, which activate ATM21 and triggers a robust activation of γ-H2AX.22 Cisplatin forms DNA adducts, primarily intra-strand crosslinks, which lead to replication fork stalling and ATR activation.23
We injected 20 ng neocarzinostatin intra-ocularly in ovo in st25 embryos and γ-H2AX immunoreactivity and TUNEL were analyzed. The injection gave a robust labeling for γ-H2AX after 30 min with few TUNEL cells (Fig. 2A). Interestingly, the γ-H2AX immunoreactivity was not evenly distributed over the retina. Cells on the basal side of the retina, in the prospective ganglion cell and inner nuclear layers, were intensely labeled, while cells on the apical side were not labeled (Fig. 2A). This indicated that retinal progenitor cells respond differently to the DNA damage-inducing agent. It should be noted that γ-H2AX immunoreactivity was seen at the outer limiting membrane of the retina and pigment epithelium (Fig. 2A and B), and staining was not associated with nuclei or DNA of retinal progenitor cells, and we considered it not to be relevant for the present analysis. A similar but weaker pattern was seen in the vehicle-treated retinas (Fig. 2C and D). Neocarzinostatin treatment for 2 h showed fewer γ-H2AX-stained cells, and in contrast to the 30 min time point, there was massive TUNEL staining in the retina (Fig. 2B). The number of γ-H2AX, TUNEL double-positive cells was higher at 2 h compared with 30 min (Fig. 2E). The cytotoxic effects of neocarzinostatin were also seen on the retinal structure after 2 h, and the retinal lamination was affected with initial signs of cell loss (Fig. 2B). Neocarzinostatin treatment for 30 min showed Lim1+ HPCs intensely labeled for γ-H2AX+ (Fig. 2F). Despite the cell loss following 2 h neocarzinostatin treatment, were some of the remaining Lim1+ HPCs γ-H2AX+ (Fig. 2G). Lim1, C-casp-3 double-positive cells were not seen after 30 min treatment (Fig. 2H). However, Lim1, C-casp-3 double-positive cells were seen after 2 h treatment (Fig. 2I). Weak Rad51 staining was seen in cells on the basal side of the retina (Fig. 2K, arrow) after 2 h treatment. The Lim1+ cells were Rad51-negative (Fig. 2J and K, arrowhead). The results showed that γ-H2AX and C-casp-3 can be induced in Lim1+ HPCs by DNA double-strand breaks.

Figure 2. γ-H2AX, cleaved caspase-3 and Rad51 foci in Lim1+ horizontal progenitor cells after DNA damage. Fluorescence micrographs of γ-H2AX, TUNEL double-positive cells in st25 retinas after (A and B) neocarzinostatin (NCS) or (C and D) vehicle intra-ocular in ovo treatment. (E) Bar graph summarizing the density of γ-H2AX and TUNEL cells after 30 min and 2 h treatment with neocarzinostatin or vehicle of st25 retina. γ-H2AX and Lim1 (F), cleaved caspase-3 (C-casp-3) and Lim1 (H) and Rad51 and Lim1 (J) in st25 retina 30 min after intra-ocular injection with 20 ng neocarzinostatin. γ-H2AX and Lim1 (G), C-casp-3 and Lim1 (I) and Rad51 and Lim1 (K) in st25 retina 2 h after intra-ocular injection with 20 ng neocarzinostatin. Fluorescence micrographs of γ-H2AX, Lim1 and EdU (L) and Rad51 foci, Lim1, and EdU (M) in in ovo st25 cisplatin- and EdU-treated retinas. Arrowhead, positive cell; arrow, Rad51-positive cell; gcl, ganglion cell layer; st, Hamburger and Hamilton stages. Scale bar is 10 µm.
Increased γ-H2AX and Rad51 in Lim1+ HPCs after cisplatin treatment
We induced DNA damage using the cytotoxic drug cisplatin, which produces a milder damage to DNA than neocarzinostatin. Cisplatin blocks replication and activates the DNA damage response pathway without directly triggering apoptosis.24 Cisplatin was injected intra-ocularly with EdU at st25 and γ-H2AX or Rad51 immunoreactivity was analyzed after 3 h. The EdU incorporation for 3 h allowed identification of cells that are in S-phase or have subsequently passed into G2-phase. Cisplatin induced γ-H2AX and generation of Rad51 foci both in Lim1+ and Lim1-negative cells (Fig. 2L and M), while clear Rad51 staining was seen in EdU+ cells (both Lim1+ and Lim1 negative cells). Rad51 staining was observed in some EdU-negative cells, indicating that they had been in G2-phase for longer time than the 3 h EdU pulse. The results are consistent with Rad51 being expressed during the S- and G2-phase of the cell cycle.18 We concluded that the Lim1+ HPCs have the capacity to respond to DNA damage with increased γ-H2AX and Rad51 foci-expression.
Inhibition of ATM with KU55933 does not promote M-phase entry
In the presence of DNA damage, mainly double-strand breaks, ATM becomes activated and phosphorylates downstream targets, including Chk1/2.25 The ATM-specific inhibitor KU5593326 has been shown to abrogate a G2-phase arrest caused by irradiation.27 To investigate if we are able to induce M-phase entry in the Lim1+ HPCs, we cultured st25 and st27 retinal explants in the presence of KU55933 or vehicle for 2 h. The dynamics and length of a normal mitosis in the retina indicated that an 2 h incubation was sufficient for an S/G2-phase arrested cell to progress into the M-phase.28 The M-phase entry was monitored by phosphorylation of histone 3 (PH3). We counted basal and apical mitoses separately in order to monitor HPCs (basal mitoses) and other retinal progenitor cells, which undergo interkinetic nuclear migration (apical mitoses). PH3+ cells were counted, and there was no difference between inhibitor- or vehicle-treated retinas either at st25 or st27 (Fig. 3A and B). The results suggest that the cell cycle progression of Lim1+ HPCs with or without basal mitosis were independent of active ATM kinase.

Figure 3. Effects on basal (HC) and apical mitoses in developing retina by inhibitors of ATM and ATR. Bar graphs summarizing the relative density of mitoses (PH3+ cells/mm2) in the central region of inhibitor-treated (dark gray bars) compared with control (vehicle, light gray bars) st25 and st27 retinas. Retinas were grown as whole retinal explants and treated with inhibitors for 2 h. The basal mitoses are terminally dividing HCs. Relative density of (A and C) basal (HCs) PH3+ and (B and D) apical PH3+ cells after treatment with (A and B) the ATM inhibitor KU55933, (C and D) the ATM/ATR inhibitor CGK733 compared with control (vehicle). (E) Bar graph showing the relative density of γ-H2AX, Lim1 double-positive cells in the central region of st25 retinal explants treated with CGK733 or vehicle followed by cisplatin. Fluorescence micrographs of γ-H2AX, Lim1 double-positive cells in st25 retinal explants after (F) vehicle and cisplatin or (G) CGK733 and cisplatin treatment. Arrowhead, double-positive cell; gcl, ganglion cell layer; st, Hamburger and Hamilton stages. Scale bar is 10 µm. Student t test, * P < 0.05, n ≥ 4 treated eyes, 4 sections per eye.
ATM/ATR inhibitor CGK733 does not promote M-phase entry
The DNA damage response activates both the ATM/ATR kinases, which, in turn, activate Chk1/2.25 Inactivation of only ATM may not be sufficient to abrogate an S/G2-phase arrest ,and we therefore used CGK733 that inhibit the activity of both ATM and ATR.29 Stage 25 and st27 retinal explants were treated for 2 h, and PH3+ cells were counted. There was no difference between the ATM/ATR inhibitor and vehicle-treated st25 or st27 retinas with regard to Lim1+ HPCs or apical mitoses (Fig. 3C and D). Shorter incubation times were tested, and the results were similar to the results from the longer incubation (data not shown). Phosphorlylation of H2AX is mediated by the kinases ATM/ATR and inhibition of ATM/ATR activity is known to reduce phosphorylation of H2AX.15 To control that the CGK733 treatment was effective, γ-H2AX+ cells were counted. The total number of γ-H2AX+ cells was lower (P < 0.05) with inhibitor compared with wild type (23 ± 5 vs. 48 ± 4 cells, n = 4). We also checked if sustained inhibition of ATM/ATR using CGK733 for 6 h on cultured st25 retinal explants had any effect on caspase-3 activation. Inhibition of ATM/ATR did not lead to activation of caspase-3 in Lim1+ cells (> 200 Lim1+ cells counted, n = 4) or in other cells (data not shown).
Inhibition of ATM/ATR with CGK733 in combination with cisplatin reduces γ-H2AX in Lim1+ HPCs
The specificity of CGK733 has been questioned.29-31 To verify the activity of CGK733 on the ATM/ATR response pathway, we exposed retinas to CGK733 followed by induction of DNA damage using cisplatin. Stage 25 retinal explants were cultured in the presence of CGK733 or vehicle for 2 h followed by administration of cisplatin for 2 h. The percentage of γ-H2AX, Lim1 double-positive cells was calculated and compared with vehicle treatment. A clear reduction of the percentage of Lim1, γ-H2AX double-positive cells was observed with CGK733, compared with vehicle (Fig. 3E–G), confirming that CGK733 reduces the generation of γ-H2AX+ cells in this system.
Inhibition of DNA-PK does neither induce apoptosis in progenitors nor mitosis in the Lim1+ HPCs
While ATM and ATR are involved in regulation of cell cycle progression following different types of DNA damage, DNA-PK is involved in repair of the damaged DNA by mediating non-homologous end-joining repair.32 In the mouse retina, repair of DNA breaks that occur during development are dependent on DNA-PK, and inhibition of DNA-PK with NU7026 increases caspase-dependent cell death.8 To investigate if DNA-PK has a role in the repair of developmental DNA breaks, we treated retinal explants with NU7026. Stage 25 retinas were cultured in the presence of NU7026 or vehicle for 6 h followed by analysis of C-casp-3 immunoreactivity. No increase of the number of C-casp-3+ cells was observed with the inhibitor compared with vehicle (13 vs. 12 cells, n = 2). To examine if the DNA-PK inhibitor induced M-phase entry in the Lim1+ HPCs, we cultured st25 retinal explants in the presence of NU7026 or vehicle for 2 h. PH3+ cells were counted, and there was no difference between the inhibitor and vehicle-treated st25 retinas with regard to Lim1+ HPCs or apical mitoses (data not shown). These results indicate that DNA-PK does not have a central role in survival or M-phase entry of Lim1+ HPCs.
Chk1 and Chk2 inhibitors does not promote M-phase entry
Activation of the ATM/ATR kinases upon DNA damage leads to activation of downstream substrates Chk1/2.25 Chk1 inhibits cdc25C by phosphorylation, rendering the CyclinB1–Cdk1 complex inactive, and the cell will be restrained from entering M-phase.15 We blocked the activity of Chk1 by using the Chk1 kinase inhibitor SB218078.33 Inhibition of Chk1 abrogates cell cycle arrest caused by DNA damage by forcing cells to enter M-phase.33 Retina explants from st25 and st27 embryos were incubated with Chk1 inhibitor SB218078 for 2 h. The number of Lim1, PH3 double-positive HPCs was similar after treatment with inhibitor or vehicle, indicating that Chk1 did not maintain the block in S/G2-phase transition in the HPCs (Fig. 4A). In contrast, an increase (P < 0.05) of cells entering apical mitoses at both stages after treatment with Chk1 inhibitor compared with vehicle was seen (Fig. 4B). This result indicated that the inhibitor treatment was effective in the retina explants, and that Chk1 influences the regulation of the cell cycle during interkinetic nuclear migration. The activity of Chk2, a downstream target of ATM/ATR, was blocked with Chk2 inhibitor NSC 109555. We incubated st25 and st27 retinal explants with the Chk2 inhibitor for 2 h and analyzed M-phase entry by PH3+ basal HPCs and PH3+ cells on the apical side of the retina. NSC 109555 treatment neither altered the number of basal PH3+ HPCs, nor the number of apical PH3+ cells (Fig. 4C and D).
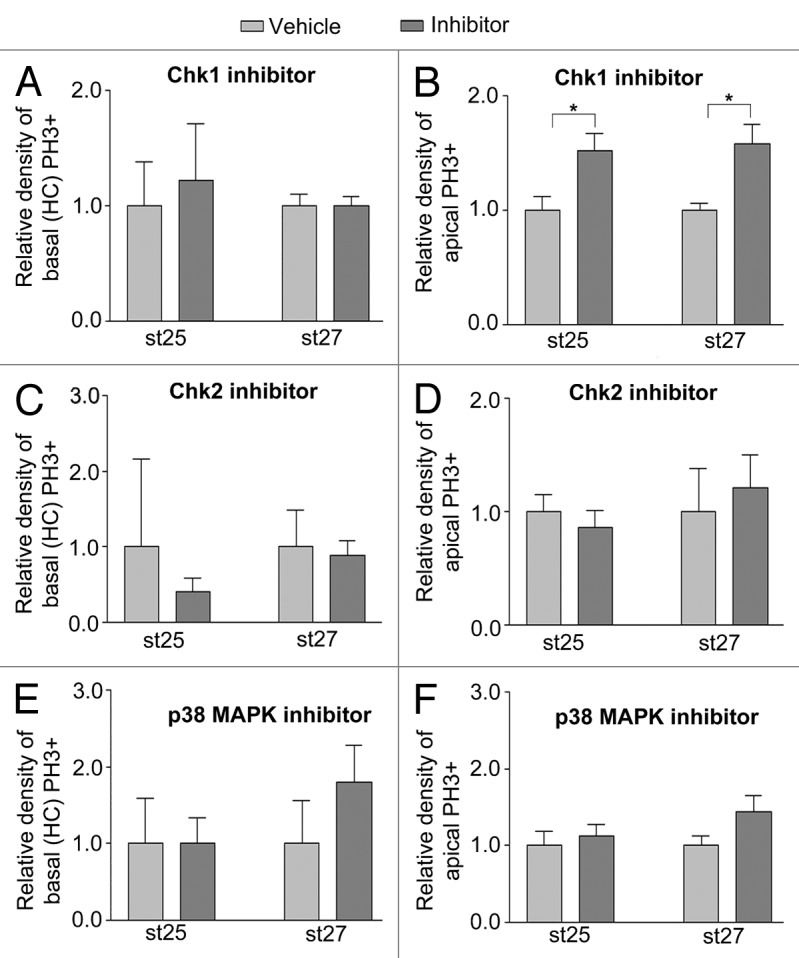
Figure 4. Effects on basal (HC) and apical mitoses in developing retina by inhibitors of Chk1, Chk2 and p38MAPK. Bar graphs summarizing the relative density of mitoses (PH3+ cells/mm2) in the central region of inhibitor-treated (dark gray bars) compared with control (vehicle, light gray bars) st25 and st27 retinas. Retinas were grown as whole retinal explants and treated with inhibitors for 2 h. The basal mitoses were done by terminally dividing HCs. Relative density of (A, C, and E) basal (HCs) PH3+ and (B, D, and F) apical PH3+ cells after treatment with (A and B) the Chk1 inhibitor SB 218078, (C and D) the Chk2 inhibitor NSC 109555 and (E and F) the p38MAPK inhibitor SB 239063 compared with control (vehicle). Note that the Chk1 inhibitor has effects on apical but not on the basal (HCs) mitoses. St, Hamburger and Hamilton stages. Student t test, * P < 0.05, n ≥ 4 treated eyes, 4 sections per eye.
p38MAPK inhibitor SB239063 does not promote M-phase entry
p38MAPK is a pivotal regulator of stress-induced pathways, and it transmits stress signals to the cell cycle by interacting with checkpoint kinase MK2,34 which phosphorylates and inactivates cdc25 family phosphatases, leading to inactivation of CyclinB1-Cdk1 and block in G2/M-phase transition.15,35 We incubated st25 and st27 retinal explants with the selective p38MAPK inhibitor SB23906336 for 2 h and analyzed M-phase entry by PH3+ basal HPCs and PH3+ cells on the apical side of the retina. SB239063 did not alter the number of basal PH3+ HPCs, nor was the number of apical PH3+ cells altered by the treatment (Fig. 4E and F). Activated p38MAPK can be detected by immunohistochemistry in chicken retinal neurons,37 and a positive control experiment with N-methyl-D-aspartate (NMDA)-treated retinas gave robust nuclear labeling for p38MAPK in cells (data not shown). We analyzed st25 and st27 retinas for p38MAPK but did not detect any immunoreactivity in the HPCs. These results argue against activation of p38MAPK in HPCs during this period of development.
Discussion
We have studied the DNA damage response during Lim1+ HC neurogenesis in the chicken retina. Our previous results show that Lim1+ HPCs are heterogenic with regards to when and during what cell cycle phase they leave the cell cycle. There are some Lim1+ HPCs that undergo an S-phase that is not followed by any mitosis. Such cells remain with a fully or partially replicated genome.2 Neurons with aneuploidy have been described as being functional and active and part of the normal organization of the mammalian brain.1,5 However, it is clear that cell death is the most common fate for aneuploid cells, both in the CNS and in other systems.4,38-40 We show that the Lim1+ HPCs, in spite of having a heterogenic final cell cycle, do not seem to be regulated by an active DNA damage response. However, the cells are capable of responding to DNA damage with phosphorylation of H2AX and subsequent apoptosis. In line with that, the DNA damage response is disengaged in the regulation of the final HPC cell cycle, inhibition of the DNA damage pathway regulators: ATM/ATR kinases, Chk1/2, or p38MAPK did not have any detectable effects on the final cell cycle behavior.
We initially hypothesized that the DNA damage response pathway would be activated during the heterogenic final cell cycle of the HPCs that gives rise to heteroploid cells. However, the Lim1+ cells showed no presence of γ-H2AX or Rad51 foci. In parallel, we analyzed cells during the course of retinal development for γ-H2AX and TUNEL. There were more γ-H2AX+ cells than γ-H2AX, TUNEL double-positive cells. However, the temporal distribution was similar (Fig. 1C), indicating that there is a robust number of cells in the normal developing retina with a triggered DNA damage response. These results are consistent with an active double-strand break repair being critical for retinal development, as shown by the dependence of the DNA damage response, including ATM, ATR, or DNA-PK, for the survival of newly formed retinal neurons.8 When DNA damage was inflicted, with cisplatin or neocarzinostatin, to the Lim1+ HPCs they presented γ-H2AX and Rad51 foci, showing that HPCs have the machinery and the capacity to trigger a DNA damage response. Interestingly, it seems that retinal progenitor cells have different propensity to respond with γ-H2AX or Rad51 foci to the DNA damaging drugs; however, additional studies are required to confirm this.
It has been demonstrated that CNS aneuploid cells are under strict regulation of caspase-mediated cell death. Removal of caspase-3 or caspase-9 increased the number, the range, and the form of aneuploid cells observed.4 Therefore, the presence of an aneuploid population in the developing cortex is due to reductions in caspase activation, and this observation may be related to the fact that at no time during retinal development have the HCs been shown to undergo developmental/programmed cell death11 (also our own observation). We show that the Lim1+ HPCs have the capacity to activate caspase-3 after DNA damage. However, further investigation is needed to clarify if the absence of apoptosis during the heterogenic final cell cycle in Lim1+ HPCs is related to an endogenous resistance to apoptosis.
Phosphorylation of H2AX is early and transient, and we analyzed downstream events in the DNA damage pathway by blocking ATM, ATR, DNA-PK, and Chk1/2 activity. The ATM/ATR kinases are part of the immediate response to DNA-damage and trigger S/G2-phase arrest while Chk1/2 prolongs the arrest.13,41 Inhibition of either of these kinases will result in M-phase entry even after artificial DNA damage.33,42 Treating retinal explants with ATM/ATR inhibitors or with Chk1/2 inhibitors, neither triggered apoptosis, nor increased the number of basal PH3+ HPCs at st25, as would have been expected if the DNA damage response was responsible for the S/G2-phase transition blockage. These results imply that the block in S/G2-phase transition was both ATM/ATR- and Chk1/2-independent. Unlike the mitoses by HPCs on the basal side of the retina, the number of apical mitoses increased after 2 h treatment with the Chk1 inhibitor, indicating that the cell cycle progression during interkinetic nuclear migration is to some degree under Chk1-kinase control. This is consistent with a recently identified Chk1-dependent S/G2-phase arrest in retinal progenitors during interkinetic nuclear migration. The Chk1-dependent S/G2-phase arrest was not induced by DNA damage; instead, it was under the influence of platelet-activating factor.43 The Chk1 activity has been proposed to have an essential role that cannot be substituted by Chk2.44,45
The absence of p38MAPK during the heterogenic final cell cycle of Lim1+ HPC further strengthens the results that the Lim1+ HPCs are not under stress while remaining in an S/G2-phase. The DNA damage response interferes with Cdc25, which is necessary for the CyclinB1–cdk1 complex to initiate mitosis.46 We have previously shown that there was no detectable Cdc25C immunoreativity in the Lim1+, PH3-negative HPCs. However, Cdc25C immunoreativity was detected in the Lim, PH3 double-positive HPCs, which are in M-phase or in the transition into M-phase.2 This indicated that cdc25C expression is low or even absent in the basal HPCs during the G2-phase, which is consistent with the Chk1/2- and p38MAPK-independent G2-phase in HPCs, since cdc25 phosphatases are the main targets and mediators of Chk1/2 or p38MAPK-MK2 actions on the cell cycle15 (Fig. 5).
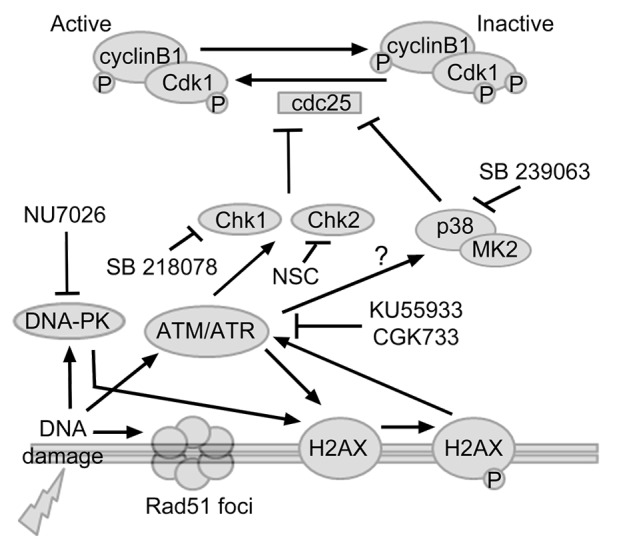
Figure 5. Schematic figure of the analyzed interactions that regulate the DNA damage response. DNA damage will trigger the activation of ATM, ATR and DNA-PK, which, in turn, phosphorylates H2AX. ATM/ATR will activate Chk1, Chk2, or p38MAPK, leading to S/G2-phase arrest. Chk1, Chk2, and p38MAPK inhibits cdc25C by phosphorylation, rendering the CyclinB1-Cdk1 complex inactive, and the cell will be restrained from entering M-phase. The repair mechanisms during S/G2-phase include DNA-PK and recruitment of the recombinase Rad51. Inhibiting ATM/ATR reduced the number of γ-H2AX immunoreactive Lim1+ HPCs. However, blocking the DNA damage response with ATM/ATR or DNA-PK inhibitors did not induce apoptosis. Furthermore, blocking ATM/ATR, DNA-PK, Chk1/2, or p38MAPK did not alter the number of Lim1+ HPCs cells entering M-phase. Our data indicate that the Lim1+ HPCs do not initiate the DNA damage response pathway during the heterogenic final cell cycle.
Regulation of proliferation is managed during the G1-phase, and this is associated with differentiation and cell cycle arrest. However, it is shown that neural stem and progenitor cells lack a G1 checkpoint when affected by DNA damage.47,48 Furthermore, the S/G2 cell cycle checkpoint is also known for its role in the maintenance of chromatin structural integrity. The S/G2 checkpoint can be induced in the absence of cytotoxic stress by overexpression of p53 and subsequent p21CIP/Waf1 inactivation of Cdks, but also by the ability of pRb1 to modulate the cell cycle through inhibition of the E2f transcription factors and the transcriptional repression of genes encoding cell cycle regulators.49 E2f family members have recently been discovered to switch from being activators in progenitor cells to repressors in differentiating cells.50 Moreover, the pRb-family members appear to be dispensable for normal cell cycling in many cells. They have roles in triggering permanent cell cycle exit particularly associated with differentiation and senescence,51 results that are consistent with the implied role for pRb during late G2-phase in assembly and stabilization of mitotic chromosomes at the entry into M-phase.52 We have previously demonstrated that pRb is present and functional during the heterogenic final cell cycle of Lim1+ HPCs. However, transcription of downstream gene CyclinB1 is absent.2 A more detailed analysis of the E2f transcription factors and the transcription of downstream genes encoding cell cycle regulators is required.
The results indicate that the heterogenic final cell cycle is not directly under the regulation of the DNA damage response pathway. The occurrence of heteroploid cells without triggering of apoptosis is of interest, since the HCs have been suggested to be potential “cell of origin” for retinoblastoma. HCs in the retina of Rb-family knockout mice defy signals to become post-mitotic and instead continue to proliferate and develop a malignant tumor.53 Our results suggest that the heterogeneity seen in the final cell cycle of chicken Lim1+ HPCs is independent of the DNA damage response pathway, and manipulation of the pathway does neither induce apoptosis nor M-phase entry. However, the DNA damage response pathway machinery is present and can be activated as shown by the γ-H2AX and Rad51 response when the HPCs were exposed to DNA damaging agents.
Materials and Methods
Animals and immunohistochemistry
Fertilized white Leghorn eggs (Gallus gallus) were obtained from Ova Production AB and incubated at 38 °C in a humidified incubator. Embryos were classified into st according to Hamburger and Hamilton.54
The Lim1+ HPCs exhibit 3 different behaviors during their development. They are generated by: (1) an interkinetic nuclear migration with an apical mitosis (between st19–31); (2) by a final cell cycle with an S-phase that is not followed by any mitosis (between st19–31), such cells remain with a replicated genome; or (3) by non-apical (basal) mitoses (between st26–31).2 We have used 2 developmental stages for our experiments, either st25 (before the first basal mitoses) or st27 (period with basal mitoses). The st25 and st27 correspond to E4.5 and 5, respectively.
Immunohistochemistry was performed as described previously.55 Tissue was fixed in 4% paraformaldehyde in PBS. The following antibodies were used: the transcription factor Lim1/2 (1:20, mouse, 4F2 s, developmental studies hybridoma bank [DSHB]), phospho-histone 3 (PH3) (1:4000, rabbit, 06–570, Millipore; 1:400, goat, sc-12927, Santa Cruz), γ-H2AX (1:4000, rabbit, ab11174, Abcam), Rad51 (1:4000, rabbit, ab63801, Abcam), Caspase-3, cleaved (1:4000, rabbit, #9661, Cell Signaling), and Phospho-p38 MAPK (1:500, rabbit, #4511, Cell Signaling). Secondary antibodies were obtained from Invitrogen. Samples were analyzed using a Zeiss Axioplan 2 microscope or a Zeiss LSM 510 confocal microscope, equipped with an AxioCam C camera and Axiovision software. Images were formatted, resized, enhanced, and arranged using Axiovision and Adobe Photoshop CS4.
5-ethynyl-20-deoxyuridine (EdU) injections
Fifty microgram of the thymidine analog EdU56 (Click iT EdU imaging kit C10084, Invitrogen) was injected into the yolk of st25 or st27 embryos to visualize cells in S-phase. Three hours after the injection, the eyes were collected and analyzed using immunohistochemistry. The time was selected to be long enough to allow a substantial number of cells to be labeled and short enough to not label 2 subsequent S-phases by the same cell.
Neocarzinostatin injections and TUNEL
Stage 25 eyes were injected with 1 µl (20 ng/µl) neocarzinostatin (N9162, Sigma-Aldrich) (Table 1) followed by either a 30 min or a 2 h incubation prior to analysis.21 The retinas were treated according to the immunohistochemical protocol. First they were incubated with anti-γ-H2AX antibody, as an indicator of double-strand DNA breaks, followed by TUNEL (TdT-mediated-dUTP-nick-end-labeling, G3250, Promega) to visualize DNA fragmentation, which indicates apoptotic cell death according to the manufacturer’s protocol. The number of γ-H2AX+ and γ-H2AX, TUNEL double-positive cells were counted in the central part of the retina and the cell density (cells/mm2) was calculated.
Table 1. The chemicals used in the study and their modes of action.
| Target | Chemical | Action |
|---|---|---|
| ATM | KU55933 | Small-molecule inhibitor26 |
| ATM/ATR | CGK733 | Kinase inhibitor29 |
| Chk1 | SB 218078 | Kinase inhibitor; ATP-competitive inhibitor of Chk133 |
| Chk2 | NSC 109555 ditosylate | Selective, reversible, ATP-competitive Chk2 inhibitor57 |
| DNA | Cisplatin | DNA adduct formation and blockage in DNA synthesis23 |
| DNA | Neocarzinostatin | Induces double-strand breaks21 |
| DNA-PK | NU7026 | Competitive selective inhibitor58 |
| p38MAPK | SB 239063 | Binds to p38MAPK blocking catalytic activity36 |
Analysis of γ-H2AX and TUNEL
For the γ-H2AX series with or without neocarzinostatin, 4 sections per eye from 2 embryos per treatment and antibody combination were used. Only the central part of the retina was analyzed to avoid bias imposed by the temporal and centro-peripherial aspects of retinal development.55
Cisplatin and EdU injections
Cisplatin (2251, Tocris) induces blockage in DNA synthesis and activation of the DNA damage response pathway.23 Stage 25 eyes were injected in ovo with 1 µl (5 mM) cisplatin simultaneously as 50 µg of EdU was injected into the yolk to visualize cells in S-phase. Three hours after the injections, the eyes were collected and analyzed using immunohistochemistry.
Whole retinal explants
Eyes from st25 and st27 embryos were dissected in 37 °C PBS. The pigment epithelium was removed, leaving the lens and the whole neuronal retina attached to the vitreous body. The eyes were cultured at 37 °C in 35-mm dishes on a rotator shaker with a constant speed of 50 rpm inside an incubator with 5% CO2. The retinas were cultured 90 min before adding the inhibitors or vehicle. Medium was 1:1 DMEM:F12 Nutrient mix, 10% FCS, 10 U/ml penicillin streptomycin, 5 µg/ml insulin, and 2 mM l-glutamin. The control eye and the treated eye were dissected from the same embryo. After treatment for 2–6 h (dependent on the experiment) the eyes were analyzed by immunohistochemistry.
Seven different chemicals were administered to the retinal explants (Table 1). Chemicals were resuspended in either DMSO or EtOH and their final concentration in the medium is given. KU55933 (3544, Tocris), a small-molecule inhibitor of ATM, was uses at a concentration of 10 µM in 0.1% DMSO. CGK733 (2639, Tocris), a selective inhibitor of ATM and ATR kinases, was used at a concentration of 10 µM in 0.4% DMSO. An experiment with a combination of CGK733 and cisplatin (2251, Tocris) was conducted where retinal explants were cultured either in the presence of CGK733 at 10 µM or vehicle for 2 h, followed by cisplatin at 0.5 mM for 2 h in 1% DMSO. NU7026 (2828, Tocris), a competitive and specific inhibitor of DNA-PK, was used at a concentration of 10 µM in 0.1% DMSO. SB 218078 (2560, Tocris), an inhibitor of Chk1, was used at a concentration of 1 µM in 0.09% DMSO. NSC 109555 ditosylate (3034, Tocris), a selective ATP-competitive Chk2 inhibitor, was used at a concentration of 0.5 µM in <0.01% DMSO. SB 239063 (1962, Tocris), a p38MAP kinase inhibitor, was used at a concentration of 20 µM in 0.1% DMSO. The concentration of DMSO and EtOH in the vehicle treated controls was always identical to the experimental eye.
NMDA control experiment
Eyes were injected with 0.3 μg of NMDA (M3262, Sigma-Aldrich) in sterile saline (0.15 M NaCl) and analyzed 24 h later. Activation of p38MAP kinase was investigated using immunohistochemistry.
Quantification of mitoses
Cells in late G2/M-phase were identified using a PH3 antibody and at least 4 sections per eye from 4 different embryos per treatment and antibody combination were used for cell counting. Only the central part of the retina was analyzed to avoid bias imposed by the temporal and centro-peripherial aspects of retinal development. Both ventricular (apical) and vitreal (basal) mitotic cells were counted. The mean percentage (+/− SD) for each combination of labeling and stage was calculated and the data analyzed in GraphPad Prism (v3.02, GraphPad software Inc.). Analysis of variance was done with Student t test, and statistical significance was set to P < 0.05.
Ethics statement
This study was performed in accordance with the recommendations in the “Guide for the Care and Use of Laboratory Animals of the Association for research in vision and ophthalmology”. The protocol was approved by the Committee on the Ethics of Animal Experiments by Uppsala djurförsöksetiska nämnd (Permit number C14/9).
Acknowledgments
We thank Florian Frohns for thoughtful comments and discussion. The work was supported by Barncancerfonden (PROJ09/038), Swedish Research Council (20859-01-3, 12187-15-3), Ögonfonden, Kronprinsessan Margaretas arbetsnämnd för synskadade, Synfrämjandets forskningsfond, and St Eriks Ögonsjukhus forskningsstipendier. Authors declare no conflict of interest. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Glossary
Abbreviations:
- ATM
ataxia telangiectasia mutated
- ATR
ataxia telangiectasia Rad-3 related protein
- C-casp-3
cleaved caspase 3
- Chk1
checkpoint kinase 1
- Chk2
checkpoint kinase 2
- DNA-PK
DNA-dependent protein kinase
- E
Embryonic day
- γ-H2AX
phosphorylated histone H2AX
- HCs
horizontal cells
- HPCs
horizontal progenitor cells
- p38MAPK
p38 mitogen-activated protein kinase
- PCD
programmed cell death
- PH3
phospho-histone 3
- st
stage
- TUNEL
TdT-mediated-dUTP-nick-end-labeling
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27200
References
- 1.Rehen SK, McConnell MJ, Kaushal D, Kingsbury MA, Yang AH, Chun J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc Natl Acad Sci U S A. 2001;98:13361–6. doi: 10.1073/pnas.231487398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shirazi Fard S, Jarrin M, Boije H, Fillon V, All-Eriksson C, Hallböök F. Heterogenic final cell cycle by chicken retinal Lim1 horizontal progenitor cells leads to heteroploid cells with a remaining replicated genome. PLoS One. 2013;8:e59133. doi: 10.1371/journal.pone.0059133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morillo SM, Escoll P, de la Hera A, Frade JM. Somatic tetraploidy in specific chick retinal ganglion cells induced by nerve growth factor. Proc Natl Acad Sci U S A. 2010;107:109–14. doi: 10.1073/pnas.0906121107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterson SE, Yang AH, Bushman DM, Westra JW, Yung YC, Barral S, Mutoh T, Rehen SK, Chun J. Aneuploid cells are differentially susceptible to caspase-mediated death during embryonic cerebral cortical development. J Neurosci. 2012;32:16213–22. doi: 10.1523/JNEUROSCI.3706-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kingsbury MA, Friedman B, McConnell MJ, Rehen SK, Yang AH, Kaushal D, Chun J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc Natl Acad Sci U S A. 2005;102:6143–7. doi: 10.1073/pnas.0408171102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boya P, de la Rosa EJ. Cell death in early neural life. Birth Defects Res C Embryo Today. 2005;75:281–93. doi: 10.1002/bdrc.20054. [DOI] [PubMed] [Google Scholar]
- 7.Vecino E, Hernández M, García M. Cell death in the developing vertebrate retina. Int J Dev Biol. 2004;48:965–74. doi: 10.1387/ijdb.041891ev. [DOI] [PubMed] [Google Scholar]
- 8.Baleriola J, Suárez T, de la Rosa EJ. DNA-PK promotes the survival of young neurons in the embryonic mouse retina. Cell Death Differ. 2010;17:1697–706. doi: 10.1038/cdd.2010.46. [DOI] [PubMed] [Google Scholar]
- 9.de la Rosa EJ, de Pablo F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci. 2000;23:454–8. doi: 10.1016/S0166-2236(00)01628-3. [DOI] [PubMed] [Google Scholar]
- 10.Karlsson M, Mayordomo R, Reichardt LF, Catsicas S, Karten H, Hallböök F. Nerve growth factor is expressed by postmitotic avian retinal horizontal cells and supports their survival during development in an autocrine mode of action. Development. 2001;128:471–9. doi: 10.1242/dev.128.4.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cook B, Portera-Cailliau C, Adler R. Developmental neuronal death is not a universal phenomenon among cell types in the chick embryo retina. J Comp Neurol. 1998;396:12–9. doi: 10.1002/(SICI)1096-9861(19980622)396:1<12::AID-CNE2>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 12.Donovan SL, Corbo JC. Retinal horizontal cells lacking Rb1 sustain persistent DNA damage and survive as polyploid giant cells. Mol Biol Cell. 2012;23:4362–72. doi: 10.1091/mbc.E12-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 14.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 15.Reinhardt HC, Yaffe MB. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr Opin Cell Biol. 2009;21:245–55. doi: 10.1016/j.ceb.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–27. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Park EJ, Chan DW, Park JH, Oettinger MA, Kwon J. DNA-PK is activated by nucleosomes and phosphorylates H2AX within the nucleosomes in an acetylation-dependent manner. Nucleic Acids Res. 2003;31:6819–27. doi: 10.1093/nar/gkg921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Henning W, Stürzbecher HW. Homologous recombination and cell cycle checkpoints: Rad51 in tumour progression and therapy resistance. Toxicology. 2003;193:91–109. doi: 10.1016/S0300-483X(03)00291-9. [DOI] [PubMed] [Google Scholar]
- 19.West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4:435–45. doi: 10.1038/nrm1127. [DOI] [PubMed] [Google Scholar]
- 20.Edqvist PH, Lek M, Boije H, Lindbäck SM, Hallböök F. Axon-bearing and axon-less horizontal cell subtypes are generated consecutively during chick retinal development from progenitors that are sensitive to follistatin. BMC Dev Biol. 2008;8:46. doi: 10.1186/1471-213X-8-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674–7. doi: 10.1126/science.281.5383.1674. [DOI] [PubMed] [Google Scholar]
- 22.Yuan SS, Yang YK, Chen HW, Chung YF, Chang HL, Su JH. Neocarzinostatin-induced Rad51 nuclear focus formation is cell cycle regulated and aberrant in AT cells. Toxicol Appl Pharmacol. 2003;192:231–6. doi: 10.1016/S0041-008X(03)00013-9. [DOI] [PubMed] [Google Scholar]
- 23.Siddik ZH. Cisplatin: mode of cytotoxic action and molecular basis of resistance. Oncogene. 2003;22:7265–79. doi: 10.1038/sj.onc.1206933. [DOI] [PubMed] [Google Scholar]
- 24.Jordan P, Carmo-Fonseca M. Molecular mechanisms involved in cisplatin cytotoxicity. Cell Mol Life Sci. 2000;57:1229–35. doi: 10.1007/PL00000762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walworth NC, Bernards R. rad-dependent response of the chk1-encoded protein kinase at the DNA damage checkpoint. Science. 1996;271:353–6. doi: 10.1126/science.271.5247.353. [DOI] [PubMed] [Google Scholar]
- 26.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 27.Momcilović O, Choi S, Varum S, Bakkenist C, Schatten G, Navara C. Ionizing radiation induces ataxia telangiectasia mutated-dependent checkpoint signaling and G(2) but not G(1) cell cycle arrest in pluripotent human embryonic stem cells. Stem Cells. 2009;27:1822–35. doi: 10.1002/stem.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alexiades MR, Cepko C. Quantitative analysis of proliferation and cell cycle length during development of the rat retina. Dev Dyn. 1996;205:293–307. doi: 10.1002/(SICI)1097-0177(199603)205:3<293::AID-AJA9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 29.Won J, Kim M, Kim N, Ahn JH, Lee WG, Kim SS, Chang KY, Yi YW, Kim TK. Small molecule-based reversible reprogramming of cellular lifespan. Nat Chem Biol. 2006;2:369–74. doi: 10.1038/nchembio800. [DOI] [PubMed] [Google Scholar]
- 30.Won J, Kim M, Kim N, Ahn JH, Lee WG, Kim SS, Chang KY, Yi YW. Retraction: small molecule-based reversible reprogramming of cellular lifespan. Nat Chem Biol. 2008;4:431. doi: 10.1038/nchembio0708-431. [DOI] [PubMed] [Google Scholar]
- 31.Choi S, Toledo LI, Fernandez-Capetillo O, Bakkenist CJ. CGK733 does not inhibit ATM or ATR kinase activity in H460 human lung cancer cells. DNA Repair (Amst) 2011;10:1000–1, author reply 1002. doi: 10.1016/j.dnarep.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008;18:114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 33.Jackson JR, Gilmartin A, Imburgia C, Winkler JD, Marshall LA, Roshak A. An indolocarbazole inhibitor of human checkpoint kinase (Chk1) abrogates cell cycle arrest caused by DNA damage. Cancer Res. 2000;60:566–72. [PubMed] [Google Scholar]
- 34.Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007;11:175–89. doi: 10.1016/j.ccr.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rieder CL. Mitosis in vertebrates: the G2/M and M/A transitions and their associated checkpoints. Chromosome Res. 2011;19:291–306. doi: 10.1007/s10577-010-9178-z. [DOI] [PubMed] [Google Scholar]
- 36.Underwood DC, Osborn RR, Kotzer CJ, Adams JL, Lee JC, Webb EF, Carpenter DC, Bochnowicz S, Thomas HC, Hay DW, et al. SB 239063, a potent p38 MAP kinase inhibitor, reduces inflammatory cytokine production, airways eosinophil infiltration, and persistence. J Pharmacol Exp Ther. 2000;293:281–8. [PubMed] [Google Scholar]
- 37.Fischer AJ, Scott MA, Ritchey ER, Sherwood P. Mitogen-activated protein kinase-signaling regulates the ability of Müller glia to proliferate and protect retinal neurons against excitotoxicity. Glia. 2009;57:1538–52. doi: 10.1002/glia.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Voullaire L, Slater H, Williamson R, Wilton L. Chromosome analysis of blastomeres from human embryos by using comparative genomic hybridization. Hum Genet. 2000;106:210–7. doi: 10.1007/s004390051030. [DOI] [PubMed] [Google Scholar]
- 39.Zupanc GK, Wellbrock UM, Sîrbulescu RF, Rajendran RS. Generation, long-term persistence, and neuronal differentiation of cells with nuclear aberrations in the adult zebrafish brain. Neuroscience. 2009;159:1338–48. doi: 10.1016/j.neuroscience.2009.02.014. [DOI] [PubMed] [Google Scholar]
- 40.Rajendran RS, Wellbrock UM, Zupanc GK. Apoptotic cell death, long-term persistence, and neuronal differentiation of aneuploid cells generated in the adult brain of teleost fish. Dev Neurobiol. 2008;68:1257–68. doi: 10.1002/dneu.20656. [DOI] [PubMed] [Google Scholar]
- 41.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–96. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 42.Gogineni VR, Nalla AK, Gupta R, Dinh DH, Klopfenstein JD, Rao JS. Chk2-mediated G2/M cell cycle arrest maintains radiation resistance in malignant meningioma cells. Cancer Lett. 2011;313:64–75. doi: 10.1016/j.canlet.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fragel-Madeira L, Meletti T, Mariante RM, Monteiro RQ, Einicker-Lamas M, Bernardo RR, Lopes AH, Linden R. Platelet activating factor blocks interkinetic nuclear migration in retinal progenitors through an arrest of the cell cycle at the S/G2 transition. PLoS One. 2011;6:e16058. doi: 10.1371/journal.pone.0016058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–9. doi: 10.1016/S1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- 45.Stark GR, Taylor WR. Control of the G2/M transition. Mol Biotechnol. 2006;32:227–48. doi: 10.1385/MB:32:3:227. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez Y, Wong C, Thoma RS, Richman R, Wu Z, Piwnica-Worms H, Elledge SJ. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497–501. doi: 10.1126/science.277.5331.1497. [DOI] [PubMed] [Google Scholar]
- 47.Burdon T, Smith A, Savatier P. Signalling, cell cycle and pluripotency in embryonic stem cells. Trends Cell Biol. 2002;12:432–8. doi: 10.1016/S0962-8924(02)02352-8. [DOI] [PubMed] [Google Scholar]
- 48.Roque T, Haton C, Etienne O, Chicheportiche A, Rousseau L, Martin L, Mouthon MA, Boussin FD. Lack of a p21waf1/cip -dependent G1/S checkpoint in neural stem and progenitor cells after DNA damage in vivo. Stem Cells. 2012;30:537–47. doi: 10.1002/stem.1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci U S A. 1995;92:8493–7. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chong JL, Wenzel PL, Sáenz-Robles MT, Nair V, Ferrey A, Hagan JP, Gomez YM, Sharma N, Chen HZ, Ouseph M, et al. E2f1-3 switch from activators in progenitor cells to repressors in differentiating cells. Nature. 2009;462:930–4. doi: 10.1038/nature08677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466:1110–4. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 2010;24:1377–88. doi: 10.1101/gad.580710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ajioka I, Martins RA, Bayazitov IT, Donovan S, Johnson DA, Frase S, Cicero SA, Boyd K, Zakharenko SS, Dyer MA. Differentiated horizontal interneurons clonally expand to form metastatic retinoblastoma in mice. Cell. 2007;131:378–90. doi: 10.1016/j.cell.2007.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hamburger V, Hamilton HL. A series of normal stages in the development of the chick embryo. J Morphol. 1951;88:49–92. doi: 10.1002/jmor.1050880104. [DOI] [PubMed] [Google Scholar]
- 55.Edqvist PH, Hallböök F. Newborn horizontal cells migrate bi-directionally across the neuroepithelium during retinal development. Development. 2004;131:1343–51. doi: 10.1242/dev.01018. [DOI] [PubMed] [Google Scholar]
- 56.Buck SB, Bradford J, Gee KR, Agnew BJ, Clarke ST, Salic A. Detection of S-phase cell cycle progression using 5-ethynyl-2′-deoxyuridine incorporation with click chemistry, an alternative to using 5-bromo-2′-deoxyuridine antibodies. Biotechniques. 2008;44:927–9. doi: 10.2144/000112812. [DOI] [PubMed] [Google Scholar]
- 57.Lountos GT, Tropea JE, Zhang D, Jobson AG, Pommier Y, Shoemaker RH, Waugh DS. Crystal structure of checkpoint kinase 2 in complex with NSC 109555, a potent and selective inhibitor. Protein Sci. 2009;18:92–100. doi: 10.1002/pro.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA, Durkacz BW. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood. 2004;103:4659–65. doi: 10.1182/blood-2003-07-2527. [DOI] [PubMed] [Google Scholar]