

Abstract
β-glucogallin (BGG), a major component of the Emblica officinalis medicinal plant, is a potent and selective inhibitor of aldose-reductase (AKR1B1). New linkages (ether/triazole/amide) were introduced via high yielding, efficient syntheses to replace the labile ester, and an original 2-step (90%) preparation of BGG was developed. Inhibition of AKR1B1was assessed in vitro and using transgenic lens organ cultures, which identified the amide linked glucoside (BGA) as a stable, potent and selective lead therapeutic toward the treatment of diabetic eye disease.
Introduction
Human aldose reductase (AKR1B1) is a member of the aldo-keto reductase superfamily, which consists of 15 families, 3 of which are mammalian containing the thirteen human aldo-keto reductase enzymes currently identified.1 AKR1B1 functions in the polyol pathway as an NADPH-dependent enzyme, catalyzing the reduction of glucose to sorbitol, which is then converted to fructose by sorbitol dehydrogenase.2 The increased reduction of glucose to sorbitol under hyperglycemic conditions has been implicated in tissue injury and the progression of a wide variety of diabetic complications, including neuropathy and retinopathy.3, 4 Inhibition of AKR1B1 has been shown to both prevent and reverse diabetic tissue injury that arises from the accumulation of sorbitol.3, 5–7
Diabetes Mellitus has become a pandemic affecting both affluent countries and the developing world, with prevalence expected to double by 2030.8 Currently, there is no medical treatment that prevents the onset and progression of diabetic eye diseases like cataracts and retinopathy, which account for the majority of vision loss in diabetics.9 Surgical procedures for diabetic eye diseases are expensive and diabetic patients have significantly higher complication rates.10
In general, aldose reductase inhibitors (ARIs) developed to target AKR1B1 are non-selective and inhibit other members of the aldo-keto reductase superfamily such as AKR1B10 (small intestine reductase) and AKR1A1 (aldehyde reductase), which may contribute to toxicity and adverse effects.1 Despite the failure of ARIs such as sorbinil, zopalrestat, and tolrestat in clinical trials11, the role of AKR1B1 in diabetic tissue damage has been thoroughly substantiated.12–14 Thus, the discovery of selective AKR1B1 inhibitors that can both prevent and reverse complications of diabetes remains of paramount clinical importance.
Our previous research identified 1-O-galloyl-β-D-glucose (β-glucogallin or BGG or 1) shown in Scheme 1, purified from Indian gooseberry (Emblica officinalis), as a non-cytotoxic, selective, and relatively potent AKR1B1 inhibitor that reduces sorbitol accumulation in vitro and in organ culture assays of transgenic mouse lenses.15, 16 Thus, BGG is a viable lead compound to develop novel therapies for inflammatory diseases, particularly diabetic eye disease.
Scheme 1.
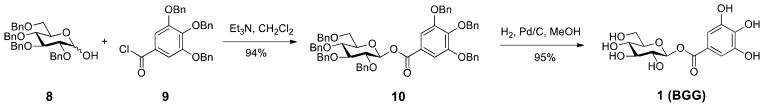
The Synthesis of β-Glucogallin (BGG), 1
BGG belongs to one of the simplest classes of hydrolyzable tannins, the gallotannins, and consists of a polyphenol monomer (gallic acid) linked to a β-D-glucose ring by an ester functionality. During our biological evaluation of BGG we observed that the glycosyl-1-ester is labile in aqueous solution. Therefore, our initial goal in developing novel inhibitors of AKR1B1, based on the BGG pharmacophore, was to design an optimal stable linkage between the sugar moiety and the gallate ring while maintaining or improving potency and specificity for AKR1B1 over other aldo-keto reductases. Using this rationale, new linkages between the sugar moiety and the gallate ring were introduced to replace the labile ester, including: ether, triazol, and amide functional groups. High yielding efficient syntheses were developed to prepare BGG derivatives, including an original 2-step ~90% yield preparation of BGG (Scheme 1).17 Derivatives were compared to BGG for their ability to inhibit AKR1B1 using recombinant enzyme, cell-based, and ex vivo lens organ cultures.
Results and Discussion
Chemistry
The first modification entailed the bioisosteric replacement of the ester with an amide linkage. A PMe3-mediated Staudinger reaction with glucosyl azide and benzoyl chloride resulted in the formation of N-glycosylamide,18 which was smoothly converted to the β-Glucogallin N-glycoside amide (BGA or 2) by exposure to NaOMe followed by debenzylation (Scheme 2, 3 steps, 79% yield). The amide linkage was confirmed by HMBC 2D NMR spectroscopy, which proved connectivity with correlations between the amide carbonyl, the sugar moiety, and the gallate ring (Supporting Information).
Scheme 2.
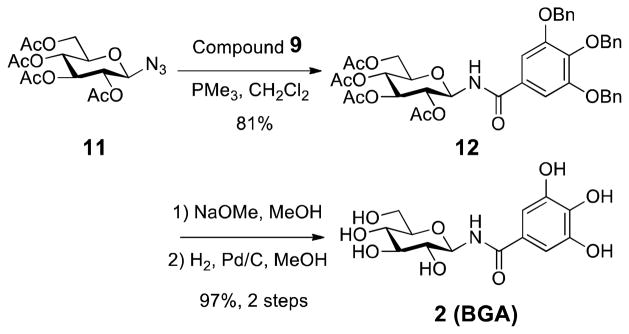
The Synthesis of β-Glucogallin amide BGA, 2
We then replaced the ester/amide functionality with a triazole linkage 3, which mimics amide functionality but would not be substrates for proteases in vivo and thus may be potentially much more stable. Coupling of substituted phenylacetylene 13 and glucosyl azide 11 using click chemistry19 generated 3 in greater than 86% yield overall after deprotection (Scheme 3).
Scheme 3.

The Synthesis of Triazole, 3
In addition, a vast majority of biologically and therapeutically active carbohydrates exist as monosaccharide units joined via glycosidic bonds.20 Hence, we set our sights on glycoside BGG derivatives where we varied the carbon tether length. We first attempted to prepare the phenolic ether (no carbon) and benzyl type (1-carbon) glycosides but quickly dismissed these linkages, due to their instability at room temperature. However, glycosides 4 (2-carbon) and 5 (3-carbon) were stable and were prepared accordingly. Silver carbonate promoted Koenigs-Knorr coupling of glucosyl bromide 16 and respective alcohol acceptor,21 followed by the removal of acetyl and benzyl ether protecting groups, led to the formation of β-glycosides (Scheme 4A and B). In addition, we prepared 6, which contains a mixed ether and triazole linkage. Starting from readily available benzyl azide 20, triazole 6 was assembled in three convenient steps using copper-catalysed azide–alkyne cycloaddition reaction described above (Scheme 4C). Importantly, the anomeric configuration of the glucoside products were readily assigned by the NMR coupling constant (~7–8 Hz) between H-1 and H-2 based on the Karplus equation.22
Scheme 4.

The Synthesis of Glucosides 4, 5, and 6
Biological Evaluation and Stability Studies
Previously, we have shown that BGG is a potent and selective inhibitor of AKR1B1 in vitro and in ex vivo lens organ cultures.16 BGG appears to act via non-competitive inhibition through binding the active site of AKR1B1, and occupies both the “anionic” and the “specificity” pockets.23 This non-competitive inhibition and active site binding is not uncommon and has been reported for many AKR1B1 inhibitors.24–26 Thus, BGG derivatives were first assessed using enzyme inhibition studies with recombinant human AKR1B1 in the presence of the natural substrate glyceraldehyde (Figure 1). Interestingly, the N-glycoside BGA was the only derivative that maintained inhibitory activity against AKR1B1 (Figure 1A). In fact, BGA potency (IC50 = 9 μM) was virtually identical to that of BGG (IC50 = 8 μM). BGA also replicated BGG in specificity for AKR1B1 over AKR1B10 and AKR1A1 (Figure 1B). Based on these results it appears that by extending the linker chain length by just one carbon (e.g. 2 carbon ether) compared to BGG or BGA, all AKR1B1 inhibitor activity is lost.
Figure 1.

Aldose reductase inhibitor studies, including: (A) Compounds 3–7 show no inhibitory activity, and the IC50 of BGG and BGA were determined to be 8±1 μM and 9±2 μM, respectively, in the presence of the natural substrate glyceraldehyde; and (B) BGG and BGA showed no activity against AKR1B10 and AKR1A1.
However, the loss of activity for the ether or triazole derivatives 3–6 may also be due to the absence of the carbonyl group and resulting electronic effect. To further investigate the role of the carbonyl functionality, we prepared amide 7, a BGA analogue, which contains an extra carbon between the carbonyl group and the gallate ring. In a similar approach, N-phenylacetyl β-D-glucopyranosylamine 7 was constructed in 3 steps from acid 23 and azide 11 (Scheme 4). The loss of activity for amide 7 (Figure 1A) indicates the impact of linker chain length on AKR1B1 enzyme inhibition. Thus, we hypothesize that the linker group cannot exceed 1 carbon in order to maintain inhibitory activity against AKR1B1.
Scheme 4.
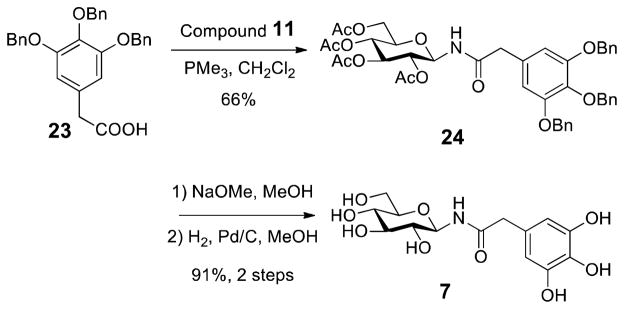
The Synthesis of Amide, 7
Previously, we utilized molecular modeling to help explain the enzyme inhibition profile of BGG. Likewise, to understand this profound structure activity relationship, we turned to molecular modeling. Using Discovery Studio software (Accelrys) BGG derivatives were docked into the crystal structure of human AKR1B1 (PDB: 1US0). Although we observed comparable binding energies to BGG, BGA was the only derivative that minimized in the AKR1B1 site with a similar pose to BGG (Figure 2A). Specifically, in addition to binding energies we considered and analyzed the top 20 poses for all compounds (based on dock score). Interestingly, we observed a trend where BGG and BGA favorably bind with the sugar moiety in the “anionic” pocket and the gallate ring extending into the “specificity” pocket. However, all the other derivatives 3–7 bound opposite to this configuration with the sugar moiety in the “specificity” pocket (Figure 2B and Figure S1, see supporting information). Given that glucose is a natural substrate that binds in the “anionic” site and the sugar moiety of BGG and BGA appear to mimic this, it seems logical that this type of binding pose would be preferential and may explain the loss of activity for 3–7, which exceed our hypothesized 1-carbon linker limit. As a result of their inactivity, 3–7 were no longer investigated as inhibitors of AKR1B1
Figure 2.
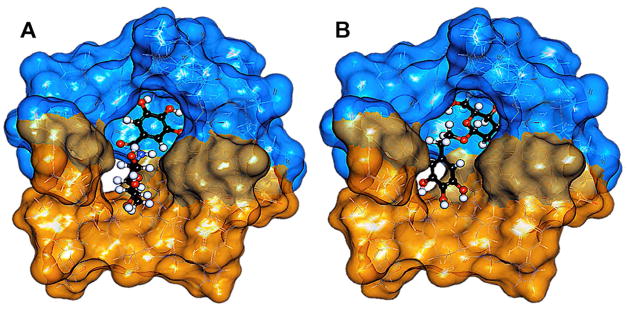
Representative binding poses for BGG derivatives 2–7 bound to AKR1B1 using the top 20 poses ranked by docking score. (A) A favorable inhibitory binding pose, represented with BGA, where the sugar moiety is positioned in the “anionic” pocket (orange surface) and the gallate ring is positioned in the “specificity” pocket (blue surface). (B) An unfavorable binding pose represented by glycoside 3 depicting an opposite configuration to BGG and BGA.
The stability of BGG and BGA were investigated using quantitative HPLC (see calibration curves in the supporting information) after treatment in aqueous sulfuric acid solutions (pH = 0.33) while heating at 80 °C (Figure 3). As expected from our previous observations, and from literature reports regarding glycosyl-1-ester stability,27, 28 we found that BGG degraded by 76% after 15 minutes and completely decomposed within 30 minutes. The major degradation product was determined to be gallic acid based on HPLC retention time. In stark contrast and to our surprise, 96% of BGA remained intact after 6 days under the same conditions. BGA proves to be significantly more stable than BGG even under extreme conditions.
Figure 3.

Stability studies with BGG and BGA under thermal acidic conditions showing the % compound remaining (mean concentration) over time. Statistical analysis was done using the student’s t-test, where, ***P ≤ 0.0001.
Once the stability of BGA was determined we then evaluated whether or not BGA could effectively inhibit AKR1B1 using a Raw264.7 murine macrophage cell based assay. Previously, we found that BGG exhibited low cytotoxicity in Raw264.7 cells and effectively inhibited sorbitol accumulation.15 In this same study, sorbinil, a reference aldose reductase inhibitor gave similar results reducing sorbitol accumulation by 44%. Likewise, BGG and BGA inhibited AKR1B1 activity in Raw264.7 cells blocking sorbitol accumulation by approximately 50% (Figure 4A).
Figure 4.

(A) Macrophages were incubated with either BGG or BGA for 24 h before they were harvested. (B) Lenses extracted from wild type or AR transgenic mice were incubated with hyperglycemic conditions in the presence or absence of BGA for 72 h. The sorbitol levels in the macrophages or lenses were measured using a sorbitol colorimetric assay. The amount of sorbitol was normalized to total protein. Statistical analysis was done using the student’s t-test, where *P ≤ 0.05; **P ≤ 0.01.
In addition to cell based assays we assessed AKR1B1 inhibitory activity of BGA using a transgenic lens organ culture model measuring sorbitol accumulation under hyper-glycemic conditions. This mouse model overexpresses human AKR1B1 in lenses, which express low levels of endogenous aldose-reductase. Thus, this model is a direct measure of AKR1B1 activity. Furthermore, these animals develop cataracts as a result of sorbitol accumulation.16 Briefly, lenses are extracted from the eyes of transgenic mice and cultured ex vivo supplemented with 27.5 mM glucose. Lenses were treated with vehicle controls (H2O) or BGA for 72 hours. At both 25 and 50 μM concentrations BGA blocked sorbitol accumulation by 80% (Figure 4B). Similarly, we have shown that BGG blocks sorbitol accumulation in this lens organ culture model by 74%.16
CONCLUSION
In summary, this report demonstrates the design, synthesis and biological evaluation of a new series of glycosides based on β-glucogallin pharmacophore, a relatively potent but specific inhibitor of AKR1B1. We have determined that the chain length between the glucose moiety and the gallate ring of BGG is crucial and may not exceed 1 carbon. We observed a major flaw in the natural product pharmacophore to be the ester linkage, which decomposes under neutral aqueous solutions slowly and rapidly decomposed under extreme conditions within 30 minutes. By design, we overcame this instability through isosteric replacement of the ester for an amide to yield the novel derivative BGA. BGA proves to be stable under extreme heat and strong acid for an extended time course of 6 days. Furthermore, BGA maintained both potency and specificity for AKR1B1 in vitro and in a transgenic ex vivo organ culture model. With compatible activity and greatly improved stability, BGA becomes an attractive therapeutic lead toward the treatment of diabetic complications. Further structure based drug design is presently ongoing to improve the pharmacological profile of BGA, and more sophisticated animal models will be used to test BGA efficacy in vivo.
Experimental Section
General Procedures
All commercial chemicals were used as supplied unless otherwise indicated. All reactions were performed under an inert atmosphere of ultrapure nitrogen with oven-dried glassware. 1H and 13C NMR spectra were recorded on a Varian 500 MHz spectrometer. High-resolution mass spectra data were acquired on a Bruker Q-TOF-2 capable of ESI ion source. Analysis of sample purity was performed on a Shimadzu Prominence HPLC system with a Phenomenex Kinetex C18 reversed phase column (5 μm, 100 A, 250 × 4.6 mm). HPLC conditions: solvent A = H2O, solvent B = MeCN; flow rate = 1.0 mL/min; compounds were eluted with a gradient of Water to MeCN over 30 min. All tested compounds have a purity ≥95%.
General Deacetylation Procedure
To a stirred solution of the acetyl protected compound in dry MeOH (0.1 M) was added 1–2 drops of 1 M methanolic NaOMe solution and the resulting mixture was stirred for 1 h at room temperature. The reaction mixture was neutralized with solid Amberlyst-15 (H+ form) ion-exchange resin, filtered and concentrated under reduced pressure to yield the product.
General Hydrogenolysis Procedure
A 0.05 M solution of the substrate and 10% mol Pd/C in a mixture of MeOH/EtOAc (v/v, 5:1) were shaken in a Parr hydrogenator for 5 h under 50 psi of H2. After filtration of the catalyst, evaporation of the filtrate under reduced pressure gave a solid or syrup.
2,3,4,6-Tetra-O-benzyl-1-O-(tri-O-benzylgalloyl) β-D-glucopyranose (10)
To a stirred solution of 8 (301.5 mg, 0.557 mmol) and 3 A molecular sieves in CH2Cl2 was added triethylamine (805 μL, 5.77 mmol) dropwise. After 10 min of stirring at room temperature, benzoyl chloride 9 29 (383.9 mg, 0.836 mmol) was then added to the resulting solution in four portions over 20 min, and the stirring was continued for 3 h. The reaction mixture was filtered through a pad of Celite and washed with CH2Cl2, after which the filtrate was washed with brine and extracted with CH2Cl2. The combined organic layer was separated, dried over Na2SO4, and concentrated. Purification by column chromatography on silica gel, eluting with 15% ethyl acetate in hexane, afforded the corresponding ester 10 (505.2 mg, 0.524 mmol, 94%) as a clear oil. [α]22D -229.9 (c = 0.42, CHCl3); IR (neat) νmax 3062, 3033, 2926, 2862, 1732, 1590, 1496, 1433, 1204 cm−1; TLC (20% ethyl acetate in hexane) Rf = 0.55; 1H NMR (500 MHz, CDCl3) δ 7.41–7.12 (m, 37H), 5.85 (d, J = 7.5 Hz, 1H), 5.15–5.06 (m, 6H), 4.92–4.82 (m, 3H), 4.69–4.60 (m, 3H), 4.55 (d, J = 10.5 Hz, 1H), 4.48 (d, J = 12.5 Hz, 1H), 3.84–3.62 (m, 6H); 13C NMR (125.7 MHz, CDCl3) δ 164.5, 152.7, 143.0, 138.5, 138.1, 137.9, 137.8, 137.4, 136.7, 128.7, 128.5, 128.4, 128.3, 128.1, 127.98, 127.96, 127.9, 127.5, 124.3, 109.7, 94.9, 85.0, 81.1, 77.3, 75.8, 75.7, 75.2, 75.1, 73.7, 71.3, 68.1. ESI-HRMS calcd. for C62H58O10Na [M + Na]+ 985.3922, found 985.3934.
1-O-galloyl β-D-glucopyranoside (BGG, 1)
General hydrogenolysis procedure with ester 10 (128.9 mg, 0.134 mmol) afforded 1 (41.1 mg, 0.127 mmol, 95%) as a pale yellow oil that when solidifies becomes off-white in color. All spectral analyses were in accordance with our previously reported values for BGG.16
N-(3,4,5-Tri-O-benzylgalloyl)-2,3,4,6-tetra-O-acetyl β-D-glucopyranosylamine (12)
To a stirred solution of azide 10 (240.0 mg, 0.643 mmol) in CH2Cl2 (3.2 mL) was added PMe3 (707 μL, 1.0 M solution in THF, 0.707 mmol) dropwise. The mixture was stirred at room temperature until nitrogen evolution had ceased and TLC had indicated the complete transformation of 11 (~15 min). Benzoyl Chloride 9 (354.1 mg, 0.772 mmol) was added and stirring continued for 48 h at room temperature. The organic solvent was removed under reduced pressure and chromatographic purification on silica gel (40% ethyl acetate in hexane) afforded 12 (398.6 mg, 0.518 mmol, 81%) as a white solid. M.p. 171–172 °C; [α]23D -32.3 (c = 0.2, CHCl3); IR (neat) νmax 3297, 3029, 2945, 1748, 1662, 1583, 1532, 1497, 1204 cm−1; TLC (35% ethyl acetate in hexane) Rf = 0.30; 1H NMR (500 MHz, CDCl3) δ 7.44–7.23 (m, 15H), 7.08 (s, 2H), 6.99 (d, J = 9.0 Hz, 1H), 5.389 (t, J = 9.5 Hz, 1H), 5.386 (t, J = 9.0 Hz, 1H), 5.12 (s, 4H), 5.11 (t, J = 9.5 Hz, 1H), 5.10 (s, 2H), 5.03 (t, J = 10.0 Hz, 1H), 4.36–4.33 (dd, J = 4.5, 12.5 Hz, 1H), 4.11–4.08 (dd, J = 2.0, 12.5 Hz, 1H), 3.92–3.88 (ddd, J = 2.0, 4.0, 10.0 Hz, 1H), 2.07 (s, 3H), 2.04 (s, 6H), 2.00 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 171.7, 170.7, 169.9, 169.7, 166.8, 152.9, 141.9, 137.5, 136.6, 128.7, 128.6, 128.3, 128.1, 128.1, 127.6, 106.9, 79.2, 75.2, 73.7, 72.7, 71.3, 71.0, 68.3, 61.7, 20.9, 20.8, 20.7; ESI-HRMS calcd. for C42H43NO13Na [M + Na]+, 792.2627; found, 792.2632.
N-galloyl β-D-glucopyranosylamine (BGA, 2)
General deacetylation procedure followed by general hydrogenolysis procedure with 12 (114.2 mg, 0.148 mmol) provided amide 2 (47.3 mg, 0.143 mmol, 96%) as a yellow oil. [α]24D −20.2 (c = 0.1, CH3OH); IR (neat) νmax 3264, 2926, 1642, 1604, 1522, 1332, 1210 cm−1; 1H NMR (500 MHz, CD3OD) δ 6.97 (s, 2H, H-10/H-14), 5.10 (d, J = 9.0 Hz, 1H, H-1), 3.89–3.86 (dd, J = 2.0, 12.0 Hz, 1H, H-6a), 3.73–3.69 (dd, J = 5.5, 12.0 Hz, 1H, H-6b), 3.49–3.46 (m, 2H, H-2/H-4), 3.45–3.41 (m, 1H, H-5), 3.39–3.37 (m, 1H, H-3); 13C NMR (125.7 MHz, CD3OD) δ 171.2 (C-8), 146.6 (C-11/C-13), 138.7 (C-12), 125.5 (C-9), 108.3 (C-10/C-14), 81.8 (C-1), 79.7 (C-5), 79.0 (C-2), 73.7 (C-4), 71.4 (C-3), 62.7 (C-6); ESI-HRMS calcd. for C13H17NO9 [M + H]+ 332.0976, found 332.0977.
1-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-4-(3,4,5-tribenzyloxyphenyl)-1,2,3-triazole (14)
To a stirred solution of azide 11 (53.4 mg, 0.143 mmol) and alkyne 13 (60.1 mg, 0.143 mmol) in EtOH/H2O (6 mL, v/v, 1:1) was added CuSO4·5H2O (5.4 mg, 21.5 μmol) and sodium ascorbate (12.7 mg, 64.4 μmol). The heterogeneous mixture was stirred vigorously for 24 h at 70 °C. The reaction mixture was diluted with H2O, extracted with CH2Cl2. The organic layer was dried over Na2SO4, filtered and concentrated. Chromatographic purification on silica gel (10% ethyl acetate in dichloromethane) gave 14 (100.2 mg, 0.129 mmol, 90%) as a white solid; M.p. 232 °C (decompose); [α]24D −3.5 (c = 0.92, CHCl3); IR (neat) νmax 3033, 2926, 2854, 1750, 1584, 1369, 1210 cm−1; TLC (35% ethyl acetate in hexane) Rf = 0.25; 1H NMR (500 MHz, CDCl3) δ 7.94 (s, 1H), 7.46-7.18 (m, 17H), 5.93 (d, J = 9.5 Hz, 1H), 5.53 (t, J = 9.5 Hz, 1H), 5.44 (t, J = 9.5 Hz, 1H), 5.28 (t, J = 10.0 Hz, 1H), 5.16 (s, 4H), 5.08 (s, 2H), 4.36–4.32 (dd, J = 5.0, 12.5 Hz, 1H), 4.16 (d, J = 12.5 Hz, 1H), 4.05–4.01 (ddd, J = 2.0, 5.0, 10.0 Hz, 1H), 2.09 (s, 3H), 2.08 (s, 3H), 2.04 (s, 3H), 1.89 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 170.6, 170.0, 169.5, 169.1, 153.4, 148.4, 139.0, 137.8, 137.0, 128.7, 128.6, 128.3, 128.0, 127.9, 127.6, 125.6, 117.7, 105.8, 85.9, 75.4, 75.3, 72.9, 71.5, 70.4, 67.9, 61.7, 20.8, 20.6, 20.3; ESI-HRMS calcd. for C43H43N3O12Na [M + Na]+ 816.2739, found 860.2726.
1-(β-D-glucopyranosyl)-4-(3,4,5-trihydroxyphenyl)-1,2,3-triazole (3)
General procedures for deacetylation followed by hydrogenolysis with 14 (84.1 mg, 0.106 mmol) provied 3 (36.4 mg, 0.102 mmol, 96%) as a yellow oil. [α]23D −0.46 (c = 0.6, CH3OH); IR (neat) νmax 3424, 2927, 2854, 1590, 1501, 1367, 1036 cm−1; 1H NMR (500 MHz, CD3OD) δ 8.20 (s, 1H), 6.75 (s, 2H), 5.52 (d, J = 9.5 Hz, 1H), 3.85 (t, J = 9.5 Hz, 1H), 3.81–3.42 (m, 5H); 13C NMR (125.7 MHz, CD3OD) δ 149.4, 147.4, 134.9, 122.5, 120.4, 106.1, 89.6, 81.1, 78.5, 74.0, 70.9, 62.4; ESI-HRMS calcd. for C14H17N3O8Na [M + Na]+ 378.0908, found 378.0900.
2-(3,4,5-Tribenzyloxyphenyl)ethyl β-D-glucopyranoside (17)
A mixture of alcohol 15 (188.2 mg, 0.441 mmol) and Ag2CO3 (139.0 mg, 0.504 mmol) in CH2Cl2 (2 ml) was stirred over 3 A molecular sieves for 15 min before a solution of bromide 16 (172.8 mg, 0.420 mmol) in CH2Cl2 (2 ml) was added dropwise. The reaction mixture was covered in aluminum foil and stirred at room temperature for 24 h. The reaction mixture was filtered through a pad of Celite and washed with ethyl acetate, the filtrate was then concentrated under reduced pressure. The crude mixture was dissolved in CH3OH (4 ml) and 2 drops of 1 M methanolic NaOMe solution was added, the resulting mixture was stirred for 1 h at room temperature. The reaction mixture was neutralized with solid Amberlyst-15 (H+ form) ion-exchange resin, filtered and concentrated. Chromatographic purification on silica gel (35% ethyl acetate in hexane to 5% CH3OH in CH2Cl2) provided 17 (139.1 mg, 0.231 mmol) in 55% yield. White solid; M.p. 76–77 °C; [α]23D −75.7 (c = 0.148, CH3OH); IR (neat) νmax 3348, 2924, 2880, 1590, 1507, 1431, 1145 cm−1; TLC (10% CH3OH in CH2Cl2) Rf = 0.45; 1H NMR (500 MHz, CD3OD) δ 7.49–7.22 (m, 15H), 6.73 (s, 2H), 5.11 (s, 4H), 4.95 (s, 2H), 4.34 (d, J = 7.5 Hz, 1H), 4.13–4.09 (dt, J = 7.0, 9.5 Hz, 1H), 3.93–3.90 (dd, J = 2.0, 12.5 Hz, 1H), 3.80–3.75 (dt, J = 7.0, 9.5 Hz, 1H), 3.73–3.69 (dd, J = 5.5, 12.5 Hz, 1H), 3.41 (t, J = 9.0 Hz, 1H), 3.36–3.29 (m, 2H), 3.25 (t, J = 9.0 Hz, 1H), 2.90 (t, J = 7.0 Hz, 2H); 13C NMR (125.7 MHz, CD3OD) δ 153.9, 139.1, 138.7, 137.4, 136.6, 129.4, 129.1, 128.9, 128.8, 109.5, 104.3, 78.1, 78.0, 76.3, 75.1, 72.1, 71.7, 71.4, 62.8, 37.3; ESI-HRMS calcd. for C35H38O9Na [M + Na]+ 625.2408, found 625.2411.
2-(3,4,5-Trihydroxyphenyl)ethyl β-D-glucopyranoside (4)
General hydrogenolysis procedure with 17 (54.3 mg, 90.1 μmol) afforded 4 (27.1 mg, 83.8 μmol, 93%) as a yellow oil. [α]24D −10.1 (c = 0.3, CH3OH); IR (neat) νmax 3332, 2942, 2829, 1608, 1449, 1076, 1022 cm−1; 1H NMR (500 MHz, CD3OD) δ 6.21 (s, 2H), 4.25 (d, J = 7.5 Hz, 1H), 4.00–3.94 (dd, J = 8.0, 9.0 Hz, 1H), 3.84–3.81 (dd, J = 2.0, 12.0 Hz, 1H), 3.67–3.60 (m, 2H), 3.34–3.20 (m, 3H), 3.15 (t, J = 8.5 Hz, 1H), 2.78–2.65 (m, 2H); 13C NMR (125.7 MHz, CD3OD) δ 146.8, 132.4, 130.9, 108.9, 104.3, 78.0, 77.8, 75.1, 72.0, 71.6, 62.7, 36.7. ESI-HRMS calcd. for C14H20O9Na [M + Na]+ 355.1000, found 355.0990.
3,4,5-Tribenzyloxycinnamyl β-D-glucopyranoside (19)
Following the general procedure for making 17, E-(3,4,5-tribenzyloxy)cinnamyl alcohol30 (171.5 mg, 0.379), Ag2CO3 (119.5 mg, 0.433 mmol) and bromide 16 (148.4 mg, 0.361 mmol) were converted to 19 (100.2 mg, 0.163 mmol, 45%). White solid; M.p. 119–120 °C; [α]23D −29.5 (c = 0.02, CH3OH); IR (neat) νmax 3309, 3029, 2938, 2856, 1581, 1505, 1427, 1126 cm−1; TLC (10% CH3OH in CH2Cl2) Rf = 0.45; 1H NMR (500 MHz, CD3OD) δ 7.49–7.23 (m, 15H), 6.85 (s, 2H), 6.62 (d, J = 15.5 Hz, 1H), 6.33–6.26 (dt, J = 6.5, 16.0 Hz, 1H), 5.14 (s, 4H), 5.00 (s, 2H), 4.56–4.52 (ddd, J = 1.5, 6.0, 13.0 Hz, 1H), 4.40 (d, J = 8.0 Hz, 1H), 4.36–4.32 (dt, J = 1.0, 6.5, 12.5 Hz, 1H), 3.94–3.90 (dd, J = 1.5, 12.0 Hz, 1H), 3.73–3.70 (dd, J = 5.5, 12.0 Hz, 1H), 3.40 (t, J = 9.0 Hz, 1H), 3.55–3.25 (m, 3H); 13C NMR (125.7 MHz, CD3OD) δ 154.1, 139.0, 138.8, 138.6, 134.4, 133.6, 129.8, 129.5, 129.1, 128.9, 128.8, 126.4, 107.1, 103.3, 78.1, 78.0, 76.3, 75.1, 72.1, 71.7, 70.7, 62.8; ESI-HRMS calcd. for C36H38O9Na [M + Na]+ 637.2408, found 637.2405.
3-(3,4,5-Trihydroxyphenyl)propyl β-D-glucopyranoside (5)
General hydrogenolysis procedure with 19 (59.8 mg, 97.0 μmol) afforded 5 (32.3 mg, 93.2 μmol, 96%) as a yellow oil. [α]24D −20.3 (c = 0.175, CH3OH); IR (neat) νmax 3359, 2938, 2835, 1615, 1452, 1078, 1024 cm−1; 1H NMR (500 MHz, CD3OD) δ 6.18 (s, 2H), 4.21 (d, J = 7.5, 1H), 3.87–3.81 (m, 2H), 3.66–3.62 (dd, J = 5.5, 12.0 Hz, 1H), 3.50–3.45 (dt, J = 6.5, 9.5 Hz, 1H), 3.39–3.19 (m, 3H), 3.16 (t, J = 8.0 Hz, 1H), 2.46 (t, J = 7.5 Hz, 2H), 1.80 (p, J = 7.0 Hz, 2H); 13C NMR (125.7 MHz, CD3OD) δ 146.8, 134.3, 132.0, 108.5, 104.4, 78.1, 77.9, 75.1, 71.6, 69.9, 62.7, 32.6; ESI-HRMS calcd. for C15H22O9Na [M + Na]+ 369.1156, found 369.1144.
[1-(3,4,5-tribenzyloxybenzyl)-1,2,3-triazole-4-yl]methyl 2,3,4,6-tetra-O-acetyl β-D-glucopyranoside (22)
To a stirred solution of azide 20 (93.1 mg, 0.241 mmol) and alkyne 21 31 (108.8 mg, 0.241 mmol) in CH2Cl2/H2O (4 mL, v/v, 1:1) was added CuSO4·5H2O (9.0 mg, 36.2 μmol) and sodium ascorbate (21.5 mg, 0.108 mmol). The heterogeneous mixture was stirred vigorously for 12 h at room temperature. The reaction mixture was diluted with H2O, extracted with CH2Cl2. The combined organic layer was dried over Na2SO4 and concentrated to give 22 (199.5 mg, 0.238 mmol, 99%) as a clear oil. [α]23D −29.1 (c = 0.65, CHCl3); IR (neat) νmax 3062, 2946, 2843, 1754, 1597, 1433, 1229, 1038 cm−1; TLC (ethyl acetate) Rf = 0.65; 1H NMR (500 MHz, CDCl3) δ 7.41–7.26 (m, 16H), 6.55 (s, 2H), 5.38 (d, J = 15.0 Hz, 1H), 5.34 (d, J = 15.0 Hz, 1H), 5.19 (t, J = 9.5 Hz, 1H), 5.09 (t, J = 10.0 Hz, 1H), 5.07 (s, 4H), 5.04 (s, 2H), 5.00 (t, J = 8.0 Hz, 1H), 4.90 (d, J = 12.5 Hz, 1H), 4.78 (d, J = 12.5 Hz, 1H), 4.68 (d, J = 8.0 Hz, 1H), 4.28–4.24 (dd, J = 5.0, 12.5 Hz, 1H), 4.16–4.13 (dd, J = 2.0, 12.5 Hz, 1H), 3.74–3.71 (m, 1H), 2.06 (s, 3H), 2.02 (s, 3H), 1.99 (s, 3H), 1.88 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 170.7, 170.3, 169.5, 169.4, 153.3, 144.6, 139.0, 137.7, 136.7, 129.9, 128.6, 128.3, 128.1, 128.0, 127.6, 122.7, 108.1, 100.1, 75.3, 72.8, 72.0, 71.4, 71.3, 68.4, 63.1, 61.9, 54.4, 20.7; ESI-HRMS calcd. for C45H47N3O13Na [M + Na]+ 860.3001, found 860.3017.
[1-(3,4,5-trihydroxybenzyl)-1,2,3-triazole-4-yl]methyl β-D-glucopyranoside (6)
General procedures for deacetylation followed by hydrogenolysis with 22 (169.6 mg, 0.202 mmol) provied 6 (77.6 mg, 0.194 mmol, 96%) as a yellow oil. [α]23D −19.3 (c = 0.1, CH3OH); IR (neat) νmax 3324, 2945, 2835, 1600, 1457, 1352, 1020 cm−1; 1H NMR (500 MHz, CD3OD) δ 7.84 (s, 1H), 6.29 (s, 2H), 5.29 (s, 2H), 4.90 (d, J = 12.5 Hz, 1H), 4.70 (d, J = 12.0 Hz, 1H), 4.33 (d, J = 8.0 Hz, 1H), 3.83 (d, J = 11.5 Hz, 1H), 3.65–3.62 (dd, J = 4.0, 11.5 Hz, 1H), 3.21–3.14 (m, 3H), 3.17 (t, J = 8.5 Hz); 13C NMR (125.7 MHz, CD3OD) δ 147.4, 145.8, 134.7, 127.1, 125.1, 108.3, 103.5, 77.91, 77.85, 74.9, 71.5, 63.0, 62.8, 55.0; ESI-HRMS calcd. for C16H21N3O9Na [M + Na]+ 422.1170, found 422.1182.
N-(3,4,5-Tri-O-benzylphenylacetyl)-2,3,4,6-tetra-O-acetyl β-D-glucopyranosylamine (24)
To a stirred solution of azide 11 (197.7 mg, 0.530 mmol) in CH2Cl2 (3.2 mL) was added PMe3 (557 μl, 1.0 M solution in THF, 0.557 mmol) dropwise. The mixture was stirred at room temperature until nitrogen evolution had ceased and TLC had indicated the complete transformation of 11 (~15 min). 3,4,5-Tri-O-benzylphenylacetic acid (361.3 mg, 0.795 mmol) was added and stirring continued for 48 h at room temperature. The organic solvent was removed under reduced pressure and chromatographic purification on silica gel (40% ethyl acetate in hexane) afforded 24 (274.8 mg, 0.351 mmol, 66%) as a white solid. M.p. 154 °C; [α]24D −19.0 (c = 0.23, CHCl3); IR (neat) νmax 3309, 3062, 2941, 1749, 1698, 1589, 1503, 1435, 1221 cm−1; TLC (50% ethyl acetate in hexane) Rf = 0.48; 1H NMR (500 MHz, CDCl3) δ 7.44–7.24 (m, 15H), 6.54 (s, 2H), 6.35 (d, J = 9.0 Hz, 1H), 5.27 (t, J = 9.5 Hz, 1H), 5.18 (t, J = 9.5 Hz, 1H), 5.11 (s, 4H), 5.06–5.03 (m, 3H), 4.82 (t, J = 9.5 Hz, 1H), 4.33–4.30 (dd, J = 4.0, 12.5 Hz, 1H), 4.09–4.06 (dd, J = 2.0, 12.5 Hz, 1H), 3.82–3.79 (ddd, J = 2.0, 4.5, 10.0 Hz, 1H), 3.46 (d, J = 15.5 Hz, 1H), 3.38 (d, J = 15.0 Hz, 1H), 2.05 (s, 3H), 2.01 (s, 3H), 1.98 (s, 3H), 1.81 (s, 3H); 13C NMR (125.7 MHz, CDCl3) δ 171.2, 170.6, 170.6, 169.8, 169.6, 153.3, 138.0, 137.8, 137.0, 129.3, 128.6, 128.2, 128.0, 127.9, 127.5, 108.9, 78.5, 75.3, 73.7, 72.6, 71.2, 70.3, 68.2, 61.7, 44.1, 20.8, 20.6, 20.3; ESI-HRMS calcd. for C43H45NO13Na [M + Na]+, 806.2783; found, 806.2769.
N-Phenylacetyl β-D-glucopyranosylamine (7)
General deacetylation procedure followed by general hydrogenolysis procedure with 24 (88.2 mg, 0.112 mmol) provided 7 (35.3 mg, 0.102 mmol, 91%) as a yellow oil. [α]25D −3.4 (c = 0.22, CH3OH); IR (neat) νmax 3410, 1637, 1604, 1532, 1448, 1333 cm−1; 1H NMR (500 MHz, CD3OD) δ 6.28 (s, 2H), 4.85 (d, J = 9.0 Hz, 1H), 3.79–3.76 (dd, J = 2.0, 12.0 Hz, 1H), 3.61–3.58 (dd, J = 5.0, 12.0 Hz, 1H), 3.37–3.20 (m, 6H); 13C NMR (125.7 MHz, CD3OD) δ 175.5, 147.0, 133.2, 127.0, 109.4, 81.2, 79.6, 79.0, 73.9, 71.3, 62.6, 43.5; ESI-HRMS calcd. for C14H15NO9Na [M + Na]+, 368.0952; found, 368.0958.
Supplementary Material
Acknowledgments
This study is supported by NIH National Eye Institute grants EY021498 (MPI: DVL and JMP), EY005856 (JMP), and a diversity supplement to EY021498. This research utilized services of the Medicinal Chemistry and the Mass Spectrometry Core facilities housed within the Skaggs School of Pharmacy and Pharmaceutical Sciences in the Department of Pharmaceutical Sciences. The Medicinal Chemistry Core facility is funded in part by the Colorado Clinical and Translational Sciences Institute grant 5UL1RR025780 from NIH NCRR.
ABBREVIATIONS USED
- AKR1B1
Human aldose reductase
- AKR1B10
small intestine reductase
- AKR1A1
aldehyde reductase
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- BGG
β-glucogallin
- BGA
β-Glucogallin N-glycoside amide
Footnotes
The authors declare no competing financial interest.
Supporting Information. The experimental methods and spectroscopic data for all synthetic compounds, quantitative HPLC analysis, molecular modeling, and biological assays are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Barski OA, Tipparaju SM, Bhatnagar A. The aldo-keto reductase superfamily and its role in drug metabolism and detoxification. Drug Metab Rev. 2008;40:553–624. doi: 10.1080/03602530802431439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrash JM, Tarle I, Wilson DK, Quiocho FA. Aldose reductase catalysis and crystallography: insights from recent advances in enzyme structure and function. Diabetes. 1994;43:955–959. doi: 10.2337/diab.43.8.955. [DOI] [PubMed] [Google Scholar]
- 3.Gabbay K. Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Curr Diab Rep. 2004;4:405–408. doi: 10.1007/s11892-004-0047-z. [DOI] [PubMed] [Google Scholar]
- 4.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 5.Dvornik D, Simard-Duquesne N, Krami M, Sestanj K, Gabbay KH, Kinoshita JH, Varma SD, Merola LO. Polyol accumulation in galactosemic and diabetic rats: control by an aldose reductase inhibitor. Science. 1973;182:1146–1148. doi: 10.1126/science.182.4117.1146. [DOI] [PubMed] [Google Scholar]
- 6.Nicolucci A, Carinci F, Cavaliere D, Scorpiglione N, Belfiglio M, Labbrozzi D, Mari E, Benedetti MM, Tognoni G, Liberati A. A meta-analysis of trials on aldose reductase inhibitors in diabetic peripheral neuropathy. Diabet Med. 1996;13:1017–1026. doi: 10.1002/(SICI)1096-9136(199612)13:12<1017::AID-DIA252>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 7.Bril V, Buchanan RA. Long-term effects of ranirestat (AS-3201) on peripheral nerve function in patients with diabetic sensorimotor polyneuropathy. Diabetes Care. 2006;29:68–72. doi: 10.2337/diacare.29.01.06.dc05-1447. [DOI] [PubMed] [Google Scholar]
- 8.Ginter E, Simko V. Type 2 diabetes mellitus, pandemic in 21st century. Adv Exp Med Biol. 2012;771:42–50. doi: 10.1007/978-1-4614-5441-0_6. [DOI] [PubMed] [Google Scholar]
- 9.Fong DS, Aiello L, Gardner TW, King GL, Blankenship G, Cavallerano JD, Ferris FL, Klein R. Retinopathy in diabetes. Diabetes Care. 2004;27:s84–s87. doi: 10.2337/diacare.27.2007.s84. [DOI] [PubMed] [Google Scholar]
- 10.Pollreisz A, Schmidt-Erfurth U. Diabetic cataract-pathogenesis, epidemiology and treatment. J Ophthalmol. 2010;2010:608751. doi: 10.1155/2010/608751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu C. Aldose Reductase Inhibitors as Potential Therapeutic Drugs of Diabetic Complications. In: Oguntibeju OO, editor. Diabetes Mellitus - Insights and Perspectives. InTech; 2013. pp. 17–46. [Google Scholar]
- 12.Kinoshita JH. A thirty year journey in the polyol pathway. Exp Eye Res. 1990;50:567–573. doi: 10.1016/0014-4835(90)90096-d. [DOI] [PubMed] [Google Scholar]
- 13.Ramunno A, Cosconati S, Sartini S, Maglio V, Angiuoli S, La Pietra V, Di Maro S, Giustiniano M, La Motta C, Da Settimo F, Marinelli L, Novellino E. Progresses in the pursuit of aldose reductase inhibitors: the structure-based lead optimization step. Eur J Med Chem. 2012;51:216–226. doi: 10.1016/j.ejmech.2012.02.045. [DOI] [PubMed] [Google Scholar]
- 14.Calcutt NA, Cooper ME, Kern TS, Schmidt AM. Therapies for hyperglycaemia-induced diabetic complications: from animal models to clinical trials. Nat Rev Drug Discov. 2009;8:417–429. doi: 10.1038/nrd2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang KC, Laffin B, Ponder J, Enzsoly A, Nemeth J, LaBarbera DV, Petrash JM. Beta-glucogallin reduces the expression of lipopolysaccharide-induced inflammatory markers by inhibition of aldose reductase in murine macrophages and ocular tissues. Chem Biol Interact. 2013;202:283–287. doi: 10.1016/j.cbi.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Puppala M, Ponder J, Suryanarayana P, Reddy GB, Petrash JM, LaBarbera DV. The isolation and characterization of β-glucogallin as a novel aldose reductase inhibitor from Emblica officinalis. PLoS One. 2012;7:e31399. doi: 10.1371/journal.pone.0031399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bols M, Hansen HC. Simple synthesis of β-D-glucosyl esters. Acta Chem Scand. 1993;47:818–822. [Google Scholar]
- 18.Gyorgydeak Z, Hadady Z, Felfoldi N, Krakomperger A, Nagy V, Toth M, Brunyanszki A, Docsa T, Gergely P, Somsak L. Synthesis of N-(β-D-Glucopyranosyl)-and N-(2-acetamido-2-deoxy-β-D-glucopyranosyl) amides as inhibitors of glycogen phosphorylase. Biorg Med Chem. 2004;12:4861–4870. doi: 10.1016/j.bmc.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 19.Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 20.Davis BG. Synthesis of glycoproteins. Chem Rev. 2002;102:579–602. doi: 10.1021/cr0004310. [DOI] [PubMed] [Google Scholar]
- 21.Koenigs W, Knorr E. Ueber einige derivate des traubenzuckers und der galactose. Ber Dtsch Chem Ges. 1901;34:957–981. [Google Scholar]
- 22.Brito-Arias M. Nuclear Magnetic Resonance of Glycosides. In: Brito-Arias M, editor. Synthesis and Characterization of Glycosides. Springer; 2007. pp. 314–329. [Google Scholar]
- 23.Klebe G, Kramer O, Sotriffer C. Strategies for the design of inhibitors of aldose reductase, an enzyme showing pronounced induced-fit adaptations. Cell Mol Life Sci. 2004;61:783–793. doi: 10.1007/s00018-003-3406-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ehrig T, Bohren KM, Prendergast FG, Gabbay KH. Mechanism of aldose reductase inhibition: binding of NADP+/NADPH and alrestatin-like inhibitors. Biochemistry. 1994;33:7157–7165. doi: 10.1021/bi00189a019. [DOI] [PubMed] [Google Scholar]
- 25.El-Kabbani O, Rogniaux H, Barth P, Chung RP, Fletcher EV, Van Dorsselaer A, Podjarny A. Aldose and aldehyde reductases: correlation of molecular modeling and mass spectrometric studies on the binding of inhibitors to the active site. Proteins. 2000;41:407–414. doi: 10.1002/1097-0134(20001115)41:3<407::aid-prot120>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 26.Kador PF, Kinoshita JH, Sharpless NE. Aldose reductase inhibitors: a potential new class of agents for the pharmacological control of certain diabetic domplications. J Med Chem. 1985;28:841–849. doi: 10.1021/jm00145a001. [DOI] [PubMed] [Google Scholar]
- 27.Wood HB, Fletcher HG. Migration of a mesitoyl group from C1 to C2 in α-D-glucopyranose. Derivatives of 2-O-mesitoyl-D-glucose. J Am Chem Soc. 1956;78:2849–2851. [Google Scholar]
- 28.Yoshimoto K, Tsuda Y. Utilization of sugars in organic synthesis. XI. general path of O-acyl migration in D-glucose derivatives: acyl migration of methyl mono-O-myristoyl-α- and β-D-glucopyranosides and mono-O-myristoyl-D-glucopyranoses. Chem Pharm Bull. 1983;31:4324–4334. [Google Scholar]
- 29.Dodo K, Minato T, Noguchi-Yachide T, Suganuma M, Hashimoto Y. Antiproliferative and apoptosis-inducing activities of alkyl gallate and gallamide derivatives related to (−)-epigallocatechin gallate. Bioorg Med Chem. 2008;16:7975–7982. doi: 10.1016/j.bmc.2008.07.063. [DOI] [PubMed] [Google Scholar]
- 30.Li L, Chan TH. Enantioselective synthesis of epigallocatechin-3-gallate (EGCG), the active polyphenol component from green tea. Org Lett. 2001;3:739–741. doi: 10.1021/ol000394z. [DOI] [PubMed] [Google Scholar]
- 31.Beckmann HSG, Wittmann V. One-pot procedure for diazo transfer and azide–alkyne cycloaddition: triazole linkages from amines. Org Lett. 2007;9:1–4. doi: 10.1021/ol0621506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.