

Abstract
In neural stem cells (NSCs), glycoconjugates and carbohydrate antigens are known not only to serve as excellent cell surface biomarkers for cellular differentiation and development but also to play important functional roles in determining cell fate. O-linked β-N-acetylglucosamine (O-GlcNAc), which modifies nuclear and cytoplasmic proteins on the serine and threonine residues, is also expected to play an important regulatory role. It is not known, however, whether O-GlcNAc is expressed in NSCs or what the function of this expression is. In this study, we evaluated the patterns and possible functions O-GlcNAcylation in mouse embryonic neuroepithelial cells (NECs), which are known to be rich in NSCs. We confirmed the expression of O-GlcNAc transferase, O-GlcNAcase, and several O-GlcNAcylated proteins in NECs. Treatment of NECs with O-GlcNAcase inhibitors, PUGNAc and streptozotocin, induced robust accumulation of O-GlcNAc in NECs and reduction of number of NECs. In O-GlcNAcase inhibitor-treated NECs, the Rasmitogen-activated protein kinase pathway and the phosphoinositide 3-kinase-Akt pathway, important for proliferation and survival, respectively, were intact, but caspase-3, an executioner for cell death, was activated. These results suggest the possibility that O-GlcNAc is involved in cell death signaling in NECs. Furthermore, for NECs, we identified an O-GlcNAc-modified protein, Sp1 transcription factor. Our study is the first to evaluate expression and functions of O-GlcNAc in NECs.
Keywords: development, carbohydrate, signal transduction
A neural stem cell (NSC) is an undifferentiated neural cell that is endowed with a high potential for proliferation and the capacity for self-renewal, with retention of multipotency to differentiate into neurons and glial cells (Weiss et al., 1996; McKay, 1997; Gage, 2000; Zhao et al., 2008). Because of their biological potential in neurogenesis and neural repair, there has been tremendous interest in the basic biology and clinical use of NSCs. The fate of NSCs is largely defined by extracellular cues, including cytokine signaling generated by the niche and intracellular programs such as epigenetic modifications (Fukuda and Taga, 2005; Zhao et al., 2008; Namihira et al., 2008). Not only cytokine signaling and epigenetic modifications but also “glycosignaling” mediated or modulated by carbohydrate antigens and glycoconjugates, however, is involved in NSC fate regulation (Yu and Yanagisawa, 2007). Glycoconjugates, including proteoglycans, glycoproteins, and glycolipids, are known as useful neural cell-lineage-specific markers, and emerging data indicate that glycoconjugates also mediate cell fate-regulating signals in NSCs (Yanagisawa and Yu, 2007). O-linked β-N-acetylglucosamine (O-GlcNAc) is also expected to have important roles mediating glycosignaling in NSCs.
O-GlcNAc is known to modify posttranslationally the serine and threonine residues of nuclear and cytoplasmic proteins (Wells et al., 2003; Kudlow, 2006; Zachara and Hart, 2006; Hart et al., 2007; Rexach et al., 2008); this modification is referred to as O-N-acetylglucosaminylation (O-GlcNAcylation). O-GlcNAcylation is catalyzed by O-linked N-acetylglucosamine transferase (O-GlcNAcT), which catalyzes the addition of a single O-GlcNAc residue from the donor UDP-GlcNAc to the carrier protein. Conversely, the O-GlcNAc is removed from the carrier protein by O-linked N-acetylglucosaminidase (O-GlcNAcase). O-GlcNAc and the enzymes regulating O-GlcNAcylation have been shown to play various functional roles in cellular processes, including transcription, cell cycle regulation, signal transduction, stress response, apoptosis, glucose sensing, vesicular trafficking, and proteasome degradation. Deletion of O-GlcNAcT in mouse embryonic stem cells is lethal, indicating the essential role of O-GlcNAc for cell integrity (Shafi et al., 2000). Because O-GlcNAcylation occurs in a manner similar to phosphorylation, and both appear at the same or adjacent sites with serine/threonine residues, it has been proposed that O-GlcNAc has a reciprocal relationship with phosphorylation in modulating protein function.
O-GlcNAcT and O-GlcNAcase were ubiquitously expressed but most abundant in brain tissues (Kreppel et al., 1997; Gao et al., 2001), suggesting the importance of O-GlcNAc in brain tissues (Rexach et al., 2008). A number of reports have appeared on the occurrence of O-GlcNAc in brain tissues and cells (Griffith and Schmitz, 1999; Rex-Mathes et al., 2001; Khidekel et al., 2004; Khidekel et al., 2007; Rengifo et al., 2007). O-GlcNAc in brain tissues is found on proteins important for gene expression, neuronal signaling, and synaptic plasticity (Khidekel et al., 2004; Vosseller et al., 2006). Interestingly, it has been reported that O-GlcNAcylation is dynamically modulated by excitatory stimulation of the brain, suggesting the involvement of O-GlcNAcylation in neural circuitry (Khidekel et al., 2007). It has also been reported that neuron-specific deletion of the O-GlcNAcT gene in mice leads to abnormal development, locomotor defects, and postnatal death (O'Donnell et al., 2004). These reports clearly indicate that O-GlcNAc has significant roles in normal brain functions. In addition, O-GlcNAc is suggested to play roles in pathogenesis of Alzheimer's disease (O'Donnell et al., 2004; Hart et al., 2007; Rexach et al., 2008) and aging (Fülöp et al., 2008). It is likely that such O-GlcNAcylation also has important regulatory roles in NSCs. The expression of O-GlcNAc in NSCs, however, is not clearly understood at present. In this study, we investigated the expression and the possible functions of O-GlcNAc in mouse embryonic neuroepithelial cells (NECs).
MATERIALS AND METHODS
Chemical Inhibitors
O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenyl carbamate (PUGNAc; an inhibitor of O-GlcNAcase; Dong and Hart, 1994; Haltiwanger et al., 1998) was purchased from Toronto Research Chemicals (North York, Ontario, Canada). U0126 (an inhibitor of mitogen-activated protein kinase kinase; MEK), tunicamycin (an inhibitor of N-linked glycosylation), streptozotocin (Stz; another inhibitor of O-GlcNAcase; Roos et al., 1998), and alloxan (Alx; an inhibitor of O-GlcNAcT; Konrad et al., 2002) were purchased from Sigma-Aldrich (St. Louis, MO). LY294002, an inhibitor of phosphoinositide 3-kinase (PI3K), was purchased from Cell Signaling Technology (Danvers, MA).
NEC Culture
NECs, which are known to be rich in NSCs (Fukuda et al., 2007), were isolated from telencephalons of ICR mouse embryos (embryonic day 14.5) as previously described (Nakashima et al., 1999; Fukuda et al., 2007). The NECs were cultured in N2-supplemented Dulbecco's modified Eagle's medium/F12 medium (DMEM/F12) containing 10 ng/ml of bFGF (Peprotech, Rocky Hill, NJ) on dishes coated with poly-L-ornithine (Sigma-Aldrich) and bovine fibronectin (Sigma-Aldrich) at 37°C in a humidified 5% CO2 atmosphere. Neurospheres, floating aggregates formed by NSCs in vitro, were prepared by culturing mechanically triturated telencephalon cells on noncoated dishes according to the method previously reported (Yanagisawa et al., 2005a,b), with minor modifications. Mice used for cell preparation were treated according to the guidelines of the Laboratory Animal Service Committee of the Medical College of Georgia.
RT-PCR
RT-PCR was performed as previously described (Ngamukote et al., 2007). In brief, total RNA samples were isolated from mouse brains, neurospheres, or NECs using a Trizol reagent (Invitrogen, Carlsbad, CA). cDNAs were synthesized from the total RNAs as templates using Superscript III reverse transcriptase (Invitrogen). PCR was performed using JumpStart REDTaq (Sigma-Aldrich) with the following settings: 94°C for 2 min; 26–30 cycles of 94°C for 20 sec, 60°C or 58°C for 20 sec, and 72°C for 0.5–1.5 min; and 72°C for 2 min. Primer sets used for PCR were as follows: 5′-GTT AGC TGA GTT GGC ACA TCG-3′, 5′-AGG TCA CTG CGA ACA CAG TAC-3′ [for O-GlcNAcT (NM_139144); PCR product, 420 bp]; 5′-GGA AGA CCT TGG GTT ATG GAG-3′, 5′-CAT TGG TGA TGG AAA CTT GTG C-3′ [for O-GlcNAcase (AF132214); PCR product, 394 bp]; 5′-ATG TAT AAC ATG ATG GAG ACG GAG C-3′, 5′-TCA CAT GTG CGA CAG GGG CAG TGT-3′ (for Sox2; PCR product, 960 bp); 5′-ACC ACA GTC CAT GCC ATC AC-3′, 5′-TCC ACC ACC CTG TTG CTG TA-3′ [for glyceraldehyde-3-phosphate dehydrogenase (G3PDH); PCR product, 452 bp]; 5′-AAG TGC CTG GTC ATC AAG TAT G-3′, 5′-CTG AAG ACT TCT TAA AGT AGC AC-3′ (for thymidine kinase; PCR product, 419 bp). Ten percent dimethyl sulfoxide was added in the PCR mixture to detect Sox2. PCR products were analyzed by agarose gel electrophoresis with 2% agarose gels containing SYBR Safe DNA Gel stain (Invitrogen).
Western Blot Analysis
Mouse brains, neurospheres, or NECs treated with or without a chemical inhibitor in N2-supplemented DMEM/F12 were lyzed in a lysis buffer containing 1% Triton X-100, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, and a protease inhibitor cocktail (Sigma-Aldrich). The protein concentration in the lysates was measured by Bradford method using a Protein Assay Kit (Bio-Rad Laboratories, Hercules, CA). The same amount of protein present in the lysates denatured in Laemmli sample buffer by boiling was applied to SDS-PAGE (4–20% gradient polyacryl-amide gels; Bio-Rad Laboratories) and then transferred to polyvinylidene fluoride membranes (GE Healthcare Life Sciences, Piscataway, NJ). Western blot analysis was performed using subclass control IgM (BD Biosciences, San Jose, CA), anti-O-GlcNAc monoclonal antibody (CTC110.6; IgM; Sigma-Aldrich; Comer et al., 2001), antinestin monoclonal antibody (Rat401; BD Biosciences), anti-β-actin monoclonal antibody (AC-15; Sigma-Aldrich), antiphospho-extracellular signal-regulated kinase (ERK; Thr202/Tyr204) monoclonal antibody (E10; Cell Signaling Technology), anti-ERK polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA), antiphospho-Akt(Thr308) rabbit monoclonal antibody (C31E5; Cell Signaling Technology), antiphospho-Akt(Ser473) rabbit monoclonal antibody (Cell Signaling Technology), anti-Akt polyclonal antibody (Cell Signaling Technology), and anticaspase-3 rabbit monoclonal antibody (8G10; Cell Signaling Technology). Horseradish peroxidase-conjugated anti-mouse IgM antibody (Jackson Immunoresearch, West Grove, PA), anti-mouse IgG antibody, or anti-rabbit IgG antibody (GE Healthcare Life Sciences) was used as the secondary antibody. Protein bands that reacted with the antibodies were detected by using WesternLightning Chemiluminescence Reagent (Perkin Elmer Life And Analytical Sciences, Waltham, MA).
Immunocytochemistry
Immunocytochemistry of NECs was performed as previously described (Yanagisawa et al., 2005a). In brief, NECs cultured in N2-supplemented DMEM/F12 with or without a chemical inhibitor (PUGNAc or Stz) on poly-L-ornithine and fibronectin-coated chamber slides (Nalge Nunc International, Naperville, IL) were fixed with phosphate-buffered saline containing 4% paraformaldehyde. After permeabilization and blocking with phosphate-buffered saline containing 0.1% Triton X-100 and 3% fetal calf serum for 1 hr, the cells were stained with CTC110.6 anti-O-GlcNAc antibody overnight and then with Alexa Fluor 488-conjugated anti-mouse IgM antibodies (Invitrogen) for 2 hr. Hoechst 33258 (Sigma-Aldrich) was used to stain the nuclei. Stained NECs were photographed under a Nikon Eclipse TE300 fluorescent microscope (Nikon Instruments, Melville, NY) equipped with a Magnafire digital charge-coupled device camera (Optronics, Goleta, CA).
WST-8 Assay
The number of cultured NECs was measured by WST-8 assay using a Cell Counting Kit-8 (Dojindo, Kumamoto, Japan; Kanemura et al., 2002; Yu and Yanagisawa, 2007). In brief, NECs were plated onto poly-L-ornithine- and fibronectin-coated 96-well plates and cultured in N2-supplemented DMEM/F12 with or without basic fibroblast growth factor (bFGF; 5 ng/ml) and a chemical inhibitor for 4 days. Then, the cells were incubated with WST-8 [2-(2-methoxy-4-nitrophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium] solution at 37°C for 3 hr. The spectrophotometric absorbance of WST-8-formazan produced by the dehydrogenase activity in the living NECs was measured at the wavelength of 450 nm (reference, 650 nm) using a Benchmark Plus microplate spectrophotometer (Bio-Rad Laboratories).
Caspase-3 Activity Assay
Caspase-3 activity in NECs was measured with a caspase-3/CPP32 Colorimetric Kit (BioVision, Mountain View, CA). In brief, NECs treated with or without a chemical inhibitor (PUGNAc, tunicamycin, or Stz) in N2-supplemented DMEM/F12 were lysed in a cell lysis buffer, and then the NEC lysates containing the same amount of protein were incubated with 200 μM of chromophore p-nitroanilide-conjugated caspase-3 substrate tetrapeptide (Asp-Glu-Val-Asp-pNA) in the reaction buffer at 37°C for 4 hr. The spectrophotometric absorbance of p-nitroanilide cleaved from the Asp-Glu-Val-Asp-pNA by active caspase-3 was measured at the wavelength of 405 nm using a Benchmark Plus microplate spectrophotometer.
Proteasome Activity Assay
The proteasome activity in NECs was measured using a 20S Proteasome Assay Kit (Cayman Chemical Company, Ann Arbor, MI). In brief, NECs treated with or without a chemical inhibitor (PUGNAc or Stz) in N2-supplemented DMEM/F12 were lysed in a lysis buffer, and then the cell lysates containing the same amount of protein were incubated with the fluorogenic substrate succinyl-Leu-Leu-Val-Tyr-4-methylcoumaryl-7-amide in the assay buffer at 37°C for 2 hr. The fluorescence intensity of 4-methylcoumaryl-7-amide cleaved from the substrate by proteasomal activity in the NEC lysates was measured by using a Victor3 V multilabel plate reader (Perkin Elmer Life and Analytical Sciences) equipped with a 360-nm excitation filter and a 460-nm emission filter. Epigallocatechin gallate, a proteasome inhibitor, was added to the reaction mixtures as a negative control.
Immunoprecipitation
Immunoprecipitation was performed to detect O-GlcNAcylation of Sp1 protein. In brief, NECs treated with or without PUGNAc (100 μM) in N2-supplemented DMEM/F12 were lysed in a lysis buffer containing 1% Triton X-100, 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 2 mM Na3VO4, and a protease inhibitor cocktail, and the lysates containing the same amount of protein were gently agitated in the presence of anti-Sp1 polyclonal antibody (PEP 2; Santa Cruz Biotechnology) and protein A-Sepharose (GE Healthcare Life Sciences) at 4°C overnight. After washing with the lysis buffer three times, immunoprecipitates were denatured in Laemmli sample buffer by boiling and subjected to SDS-PAGE followed by Western blot analysis. Detection of Sp1 and O-GlcNAc was performed with anti-Sp1 antibody and CTC110.6 anti-O-GlcNAc antibody, respectively.
RESULTS
Expression of O-GlcNAc in Mouse Embryonic NECs
First, we analyzed the expression of enzymes catalyzing O-GlcNAc turnover, O-GlcNAcT and O-GlcNAcase, in developing mouse brains and mouse NECs that are known to be rich in NSCs (Fukuda et al., 2007). As shown in Figure 1A, both O-GlcNAcT and O-GlcNAcase were expressed in mouse embryonic, postnatal, and adult brains. In addition, these enzymes were found to be expressed in NECs as well as in neurospheres, floating aggregates formed by NSCs in vitro prepared from E14 embryonic brains (Fig. 1B). This result indicates that proteins expressed in mouse embryonic NSCs are O-GlcNAcylated. To confirm this finding further, we evaluated O-GlcNAcylation of proteins expressed in neurospheres and NECs by Western blot analysis with anti-O-GlcNAc antibody. As shown in Figure 1C, several protein bands positive for O-GlcNAc were detected in NEC and neurosphere lysates. The expression pattern of O-GlcNAc-positive proteins in NECs was quite different from that in adult brain tissues but was identical to that in neurospheres. Therefore, we evaluated the functional roles of O-GlcNAc in NSCs using NECs.
Fig. 1.
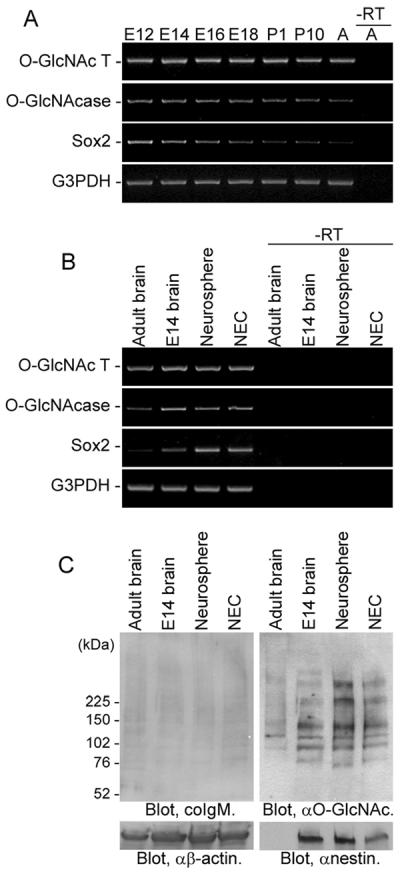
O-GlcNAcylation in developing mouse brains and mouse NSCs. A: Expression of O-GlcNAcT and O-GlcNAcase in developing mouse brains [embryonic day (E) 12, E14, E16, E18, postnatal day (P) 1, P10] and adult mouse brains (A) was examined by RTPCR. B: Expression of O-GlcNAcT and O-GlcNAcase in adult mouse brains, E14 brains, neurospheres (floating cell aggregates formed by NSCs in vitro) and NECs was examined. Sox2 is a marker gene of undifferentiated NSCs. “–RT” indicates negative controls without reverse transcription. C: Expression patterns of O-GlcNAcylated proteins in adult mouse brains, E14 brains, neurospheres, and NECs were analyzed by Western blot. A mouse IgM (coIgM) was used as a subclass control for anti-O-GlcNAc antibody. Nestin is a marker protein of undifferentiated NSCs.
Effects of an O-GlcNAcase Inhibitor, PUGNAc, on NECs
First, we evaluated the functional roles of O-GlcNAc in NECs by treating the cells with an O-GlcNAcase inhibitor, PUGNAc (Dong and Hart, 1994; Haltiwanger et al., 1998). As expected, a robust accumulation of O-GlcNAc in NECs treated with PUGNAc was detected by immunocytochemistry with an anti-O-GlcNAc antibody (Fig. 2A). Also, it was confirmed by Western blot analysis that increasing concentrations of PUGNAc resulted in an increase in detectable bands of O-GlcNAc-positive proteins in the NECs (Fig. 2B). To evaluate effects of O-GlcNAc accumulation, we examined the growth ability of the NECs treated by PUGNAc. To measure the number of NECs, we utilized a highly sensitive and reproducible method, the WST-8 assay (Kanemura et al., 2002; Yu and Yanagisawa, 2007). As shown in Figure 2C, the spectrophoto-metric absorbance measured by this assay is highly correlated with the number of living NECs. The result of the WST-8 assay showed that PUGNAc treatment significantly reduced the number of NECs during culture (Fig. 2D).
Fig. 2.

Effects of the O-GlcNAcase inhibitor, PUGNAc, on NECs. A:O-GlcNAcylation in NECs treated with or without PUGNAc (100 μM) for 24 hr was examined by immunocytochemistry. Nuclei and O-GlcNAc were stained with Hoechst 33258 and CTC110.6 anti-O-GlcNAc antibody, respectively. B: Dose-dependent increases of O-GlcNAcylation in NECs treated with PUGNAc for 24 hr was examined by Western blot analysis. C: Correlation between the NEC number and the spectrophotometric absorbance (Abs) of WST-8-formazan produced by living NECs. The absorbance was measured at the wavelength of 450 nm (reference, 650 nm). D: The NEC number reduction by treatment with PUGNAc for 0–4 days was traced by the WST-8 assay. bFGF was used as a mitogen.
Molecular Mechanisms Underlying Reduction of the Number of NECs by PUGNAc
As the molecular mechanism causing the reduction of NEC number, inhibition of the proliferation signaling, inhibition of the survival signaling, induction of the cell death signaling, or induction of the cell differentiation signaling was expected. These possibilities were evaluated as follows.
The proliferation and survival of NECs are regulated mainly by signaling pathways mediated by serine/threonine kinases, mitogen-activated protein kinase (MAPK), and Akt. As shown in Figure 3A, the proliferation of NECs was inhibited in the presence of U0126, a chemical inhibitor of MEK (MAPK kinase), demonstrating that proliferation of NECs is highly dependent on the Ras-MAPK pathway as previously reported (Yanagisawa et al., 2005b). This confirmation suggests the possibility that activation of this pathway might be inhibited in PUGNAc-treated NECs. Results of Western blot analysis of NEC lysates, however, showed that bFGF-induced activation of ERK (MAPK) was inhibited by U0126 but not by PUGNAc (Fig. 3B). This result suggests that a phosphorylation site of ERK (Thr203 of ERK1 and Thr183 of ERK2) is not O-GlcNAcylated in NECs and that O-GlcNAc accumulation did not inhibit proliferation signaling in NECs, at least at this signaling step. There is a possibility that O-GlcNAc accumulation in NECs is involved in cell cycle regulation at the downstream step such as at cyclin protein expression and mitotic phosphorylation, as previously reported (Slawson et al., 2005).
Fig. 3.
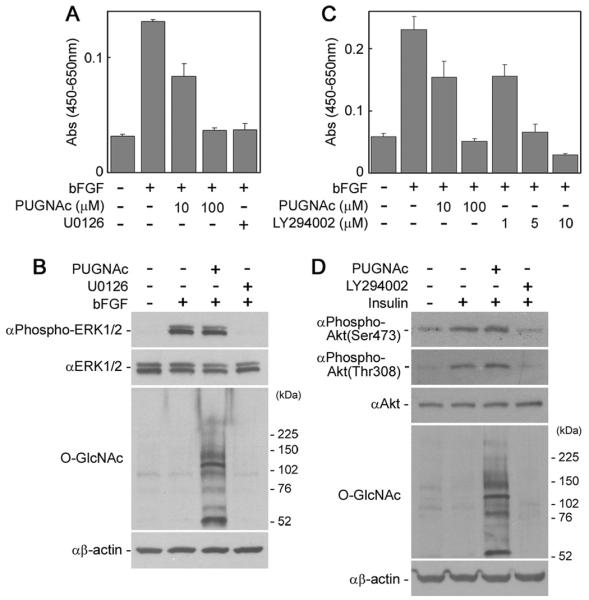
Proliferation and survival signalings in PUGNAc-treated NECs. A: The numbers of NECs cultured with or without PUGNAc or a MEK inhibitor, U0126 (8 lM), in the presence of bFGF (5 ng/ml) for 4 days were measured by the WST-8 assay. B: The activation of ERK MAPK in PUGNAc-treated NECs was analyzed by Western blot. After starvation and treatment with or without PUGNAc (100 μM) for 24 hr or U0126 (4 μM) for 2 hr, the NECs were stimulated with bFGF (20 ng/ml) for 10 min and then lyzed for Western blot analysis. C: The numbers of NECs cultured with or without PUGNAc or a PI3K inhibitor, LY294002, in the presence of bFGF for 4 days were measured by the WST-8 assay. D: Activation of Akt in PUGNAc-treated NECs was analyzed by Western blot. After starvation and treatment with or without PUGNAc for 24 hr or LY294002 (40 μM) for 2 hr, NECs were stimulated with insulin (20 μg/ml) for 10 min and then lysed for Western blot analysis.
Next, we evaluated the survival signaling in PUGNAc-treated NECs. As shown in Figure 3C, the number of NECs was reduced in the presence of a chemical inhibitor of PI3K, LY294006, demonstrating that the survival of NECs is dependent on activation of the PI3K-Akt pathway, as previously reported (Chang et al., 2004). This reduction suggests the possibility that activation of this pathway is inhibited in the presence of PUGNAc. In fact, it has been reported that PUGNAc treatment attenuates Akt phosphorylation at Thr308 in adipocytes (Vosseller et al., 2002; Yang et al., 2008). Western blot analysis of NEC lysates, however, showed that phosphorylation of Akt at Ser473 and Thr308 induced by insulin, a major activator of the survival signaling in NECs in vitro, was attenuated by LY294006 but not by PUGNAc (Fig. 3D). This result suggests that phosphorylation sites of Akt (Thr308 and Ser473) are not O-GlcNAcylated in NECs and that PUGNAc did not affect survival signaling via the PI3K-Akt pathway in NECs. It has been reported that attenuation of Akt phosphorylation by PUGNAc was not found in either neuroblastoma cells (Gandy et al., 2006) or skeletal muscles (Arias et al., 2004). The inhibition of Akt phosphorylation by PUGNAc treatment may be cell type specific. In addition, the expression levels of genes such as Sox2, Pax6, Mash1, Neurogenin, microtubule-associated protein 2, and Tau were not significantly changed in the NECs treated with PUGNAc (data not shown), indicating that the lineage of the NECs is not modified by PUGNAc and O-GlcNAcylation. These results suggest that the cell number reduction by PUGNAc was not caused by inhibition of the proliferation signaling, inhibition of the survival signaling, or induction of the cell differentiation signaling.
In adult mouse hippocampal neurons, O-GlcNAc accumulation has been reported to activate cell death signaling (Liu et al., 2004). As shown in Figure 4A, treatment of NECs with tunicamycin, an inhibitor of N-linked glycosylation and inducer of stress-mediated NSC death (Ohashi et al., 2007), reduced the number of cells. This result suggests that PUGNAc treatment and O-GlcNAc accumulation reduced the number of NECs by activating cell death signaling, so we evaluated that possibility. Caspase-3 is a cysteine protease with aspartate specificity and a critical executioner of apoptosis or programmed cell death signaling, which is activated by proteolytic processing of its inactive precursor. In NECs treated with PUGNAc, both activation of caspase-3 (Fig. 4B) and an increase of the proteolytic activity (Fig. 4C) were confirmed. Therefore, PUGNAc-induced O-GlcNAc accumulation in NECs is considered to activate the cell death signaling mediated by caspase-3 and, at least partially, to reduce the number of cells.
Fig. 4.
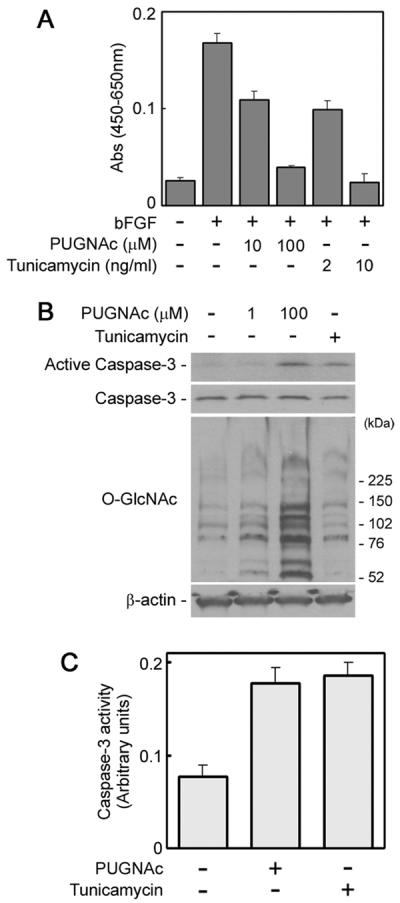
Cell-death signaling in PUGNAc-treated NECs. A: The numbers of NECs cultured with or without PUGNAc or tunicamycin, an inhibitor of N-linked glycosylation and inducer of stress-mediated cell death, in the presence of bFGF for 4 days were measured by the WST-8 assay. B: The activation of caspase-3 in NECs treated with PUGNAc for 24 hr or tunicamycin (0.5 μg/ml) for 8 hr was analyzed by Western blot. C: Caspase-3 activity in NECs treated with PUGNAc for 24 hr or tunicamycin for 8 hr was measured by detecting the absorbance of p-nitroanilide after cleavage from Asp-Glu-Val-Asp-pNA by caspase-3 at the wavelength of 405 nm.
Effects of Another O-GlcNAcase Inhibitor, Streptozotocin, on NECs
As described above, O-GlcNAc accumulation by PUGNAc treatment led to reduction of the numbers of NECs during culture. However, a possibility that reduction of the number of NECs by PUGNAc is not due to O-GlcNAcase inhibition and O-GlcNAc accumulation cannot be eliminated, because PUGNAc is a chemical inhibitor that may have other side effects. To eliminate this possibility, we treated NECs with another O-GlcNA-case inhibitor, streptozotocin (Stz; Roos et al., 1998). As was true with PUGNAc, Stz led to robust accumulation of O-GlcNAc in NECs (Fig. 5A,B). Furthermore, as was true with PUGNAc, Stz reduced the number of NECs during culture (Fig. 5C,D). Although Stz also failed to inhibit bFGF-induced ERK activation and insulin-induced Akt activation (data not shown), both activation of caspase-3 and an increase of caspase-3 activity were detected (Fig. 5E,F). These results suggest that reduction of NECs by PUGNAc and Stz possibly is due to the O-GlcNAcase inhibition, O-GlcNAc accumulation, and activation of cell death signaling. The molecular mechanism underlying the activation of the cell death signaling by PUGNAc and Stz is still unknown. On the other hand, the possibility that the cell death signaling was not directly induced by O-GlcNAc accumulation cannot be completely eliminated (see Discussion).
Fig. 5.

Effects of another O-GlcNAcase inhibitor, Stz, on NECs. A: The increase of O-GlcNAc in NECs treated with Stz (10 mM) for 6 hr was examined by immunocytochemistry. Nuclei and O-GlcNAc were stained with Hoechst 33258 and CTC110.6 anti-O-GlcNAc antibody, respectively. B: Dose-dependent increases of O-GlcNAc in NECs treated with Stz for 6 hr were examined by Western blot analysis. C: The NEC number reduction by treatment with Stz for 0–4 days was traced with the WST-8 assay. D: The NEC number reduction by treatment with Stz for 4 days was compared with that of an MEK inhibitor, U0126 (4 μM). E: The activation of caspase-3 in NECs treated with or without Stz (10 mM) for 8 hr was analyzed by Western blot. F: Caspase-3 activity in NECs treated with or without Stz (10 mM) for 8 hr was assayed by measuring the absorbance of p-nitroanilide after cleavage from Asp-Glu-Val-Asp-pNA by caspase-3 at a wavelength of 405 nm.
Effects of O-GlcNAcase Inhibitors on Proteasome Activity in NECs
The ubiquitin-proteasome pathway is an essential system to select and degrade its substrates tagged with ubiquitin. The proteolytic activity of the proteasomes is reported to be negatively regulated by O-GlcNAcylation (Zhang et al., 2003). In addition, it has been reported that, in hippocampal neurons in adult mice treated with Stz, O-GlcNAc accumulation-caused caspase-3 activation and apoptosis were induced via inhibition of proteasome function (Liu et al., 2004). Therefore, we examined the proteolytic activity of proteasomes in O-GlcNAc-accumulated NECs. Interestingly, in NECs treated with Stz, the proteolytic activity of proteasomes was not severely reduced (just 20–30% reduction in NECs treated with 10 mM Stz; Fig. 6A). In NECs treated with PUGNAc, the proteolytic activity of proteasomes was not reduced at all (Fig. 6B). These results suggest that the proteasomes in NECs are not affected by O-GlcNAc accumulation. Furthermore, the result also suggests that O-GlcNAc accumulation-caused caspase-3 activation in NECs treated with PUGNAc or Stz was not induced via inhibition of proteasome function, which was at variance with the case of the adult mouse hippocampal neurons (Liu et al., 2004). It is likely that inhibition of proteolytic activity of proteasomes by O-GlcNAcase inhibitors is cell type specific.
Fig. 6.

Proteasome activity in NECs treated with O-GlcNAcase inhibitors. Lysates of NECs treated with or without Stz for 8 hr (A) or PUGNAc for 24 hr (B) were incubated with a fluorogenic substrate of the proteasome succinyl-Leu-Leu-Val-Tyr-4-methylcoumaryl-7-amide. After 4 hr, the fluorescence intensity of 4-methylcoumaryl-7-amide cleaved from the substrate by the proteasome activity was measured by using a multilabel plate reader equipped with a 360-nm excitation filter and a 460-nm emission filter. Epigallocatechin gallate (EGCG), a proteasome inhibitor, was used as a negative control.
Effects of an O-GlcNAc Transferase Inhibitor, Alloxan, on NECs
Because of their biological potential, the usefulness of NSCs for cell therapy by transplantation to treat neurodegenerative diseases and spinal cord injury has been much anticipated. Because cell therapy by transplantation requires a number of NSCs, enrichment of the cells after isolation is important. As we found, O-GlcNAc accumulation reduced the number of NECs. Thus, we hypothesized that it is possible to increase NSC numbers by inhibiting O-GlcNAc accumulation. To test this hypothesis, we utilized alloxan (Alx), which has been reported to inhibit O-GlcNAcylation of carrier proteins by O-GlcNAcT (Konrad et al., 2002). First, we treated NECs with Alx. The NEC numbers were not significantly changed during culture, however, in the presence or absence of Alx (Fig. 7A). We assumed that the basal O-GlcNAc level in NECs is already quite low and does not further inhibit proliferation. We then treated NECs with PUGNAc and Alx simultaneously to examine whether Alx can counteract the effect of PUGNAc. We found that Alx did not counteract the effect of PUGNAc at any concentration (Fig. 7B). Western blot analysis of the lysate of NECs treated with Alx revealed that Alx reduced neither the basal O-GlcNAc level (Fig. 7C) nor the accumulated O-GlcNAc level by PUGNAc (Fig. 7D) in NECs. These results indicate that Alx has no inhibitory effect on O-GlcNAcT or inhibits not only O-GlcNAcT but also O-GlcNAcase (Lee et al., 2006) in mouse embryonic NECs.
Fig. 7.

Treatment of NECs with an O-GlcNAcT inhibitor, Alx. The numbers of NECs treated with Alx (A) or Alx and PUGNAc (B) in the presence of bFGF (5 ng/ml) for 4 days were analyzed by the WST-8 assay. The effect of Alx on O-GlcNAcylation in NECs in the absence (C) or presence (D) of PUGNAc was analyzed by Western blot.
Identification of Sp1 Transcription Factor as an O-GlcNAcylated Protein
In this study, Western blot analysis of NEC lysates with anti-O-GlcNAc antibody clearly showed that there are at least 10 protein bands positive for O-GlcNAc, and we undertook a study to identify the O-GlcNAcylated protein. As an O-GlcNAcylated protein, Sp1 transcription factor is well known (Jackson and Tjian, 1988). To examine whether Sp1 is O-GlcNAcylated in NECs, we first analyzed expression of thymidine kinase, a gene whose expression is highly dependent on Sp1 transcription factor (Marin et al., 1997), in NECs treated with or without PUGNAc. As shown in Figure 8A, expression of thymidine kinase was down-regulated in NECs treated with PUGNAc. Because O-GlcNAcylation of Sp1 was reported to decrease transcription activity (Yang et al., 2001), we expected to find that Sp1 is O-GlcNA-cylated in NECs. To evaluate this hypothesis, we immunoprecipitated Sp1 in lysates of NECs treated with or without PUGNAc and analyzed the O-GlcNAcylation. As shown in Figure 8B, immunoprecipitated Sp1 in a lysate of PUGNAc-treated NECs was recognized by anti-O-GlcNAc antibody. Although O-GlcNAcylation of Sp1 is reported to be cell type specific (Wells et al., 2003), this result indicates that Sp1 in NECs can be O-GlcNAcylated. The increase of Sp1 O-GlcNAcylation in PUGNAc-treated NECs may also be involved in the reduction of the number of NECs (see Discussion).
Fig. 8.

Identification of Sp1 transcription factor as an O-GlcNAcylated protein. A: Expression of thymidine kinase, a target gene of Sp1, in NECs treated with PUGNAc (100 μM) for 24 hr was analyzed by RT-PCR. “–RT” indicates negative controls without reverse transcription. B: Lysates prepared from PUGNAc-treated NECs were immunoprecipitated with anti-Sp1 polyclonal antibody and protein A-Sepharose. The immunoprecipitates (IP) were analyzed by Western blot with anti-Sp1 antibody and then reprobed with anti-O-GlcNAc antibody.
DISCUSSION
In this study, we investigated the expression and the functional roles of O-GlcNAc in mouse embryonic NECs and found that accumulation of O-GlcNAc in NECs caused by treatment with O-GlcNAcase inhibitors, PUGNAc and Stz, activated cell death signaling and severely reduced the number of NECs. Previous research has indicated that glycoconjugates to mediate or modulate glycosignaling in NSCs are confined to the cell surface, outside the cell, or within the lumen of intracellular organelles (Yu and Yanagisawa, 2007; Yanagisawa and Yu, 2007). In contrast, O-GlcNAc is found on nuclear and cytoplasmic proteins (Wells et al., 2003; Kudlow, 2006; Zachara and Hart, 2006; Hart et al., 2007; Rexach et al., 2008). The current report presents the first evidence that O-GlcNAc is a glycosignaling molecule functioning in the intracellular space of NSCs. On the other hand, the possibility that the cell death signaling is not directly induced by O-GlcNAc accumulation cannot be completely eliminated. It has been reported that Stz caused genomic DNA fragmentation and cell death in Min6 β-cells in an O-GlcNAc-independent manner, but PUGNAc did not result in any apparent cell damage (Gao et al., 2000). A contaminant in PUGNAc and Stz or a side effect of these inhibitors might have induced the cell death signaling in NECs. This point remains to be tested.
Our study has demonstrated that O-GlcNAc accumulation caused a reduction in the number of NECs in vitro. It is still not clear, however, whether O-GlcNAc and O-GlcNAc-activated cell death signaling occur in NSCs under physiological conditions. There are reports suggesting the involvement of O-GlcNAc accumulation in NSC death in vivo. In adult mammal brains, NSCs are located in the subventricular zone of the lateral ventricles and the subgranular layer of the dentate gyrus in the hippocampus. These NSCs can be identified by administering 5-bromo-2′-deoxyuridine (BrdU) to the animal and then detecting incorporated BrdU in the nuclei of proliferating NSCs. It has been reported that the BrdU-positive cell numbers were significantly reduced in the dentate gyrus and the subventricular zone in rodents administered Stz (Jackson-Guilford et al., 2000; Saravia et al., 2004). Unfortunately, O-GlcNAc accumulation in the brains of these rodents given Stz was not confirmed in those studies, but there is a strong possibility that Stz reduced the number of NSCs via induction of O-GlcNAc accumulation. At present, it is not clear how Stz administration reduced the BrdU-positive numbers of NSCs in vivo. In the hippocampal neurons of mice given Stz, the O-GlcNAcase inhibitor was reported to induce O-GlcNAc accumulation, activation of caspase-3, and subsequently apoptosis (Liu et al., 2004). In a similar manner, Stz may induce O-GlcNAc accumulation and cell death signaling in NSCs in vivo.
On the other hand, we do not consider that the reduction of the number of NECs we found in this study was caused exclusively by activation of cell death signaling. As shown in Figure 8, in PUGNAc-treated NECs, increased O-GlcNAcylation of Sp1 transcription factor was found. This is consistent with the fact that a thymidine kinase gene, a target gene of Sp1 transcription factor, is down-regulated in PUGNAc-treated NECs. Because the O-GlcNAcylation of Sp1 was reported to decrease the transcription activity (Yang et al., 2001), it is possible that Sp1 with excess O-GlcNAcylation malfunctioned in the PUGNAc-treated NECs. It has been reported that Sp1-deficient embryos are retarded in development and die at about day 11 of gestation (Marin et al., 1997). Consequently, the malfunctioning of Sp1 with excess O-GlcNAcylation in NECs treated with OGlcNAcase inhibitors may also be involved in reduction of the number of NECs by disturbing the cells' integrity. Also, the possibility should be considered that OGlcNAc accumulation in NECs is involved in cell cycle regulation, as previously reported (Slawson et al., 2005).
In addition to Sp1, other O-GlcNAcylated proteins may be involved in the integrity of NECs. As we showed by Western blot analysis, at least 10 protein bands positive for O-GlcNAc were detected in NECs. It is likely that these O-GlcNAcylated proteins had multiple defects caused by excess O-GlcNAcylation. It is important, therefore, to identify these O-GlcNAcylated proteins, not only for elucidating the molecular mechanism of the reduction in NECs by O-GlcNAc accumulation but also for better understanding the functional roles of O-GlcNAc in NECs. At this point, a number of new techniques to detect and study O-GlcNAcylation, such as chemoenzymatic tagging methods and quantitative proteomics strategies, have been developed (Rexach et al., 2008). At present, our understanding of the functional roles of O-GlcNAc in NSCs is still fragmentary and incomplete. Further studies using those techniques should reveal more thoroughly the biological functions of O-GlcNAc.
In cell therapy by transplantation of NSCs, glycoconjugate antigens are considered useful for 1) defining NSCs by staining with anticarbohydrate probes, 2) isolating NSCs by cell staining and sorting, 3) enriching NSCs, and 4) modulating NSCs to differentiate into beneficial neuronal or oligodendroglial lineage by modifying the glycosignaling pathways (Yanagisawa and Yu, 2007). In this study, our attempt to enrich NECs by inhibiting O-GlcNAc accumulation with Alx has unfortunately at this point not been successful. A thorough understanding of the biological functions of O-GlcNAc will enable efficient enrichment of NSCs by modifying the glycosignaling.
ACKNOWLEDGMENTS
The authors thank Ms. Donna Li, Dr. Fung-Chow Chiu, and Dr. Guichao Zeng for their help.
Contract grant sponsor: National Institutes of Health; Contract grant number: NS11853; Contract grant number: NS26994; Contract grant number: AG027199; Contract grant sponsor: Childrens' Medical Research Foundation.
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Arias EB, Kim J, Cartee GD. Prolonged incubation in PUGNAc results in increased protein O-linked glycosylation and insulin resistance in rat skeletal muscle. Diabetes. 2004;53:921–930. doi: 10.2337/diabetes.53.4.921. [DOI] [PubMed] [Google Scholar]
- Chang MY, Park CH, Son H, Lee YS, Lee SH. Developmental stage-dependent self-regulation of embryonic cortical precursor cell survival and differentiation by leukemia inhibitory factor. Cell Death Differ. 2004;11:985–996. doi: 10.1038/sj.cdd.4401426. [DOI] [PubMed] [Google Scholar]
- Comer FI, Vosseller K, Wells L, Accavitti MA, Hart GW. Characterization of a mouse monoclonal antibody specific for O-linked N-acetylglucosamine. Anal Biochem. 2001;293:169–177. doi: 10.1006/abio.2001.5132. [DOI] [PubMed] [Google Scholar]
- Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-β-D-glucosaminidase from rat spleen cytosol. J Biol Chem. 1994;269:19321–19330. [PubMed] [Google Scholar]
- Fukuda S, Taga T. Cell fate determination regulated by a transcriptional signal network in the developing mouse brain. Anat Sci Int. 2005;80:12–18. doi: 10.1111/j.1447-073x.2005.00097.x. [DOI] [PubMed] [Google Scholar]
- Fukuda S, Abematsu M, Mori H, Yanagisawa M, Kagawa T, Nakashima K, Yoshimura A, Taga T. Potentiation of astrogliogenesis by STAT3-mediated activation of bone morphogenetic protein-Smad signaling in neural stem cells. Mol Cell Biol. 2007;27:4931–4937. doi: 10.1128/MCB.02435-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fülöp N, Feng W, Xing D, He K, Nöt LG, Brocks CA, Marchase RB, Miller AP, Chatham JC. Aging leads to increased levels of protein O-linked N-acetylglucosamine in heart, aorta, brain and skeletal muscle in brown-Norway rats. Biogerontology. 2008;9:139–151. doi: 10.1007/s10522-007-9123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage FH. Mammalian neural stem cells. Science. 2000;287:1433–1438. doi: 10.1126/science.287.5457.1433. [DOI] [PubMed] [Google Scholar]
- Gandy JC, Rountree AE, Bijur GN. Akt1 is dynamically modified with O-GlcNAc following treatments with PUGNAc and insulin-like growth factor-1. FEBS Lett. 2006;580:3051–3058. doi: 10.1016/j.febslet.2006.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Parker GJ, Hart GW. Streptozotocin-induced β-cell death is independent of its inhibition of O-GlcNAcase in pancreatic Min6 cells. Arch Biochem Biophys. 2000;383:296–302. doi: 10.1006/abbi.2000.2094. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wells L, Comer FI, Parker GJ, Hart GW. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic β-N-acetylglucosaminidase from human brain. J Biol Chem. 2001;276:9838–9845. doi: 10.1074/jbc.M010420200. [DOI] [PubMed] [Google Scholar]
- Griffith LS, Schmitz B. O-linked N-acetylglucosamine levels in cerebellar neurons respond reciprocally to pertubations of phosphorylation. Eur J Biochem. 1999;262:824–831. doi: 10.1046/j.1432-1327.1999.00439.x. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Grove K, Philipsberg GA. Modulation of O-linked N-acetylglucosamine levels on nuclear and cytoplasmic proteins in vivo using the peptide O-GlcNAc-β-N-acetylglucosaminidase inhibitor O-(2-acetamido-2-deoxy-D-glucopyranosylidene)amino-N-phenylcarbamate. J Biol Chem. 1998;273:3611–3617. doi: 10.1074/jbc.273.6.3611. [DOI] [PubMed] [Google Scholar]
- Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–1022. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Tjian R. O-glycosylation of eukaryotic transcription factors: implications for mechanisms of transcriptional regulation. Cell. 1988;55:125–133. doi: 10.1016/0092-8674(88)90015-3. [DOI] [PubMed] [Google Scholar]
- Jackson-Guilford J, Leander JD, Nisenbaum LK. The effect of streptozotocin-induced diabetes on cell proliferation in the rat dentate gyrus. Neurosci Lett. 2000;293:91–94. doi: 10.1016/s0304-3940(00)01502-0. [DOI] [PubMed] [Google Scholar]
- Kanemura Y, Mori H, Kobayashi S, Islam O, Kodama E, Yamamoto A, Nakanishi Y, Arita N, Yamasaki M, Okano H, Hara M, Miyake J. Evaluation of in vitro proliferative activity of human fetal neural stem/progenitor cells using indirect measurements of viable cells based on cellular metabolic activity. J Neurosci Res. 2002;69:869–879. doi: 10.1002/jnr.10377. [DOI] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O-GlcNAc proteome: direct identification of O-GlcNAc-modified proteins from the brain. Proc Natl Acad Sci USA. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, Hsieh-Wilson LC. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nat Chem Biol. 2007;3:339–348. doi: 10.1038/nchembio881. [DOI] [PubMed] [Google Scholar]
- Konrad RJ, Zhang F, Hale JE, Knierman MD, Becker GW, Kudlow JE. Alloxan is an inhibitor of the enzyme O-linked N-acetylglucosamine transferase. Biochem Biophys Res Commun. 2002;293:207–212. doi: 10.1016/S0006-291X(02)00200-0. [DOI] [PubMed] [Google Scholar]
- Kreppel LK, Blomberg MA, Hart GW. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J Biol Chem. 1997;272:9308–9315. doi: 10.1074/jbc.272.14.9308. [DOI] [PubMed] [Google Scholar]
- Kudlow JE. Post-translational modification by O-GlcNAc: another way to change protein function. J Cell Biochem. 2006;98:1062–1075. doi: 10.1002/jcb.20926. [DOI] [PubMed] [Google Scholar]
- Lee TN, Alborn WE, Knierman MD, Konrad RJ. Alloxan is an inhibitor of O-GlcNAc-selective N-acetyl-β-D-glucosaminidase. Biochem Biophys Res Commun. 2006;350:1038–1043. doi: 10.1016/j.bbrc.2006.09.155. [DOI] [PubMed] [Google Scholar]
- Liu K, Paterson AJ, Zhang F, McAndrew J, Fukuchi K, Wyss JM, Peng L, Hu Y, Kudlow JE. Accumulation of protein O-GlcNAc modification inhibits proteasomes in the brain and coincides with neuronal apoptosis in brain areas with high O-GlcNAc metabolism. J Neurochem. 2004;89:1044–1055. doi: 10.1111/j.1471-4159.2004.02389.x. [DOI] [PubMed] [Google Scholar]
- Marin M, Karis A, Visser P, Grosveld F, Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997;89:619–628. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- McKay R. Stem cells in the central nervous system. Science. 1997;276:66–71. doi: 10.1126/science.276.5309.66. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Wiese S, Yanagisawa M, Arakawa H, Kimura N, Hisatsune T, Yoshida K, Kishimoto T, Sendtner M, Taga T. Developmental requirement of gp130 signaling in neuronal survival and astrocyte differentiation. J Neurosci. 1999;19:5429–5434. doi: 10.1523/JNEUROSCI.19-13-05429.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Abematsu M, Nakashima K. Epigenetic mechanisms regulating fate specification of neural stem cells. Philos Trans R Soc Lond B Biol Sci. 2008;363:2099–2109. doi: 10.1098/rstb.2008.2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngamukote S, Yanagisawa M, Ariga T, Ando S, Yu RK. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J Neurochem. 2007;103:2327–2341. doi: 10.1111/j.1471-4159.2007.04910.x. [DOI] [PubMed] [Google Scholar]
- O'Donnell N, Zachara NE, Hart GW, Marth JD. Ogt-dependent X-chromosome-linked protein glycosylation is a requisite modification in somatic cell function and embryo viability. Mol Cell Biol. 2004;24:1680–1690. doi: 10.1128/MCB.24.4.1680-1690.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi H, Nishikawa K, Ayukawa K, Hara Y, Nishimoto M, Kudo Y, Abe T, Aoki S, Wada K. Alpha 1-adrenoceptor agonists protect against stress-induced death of neural progenitor cells. Eur J Pharmacol. 2007;573:20–28. doi: 10.1016/j.ejphar.2007.06.060. [DOI] [PubMed] [Google Scholar]
- Rengifo J, Gibson CJ, Winkler E, Collin T, Ehrlich BE. Regulation of the inositol 1,4,5-trisphosphate receptor type I by O-GlcNAc glycosylation. J Neurosci. 2007;27:13813–13821. doi: 10.1523/JNEUROSCI.2069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex-Mathes M, Werner S, Strutas D, Griffith LS, Viebahn C, Thelen K, Schmitz B. O-GlcNAc expression in developing and ageing mouse brain. Biochimie. 2001;83:583–590. doi: 10.1016/s0300-9084(01)01305-0. [DOI] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Hsieh-Wilson LC. Chemical approaches to understanding O-GlcNAc glycosylation in the brain. Nat Chem Biol. 2008;4:97–106. doi: 10.1038/nchembio.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos MD, Xie W, Su K, Clark JA, Yang X, Chin E, Paterson AJ, Kudlow JE. Streptozotocin, an analog of N-acetylglucosamine, blocks the removal of O-GlcNAc from intracellular proteins. Proc Assoc Am Physicians. 1998;110:422–432. [PubMed] [Google Scholar]
- Saravia F, Revsin Y, Lux-Lantos V, Beauquis J, Homo-Delarche F, De Nicola AF. Oestradiol restores cell proliferation in dentate gyrus and subventricular zone of streptozotocin-diabetic mice. J Neuroendocrinol. 2004;16:704–710. doi: 10.1111/j.1365-2826.2004.01223.x. [DOI] [PubMed] [Google Scholar]
- Shafi R, Iyer SP, Ellies LG, O'Donnell N, Marek KW, Chui D, Hart GW, Marth JD. The O-GlcNAc transferase gene resides on the X chromosome and is essential for embryonic stem cell viability and mouse ontogeny. Proc Natl Acad Sci USA. 2000;97:5735–5739. doi: 10.1073/pnas.100471497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, Hart GW. Perturbations in O-linked β-N-acetylglucosamine protein modification cause severe defects in mitotic progression and cytokinesis. J Biol Chem. 2005;280:32944–32956. doi: 10.1074/jbc.M503396200. [DOI] [PubMed] [Google Scholar]
- Vosseller K, Wells L, Lane MD, Hart GW. Elevated nucleocytoplasmic glycosylation by O-GlcNAc results in insulin resistance associated with defects in Akt activation in 3T3-L1 adipocytes. Proc Natl Acad Sci USA. 2002;99:5313–5318. doi: 10.1073/pnas.072072399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosseller K, Trinidad JC, Chalkley RJ, Specht CG, Thalhammer A, Lynn AJ, Snedecor JO, Guan S, Medzihradszky KF, Maltby DA, Schoepfer R, Burlingame AL. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Mol Cell Proteomics. 2006;5:923–934. doi: 10.1074/mcp.T500040-MCP200. [DOI] [PubMed] [Google Scholar]
- Weiss S, Reynolds BA, Vescovi AL, Morshead C, Craig CG, van der Kooy D. Is there a neural stem cell in the mammalian forebrain? Trends Neurosci. 1996;19:387–393. doi: 10.1016/s0166-2236(96)10035-7. [DOI] [PubMed] [Google Scholar]
- Wells L, Whelan SA, Hart GW. O-GlcNAc: a regulatory post-translational modification. Biochem Biophys Res Commun. 2003;302:435–441. doi: 10.1016/s0006-291x(03)00175-x. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Yu RK. The expression and functions of glycoconjugates in neural stem cells. Glycobiology. 2007;17:57R–74R. doi: 10.1093/glycob/cwm018. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Taga T, Nakamura K, Ariga T, Yu RK. Characterization of glycoconjugate antigens in mouse embryonic neural precursor cells. J Neurochem. 2005a;95:1311–1320. doi: 10.1111/j.1471-4159.2005.03452.x. [DOI] [PubMed] [Google Scholar]
- Yanagisawa M, Nakamura K, Taga T. Glycosphingolipid synthesis inhibitor represses cytokine-induced activation of the Ras-MAPK pathway in embryonic neural precursor cells. J Biochem. 2005b;138:285–291. doi: 10.1093/jb/mvi129. [DOI] [PubMed] [Google Scholar]
- Yang X, Su K, Roos MD, Chang Q, Paterson AJ, Kudlow JE. O-linkage of N-acetylglucosamine to Sp1 activation domain inhibits its transcriptional capability. Proc Natl Acad Sci USA. 2001;98:6611–6616. doi: 10.1073/pnas.111099998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Ongusaha PP, Miles PD, Havstad JC, Zhang F, So WV, Kudlow JE, Michell RH, Olefsky JM, Field SJ, Evans RM. Phosphoinositide signalling links O-GlcNAc transferase to insulin resistance. Nature. 2008;451:964–969. doi: 10.1038/nature06668. [DOI] [PubMed] [Google Scholar]
- Yu RK, Yanagisawa M. Glycosignaling in neural stem cells: involvement of glycoconjugates in signal transduction modulating the neural stem cell fate. J Neurochem. 2007;103(Suppl 1):39–46. doi: 10.1111/j.1471-4159.2007.04710.x. [DOI] [PubMed] [Google Scholar]
- Zachara NE, Hart GW. Cell signaling, the essential role of O-GlcNAc! Biochim Biophys Acta. 2006;1761:599–617. doi: 10.1016/j.bbalip.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–725. doi: 10.1016/s0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]