

Summary
Mitis-group streptococci are ubiquitous oral commensals that can promote polybacterial biofilm virulence. Using a novel murine oral mucosal co-infection model we sought to determine for the first time whether these organisms promote the virulence of C. albicans mucosal biofilms in oropharyngeal infection and explored mechanisms of pathogenic synergy. We found that Streptococcus oralis colonization of the oral and gastrointestinal tract was augmented in the presence of C. albicans. S. oralis and C. albicans co-infection significantly augmented the frequency and size of oral thrush lesions. Importantly, S. oralis promoted deep organ dissemination of C. albicans. Whole mouse genome tongue microarray analysis showed that when compared with animals infected with one organism, the doubly infected animals had genes in the major categories of neutrophilic response/chemotaxis/inflammation significantly upregulated, indicative of an exaggerated inflammatory response. This response was dependent on TLR2 signalling since oral lesions, transcription of pro-inflammatory genes and neutrophil infiltration, were attenuated in TLR2−/− animals. Furthermore, S. oralis activated neutrophils in a TLR2-dependent manner in vitro. In summary, this study identifies a previously unrecognized pathogenic synergy between oral commensal bacteriaand C. albicans. This is the first report of the ability of mucosal commensal bacteria to modify the virulence of an opportunistic fungal pathogen.
Introduction
Oropharyngeal candidiasis (OPC) is an opportunistic infection afflicting humans in a variety of immunosuppressed states, that may also predispose them to invasive infection (Villar and Dongari-Bagtzoglou, 2008). Although Candida albicans is the primary etiologic pathogen in OPC, the microbial ecology of this infection is complex since it contains members of the endogenous mucosal bacterial flora (Dongari-Bagtzoglou et al., 2009; Nett et al., 2010; Nobile et al., 2012). Interactions of C. albicans with co-colonizing bacteria at mucosal sites can be synergistic or antagonistic in disease development, depending on the bacterial species and mucosal site (Morales and Hogan, 2010). Although synergistic interactions in biofilm development between C. albicans and oral streptococci have been demonstrated in vitro (Bamford et al., 2009; Nobbs et al., 2010; Diaz et al., 2012a), the outcome of these interactions in vivo has never been explored.
Firmicutes, including Streptococcus spp., are the predominant colonizers in healthy oral and esophageal mucosal surfaces (Pei et al., 2004; Aas et al., 2005; Dewhirst et al., 2010). Physically associated C. albicans and oral streptococci have been demonstrated in human dental plaque with cocci forming ‘corn-cob-like’ structures around C. albicans hyphae (Zijnge et al., 2004). Oral streptococci of the Mitis group (represented mainly by Streptococcus mitis, Streptococcus gordonii, Streptococcus oralis and Streptococcus sanguinis), are typically considered avirulent commensals comprising from 5% (Moore et al., 1982) to over 80% (Syed and Loesche, 1978; Diaz et al., 2012b) of the recoverable oral flora, depending on the intraoral site sampled and method of analysis. S. oralis colonizes oral mucosal surfaces (Diaz et al., 2012b) and has been reported as a cause of septicaemia in immunosuppressed patients, a population also afflicted by systemic Candida infections (Barrett, 1989; Khan and Wingard, 2001). The two organisms are frequently co-isolated from the sputum of antibiotics-treated cystic fibrosis patients, since S. oralis is notoriously resistant to penicillins and macrolides (Maeda et al., 2011). Interactions between C. albicans and S. oralis are thus likely to play a role not only during commensal colonization of healthy mucosal surfaces, but also during the course of mucosa-associated opportunistic fungal infections in immunocompromised hosts (Venkatesh et al., 2007).
Mitis-group streptococci have been termed ‘accessory pathogens’, defined by their ability to initiate multi-species biofilm assembly and promote the virulence of the mixed bacterial biofilm community in which they participate (Ramsey et al., 2011; Whitmore and Lamont, 2011). Unlike S. gordonii, which is the most studied member of the group and can form robust biofilms on its own (Bamford et al., 2009), we and others have shown that S. oralis does not form biofilms unless C. albicans or other members of the oral microbial flora are present (Rickard et al., 2006; Diaz et al., 2012a). Using a novel mucosal tissue salivary flow cell we previously showed that C. albicans and S. oralis have a mutualistic relationship, with S. oralis facilitating invasion of the oral and esophageal mucosa by C. albicans, and C. albicans enabling biofilm development by S. oralis (Diaz et al., 2012a). In this work we hypothesized that this member of the oral commensal bacterial flora exacerbates oropharyngeal candidiasis in vivo and examined inflammatory mechanisms of inter-Kingdom pathogenic synergy.
Results
Streptococci of the Mitis group enhance the frequency and severity of thrush lesions without affecting fungal burdens
To investigate the potential influence of oral streptococci of the Mitis group on the pathogenicity of C. albicans, the virulence of C. albicans in the presence of S. oralis or S. gordonii was tested in vivo, using a modification of our oral thrush mouse model (Dongari-Bagtzoglou et al., 2009; Dwivedi et al., 2011). These organisms are not known to be part of the normal mouse microbial flora, and require immunosuppression to stably colonize the GI tract (Dongari-Bagtzoglou et al., 2009; Dwivedi et al., 2011; Solis and Filler, 2012). To induce macroscopically visible oral thrush lesions in the majority of C57/BL6 mice, C. albicans requires 3 doses of cortisone over the course of 7 days (Dongari-Bagtzoglou et al., 2009; Dwivedi et al., 2011). In this modified protocol, we reduced the number of cortisone injections to 2 and shortened the observation period to 5 days. Under these conditions 46.2% of the mice infected with Candida alone developed varying degrees of tongue thrush lesions, compared with 94% of mice co-infected with S. oralis (Table 1, Fig. 1A). These lesions corresponded to the development of mixed fungal-streptococcal mucosal biofilms, as seen by immuno-FISH staining (Fig. 1B). The severity of tongue thrush lesions was also significantly enhanced by co-infection (Fig. 1C). Similar trends were observed with S. gordonii but the influence of this species on Candida virulence was not as pronounced, compared with that of S. oralis (Table 1, and data not shown).
Table 1.
Frequency of oral thrush lesions in single and mixed infection
| Number of mice with visible tongue lesion/total number of mice | |
|---|---|
| S. oralis 34 | 0/17 (0.0%) |
| S. gordonii CH1 | 0/8 (0.0%) |
| C. albicans SC5314 | 12/26 (46.2%) |
| Candida + S. oralis | 17/18 (94.4%) |
| Candida + S. gordonii | 5/8 (62.5%) |
Fig 1.

Pathogenic synergy between C. albicans and S. oralis.A. Tongues of mice were excised after five days of infection and the dorsal aspect was digitally photographed. Representative pictures are shown from 1 mouse in each group.B. Overlay images of tongue tissue sections stained with a FITC-labelled anti-Candida antibody (green), followed by FISH with an Alexa 546-labelled S. oralis-specific probe (red), and counterstained with the nucleic acid stain Hoechst 33258 (blue). The FISH signal was completely absent in biofilms formed by C. albicans only. Bars = 50 μm.C. Oral pathology score based on percent tongue surface area covered by thrush (biofilm). Image J was used to calculate the area covered by the white plaque as well as the total dorsal surface area of each tongue in order to calculate percentage of surface area covered. Each dot represents an individual mouse with 8–12 mice per group. So: S. oralis 34, Ca: C. albicans 5314, CaSo: S. oralis + C. albicans. Median values of pathology scores in each infection group are designated by horizontal lines. *P < 0.01 for a comparison between Ca and CaSo groups.D. Body weight loss in each infection group during the five day infection period, expressed as percentage of initial weight (day 1) in 17–26 animals per group from 3 independent experiments. Error bars represent SEM. *P < 0.01 for a comparison between Ca and CaSo groups.
Interestingly, quantitative culture of C. albicans from the tongue and esophageal tissues revealed that the fungal burdens at the two sites were not significantly different between the C. albicans and co-infection (i.e. C. albicans + S. oralis) groups (Fig. 2A). These results are in agreement with our previous finding that S. oralis does not affect C. albicans planktonic or mucosal biofilm growth in vitro (Diaz et al., 2012a) and show that the beneficial influence of S. oralis on C. albicans mucosal biofilms is not a consequence of growth stimulation of the latter.
Fig 2.
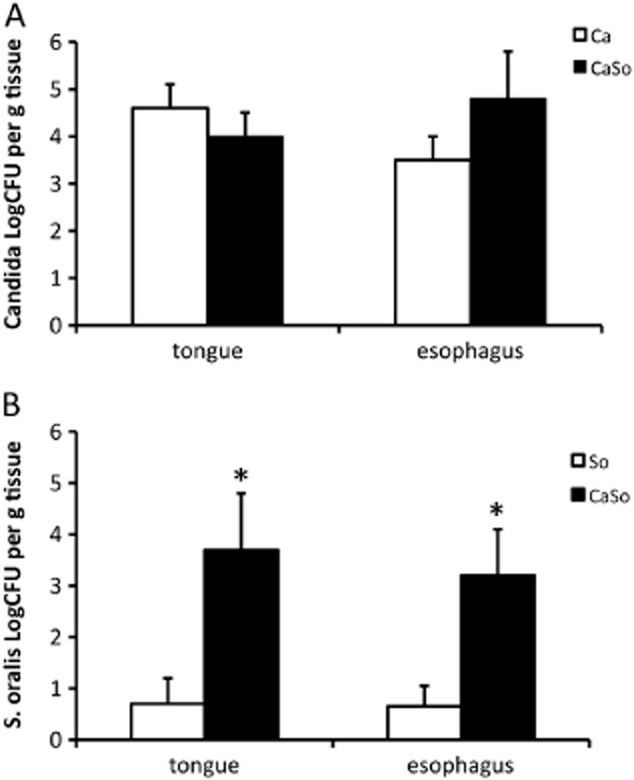
C. albicans increases S. oralis burdens on mouse tongue and esophageal tissues. Tongues and esophagi were homogenized, serially diluted and plated for cfu counts on day 5.A. C. albicans cfu.B. S. oralis cfu.Co-infection increased oroesophageal colonization of S. oralis, but not C. albicans. Results of three independent mouse experiments, with 6–8 animals per group are shown. Bars indicate SEM. *P < 0.005 compared with single, S. oralis infection.
Since painful oral mucosal lesions can adversely affect eating frequency, the extent of disease severity was also quantified by recording animal weight loss at the end of each experiment. Co-infection of C. albicans with S. oralis triggered significantly greater weight loss than single infection, in par with the more severe oral lesion development in this group (Fig. 1D). Together, these results support the hypothesis that a significant pathogenic synergy exists between the C. albicans and certain streptococci of the Mitis group in vivo.
C. albicans enhances the GI tract colonization of S. oralis in vivo
Using a salivary flow cell mucosal biofilm system we previously showed that C. albicans enhances biofilm formation of streptococci of the Mitis group (Diaz et al., 2012a). We thus hypothesized that C. albicans may also promote S. oralis GI tract colonization in vivo. Animals infected with S. oralis alone did not develop tongue lesions, and using histologic staining, bacteria were seen colonizing the tongue surface in very small numbers (Fig. 1B), and in fact most tissue sections in this group were devoid of organisms detectable by FISH. These histologic findings agreed with quantitative culture assessments for S. oralis in tongue and esophageal tissues, which showed very low levels of streptococcal recovery at both sites in singly infected animals (Fig. 2B). Similarly, when streptococcal mono-infection was performed at the cumulative dose of both organisms received during co-infection, none of the animals (n = 10) developed tongue lesions and the colony-forming units (cfu) recoveries from these tongues were very low (most animals had fewer than 10 colonies g−1 of tissue, data not shown), indicating that within this inoculum range the animals are able to efficiently clear the organism. However, quantitative culture assessments for S. oralis in tongue and esophageal tissues of co-infected animals were significantly higher at both GI tract sites in the presence of C. albicans, suggesting that fungi promote oroesophageal colonization of streptococci (Fig. 2B).
To corroborate this finding we performed fecal qPCR analysis with S. oralis 34 strain-specific primers. This showed that in the S. oralis mono-infection group, intestinal colonization was highest on the third day of the experiment (one day post-sublingual inoculation) and then it decreased sharply (Fig. 3). Low levels of intestinal colonization persisted on days 4 and 5, consistent with the daily supply of freshly cultured bacteria in the water. In contrast, in the co-infection group S. oralis cell numbers increased significantly from day 3 to 4 and were maintained at higher levels than the single infection group, through the end of the experiment (Fig. 3). These qPCR data were validated with the use of a different primer set for the S. oralis species-specific gene gtfR (Fig. S1). Taken together, these data show that C. albicans promotes the GI tract colonization of S. oralis in vivo.
Fig 3.
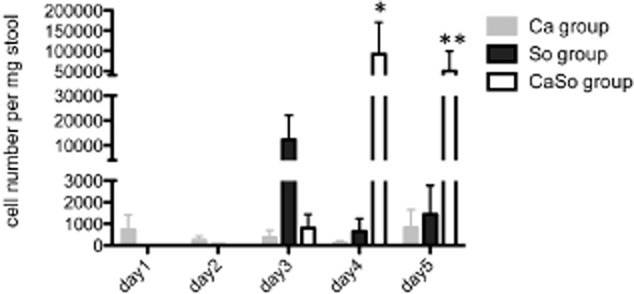
C. albicans promotes S. oralis colonization in the intestinal tract. DNA in stool samples of mice infected with C. albicans alone (Ca), S. oralis alone (So), or co-infected with C. albicans and S. oralis (CaSo) were analysed by qPCR using primers specific for the S. oralis strain 34 wefA-H gene. Cell numbers were calculated according to a standard curve using known amounts of S. oralis 34 gDNA. Results of three independent mouse experiments, with 6–8 animals per group are shown. Bars indicate standard deviations. *P < 0.005, **P < 0.05, for a comparison between the So and CaSo infection groups.
Co-infection leads to a more robust pro-inflammatory response than single infection
To determine whether the underlying mechanisms of the pathological synergy during C. albicans–S. oralis co-infection are due to differences in inflammation, activation of cell death, or inactivation of tissue repair pathways, the host transcriptional response to oral infection was analysed using whole mouse genome microarrays. A total of 21259 gene transcripts were probed and compared in control, single infection and co-infection groups. Co-infection significantly affected the expression levels of 195 genes, compared with the three other groups. Functional analysis showed that the largest percentage of these genes is involved in immune regulation (29%), and that the majority of the upregulated genes (22%) were also in this category (Fig. 4A). In fact, functional ontology analysis showed that genes that exhibited the most significant changes were genes related to cytokine and chemokine activity (Fig. 4B).
Fig 4.

C. albicans–S. oralis co-infection leads to a robust pro-inflammatory response. The oral transcriptional response to infection was analysed in tongue tissues from 3 animals per group on day 5 using whole mouse genome microarrays.A. Pie chart shows the representation of major functional categories of 195 genes differentially regulated in co-infected animals, compared with all other groups.B. molecular function tree of the differentially regulated genes in co-infected animals. The tree diagram displays the hierarchy of gene ontology terms and the levels of enrichment as indicated by FDR adjusted P-values. The genes used in the ontology analysis all exhibited greater than twofold change and adjusted P-value ≤ 0.05 in expression in the comparisons of the co-infected group against all other groups. Greater colour intensity indicates lower P-values.C. Heatmap showing relative expression levels of 31 selected genes across 4 groups of animals (3 biological replicates per group, each with technical triplicates). Normalization of each row (gene) for heatmap generation was performed by subtraction of the median value of the row followed with division by the median absolute deviation. Genes involved in similar biologic processes were grouped together in the categories shown on the right y-axis. Although there was variability in the Candida-infected animals, in the co-infected animals we found genes in the major categories of neutrophilic response/chemotaxis/inflammation to be consistently upregulated, indicative of a broad inflammatory response. Strong induction of multiple chemokines and other neutrophil-activating cytokines (e.g. IL-17C, TNF, IL-1α, IL-1β) were seen in mixed relative to single infection.
A closer examination of the immune regulatory genes upregulated in the co-infected animals showed that 31/40 of these genes could be grouped in one or more general categories of innate immune cell responses, including chemotaxis response, neutrophilic response, cytokine activity, and phagocytosis (Fig. 4C). As illustrated in the heatmap of these genes, the transcriptional response to S. oralis alone did not differ significantly from the uninfected control group. This was expected since this organism colonized oral tissues transiently and in small numbers, in the absence of C. albicans. There was significant variability in the Candida only infected animals, reflecting the variable degrees of oral thrush lesions among animals in this group, ranging from complete absence to presence of small-moderate size tongue lesions. In contrast, in the doubly infected animals, genes in all major categories of innate immune responses were consistently upregulated, indicative of a more pronounced inflammatory response (Fig. 4C).
Interestingly, strong induction of multiple neutrophil-activating cytokines (IL-17C, CXCL1, MIP-2/CXCL2, TNF, IL-1α, IL-1β) and CD177 (a neutrophil-specific protein, associated with trans-endothelial and stromal tissue migration) (Stroncek, 2007) were seen in mixed relative to single infection. Increased expression of CD177 and several neutrophil-activating cytokines (IL-17C, CXCL1, MIP-2/CXCL2) in tongue tissues of co-infected versus Candida mono-infected animals was confirmed by RT-qPCR, using the same RNA samples analysed by microarray (Fig. 5A). We also confirmed higher protein levels of MIP-2/CXCL2, the major neutrophil chemotactic cytokine in mice (Wolpe and Cerami, 1989), in C. albicans–S. oralis co-infected animals compared with C. albicans-infected animals (Fig. 5B). Mice mono-infected with S. oralis at the same total microbial dose as co-infected animals did not express significantly higher levels of these inflammatory markers, compared with uninfected mice (Fig. S2), suggesting that exposure to higher streptococcal challenge through the GI tract is not sufficient to alter the mucosal inflammatory response.
Fig 5.
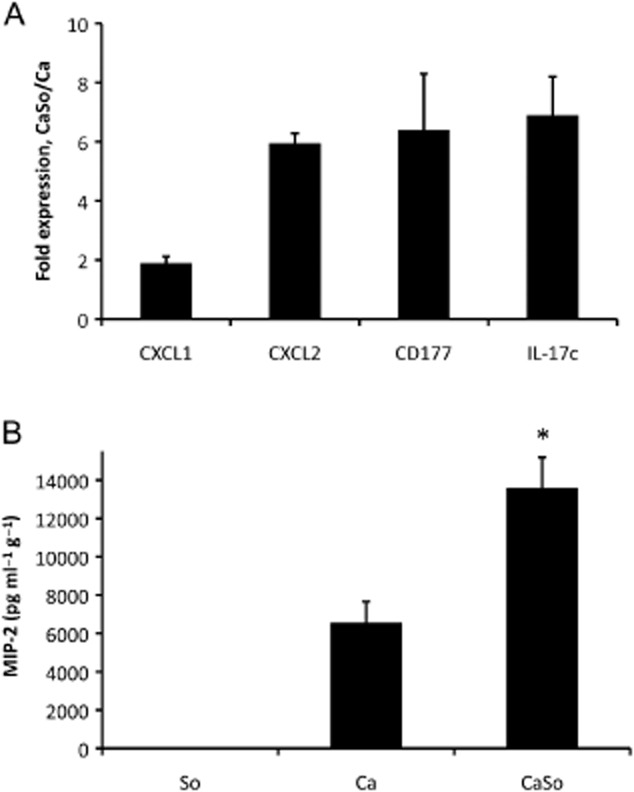
Analysis of inflammatory gene and protein expression in tongue tissues.A. Two chemokines (CXCL1, CXCL2), a neutrophil-specific antigen (CD177) and an epithelial cell-derived neutrophil-activating cytokine (IL-17C) were tested in the same RNA samples surveyed by microarray analysis on day 5 post infection. After cDNA synthesis, equal amounts from three mice per group were mixed and analysed in triplicate by RT-qPCR. All genes were significantly (P < 0.05) upregulated in co-infected animals (CaSo) compared with C. albicans alone (Ca).B. MIP-2/CXCL2 protein levels (day 5) in tongue homogenates, normalized by tissue weight (pg g−1 of tissue). MIP-2 was not detectable in uninfected mice (not shown). Results are means from three independent animal experiments, with 4–6 animals per group and error bars represent SEM. *P < 0.01 compared with C. albicans alone.
We did not find a significant representation of neutrophil-activating Th cell genes in co-infected animals (e.g. IL-17A) (Fig. 4C and RT-qPCR data not shown). This was expected, since a 5 day infection of mice immunologically naïve to these two organisms is unlikely to involve adaptive immunity. The second largest gene group, with significantly altered expression during co-infection, was epithelial cell structure genes, the overwhelming majority of which (20% versus 2%) were strongly downregulated (Fig. 4A). For example, keratin 86, keratin 34 and keratin associated protein 3-3 transcripts were downregulated by 4.8 (P < 0.0001), 7.0 (P < 0.005) and 5.3 (P < 0.0001) fold, respectively, in co-infection compared with single C. albicans infection. The structural gene data are consistent with a greater mucosal damage during co-infection.
The neutrophilic response is exaggerated during C. albicans–S. oralis co-infection
In addition to multiple chemokines, a number of genes known to be expressed by neutrophils, were found to be upregulated during co-infection by microarray analysis (CD177, CD14, MMP8, Fig. 4C), suggestive of a strong mucosal infiltration by neutrophils. To confirm a strong neutrophilic response in co-infected animals we used a two-pronged approach. First, we assessed the presence of neutrophils in histologic sections of tongue tissues of singly infected and co-infected animals by immunostaining with the monoclonal antibody NIMP-R14, highly specific for murine Ly-6G and Ly-6C, a differentiation marker primarily expressed in neutrophils (Daley et al., 2008) (Fig. 6A). Almost complete absence of neutrophils was seen in S. oralis alone-infected animals. Although some neutrophils infiltrated the tongues of C. albicans mono-infected animals, higher neutrophil infiltration was seen in co-infected animals. Many neutrophils had migrated through the entire submucosal and mucosal compartments and were localized in direct juxtaposition to the biofilm cells (Fig. 6A, third panel). Because myeloperoxidase (MPO) is a good enzymatic marker for tissue neutrophil content (Bradley et al., 1982; Hillegas et al., 1990) our second approach in assessing neutrophil presence involved measuring MPO activity in tongue tissues. As seen in Fig. 6B MPO activity was barely detectable in S. oralis-infected animals, consistent with the almost complete absence of neutrophils histologically. In contrast, both C. albicans-infected and co-infected tissues exhibited MPO activity, with co-infected tissues having the highest levels (Fig. 6B). Together these data show that significant numbers of neutrophils are recruited to the tongues of co-infected animals, suggesting that these cells may contribute to the enhanced morbidity and polymicrobial pathogenesis.
Fig 6.
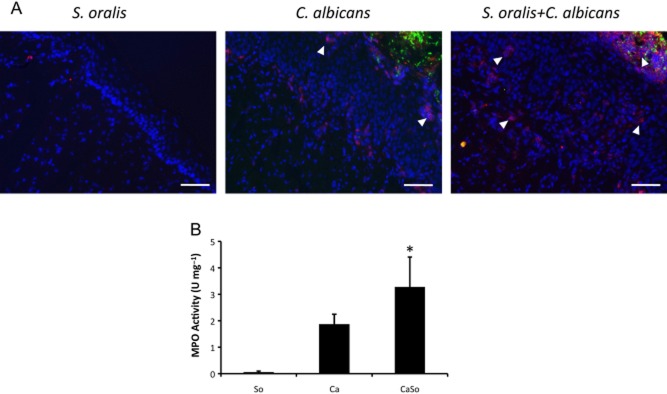
Increased neutrophil infiltration of the oral mucosa in co-infected mice.A. Immunofluorescence staining of frozen tongue sections (day 5) with monoclonal antibody NIMP-R14, highly specific for murine Ly-6G and Ly-6C. Neutrophils (red) are shown with white arrows. Note the intense staining in direct juxtaposition to the mixed bacterial-fungal biofilm in co-infected animals. Isotype control antibody did not show any staining (not shown). Bars = 50 μm.B. MPO activity in tongue homogenates on day 5 post infection. Results are means from two independent animal experiments, with 8–10 animals per group and error bars represent SEM. *P < 0.05 compared with C. albicans alone.
S. oralis transmits pro-inflammatory signals via TLR2
Since S. oralis colonized the oroesophageal and intestinal tracts in high numbers only in co-infected animals, we hypothesized that it is directly responsible for amplifying the mucosal inflammatory response in these animals. The primary signalling pathway involved in pro-inflammatory responses to a genetically related species, S. pneumonia, (> 99% homology by 16S rRNA to S. oralis, (Johnston et al., 2010)), involves TLR2 activation (Dessing et al., 2008; Beisswenger et al., 2009). We thus hypothesized that this signalling pathway is utilized by S. oralis to amplify the pro-inflammatory response. To examine the ability of S. oralis to stimulate this receptor we first utilized an in vitro reporter system consisting of HEK293 cells stably expressing TLR2, with a chemokine (IL-8) secretory response as a readout. Since streptococcal lipoteichoic acid is a strong activator of TLR2 (Dessing et al., 2008; Beisswenger et al., 2009) we expected that S. oralis would activate these cells. Indeed IL-8 reporter assays revealed that S. oralis is sensed by TLR2 in a dose-dependent manner, when added alone or in combination with C. albicans. Importantly, C. albicans alone failed to activate TLR2 under these conditions (Fig. 7). Similar findings were obtained with another reporter cell line, engineered to respond by NF-kB- and/or AP-1-mediated pro-inflammatory signalling activation to TLR2 ligands (Fig. S3).
Fig 7.
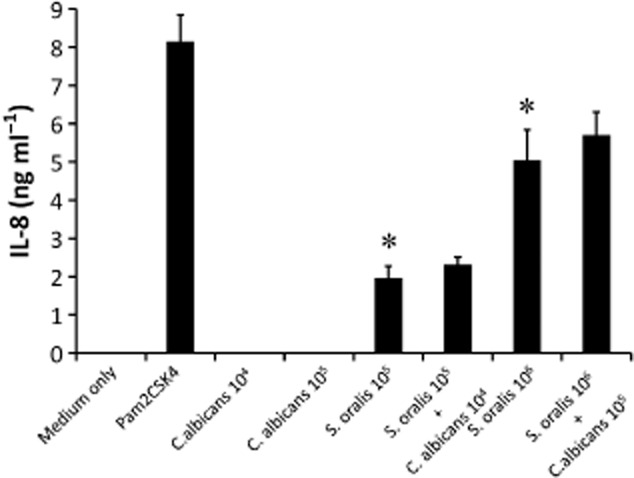
HEK293-hTLR2 expressing cells respond to S. oralis but not C. albicans with IL-8 secretion. HEK293-hTLR2 cells were challenged with live S. oralis 34 (So) and/or C. albicans (Ca) and supernatants were analysed by ELISA. Pam2CSK4 was used as a positive control. Two microbial cell doses were tested (Ca: 104, 105 cells ml−1, So: 105, 106 cells ml−1). IL-8 was undetectable with all stimulants in the HEK293-pcDNA3 negative control cells (not shown). Error bars represent SD of triplicate experiments. *P < 0.01 compared with medium only.
Since large numbers of neutrophils migrated adjacent to the mixed biofilm mass in the oral mucosa (Fig. 6A), we asked whether S. oralis can trigger direct TLR2-mediated functional activation of neutrophils in vitro. Two lines of evidence supported this conclusion. First, generation of reactive oxygen species triggered by S. oralis in the human cell line HL-60 was significantly inhibited by neutralizing anti-TLR2 antibodies, suggesting that this receptor is responsible for respiratory burst activation (Fig. 8A). In the presence of S. oralis, isotype control antibody triggered an enhanced oxidative response compared with S. oralis alone, possibly due to FcR-γ chain-dependent signalling (Otten et al., 2007), which was not statistically significant (P = 0.15). Second, freshly isolated bone marrow neutrophils from TLR2−/− mice failed to respond with an oxidative burst to S. oralis, in contrast to wild type cells, indicating that under these conditions the responsiveness to S. oralis (but not C. albicans) depended entirely on the presence of TLR2 (Fig. 8B).
Fig 8.
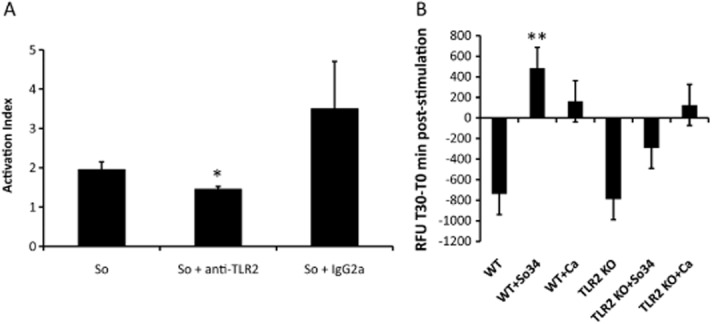
S. oralis triggers a TLR2-dependent oxidative response in neutrophils.A. ROS production was measured after 1 h incubation of HL-60 leucocytes with S. oralis 34 whole cell sonicates. TLR2 receptors on leucocytes were blocked by the addition of a neutralizing anti-human TLR2 or isotype control (IgG2a) antibody (both at 10 μg ml−1). Results represent means and standard deviations of duplicate experiments, with all conditions set up in triplicate, and are expressed as Activation Index i.e. as the ratio of fluorescence in the presence/absence of stimulus. *P < 0.05, for a comparison between S. oralis alone and S. oralis + anti-TLR2 antibody.B. Primary mouse TLR2−/− and wild type neutrophils were stimulated with live, germinated C. albicans cells (105 cells per well) or S. oralis 34 whole cell sonicates (106 cells per well) for 30 min and fluorescence was measured at 0 min and 30 min, post challenge. Results are expressed in relative fluorescence units (T30minRFU − T0minRFU) and represent means and standard deviations of neutrophil responses from 4 WT and 4 TLR2−/− animals. Negative values indicate progressive loss of activation/fluorescence during the 30 min incubation period. **P < 0.01 compared with TLR2−/− cell stimulation with S. oralis.
TLR2 plays a role in the enhanced oral inflammatory response to C. albicans-streptococcal co-infection in vivo
Since infection with the phylogenetically related pathogen S. pneumonia triggers upregulation of TLR2 mRNA in the murine alveolar mucosa (Knapp et al., 2004), we explored the possibility that S. oralis has a similar effect in the oral mucosa. C. albicans alone triggered a small, but statistically significant (P < 0.01), upregulation of TLR2 mRNA in oral tissues compared with uninfected animals, whereas neither mono-infection dose of S. oralis was able to affect TLR2 expression significantly (Fig. 9A). Compared with all groups the co-infected animals expressed the highest TLR2 transcript levels, suggesting that co-infection with S. oralis enhanced TLR2 mRNA expression (Fig. 9A). Higher transcript amounts paralleled the increased TLR2 protein levels in infected tissues of animals infected with C. albicans or the combination of C. albicans and S. oralis (Fig. 9B).
Fig 9.
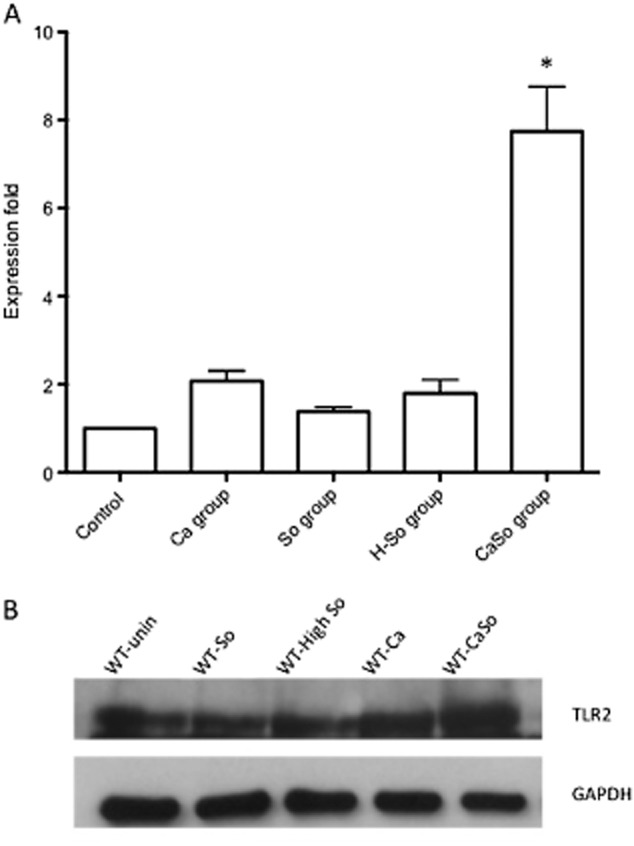
TLR2 is upregulated in the oral mucosa during co-infection.A. TLR2 transcripts were analysed in tongue homogenates on day 5 by qRT-PCR. Results represent mean fold expression levels over uninfected animals in each of the four infection groups (C. albicans alone (Ca), S. oralis alone (So), high dose of S. oralis alone (H-So) or co-infected with C. albicans and S. oralis (CaSo)) with 4–6 animals per group. Error bars are SD. *P < 0.005, compared with Ca group.B. TLR2 protein expression levels on day 5 were analysed in tongue homogenates of wild type (WT) animals by Western blotting. A representative Western is shown with tissue samples from one animal in each of the four infection groups: So: S. oralis alone, High So: high dose of S. oralis, Ca: C. albicans alone and CaSo: co-infected with C. albicans and S. oralis. GAPDH signal served as internal loading control.
Given the increased levels of TLR2 expression in co-infected animals we hypothesized that this receptor may be a determinant of the increased severity of inflammation in this group and that TLR2−/− animals will exhibit attenuated levels of lesion severity and inflammation, in response to co-infection. As expected, TLR2−/− animals had a modest but statistically significant attenuation in tongue lesion severity when co-infected with the two organisms (Fig. 10A and B), which was accompanied by reduced weight loss during the 5 day infection period (Fig. 10C), compared with wild type animals. Importantly, in co-infected animals, tongue homogenates from TLR2−/− mice showed significantly lower levels of all pro-inflammatory and neutrophil activating gene transcripts tested, compared with wild type animals (with the exception of IL-17A, P = 0.3) (Fig. 11A). Wild type and TLR2−/− animals infected with C. albicans alone did not exhibit significant differences in their oral mucosal pro-inflammatory gene expression, supporting the notion that C. albicans did not transmit TLR2-dependent pro-inflammatory signals in the oral mucosa (Fig. 11A). MIP-2/CXCL-2 protein concentrations in the tongues of TLR2−/− co-infected animals were also significantly lower than the WT animals (P < 0.0001) (Fig. 11C) whereas there was no significant difference between C. albicans-infected and C. albicans + S. oralis-infected TLR2−/− mice (P = 0.59) (not shown). In accordance, tongues from TLR2−/− mice infected with both organisms had reduced neutrophilic infiltration compared with wild type mice (Figs 6A and 11B), which paralleled the significantly reduced levels of MPO enzymatic activity in this group (Fig. 11C).
Fig 10.
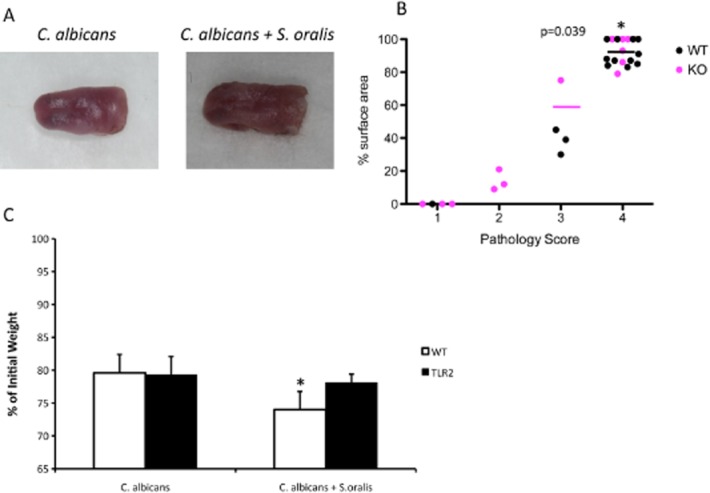
Co-infected TLR2−/− mice display reduced pathology.A and B. Wild type and TLR2−/− mice were infected with C. albicans or the combination of C. albicans and S. oralis for 5 days and tongue lesions were digitally photographed (A) and graded for pathology (B) as described in the materials and methods. Median values of pathology scores in each infection group is designated by horizontal lines. Each dot represents an individual mouse with 8–12 mice per group. Lesion scores in TLR2−/− animals co-infected with C. albicans and S. oralis were significantly lower than in wild type animals (P = 0.039).C. Body weight loss in TLR2−/− and wild type animals during the five day infection period, expressed as percentage of initial weight (day 1). Error bars represent SEM. *P = 0.04 for a comparison between TLR2−/− and WT animals.
Fig 11.
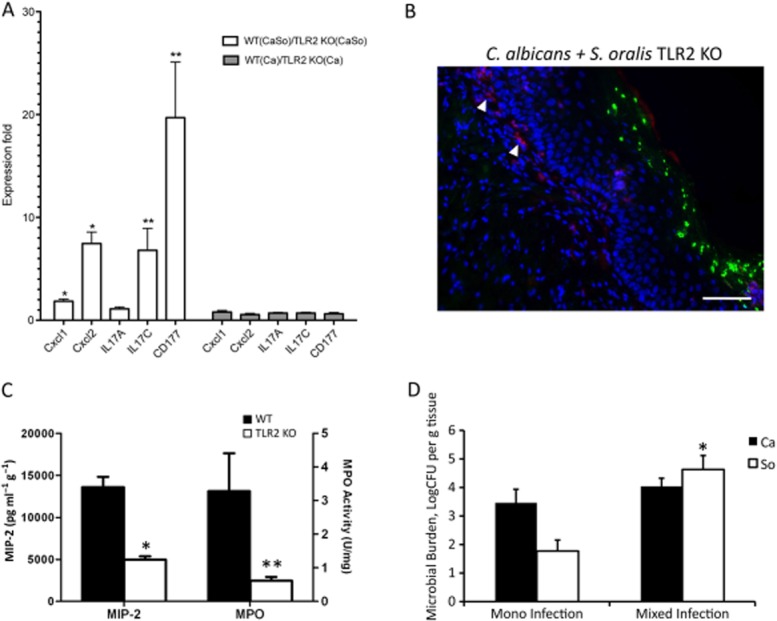
TLR2 is involved in the enhanced oral inflammatory response to C. albicans-streptococcal co-infection in vivo.A. Pro-inflammatory gene transcripts in tongue tissues of wild type and TLR2−/− animals on day 5, as assessed by RT-qPCR. Results represent mean fold expression level of wild type over TLR2−/− tissues, in 4 animals per group. Open bars: co-infection with C. albicans and S. oralis. Gray bars: C. albicans infection. *P < 0.01 and **P < 0.05 for a comparison between WT and TLR2−/− expression levels.B. Immunofluorescence staining of frozen tongue sections (day 5) with monoclonal antibody NIMP-R14, highly specific for murine Ly-6G and Ly-6C. Neutrophils (red) are shown with white arrows. Note the relative absence of neutrophils adjacent to the mucosal biofilm. Bars = 50 μm.C. Comparison of MIP-2/CXCL2 protein concentration (pg g−1 tissue) and MPO activity levels (U mg−1 tissue) in tongue homogenates between WT (closed bars) and TLR2−/− animals (open bars), co-infected with C. albicans and S. oralis. Mean of WT and TLR2−/− tongue protein levels (8–12 animals per group) ± SEM, on day 5 post infection is shown. *P < 0.001, and **P < 0.05, compared with WT.D. Tongues from TLR2−/− animals were homogenized, serially diluted and plated for C. albicans (Ca) or S. oralis (So) cfu counts on day 5. Similar to wild type animals (Fig. 12) co-infection increased oral colonization of S. oralis, but not C. albicans. Results of two independent mouse experiments, with 8–10 animals per group are shown. Bars indicate SEM. *P < 0.001 compared with single, S. oralis infection.
In co-infection S. oralis and C. albicans cfu in the oral mucosa of wild type animals did not differ significantly from those in TLR2−/− animals (Fig. 2A and B; Fig. 11D). Importantly, there was an increase in the streptococcal load in co-infected compared with singly infected TLR2−/− animals, similar to the increase observed in wild type animals (Fig. 2A and B; Fig. 11D). These findings rule out the possibility that attenuation of the pro-inflammatory response in KO animals is attributed to reduced microbial burdens. In addition, experiments with freshly isolated neutrophils from these animals revealed that, despite a trend for slightly less killing of fungal targets by TLR2−/− cells, killing was not significantly different compared with wild type cells (P = 0.088 and P = 0.12 for a comparison between TLR2−/− and WT cells, at 1:10 and 1:5 effector to target ratios, respectively) (Fig. 12). Collectively, our data support the hypothesis that attenuation of the pro-inflammatory response in TLR2−/− animals was not associated with altered microbial clearance, but more likely with absence of S. oralis signalling via this receptor.
Fig 12.
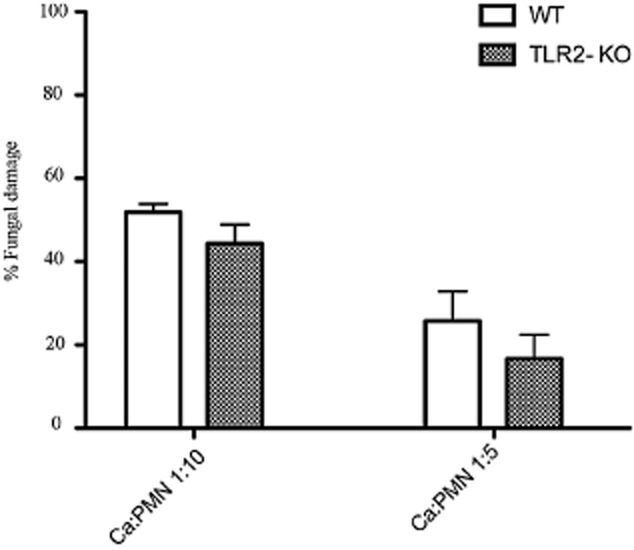
TLR2−/− neutrophils are not significantly compromised in their ability to kill C. albicans. Germinated organisms were exposed to bone marrow isolated mouse neutrophils from TLR2−/− or wild type mice, at two target to effector ratios (1:10 and 1:5), for 3 h and killing was assessed by the XTT assay. Results represent mean and SD of killing activity of neutrophils isolated from 4 mice per group.
S. oralis enhances haematogenous dissemination of C. albicans
In certain immunosuppressed mouse models of OPC C. albicans can breach the oroesophageal mucosal barrier and disseminate haematogenously into the kidneys (Conti et al., 2009). We previously reported that C. albicans acquires a more invasive phenotype in human oral and esophageal organotypic models when forming a biofilm with S. oralis under conditions of salivary flow (Diaz et al., 2012a). We thus evaluated whether S. oralis affects C. albicans dissemination into distant organs in co-infected animals, and whether TLR2 plays a role in systemic fungal spread in this model. The cfu comparisons between single infection and co-infection groups showed that S. oralis promoted deep organ dissemination of C. albicans regardless of TLR2 presence (Fig. 13). Systemic dissemination of S. oralis was rarely seen (not shown).
Fig 13.
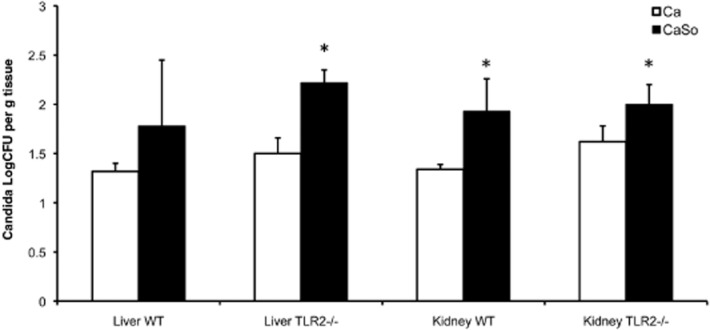
Co-infection increased dissemination of C. albicans in kidneys and livers of TLR2−/− and wild type animals. Both of the kidneys and the liver of each animal were homogenized, serially diluted and plated for cfu counts on day 5 post infection. Fungal burdens were compared in TLR2−/− and WT animals infected with C. albicans (Ca) or C. albicans plus S. oralis (CaSo); *P < 0.05 CaSo compared with Ca. Results of three independent mouse experiments, with 6–8 animals per group are shown. Bars indicate SEM.
Discussion
One principal way by which microorganisms can interact to increase pathogenicity of each other is by modulation of host responses. In this work we show for the first time that an oral commensal bacterium that colonizes the majority of healthy humans (Alam et al., 2000) can synergize with C. albicans to amplify the local pro-inflammatory response and increase the severity of oral mucosal lesions. Based on our data we propose an inter-Kingdom synergy model whereby the fungal opportunistic pathogen C. albicans promotes mucosal biofilm formation and colonization of S. oralis, which in turn signals via TLR2 to amplify the neutrophilic response to mucosal biofilms. Since S. oralis enhanced the frequency and severity of oral thrush lesions without significantly affecting the oroesophageal fungal burdens in co-infected animals, we propose that one important mechanism of pathologic synergy between the two organisms is the exaggerated host inflammatory response during co-infection. Inflammatory synergy between C. albicans and S. aureus has recently been reported in an acute peritoneal model of infection, mediated by the cyclooxygenase pathway and induction of an exaggerated neutrophilic response (Peters and Noverr, 2013). Despite the similarities in the inflammatory pathogenic synergy between these two co-infection models, ours is the first report of the ability of C. albicans to synergize with streptococcal species that are considered avirulent members of the host oral commensal microflora. Our results thus dispute the long held belief that these members of the commensal bacterial flora protect the host against mucosal candidiasis (Liljemark and Gibbons, 1973).
In OPC, the host responds with a multifocal neutrophilic inflammatory infiltrate that extends well into the mucosal biofilm mass, as shown in this and our previous work (Dongari-Bagtzoglou et al., 2009). In Candida-induced vaginitis, a similar neutrophilic infiltrate has been shown to contribute to symptomatic infection rather than fungal clearance (Yano et al., 2010). Although severe neutropenia predisposes the host to OPC, the role of neutrophils in less severe immunosuppression is not as clear. We previously showed that Candida biofilms are resistant to neutrophil killing (Xie et al., 2012), thus, similar to vaginitis, the increased neutrophilic influx and possibly activation during oral co-infection in vivo, may contribute to immunopathology rather than fungal clearance. Even with the introduction of more pathogenic species, multiple bacterial species from the commensal microbial flora can contribute to mucosal inflammation (Jergens et al., 2007; Hajishengallis et al., 2011) and there are several other examples of co-infections where infection with one organism influences the innate immune response to the other (Jamieson et al., 2010; Vonck et al., 2011; Peters and Noverr, 2013). Dysregulated immune responses to the local microbial flora may lead to inflammatory diseases such as inflammatory bowel disease and periodontitis (Jergens et al., 2007; Hajishengallis et al., 2011). This is the first report of inter-Kingdom synergy in the development of mucosal oral thrush lesions in vivo.
In this study we showed that C. albicans augments colonization of the GI tract by S. oralis which then can transmit pro-inflammatory signals via TLR2. A theoretical source of the increased TLR2 levels in oral tissues during co-infection, are the infiltrating neutrophils, which express high levels of this receptor. Another possible source of TLR2 in the oral mucosa is epithelial cells, which normally express low levels of TLR2, but can be stimulated to express higher levels by organisms such as C. albicans (Zhang et al., 2004). This may explain the upregulation of TLR2 both in the C. albicans-infected and co-infected groups. Thus, in addition to directly activating neutrophils in a TLR2-dependent manner, it is possible that oral streptococci activate TLR2-dependent epithelial innate immune response genes that augment the neutrophilic influx, as shown in other co-infection models with oral streptococci (Sibley et al., 2008). This would explain the significantly attenuated chemokine response of the oral mucosa in the TLR2−/− co-infected animals, and the dramatic reduction in IL-17C (but not IL-17A) transcripts, a neutrophil-activating cytokine mainly produced by epithelial cells (Ramirez-Carrozzi et al., 2011). The significant reduction in granulocyte presence in the oral mucosa of the TLR2−/− animals also corroborates the role of this signalling pathway in the local neutrophilic influx. The role of the oral epithelium in orchestrating the mucosal pro-inflammatory response to co-infection is currently the focus of further investigations in our laboratory.
Invasion of epithelial surfaces allows Candida to reach the bloodstream, with potential subsequent escape into deep organs. Gram (+) commensals exert opposite effects on mucosal epithelial barriers depending on their population size, ranging from promotion of homeostasis in low numbers (Rakoff-Nahoum et al., 2004) to deleterious effects in high numbers (Clarke et al., 2011). In the lower GI tract, Gram (-) anaerobes and enteric bacilli limit the growth and dissemination of Candida (Sjovall et al., 1986), and after treatment with broad-spectrum antibiotics C. albicans can invade the gut mucosa (Ponnuvel et al., 1996; Krause et al., 2001). In contrast, reports of OPC occurrence or oral mucosal invasion after antibiotic treatment are rare and only associated with severe immunosuppression (Gligorov et al., 2011). Thus, the effect that bacteria of the host microflora have on Candida invasion is likely to differ at different mucosal sites since it is affected by local host factors, such as the anatomically dictated epithelial response to infection (Yano et al., 2010). We found increased fungal liver and kidney dissemination in co-infected animals as compared with single infection. Systemic fungal dissemination has been proposed to result from an oral mucosal barrier breach in a similar model of OPC (Conti et al., 2009). While we could not reliably quantify oral mucosal tissue invasion of C. albicans in vivo, we previously compared the magnitude of fungal invasion in oral and esophageal human mucosal constructs, and found that the esophageal mucosa was more permissive to Candida invasion in both single and mixed infection with S. oralis (Diaz et al., 2012a). The ability of C. albicans to invade the esophageal but not the oral submucosa has also been documented in immunocompromised human hosts (Thom and Forrest, 2006). This suggests that the esophageal mucosa is a more plausible entry gate than the oral mucosa into the systemic circulation in co-infected mice.
Because the magnitude of dissemination was similar in TLR2−/− and wild type animals, it may be independent of TLR2 signalling. Instead, it is possible that increased tissue invasion is attributed to S. oralis-triggered increased expression of Candida virulence genes, responsible for mucosal invasion. Indeed, nearly half of the streptococcal strains of the Mitis group isolated from human sputum have pathogenic synergy with P. aeruginosa in a drosophila model, by altering Pseudomonas gene expression without affecting its growth (Sibley et al., 2008). Thus it is possible that S. oralis directly influences C. albicans virulence gene expression during co-infection.
We did not find any oral microbial burden differences in TLR2−/− and wild type animals and TLR2−/− neutrophils were not significantly compromised in their fungal killing activity ex-vivo. This was expected since, although Candida recognition by phagocytes involves TLR2, most phagocytic anti-fungal activities are mediated by other receptors (e.g. dectin-1) (reviewed in Moyes and Naglik, 2011). This is also consistent with the observation that TLR2−/− mice are not deficient in clearing intravenously (Netea et al., 2004) or intraperitoneally (Tessarolli et al., 2010) inoculated fungal organisms. Similarly, although Gram (+) bacteria-triggered epithelial inflammation is principally regulated by TLR2 (Aderem and Ulevitch, 2000), TLR2 plays a secondary role in bacterial clearance, at least in chronic models of infection (Knapp et al., 2004; Burns et al., 2006), consistent with our finding that S. oralis oral burdens were similar in TLR−/− and wild type mice.
The wide range in clinical disease manifestations of oral candidiasis (pseudomembranous, erythematous, hyperplastic) is not explicable by differences in Candida virulence or host factors. We propose that such patient to patient differences in oral disease manifestation may be explained at least in part by the interactions of C. albicans in each host with a unique resident commensal bacterial flora, which modulates the host response to infection. Elucidating the effects of oral streptococci on Candida virulence and the mucosal inflammatory response provides important new insights into the pathogenesis of fungal infection within this host niche. Finally, the discovery of a synergistic role for common oral bacterial colonizers in the pathogenesis of OPC may have important clinical implications in the treatment of candidiasis, currently based solely on antifungals.
Experimental procedures
Strains and growth conditions
Candida albicans SC5314 is a bloodstream isolate, previously used in oral biofilm mouse models (Gillum et al., 1984). Two human oral isolates of the mitis group, Streptococcus oralis 34 (a kind gift from PE Kolenbrander) and Streptococcus gordonii Challis CH1 (a kind gift from JM Tanzer), were used in mouse co-infection experiments. C. albicans was routinely maintained in yeast extract peptone dextrose (YPD) agar and grown in YPD broth, aerobically, at room temperature, on a rotor shaker. Streptococci were routinely grown in brain heart infusion (BHI) medium (Oxoid Ltd, Cambridge, UK) under aerobic, static conditions, at 37°C.
Mice
Six- to 12-week-old female C57BL/6 mice or TLR2−/− (B6.129-Tlr2tm1Kir/J) mice of the same genetic background were purchased from the Jackson Laboratory (Maine, US). Most animal experiments were repeated at least twice with 6–12 mice per group. The study was approved by the University of Connecticut Health Center Animal Care Committee (Animal protocol #100358-0215). Animals were monitored daily for distress. Given that the oral cavity is readily accessible, lesions were detected relatively early in their onset and animals were euthanized after lesion formation before visible distress/behaviour signs were observed.
Mouse oral co-infection protocol
A 5-day infection protocol was used after mice had adapted to the new housing environment for 10 days. Mice were infected with C. albicans SC5314, S. oralis 34, S. gordonii CH1 or the combination of C. albicans and streptococci. On the first and third day of the infection protocol, mice were immunosuppressed by subcutaneous injection with cortisone acetate (225 mg kg−1) dissolved in 200 ml PBS containing 0.5% Tween-20. On the second day, mice were anaesthetized by an intramuscular injection of ketamine : xylazine (90–100 and 10 mg kg−1 of body weight respectively) and a small cotton pad soaked with 100 μl of a C. albicans cell suspension (6 × 108 yeast ml−1), or 100 μl of streptococcal cell suspension (2.5 × 109 bacteria ml−1), or 50 μl of C. albicans cell suspension (1.2 × 109 yeast ml−1) combined with 50 μl of streptococcal cell suspension (5 × 109 bacteria ml−1), was used to swab the entire oral cavity. The swab was left for 2 h under the tongue and was removed before the animals awoke. During this time period animals were also given drinking water containing a daily-fresh suspension of C. albicans (6 × 108 yeast organisms ml−1) or streptococcal cell suspension (2.5 × 109 bacteria ml−1) or the combination of the two, to maintain high oral carriage loads throughout the experimental period (Dwivedi et al., 2011). In some experiments mice were infected with 100 μl of streptococcal cell suspension containing 3.1 × 109 bacteria ml−1, and the drinking water concentration was increased accordingly, to serve as a monomicrobial control for the increased microbial challenge in co-infection (Peters and Noverr, 2013).
After sacrifice on day 5, tongues were excised and digitally photographed for Image J analysis of the surface area covered with lesions, as we previously described (Dwivedi et al., 2011). Based on the percentage of the surface area affected, a pathology score (1–4) was assigned. Unmeasurable lesions were assigned a pathology score of 1, measurable lesions covering less than 25% surface area were assigned a score of 2, lesions spanning 25–75% tongue area a score of 3, and lesions covering more than 75% surface area a score of 4 (Grunwald et al., 2012).
For cfu determinations tongues, esophagi, kidneys and livers were excised and homogenized. Undiluted and diluted homogenates were plated on chloramphenicol-supplemented Sabouraud dextrose agar, or Mitis-Salivarius® agar plates for fungal and bacterial cfu counts respectively. Preliminary experiments showed that homogenates plated from uninfected animals showed no fungal or bacterial colony growth on these plates.
Bacterial DNA extraction and streptococcal quantification
Mouse stools were collected daily and bacterial DNA was purified by the QIAGENE DNA Stool Mini Kit according to the company's handbook. Briefly, stool samples from each group were mixed, homogenized and heated for 5 minutes at 70°C. InhibitEX® tablets were added into the supernatants for 1 minute at room temperature. Samples were centrifuged again and the supernatant was treated by proteinase K. After adding 200 μl ethanol to each sample the supernatant was passed through QIAamp columns and DNA was eluted. DNA quantity and quality was assessed using the NanoDrop device (ND-1000 spectrophotometer, NanoDrop Technologies).
To calculate the S. oralis 34 cell numbers in stools we assessed the number of copies of the species-specific gtfR gene in each sample by qPCR, using gtfR-specific primers (Peyyala et al., 2011). In S. oralis 34 gtfR is a single copy gene per haploid genome (Fujiwara et al., 2000). A standard curve was set up with 10-fold serial dilutions of known amounts of gDNA from a pure culture, corresponding to specific gtfR gene copy (or cell) numbers (2.2 fg gDNA per copy gtfR) as described elsewhere (Xie et al., 2011). Results with gtfR-specific primers were validated by designing S. oralis 34 strain-specific primers that spanned the adjoining sequence of two genes (wefA-wefH) in the co-aggregation receptor polysaccharide gene cluster (Yoshida et al., 2006). Real time PCR was performed with BIO-RAD CFX96 cycler and IQ™ SYBR® Green Supermix kit (BIO-RAD) was used to set up all reactions according to the manual. Primer sequences for these genes were as follows: gtfR (Forward: 5′-GCAACCTTTGGATTTGCAAC-3′, Reverse: 5′-TCCCGGTCAGCAAACTCCAGCC-3′) and wefA-wefH (Forward: 5′-CATCAAGAACTTCTCGGAGTTG-3′, Reverse: 5′-CCACAGCTCCAGAATATTTAGC-3′).
Mouse RNA extraction and cDNA synthesis
RNA was extracted from mouse tongues at the end of the infection period (day 5). To extract total RNA tissues were first homogenized using a POLYTRON® homogenizer and the supernatant was beaten by zirconia beads (Ambion). RNA was purified using the QIAgen RNeasy Mini Kit®according to the company manual. RNA concentrations and quality were determined by measuring the absorbance at 260 nm and 280 nm using the NanoDrop device. cDNA was synthesized by using SuperScript III CellsDirect cDNA Synthesis Kit® (Invitrogen), according to the manual.
Mouse whole genome microarrays and real-time RT-qPCR
Total RNA of tongue tissues (> 6 μg per animal), harvested on day 5, from 3 animals in each group (uninfected control, S. oralis, C. albicans, or co-infected) was used for microarray analysis. RNA was extracted as described above and each sample was analysed in triplicate. Microarray service was provided by Phalanx Biotech (OneArray Gene Expression Service). Briefly, cDNA microarray analysis was performed using the Mouse Whole Genome OneArray® v2 (Phalanx Biotech). RNA quality and integrity were determined utilizing an Agilent 2100 Bioanalyzer (Agilent Technologies) and a NanoDrop spectrophotometer. Only high quality RNA, having a RIN of > 6.0, and absorbance ratios A260/A280 > 1.8 and A260/A230 > 1.5, was utilized for further experimentation. RNA was converted to double-stranded cDNA and amplified using in vitro transcription that included amino-allyl UTP, and the aRNA product was subsequently conjugated with Cy5™ NHS ester (GEH Lifesciences). Fragmented aRNA was hybridized at 42°C overnight using the HybBag mixing system with 1X OneArray Hybridization Buffer (Phalanx Biotech), 0.01 mg ml−1 sheared salmon sperm DNA (Promega, Madison, WI, USA), at a concentration of 0.025 mg ml−1 labelled target.
Raw microarray data were normalized and statistical comparisons were performed using Array Studio (Omicsoft Corp.). Fold changes were calculated based on the mean values of the technical replicates for each probe and adjusted P-values were calculated using the Benjamini and Hochberg method with a false discovery rate α-value of 0.05. Genes were identified as significantly differentially expressed when intensity differences were ≥ 2-fold-change and adjusted P-value ≤ 0.05. Annotation analysis was performed to identify the regulated functional groups via Gene Ontology classifications using GOrilla and REViGO web applications (Geiss et al., 2008).
To corroborate the mouse microarray data, we analysed representative host response genes by real time RT-qPCR in the same RNA samples, using the GAPDH housekeeping gene as an internal control. Real time PCR was performed with BIO-RAD CFX96 cycler and IQ™ SYBR® Green Supermix kit (BIO-RAD) was used to set up all reactions according to the manual. Primer sequences for these genes were from PrimerBank (Wang and Seed, 2003; Spandidos et al., 2008; 2010,) and as follows: GAPDH (Forward: 5′-ATCAAGAAGGTGGTGAAGCAGG-3′, Reverse: 5′-GGAAATGAGCTTGACAAAGTTG-3′), Cd177 (Forward: 5′-TCAGCCTTCCTGGGGAGTAAAG-3′, Reverse: 5′-GACGAGGGAGGATGCTTAGAAG-3′), CXCL1 (Forward: 5′-TGGGATTCACCTCAAGAACATC-3′, Reverse: 5′-GGACAATTTTCTGAACCAAGGG-3′), CXCL2 (Forward: 5′-GCCAAGGGTTGACTTCAAGAAC-3′, Reverse: 5′-TTGGATGATTTTCTGAACCAGG-3′) IL-17C (Forward: 5′-TCTGCTGAGGAATTATCTCACGG, Reverse: 5′-GTTCCAGCTAGAGGTCCTTCA-3′), IL-17A (Forward: 5′-TTTAACTCCCTTGGCGCAAAA, Reverse: 5′-CTTTCCCTCCGCATTGACAC-3′), TLR2 (Forward: 5′-CACCACTGCCCGTAGATGAAG, Reverse: 5′-AGGGTACAGTCGTCGAACTCT-3′).
Histological staining and fluorescence in situ hybridization (FISH)
Immunofluorescence staining combined with FISH was used to visualize fungi and bacteria in the same tissue samples, as described previously (Dongari-Bagtzoglou et al., 2009). Briefly, formalin-fixed tissue sections were deparaffinized and stained with a FITC-labelled anti-Candida polyclonal antibody (Meridian Life Science). Slides were washed with PBS and permeabilized with lysozyme for 10 min at 37°C in a humid atmosphere. Samples were then dehydrated in a series of ethanol washes (50%, 80% and 100% ethanol; 3 min each) and exposed to 25 ml of hybridization buffer containing 10 ng ml−1 of streptococcal-specific probe. The oligonucleotide probe used was an Alexa 546-labelled S. oralis-specific probe (Thurnheer et al., 2001) (Eurofins MWG/Operon). Preliminary work showed that tongue tissues from uninfected mice and mice infected with C. albicans only did not hybridize with this probe. Neutrophils were visualized in frozen sections, fixed with cold 95% ethanol, and stained with the monoclonal antibody NIMP-R14, highly specific for murine Ly-6G and Ly-6C (Hycult) followed by a secondary anti–rat antibody conjugated with Alexa 555 (Invitrogen, A-21434). All cells were visualized in the same sections using the nuclear stain Hoechst 33258 (Invitrogen, H3569).
Cytokine and reporter assays
Mouse tongue tissues were homogenized in PBS and supernatants stored at −70°C until assayed. The MIP-2 /CXCL2 protein content in tissue supernatants was measured by ELISA (R&D Systems). Results were expressed as pg of protein per tissue g. To demonstrate that S. oralis can signal via the TLR2 receptor triggering a chemokine response the HEK293-hTLR2 reporter cell line with IL-8 secreted in culture supernatants as a readout. To show broader inflammatory signalling after stimulation with S. oralis the HEK-Blue™ hTLR2 was used (InvivoGen), in which TLR2 ligands induce production of NF-kB- and AP-1-dependent alkaline phosphatase secretion. To ensure the specificity of the TLR2 response vector-transfected (HEK293-pcDNA3) cells and HEK-Blue™ Null1 cells were used as negative controls for IL-8 and alkaline phosphatase responses respectively. Cells were seeded overnight in 96 well plates (105 cells per well) and the next day were challenged with increasing doses of live bacteria alone or in combination with live C. albicans, at 10:1 bacterial : fungal cell ratio. The TLR2 agonist Pam2CSK4 was used as positive control (100 ng ml−1).
MPO assay
The presence of neutrophils in tongue tissues was quantified using a myeloperoxidase (MPO) activity assay, according to a protocol by Bradley et al. (1982). Tissue samples were homogenized with HTAB buffer [50 mM potassium phosphate buffer containing 5 mg ml−1 hexadecyltrimethylammonium bromide (HTAB) pH 6.0]. MPO activity was assayed spectrophotometrically by measuring the H2O2-dependent oxidation of o-dianisidine, as the observed change in absorbance (460 nm) per minute, normalized to tissue weight.
Western blotting
TLR2 protein expression in mouse tissues was analysed by Western blot. Mouse tongue tissues were collected on day 5 post infection and homogenized manually in lysis buffer (25 mM Tris-HCL, 150 mM NaCl, 0.1% SDS, 1% Sodium deoxycholate, 5 mM EDTA, 1% Triton X-100). Total protein was quantified by BIO-RAD Quick-Start Bradford Dye Reagent according to the manual. Forty micrograms of protein was loaded in each lane and proteins were transferred to a PVDF membrane. Membranes were blocked with PBST (0.2% Tween 20), containing 5% skimmed milk powder, and probed with an anti-TLR2 rabbit polyclonal antibody (R&D) at 1:1000 dilution, followed by a secondary antibody (Invitrogen, HRP-rabbit anti-goat IgG). GAPDH was used as an internal loading control.
Neutrophils and neutrophil functional assays
Neutrophils were isolated from the bone marrow of wild type and TLR2−/− animals using a mouse neutrophil isolation kit, per manufacturer's instructions (STEMCELL Technologies). The procedure is based on negative magnetic separation and results in highly purified, functional cell populations (purity > 90% as confirmed with Wright-Giemsa stain) (Kulkarni et al., 2011). In some experiments a human promyelocytic leukaemia cell line (HL-60 cells, ATCC) was used (Xie et al., 2012). These cells are driven to granulocyte differentiation in vitro by exposure to 1.25% of dimethylsulfoxide for 7–9 days, prior to use in functional assays.
The candidacidal activity of freshly isolated bone marrow neutrophils from immunosuppressed WT and TLR2−/− mice was determined by a modified XTT [2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)2H-tetrazolium-5-carboxanilide] assay, which measures residual metabolic activity in C. albicans after exposure to leucocytes, as we described in detail previously (Dongari-Bagtzoglou et al., 2005). Briefly, leucocytes from immunosuppressed animals were added to C. albicans at target to effector cell ratios ranging from 1:5 to 1:10. After mammalian cell lysis with sterile H2O, XTT solution (0.25 mg ml−1 XTT and 40 μg ml−1 coenzyme Q) was added to each well and plates were incubated at 37°C and 5% CO2 for 2 h. Supernatants were transferred into new plates, and optical densities (OD) were measured by an Opsys Microplate Reader (Thermo Labsystems, Franklin, MA) at 450–490 nm, with a 630 nm reference filter. Antifungal activity was calculated according to the following formula: %fungal damage = (1 − x/n) × 100, where x is the OD450 of experimental wells (C. albicans with effectors) and n is the OD450 of control wells (C. albicans only).
Intracellular reactive oxygen species generation by mouse neutrophils and HL-60 cells was measured as described previously (Xie et al., 2012). Briefly, cells preloaded with CM-H2DCFDA (8 μM, Molecular Probes) were allowed to recover for 30 min at 37°C in RPMI, and were stimulated with sonicated log phase S. oralis 34 cells (equivalent of 105–106 streptococcal cells per well) for up to 1 h. To show that this activity was TLR2-dependent a neutralizing anti-TLR2 monoclonal (eBioscience, 10 μg ml−1), or isotype control antibody (IgG2a, 10 μg ml−1), was added to the cells during recovery. Reactive oxygen metabolites were quantified using a fluorescence plate reader at excitation and emission wavelength settings of 485 and 528 nm respectively. Total fluorescence was read at times 0 and 30 min post incubation. After correcting for background fluorescence results were calculated by subtracting fluorescence at time 30 from that at 0 min and expressed as relative fluorescence units (RFU).
Statistical analyses
Data were analysed for statistical differences using the Minitab® or Graph-Pad Prism® software. The fold increase of specific gene transcripts assayed by RT-qPCR and cytokine concentrations were compared among the different conditions using Student's t-tests. The Mann–Whitney asymptotic U-test, adjusted for ties, was used to analyse the non-parametrically distributed pathology scores categorized by quartiles. Statistical significance for all tests was set at P < 0.05.
Acknowledgments
This work was supported by Public Health Service grant RO1 DE013986 from the National Institute of Dental and Craniofacial Research.
Supporting Information
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
C. albicans promotes S. oralis colonization in the intestinal tract. DNA in stool samples of mice infected with C. albicans alone (Ca), S. oralis alone (So), or co-infected with C. albicans and S. oralis (CaSo) were analysed by qPCR using primers specific for the S. oralis gtfR gene.
Mono-infection with S. oralis, at the same total microbial dose as co-infected animals, did not trigger mucosal inflammatory marker gene expression. Pro-inflammatory gene transcripts in tongue tissues of wild type animals on day 5 were assessed by RT-qPCR. Results represent mean fold expression level ± SD compared with uninfected, in 4 animals per group.
S. oralis but not C. albicans activate pro-inflammatory signalling via TLR2. HEK-Blue™ -hTLR2 cells were challenged with live S. oralis 34 and/or C. albicans overnight and supernatants were analysed for alkaline phosphatase activity. Pam2CSK4 was used as a positive control. Two microbial cell doses were tested (Candida: 104, 105 cells ml−1, S. oralis: 105, 106 cells ml−1). HEK-Blue™ Null1 cells did not respond with alkaline phosphatase secretion to any stimulant (not shown). Error bars represent SD of triplicate experiments, *P < 0.001 compared with medium only.
References
- Aas JA, Paster BJ, Stokes LN, Olsen I. Dewhirst FE. Defining the normal bacterial flora of the oral cavity. J Clin Microbiol. 2005;43:5721–5732. doi: 10.1128/JCM.43.11.5721-5732.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aderem A. Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Alam S, Brailsford SR, Adams S, Allison C, Sheehy E, Zoitopoulos L, et al. Genotypic heterogeneity of Streptococcus oralis and distinct aciduric subpopulations in human dental plaque. Appl Environ Microbiol. 2000;66:3330–3336. doi: 10.1128/aem.66.8.3330-3336.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamford CV, d'Mello A, Nobbs AH, Dutton LC, Vickerman MM. Jenkinson HF. Streptococcus gordonii modulates Candida albicans biofilm formation through intergeneric communication. Infect Immun. 2009;77:3696–3704. doi: 10.1128/IAI.00438-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett AP. Recognition and management of invasive pharyngeal candidiasis in acute leukemia. Oral Surg Oral Med Oral Pathol. 1989;67:275–278. doi: 10.1016/0030-4220(89)90353-8. [DOI] [PubMed] [Google Scholar]
- Beisswenger C, Lysenko ES. Weiser JN. Early bacterial colonization induces toll-like receptor-dependent transforming growth factor beta signaling in the epithelium. Infect Immun. 2009;77:2212–2220. doi: 10.1128/IAI.01224-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley PP, Priebat DA, Christensen RD. Rothstein G. Measurement of cutaneaous inflammation: estimation of neutrophil content with an enzyme marker. J Invest Dermatol. 1982;78:206–209. doi: 10.1111/1523-1747.ep12506462. [DOI] [PubMed] [Google Scholar]
- Burns E, Bachrach G, Shapira L. Nussbaum G. Cutting edge: TLR2 is required for the innate response to Porphyromonas gingivalis: activation leads to bacterial persistence and TLR2 deficiency attenuates induced alveolar bone resorption. J Immunol. 2006;177:8296–8300. doi: 10.4049/jimmunol.177.12.8296. [DOI] [PubMed] [Google Scholar]
- Clarke TB, Francella N, Huegel A. Weiser JN. Invasive bacterial pathogens exploit TLR-mediated downregulation of tight junction components to facilitate translocation across the epithelium. Cell Host Microbe. 2011;9:404–414. doi: 10.1016/j.chom.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daley JM, Thomay AA, Connolly MD, Reichner JS. Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83:64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- Dessing MC, Florquin S, Paton JC. van der Poll T. Toll-like receptor 2 contributes to antibacterial defence against pneumolysin-deficient pneumococci. Cell Microbiol. 2008;10:237–246. doi: 10.1111/j.1462-5822.2007.01035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner AC, Yu WH, et al. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Xie Z, Sobue T, Thompson A, Biyikoglu B, Ricker A, et al. Synergistic interaction between Candida albicans and commensal oral streptococci in a novel in vitro mucosal model. Infect Immun. 2012a;80:620–632. doi: 10.1128/IAI.05896-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz PI, Dupuy AK, Abusleme L, Reese B, Obergfell C, Choquette L, et al. Using high throughput sequencing to explore the biodiversity in oral bacterial communities. Mol Oral Microbiol. 2012b;27:182–201. doi: 10.1111/j.2041-1014.2012.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Villar CC. Kashleva H. Candida albicans-infected oral epithelial cells augment the anti-fungal activity of human neutrophils in vitro. Med Mycol. 2005;43:545–549. doi: 10.1080/13693780500064557. [DOI] [PubMed] [Google Scholar]
- Dongari-Bagtzoglou A, Kashleva H, Dwivedi P, Diaz P. Vasilakos J. Characterization of mucosal Candida albicans biofilms. PLoS ONE. 2009;4:e7967. doi: 10.1371/journal.pone.0007967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwivedi P, Thompson A, Xie Z, Kashleva H, Ganguly S, Mitchell AP. Dongari-Bagtzoglou A. Role of Bcr1-activated genes Hwp1 and Hyr1 in Candida albicans oral mucosal biofilms and neutrophil evasion. PLoS ONE. 2011;6:e16218. doi: 10.1371/journal.pone.0016218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara T, Hoshino T, Ooshima T, Sobue S. Hamada S. Purification, characterization, and molecular analysis of the gene encoding glucosyltransferase from Streptococcus oralis. Infect Immun. 2000;68:2475–2483. doi: 10.1128/iai.68.5.2475-2483.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiss GK, Bumgarner RE, Birditt B, Dahl T, Dowidar N, Dunaway DL, et al. Direct multiplexed measurement of gene expression with color-coded probe pairs. Nat Biotechnol. 2008;26:317–325. doi: 10.1038/nbt1385. [DOI] [PubMed] [Google Scholar]
- Gillum AM, Tsay EY. Kirsch DR. Isolation of the Candida albicans gene for orotidine-5′-phosphate decarboxylase by complementation of S. cerevisiae ura3 and E. coli pyrF mutations. Mol Gen Genet. 1984;198:179–182. doi: 10.1007/BF00328721. [DOI] [PubMed] [Google Scholar]
- Gligorov J, Bastit L, Gervais H, Henni M, Kahila W, Lepille D, et al. Prevalence and treatment management of oropharyngeal candidiasis in cancer patients: results of the French CANDIDOSCOPE study. Int J Radiat Oncol Biol Phys. 2011;80:532–539. doi: 10.1016/j.ijrobp.2010.02.006. [DOI] [PubMed] [Google Scholar]
- Grunwald JE, Daniel E, Ying GS, Pistilli M, Maguire MG, Alexander J, et al. Photographic assessment of baseline fundus morphologic features in the comparison of age-related macular degeneration treatments trials. Ophthalmology. 2012;119:1634–1641. doi: 10.1016/j.ophtha.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, et al. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 2011;10:497–506. doi: 10.1016/j.chom.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillegas LM, Griswold DE, Brickson B. Albrightson-Winslow C. Assessment of myeloperoxidase activity in whole rat kidney. J Pharmacol Methods. 1990;24:285–295. doi: 10.1016/0160-5402(90)90013-b. [DOI] [PubMed] [Google Scholar]
- Jamieson AM, Yu S, Annicelli CH. Medzhitov R. Influenza virus-induced glucocorticoids compromise innate host defense against a secondary bacterial infection. Cell Host Microbe. 2010;7:103–114. doi: 10.1016/j.chom.2010.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jergens AE, Wilson-Welder JH, Dorn A, Henderson A, Liu Z, Evans RB, et al. Helicobacter bilis triggers persistent immune reactivity to antigens derived from the commensal bacteria in gnotobiotic C3H/HeN mice. Gut. 2007;56:934–940. doi: 10.1136/gut.2006.099242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston C, Hinds J, Smith A, van der Linden M, Van Eldere J. Mitchell TJ. Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J Clin Microbiol. 2010;48:2762–2769. doi: 10.1128/JCM.01746-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SA. Wingard JR. Infection and mucosal injury in cancer treatment. J Natl Cancer Inst Monogr. 2001;29:31–36. doi: 10.1093/oxfordjournals.jncimonographs.a003437. [DOI] [PubMed] [Google Scholar]
- Knapp S, Wieland CW, van 't Veer C, Takeuchi O, Akira S, Florquin S. van der Poll T. Toll-like receptor 2 plays a role in the early inflammatory response to murine pneumococcal pneumonia but does not contribute to antibacterial defense. J Immunol. 2004;172:3132–3138. doi: 10.4049/jimmunol.172.5.3132. [DOI] [PubMed] [Google Scholar]
- Krause R, Schwab E, Bachhiesl D, Daxbock F, Wenisch C, Krejs GJ. Reisinger EC. Role of Candida in antibiotic-associated diarrhea. J Infect Dis. 2001;184:1065–1069. doi: 10.1086/323550. [DOI] [PubMed] [Google Scholar]
- Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal. 2011;4:ra23. doi: 10.1126/scisignal.2001617. [DOI] [PubMed] [Google Scholar]
- Liljemark WF. Gibbons RJ. Suppression of Candida albicans by human oral streptococci in gnotobiotic mice. Infect Immun. 1973;8:846–849. doi: 10.1128/iai.8.5.846-849.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda Y, Elborn JS, Parkins MD, Reihill J, Goldsmith CE, Coulter WA, et al. Population structure and characterization of viridans group streptococci (VGS) including Streptococcus pneumoniae isolated from adult patients with cystic fibrosis (CF) J Cyst Fibros. 2011;10:133–139. doi: 10.1016/j.jcf.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Moore WE, Holdeman LV, Smibert RM, Hash DE, Burmeister JA. Ranney RR. Bacteriology of severe periodontitis in young adult humans. Infect Immun. 1982;38:1137–1148. doi: 10.1128/iai.38.3.1137-1148.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales DK. Hogan DA. Candida albicans interactions with bacteria in the context of human health and disease. PLoS Pathog. 2010;6:e1000886. doi: 10.1371/journal.ppat.1000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL. Naglik JR. Mucosal immunity and Candida albicans infection. Clin Dev Immunol. 2011;2011:346307. doi: 10.1155/2011/346307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Netea MG, Van der Meer JW. Kullberg BJ. Toll-like receptors as an escape mechanism from the host defense. Trends Microbiol. 2004;12:484–488. doi: 10.1016/j.tim.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Nett JE, Marchillo K, Spiegel CA. Andes DR. Development and validation of an in vivo Candida albicans biofilm denture model. Infect Immun. 2010;78:3650–3659. doi: 10.1128/IAI.00480-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobbs AH, Vickerman MM. Jenkinson HF. Heterologous expression of Candida albicans cell wall-associated adhesins in Saccharomyces cerevisiae reveals differential specificities in adherence and biofilm formation and in binding oral Streptococcus gordonii. Eukaryot Cell. 2010;9:1622–1634. doi: 10.1128/EC.00103-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, et al. A recently evolved transcriptional network controls biofilm development in Candida albicans. Cell. 2012;148:126–138. doi: 10.1016/j.cell.2011.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otten MA, Leusen JHW, Rudolph E, van der Linden JA, Beelen RHJ, van de Winkel JGJ, et al. FcR γ-Chain dependent signaling in immature neutrophils is mediated by FcαRI, but not by FcγRI. J Immunol. 2007;179:2918–2924. doi: 10.4049/jimmunol.179.5.2918. [DOI] [PubMed] [Google Scholar]
- Pei Z, Bini EJ, Yang L, Zhou M, Francois F. Blaser MJ. Bacterial biota in the human distal esophagus. Proc Natl Acad Sci USA. 2004;101:4250–4255. doi: 10.1073/pnas.0306398101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters BM. Noverr MC. Candida albicansStaphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect Immun. 2013;81:2178–2189. doi: 10.1128/IAI.00265-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyyala R, Kirakodu SS, Ebersole JL. Novak KF. Novel model for multispecies biofilms that uses rigid gas-permeable lenses. Appl Environ Microbiol. 2011;77:3413–3421. doi: 10.1128/AEM.00039-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponnuvel KM, Rajkumar R, Menon T. Sankaranarayanan VS. Role of Candida in indirect pathogenesis of antibiotic associated diarrhoea in infants. Mycopathologia. 1996;135:145–147. doi: 10.1007/BF00632335. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S. Medzhitov R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol. 2011;12:1159–1166. doi: 10.1038/ni.2156. [DOI] [PubMed] [Google Scholar]
- Ramsey MM, Rumbaugh KP. Whiteley M. Metabolite cross-feeding enhances virulence in a model polymicrobial infection. PLoS Pathog. 2011;7:e1002012. doi: 10.1371/journal.ppat.1002012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickard AH, Palmer RJ, Blehert DS, Campagna SR, Semmelhack MF, Egland PG, et al. Autoinducer 2: a concentration-dependent signal for mutualistic bacterial biofilm growth. Mol Microbiol. 2006;60:1446–1456. doi: 10.1111/j.1365-2958.2006.05202.x. [DOI] [PubMed] [Google Scholar]
- Sibley CD, Duan K, Fischer C, Parkins MD, Storey DG, Rabin HR. Surette MG. Discerning the complexity of community interactions using a Drosophila model of polymicrobial infections. PLoS Pathog. 2008;4:e1000184. doi: 10.1371/journal.ppat.1000184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjovall J, Huitfeldt B, Magni L. Nord CE. Effect of beta-lactam prodrugs on human intestinal microflora. Scand J Infect Dis Suppl. 1986;49:73–84. [PubMed] [Google Scholar]
- Solis NV. Filler SG. Mouse model of oropharyngeal candidiasis. Nat Protoc. 2012;7:637–642. doi: 10.1038/nprot.2012.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H, Dragnev S, Thurber T. Seed B. A comprehensive collection of experimentally validated primers for Polymerase Chain Reaction quantitation of murine transcript abundance. BMC Genomics. 2008;9:633. doi: 10.1186/1471-2164-9-633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spandidos A, Wang X, Wang H. Seed B. PrimerBank: a resource of human and mouse PCR primer pairs for gene expression detection and quantification. Nucleic Acids Res. 2010;38:D792–D799. doi: 10.1093/nar/gkp1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroncek DF. Neutrophil-specific antigen HNA-2a, NB1 glycoprotein, and CD177. Curr Opin Hematol. 2007;14:688–693. doi: 10.1097/MOH.0b013e3282efed9e. [DOI] [PubMed] [Google Scholar]
- Syed SA. Loesche WJ. Bacteriology of human experimental gingivitis: effect of plaque age. Infect Immun. 1978;21:821–829. doi: 10.1128/iai.21.3.821-829.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarolli V, Gasparoto TH, Lima HR, Figueira EA, Garlet TP, Torres SA, et al. Absence of TLR2 influences survival of neutrophils after infection with Candida albicans. Med Mycol. 2010;48:129–140. doi: 10.3109/13693780902964339. [DOI] [PubMed] [Google Scholar]
- Thom K. Forrest G. Gastrointestinal infections in immunocompromised hosts. Curr Opin Gastroenterol. 2006;22:18–23. doi: 10.1097/01.mog.0000196149.29077.0d. [DOI] [PubMed] [Google Scholar]
- Thurnheer T, Gmur R, Giertsen E. Guggenheim B. Automated fluorescent in situ hybridization for the specific detection and quantification of oral streptococci in dental plaque. J Microbiol Methods. 2001;44:39–47. doi: 10.1016/s0167-7012(00)00226-8. [DOI] [PubMed] [Google Scholar]
- Venkatesh MP, Pham D, Fein M, Kong L. Weisman LE. Neonatal coinfection model of coagulase-negative StaphylococcusStaphylococcus epidermidis) and Candida albicans: fluconazole prophylaxis enhances survival and growth. Antimicrob Agents Chemother. 2007;51:1240–1245. doi: 10.1128/AAC.01298-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar CC. Dongari-Bagtzoglou A. Immune defence mechanisms and immunoenhancement strategies in oropharyngeal candidiasis. Expert Rev Mol Med. 2008;10:e29. doi: 10.1017/S1462399408000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonck RA, Darville T, O'Connell CM. Jerse AE. Chlamydial infection increases gonococcal colonization in a novel murine coinfection model. Infect Immun. 2011;79:1566–1577. doi: 10.1128/IAI.01155-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmore SE. Lamont RJ. The pathogenic persona of community-associated oral streptococci. Mol Microbiol. 2011;81:305–314. doi: 10.1111/j.1365-2958.2011.07707.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolpe SD. Cerami A. Macrophage inflammatory proteins 1 and 2: members of a novel superfamily of cytokines. FASEB J. 1989;3:2565–2573. doi: 10.1096/fasebj.3.14.2687068. [DOI] [PubMed] [Google Scholar]
- Xie Z, Thompson A, Kashleva H. Dongari-Bagtzoglou A. A quantitative real-time RT-PCR assay for mature C. albicans biofilms. BMC Microbiol. 2011;11:93. doi: 10.1186/1471-2180-11-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Thompson A, Sobue T, Kashleva H, Xu H, Vasilakos J. Dongari-Bagtzoglou A. Candida albicans biofilms do not trigger reactive oxygen species and evade neutrophil killing. J Infect Dis. 2012;206:1936–1945. doi: 10.1093/infdis/jis607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano J, Lilly E, Barousse M. Fidel PL. Epithelial cell-derived S100 calcium-binding proteins as key mediators in the hallmark acute neutrophil response during Candida vaginitis. Infect Immun. 2010;78:5126–5137. doi: 10.1128/IAI.00388-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Ganguly S, Bush CA. Cisar JO. Molecular basis of L-rhamnose branch formation in streptococcal coaggregation receptor polysaccharides. J Bacteriol. 2006;188:4125–4130. doi: 10.1128/JB.01843-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Li J, Jia X. Wu Y. The expression of toll-like receptor 2 and 4 mRNA in local tissues of model of oropharyngeal candidiasis in mice. J Huazhong Univ Sci Technolog Med Sci. 24:639–641. doi: 10.1007/BF02911380. [DOI] [PubMed] [Google Scholar]
- Zijnge V, van Leeuwen MB, Degener JE, Abbas F, Thurnheer T, Gmur R. Harmsen H. Oral biofilm architecture on natural teeth. PLoS ONE. 2010;5:e9321. doi: 10.1371/journal.pone.0009321. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
C. albicans promotes S. oralis colonization in the intestinal tract. DNA in stool samples of mice infected with C. albicans alone (Ca), S. oralis alone (So), or co-infected with C. albicans and S. oralis (CaSo) were analysed by qPCR using primers specific for the S. oralis gtfR gene.
Mono-infection with S. oralis, at the same total microbial dose as co-infected animals, did not trigger mucosal inflammatory marker gene expression. Pro-inflammatory gene transcripts in tongue tissues of wild type animals on day 5 were assessed by RT-qPCR. Results represent mean fold expression level ± SD compared with uninfected, in 4 animals per group.
S. oralis but not C. albicans activate pro-inflammatory signalling via TLR2. HEK-Blue™ -hTLR2 cells were challenged with live S. oralis 34 and/or C. albicans overnight and supernatants were analysed for alkaline phosphatase activity. Pam2CSK4 was used as a positive control. Two microbial cell doses were tested (Candida: 104, 105 cells ml−1, S. oralis: 105, 106 cells ml−1). HEK-Blue™ Null1 cells did not respond with alkaline phosphatase secretion to any stimulant (not shown). Error bars represent SD of triplicate experiments, *P < 0.001 compared with medium only.