

Abstract
Although corticosteroids (CSs) affect gene expression in multiple tissues, the array of genes that are regulated by these catabolic steroids is diverse, highly tissue specific, and depends on their functions in the tissue. Liver has many important functions in performing and regulating diverse metabolic processes. Muscle, in addition to its mechanical role, is critical in maintaining systemic energy homeostasis and accounts for about 80% of insulin-directed glucose disposal. Consequently, a better understanding of CS pharmacogenomic effects in these tissues would provide valuable information regarding the tissue-specificity of transcriptional dynamics, and would provide insights into the underlying molecular mechanisms of action for both beneficial and detrimental effects.
We performed an integrated analysis of transcriptional data from liver and muscle in response to methylprednisolone (MPL) infusion, which included clustering and functional annotation of clustered gene groups, promoter extraction and putative transcription factor (TF) identification, and finally, regulatory closeness (RC) identification.
This analysis allowed the identification of critical transcriptional responses and CS-responsive functions in liver and muscle during chronic MPL administration, the prediction of putative transcriptional regulators relevant to transcriptional responses of CS-affected genes which are also potential secondary bio-signals altering expression levels of target-genes, and the exploration of the tissue-specificity and biological significance of gene expression patterns, CS-responsive functions, and transcriptional regulation.
The analysis provided an integrated description of the genomic and functional effects of chronic MPL infusion in liver and muscle.
Keywords: liver, muscle, glucocorticoids, corticosteroids, gene expression, gene regulation, promoter analysis
Introduction
Despite their potent anti-inflammatory effects,1–3 corticosteroids (CSs) are associated with numerous side-effects, particularly related to long-term treatment, eg hyperglycemia, dyslipidemia, arteriosclerosis, muscle wasting, and osteoporosis.4–7 These complex side-effects, which manifest as systematic transcriptional changes in multiple tissues,8,9 necessitate approaches to better decipher the pharmacogenomic effects of CS to properly assess the balance between their advantageous and harmful effects.
There has been an increasing number of recent studies that explored the tissue-specificity of gene expression and regulation,10,11 especially its interplay with pathology.12–14 Several databases were developed to establish a knowledge-base of large-scale data regarding tissue-specific gene expression, regulation, and disease association in a variety of human tissues.15,16 Furthermore, tissue-specificity has also been shown to link with many significant outcomes including expression-quantitative trait loci,17 evolution,14,18 and disease-association,13,19 eg tissue-specific effects of insulin signaling in diabetes.20 Consequently, in this study we placed emphasis on exploring the tissue-specificity of chronic CS administration in liver and muscle to obtain a better understanding of CS-responsive functions and its side-effects. We first identified tissue-specific transcriptional dynamics using a novel clustering approach proposed in our previous study.21 Subsequently, with the hypothesis that common functions activated across multiple tissues will be more likely to be important CS-responsive functions, the KEGG database was utilized to identify such common functions.
However, it has been noted that genes affected by CS include both immunosuppressive genes, mostly associated with therapeutic effects, and metabolic genes often associated with adverse effects whose regulation is mainly controlled by glucocorticoid receptor (GR) through gene-mediated pathways.6 However, chronic infusion of CS causes a sustained down-regulation of the receptor (mRNA and thus protein).22,23 Although several alternative mechanisms have been proposed,24–26 it is still not understood why drug effects remain strong even after the down-regulation of GR mRNA to the point of almost being eliminated. A plausible explanation is that, in addition to direct regulation through glucocorticoid response element (GRE) binding sites in the 5′ regulatory regions of genes, there are changes in expression that are also the indirect results of CS effects because of changes in expression of transcription factors (TFs) that act as secondary bio-signals directly or indirectly modulating the transcription of genes.23,27,28 Therefore, in addition to identifying critical transcriptional responses and CS-responsive functions, finding relevant TFs and understanding their relationship with CS-responsive functions is also an important aspect in this analysis.
Materials and Methods
Datasets
Adrenalectomized (ADX) male Wistar rats with body weights of 339 ± 28 g (SD) were housed and maintained under constant temperature (22 °C) and humidity with a controlled 12-hour light/dark cycle for a period of at least two weeks before surgery.22,29 Rats had free access to rat chow and 0.9% NaCl drinking water. Forty rats were given 0.3 mg/kg/hour infusions of methylprednisolone (MPL) sodium succinate for more than 168 hours via an Azlet pump. The drug solutions were prepared for each rat based on its predose body weight. Pumps were subcutaneously implanted between the shoulder blades. Animals were sacrificed at various times up to seven days, specifically at time-points 6, 10, 13, 18, 24, 36, 48, 72, 96, and 168 hours. A control group of four animals was implanted with a saline-filled pump and killed at various times throughout the seven-day study period. Liver and gastrocnemius muscle samples from each animal were ground into a fine powder in a mortar cooled by liquid nitrogen, and RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA, USA). The gene expression was obtained via the Affymetrix RAE230A array, which consists of 15,923 probesets. The data are publicly available through the GEO database under accession numbers GDS972 and GDS2688 corresponding to the liver and muscle datasets during chronic MPL infusion.
Clustering
Utilizing the concept of the agreement matrix (AM) in consensus clustering, we recently proposed a novel method to identify the core group of probesets showing the most agreement indicating they belong to the same patterns of gene expression.21 To produce the AM, a number of different clustering methods (hierarchical clustering, divisive analysis clustering, partitioning around medoids, fuzzy analysis clustering, k-means, self-organizing map, fuzzy c-means clustering, and model-based clustering) along with different metrics (Euclidean, Manhattan, and Pearson correlation) were used to reduce the bias inherent in the assumption of any specific clustering method/metric. After identifying the core set of probesets, the AM is reduced correspondingly to these selected probesets, and then the hierarchical clustering is applied on the reduced AM to obtain significant patterns of gene expression. As there should be clusters of genes located closely to other clusters in the data and the input number of clusters for the core analysis is only suggestive, these clusters may not be completely separated. As a result, some significant clusters may be not included in the selected subset because of the constraint in “clusterable” selection. Therefore, we repeated the procedure of selection and clustering on the removed domain as proposed by Nguyen et al.30 Subsequently, a trivial-cluster removal procedure and a merging process were applied to obtain final significant patterns of gene expression.30
CS-responsive function identification
We first identified the pathways from the KEGG database that were enriched in each of the gene expression patterns from each tissue (P-value < 0.05, at least five genes) using ArrayTrack.31 An intersection between the set of selected pathways in liver and those in muscle was performed to extract all common functions that were activated in both tissues during MPL infusion.
Promoter extraction and processing
The promoters of genes including all transcript-relevant alternative promoters were extracted from a rich database of promoter information with a default length (500 bp upstream and 100 bp downstream of the transcription start site) if there is no experimentally defined length suggested by Genomatix.32 To accelerate the process of identifying putative transcriptional regulators, promoters were pre-processed as in Nguyen et al.33 Specifically, MatInspector34 was applied to scan position weight matrix (PWM) matches on those promoter sequences using optimal parameters from MatBase, which ensures that the minimum number of matches found in non-regulatory sequences, ie, the false-positive matches, was minimized.32 Each gene promoter was then re-modeled to become a list of TF binding sites (TFBSs) ordered by their local positions on the promoter sequences and represented by the corresponding TF names along with their binding orientations. The conversion supports fast search for the presence of a TFBS or a cis-regulatory module (CRM) on promoter sequences.
Putative transcriptional regulator prediction
To predict putative transcriptional regulators, we utilized the context-specific CRM search technique to identify over-represented CRMs in the promoter set of a gene battery.33 Each gene battery contains a certain set of genes that are hypothetically co-regulated, ie co-expressed and co-functional in this study. With the hypothesis that common functions activated across multiple tissues may play important roles in response to CS dosing, we applied our previous tool33 to identify TFs relevant to CS transcriptional responses. In brief, we computationally defined a CRM as a list of non-overlapping TFBSs ordered by their positions on the promoter sequence and characterized with their corresponding binding strand orientation. The procedure first identified all potential TFBSs that are common in the corresponding promoter set and then searched for all possible combinations of all commonly found TFBSs above using the breadth-first search technique. Owing to the fact that a CRM can be present on promoters of many genes in the background set, we estimated the statistical significance of commonly identified CRMs for each gene battery vs. those for the background set to select those that are significantly over-represented. Subsequently, the selected CRMs are decomposed to obtain a list of TFs that are associated with the corresponding TFBSs present on CRMs.
Regulatory closeness (RC) definition
Using the hypothesis that with the more relevant transcriptional regulators, two gene batteries will share higher closeness of regulatory mechanisms, the RC was estimated to quantify the relationship between gene expression profiles, function, and transcriptional regulation. In this study, we defined the RC as the common ratio (CR) of relevant TFs shared between two gene batteries CR = c/a + c/b, where a and b are the number of TFs recognized as relevant transcriptional regulators in gene batteries A and B, and c is the number of common TFs between the two. Its range therefore is from 0 to 2 where 0.8 was selected as a cut-off value for high RC.
Results
This study involved infusion of MPL for more than 168 hours in ADX rats. Three animals were sacrificed at each of 12 time-points, and diverse tissues harvested and processed to assess gene expression using Affymetrix arrays. The liver and muscle tissues were compared in this analysis (see Materials and Methods). The pharmacokinetics of MPL for this study was reported previously35 showing steady-state concentrations in plasma at six hours and thereafter.
Critical transcriptional responses
To analyze gene expression data during chronic MPL infusion, a pre-processing step was performed by filtering differentially expressed probesets using an ANOVA test (P-value <0.05) implemented in R.36 There were 4,124 probesets in the liver dataset and 3,805 probesets in the muscle dataset selected for further analysis. All datasets were pre-processed to obtain the “true” expression profiles that incorporate the error information in replicates instead of taking the simple average expression profiles for clustering.37 The suggested number of clusters (nc*) for both datasets was 7. With the hypothesis that the more clusterable the data, the more biologically relevant they are, a more clusterable subset from each dataset was extracted and then clustered to identify significant expression patterns in each tissue during chronic MPL infusion. These significant patterns are considered as key transcriptional responses because they contain a significant number of co-expressed genes that are differentially expressed during chronic CS infusion.
For responses in liver, we obtained 12 significant patterns with 2,285 genes in total. The expression patterns as z-score vs. time of these transcriptional responses are shown in Figure 1 with the average expression patterns of all genes in each cluster. Although genes may exhibit simple or complex patterns of expression during CS administration, we only describe them with up- and/or down-regulation in this study. In brief, pattern 1 (182 genes) exhibited an early down-regulation at six hours post-MPL infusion followed by an up-regulation reaching the peak at about 18 hours. Subsequently, it was again down-regulated until 48 hours and then gradually returned to the baseline. Genes in patterns 2 (80 genes), 3 (400 genes), 4 (48 genes), and 5 (429 genes) exhibited a fast and robust enhancement in mRNA and reached their corresponding maximum peaks at 6, 10, 10, and 13 hours, respectively. Although all of them are followed by a down-regulation that reaches the baseline at 24 hours, patterns 2 and 3 seem to have a slight enhancement before returning to the baseline whereas patterns 4 and 5 showed slight sustained down-regulation until 48 hours and then may (pattern 5) or may not (pattern 4) return to the baseline. Patterns 6 (176 genes) and 7 (312 genes) show two other interesting transcriptional responses during chronic MPL infusion because they eventually converged to a new steady state in the presence of the drug, namely sustained up-regulation (pattern 6) and sustained down-regulation (pattern 7). The last five patterns show a strong down-regulation followed by a sharp enhancement. Pattern 8 (102 genes) reached a nadir at about six hours and then up-regulated to reach the peak at 24 hours, which is followed by some fluctuations before returning to the baseline. Similarly, patterns 9 (80 genes) and 10 (54 genes) exhibited down-regulation in gene expression reaching a nadir at about 10 hours followed by an up-regulation to reach the peak at about 24 hours. However, genes in pattern 9 eventually reach the baseline whereas genes in pattern 10 are stabilized to a new steady state. Additionally, the 300 genes in cluster 11 and 122 genes in cluster 12 display declines in mRNA expression at 10 and 13 hours and subsequently show an up-regulation to the peak at about 36 hours. After that, their expression gradually goes back to the baseline (which happens faster for genes in pattern 12).
Figure 1.

Critical dynamic transcriptional responses within individual tissues under chronic corticosteroid administration. Each pattern is characterized by the average gene expression profile of the corresponding cluster of genes in the liver dataset. The error bar shows the standard deviation of all probeset transcript levels at each time-point in each corresponding pattern.
In muscle responses, there are seven significant expression patterns with 2,291 genes in total. However, almost all genes follow two main transcriptional responses as characterized by patterns 1 and 2 in Figure 2. Pattern 1 including 718 genes exhibited an early down-regulation during the first 6 hours followed by a robust up-regulation to reach the peak at about 36 hours with expression levels maintained until 96 hours before returning to the baseline. In contrast, 1,012 genes in cluster 2 expressed an opposite pattern that shows a transient up-regulation in expression at six hours during MLP infusion followed by a strong decline reaching the nadir at about 36 hours. These genes show sustained repression until about 96 hours and then gradually return to the baseline. Genes in cluster 3 (261 genes) exhibited a similar pattern, but there is no early transient up-regulation in the first 6 hours, and the nadir is reached at about 18 hours. Pattern 4 (76 genes) showed a robust, fast down-regulation during the first 10 hours reaching a new steady state without returning to the baseline. Pattern 5 (63 genes) characterizes an initial up-regulation to the peak at about 10 hours followed by a down-regulation to reach the nadir at 96 hours. Patterns 6 (65 genes) and 7 (96 genes) exhibited a simple up-regulation followed by a down-regulation to the baseline. However, genes in pattern 6 reach the peak faster at about 18 hours, which was maintained until 96 hours, whereas genes in pattern 7 reach their peak at about 96 hours and then simply return to the baseline (see Supplemental Material 1).
Figure 2.

Critical dynamic transcriptional responses in muscle under chronic corticosteroid administration. Each pattern is characterized by the average gene expression profile of the corresponding cluster of genes in the muscle dataset. The error bar shows the standard deviation of all probeset transcript levels at each time-point in each corresponding pattern.
CS-responsive genes and functions
To identify specific genes and functions caused by drug dosing, we assume that co-expressed genes involved in pathways activated by MPL are more likely to be important genes and functions. Using the KEGG database and ArrayTrack,31 we searched for enriched pathways in these co-expressed clusters (P-value <0.05 with at least five genes). Significant pathways that can be considered as CS-responsive functions were divided into two types: one takes place in a tissue-specific manner in one of the two tissues (Table 1) and the other consists of those commonly activated in both tissues (Table 2). Although there are a large proportion of CS-responsive metabolic and signaling pathways activated in liver and muscle, metabolic pathways seemingly dominate in liver and signaling pathways dominate in muscle. In addition, many CS-affected metabolic and signaling functions in liver are different from those in muscle. Specifically for those tissue-specific functions shown in Table 1, liver has five enriched metabolic pathways including arginine and proline metabolism, drug metabolism—cytochrome P450, retinol metabolism, starch and sucrose metabolism, and valine, leucine, and isoleucine degradation whereas only one signaling pathway (TGF-beta signaling). In contrast, muscle genes display seven signaling pathways (eg, adipocytokine signaling, NOD-like receptor signaling, B-cell receptor signaling, calcium signaling, ErbB signaling, GnRH signaling, and phosphatidylinositol signaling) vs. five metabolic pathways (eg, fatty acid, glutathione, glycolysis/gluconeogenesis, pyrimidine, and pyruvate metabolism). In addition, there are a number of other critical liver-specific (eg, ribosome, antigen processing and presentation, and complement and coagulations cascades) and muscle-specific (eg, RNA polymerase, DNA replication, and Fc gamma R-mediated phagocytosis) functions.
Table 1.
Tissue-specific regulation by functional characterization.
| NO. | LIVER-SPECIFIC FUNCTIONS* | MUSCLE-SPECIFIC FUNCTIONS* |
|---|---|---|
| 1 | Ribosome (1)$ | Adipocytokine signaling pathway (1) |
| 2 | TGF-beta signaling pathway (3) | Fatty acid metabolism (1) |
| 3 | Ribosome (5) | Glutathione metabolism (1) |
| 4 | Arginine and proline metabolism (6) | Nod-like receptor signaling pathway (1) |
| 5 | Antigen processing and presentation (7) | Pyrimidine metabolism (1) |
| 6 | Cell adhesion molecules (7) | RNA polymerase (1) |
| 7 | Complement and coagulation cascades (7) | B cell receptor signaling pathway (2) |
| 8 | Leukocyte transendothelial migration (7) | Calcium signaling pathway (2) |
| 9 | Systemic lupus erythematosus (7) | DNA replication (2) |
| 10 | Complement and coagulation cascades (11) | ErbB signaling pathway (2) |
| 11 | Drug metabolism—cytochrome P450 (11) | Fc gamma R-mediated phagocytosis (2) |
| 12 | Retinol metabolism (11) | Gap junction (2) |
| 13 | Starch and sucrose metabolism (11) | Glycolysis/Gluconeogenesis (2) |
| 14 | Valine, leucine and isoleucine degradation (11) | GnRH signaling pathway (2) |
| 15 | Long-term potentiation (2) | |
| 16 | Melanogenesis (2) | |
| 17 | Natural killer cell mediated cytotoxicity (2) | |
| 18 | Phosphatidylinositol signaling system (2) | |
| 19 | Pyruvate metabolism (2) | |
| 20 | Regulation of actin cytoskeleton (2) |
Notes:
Functions are KEGG pathways that are enriched by sets of coexpressed genes in corresponding expression patterns. (x)$, corresponding expression patterns of functions.
Table 2.
Detailed information of selected gene batteries in liver and muscle.
| NO. | FUNCTIONS | LIVER
|
MUSCLE
|
||
|---|---|---|---|---|---|
| PATTERNS | GENES | PATTERNS | GENES | ||
| 1 | Adherens junction | 7 | actn1, ctnna1, iqgap1, ptprm, rac2 | 2 | actn3, ctnnb1, fyn, iqgap1, loc679869, met, ptprf, ptprm, pvrl2, rac1 |
|
| |||||
| 2 | Aminoacyl-tRNA biosynthesis | 5 | aars, eprs, farsa, farsb, mars2, tars, wars, yars | 1 | cars, gars, kars, lars, mars, nars, qars, sars, tars |
|
|
|||||
| 3 | ECM-receptor interaction | 7 | cd47, col1a2, col3a1, lamc1, reln | 2 | chad, col1a1, col1a2, col3a1, col4a1, col5a1, col5a2, col5a3, col6a1, col6a2, fn1, itgb1, lamb1, lamc1, tnc |
|
|
|||||
| 4 | Focal adhesion | 7 | actn1, ccnd1, col1a2, col3a1, igf1, lamc1, rac2, rap1b, reln | 2 | actn3, cav3, chad, col1a1, col1a2, col3a1, col4a1, col5a1, col5a2, col5a3, col6a1, col6a2, ctnnb1, egf, fn1, fyn, hras, itgb1, lamb1, lamc1, met, mylpf, pak1, pdgfa, ppp1r12a, prkcb, rac1, tnc |
|
|
|||||
| 3 | grb2, igf1, map2k1, ik3cd, vwf | ||||
| 5 | Insulin signaling pathway | 6 | eif4ebp1, fasn, fbp1, mtor, ppp1r3b, ppp1r3c | 2 | calm3, fbp2, flot1, fot2, hras, phka1, phkg1, prkaa2, prkab2, prkar1a, prkci, ptprf, rhoq,sh2b2 |
|
| |||||
| 6 | Lysosome | 11 | acp2, ctsh, dnase2b, fuca1, glb1, hexa, lamp2 | 1 | acp2, ap1 m1, ap3b1, ap3d1, atp6v0a1, atp6v0d1, cln5, cltb, ctsf, ctsl1, gaa, gga1, lgmn |
| 7 | MAPK signaling pathway | 3 | cdc42, gadd45a, il1r, kras, map2k1ip1, mapk14, max, pla2 g12a, srf, tp53 | 2 | cacna2d1, cacng1, dusp14, dusp6, egf, evi1, hras, map2k6, pak1, pdgfa, ppp3ca, ppp3cb, ppp3r1, prkcb, rac1, rasa1, srf, tgfb2 |
| 8 | Neurotrophin signaling pathway | 5 | akt1, arhgdia, bad, rac1, tp53, ywhae, ywhag, ywhah, ywhaz | 2 | calm3, camk2a, camk2d, camk2 g, hras, maged1, pdk1, rac1, sh2b2, ywhaq, ywhaz |
| 9 | Cell cycle relevant-processes | 5 | ppp2r1a, ppp2r5a, rbx1, ywhae, ywhag, ywhah, ywhaz | 2 | anapc1, calm3, camk2a, camk2d, camk2 g, ccnb2, cdc2, espl1, itpr1, ppp2r5a, ppp2r5c, ppp3ca, ppp3cb, ppp3r1, slk, ywhaq, ywhaz |
| 6 | atp5e, atp5l, cox6a1, cox8a, ndufa1, ndufb6, ndufs5, ndufs6, ndufv2, ndufv3, sdhd, uqcr, uqcrq | ||||
|
|
|||||
| 10 | Oxidative phosphorylation | 11 | atp5a1, atp5c1, atp5i, atp5o, cox6c, cox7a2, cox7b, ndufa11, ndufa2, ndufa4, ndufa6 | 5 | atp5 j, cox5a, cox7a2, ndufb2, uqcrh |
|
|
|||||
| 8 | atp5f1, atp5h, cox4i1, cox5b, sdhc, ndufa3, ndufb4, ndufb7, ndufb9 | ||||
|
| |||||
| 11 | Pentose phosphate pathway | 6 | aldob, fbp1, gpi, pgls, pgm1 | 2 | fbp2, g6pd, gpi, pfkp, pgm1 |
|
| |||||
| 12 | PPAR signaling pathway | 12 | acaa1, acox2, acsl1, apoa1, scp2 | 1 | acadl, acox1, acsl4, cpt1b, cpt2, ehhadh, nr1h3, slc27a1 |
|
| |||||
| 13 | Proteasome | 5 | psma3, psma5, psmb1, psmb2, psmc4, psmd13, psmd3, psmd4, psmd8 | 1 | psma1, psma2, psma3, psma4, psma5, psma7, psmb2, psmb3, psmb4, psmb7, psmc1, psmc2, psmc4, psmc5, psmd11, psmd13, psmd1, psmd3, psmd4, psmd6, psmd7, psmd8 |
|
| |||||
| 14 | Purine metabolism | 5 | adsl, ak2, gart, guk1, nt5c3, pole4, polr3d, prps2 | 1 | adcy2, ampd3, impdh2, pde2a, pde4b, pold4, polr1a, polr1b, polr1c, polr1e, polr2e, polr2f |
| 15 | Spliceosome | 3 | bat1, hnrnpa1, hnrnpk, lsm5, pcbp1, magoh, nhp2l1, plrg, prpf19, rbm17, rbm8, sf3b2, sf3b4, sfrs1, snrpb | 1 | bud31, cherp, cwc15, hnrnpu, ncbp1, pcbp1, prpf3, prpf6, sart1, thoc1, usp39, wbp11 |
| 5 | dhx15, eftud2, hnrnpc, lsm7, lsm8, prpf8, smndc1, snrpe | ||||
|
| |||||
| 16 | Tight junction | 3 | cdc42, kras, ppp2ca, ppp2cb, spna2, vapa | 2 | actn3, amotl1, cldn5, ctnnb1, cttn, epb4.1l2, exoc4, f11r, hras, myh3, mylpf, prkcb, prkci |
|
|
|||||
| 7 | actn1, amotl1, cldn3, ctnna1, exoc3, ocln, rras2 | ||||
| 17 | Ubiquitin mediated proteolysis | 3 | prpf19, uba2, ube2d3, ube2g2, ube2 j2, ube2n, ube2 s, vhl | 1 | anapc7, birc3, herc4, keap1, klhl9, pias4, ube2d3, ube2 g1, ube2 j1, ube2l3, ube4a |
|
|
|||||
| 5 | cdc34, rbx1, rchy1, syvn1, ube2d3, ube2e3, ube2f, ube2 j1 | ||||
| 18 | Wnt signaling pathway | 3 | ppp2ca, ppp2cb, psen1, ruvbl1, senp2, smad4, tp53 | 2 | camk2a, camk2d, camk2 g, ctnnb1, ctnnbip1, daam1, loc679869, ppp2r5a, ppp2r5c, ppp3ca, ppp3cb, ppp3r1, prkcb, rac1 |
Notes: +genes shown in bold type are those whose promoters contain GRE (Glucocorticoid Receptor Element) binding sites. Details are listed in supplemental material 2.
A number of pathways are significantly enriched in both tissues. Because these co-expressed genes reflect critical transcriptional responses during MPL infusion, we hypothesize that common functions activated across multiple tissues are important CS-responsive functions. Furthermore, along with the concept of “gene battery,”38,39 we define gene sets associated with each function in each corresponding cluster as gene batteries. Table 2 shows 18 common functions activated in both tissues during MPL infusion, which include 23 gene batteries in liver and 19 in muscle. These functions mainly belong to four pathway categories including signaling pathways (insulin signaling, MAPK signaling, neurotrophin signaling, PPAR signaling, and Wnt signaling), metabolic pathways (oxidative phosphorylation, pentose phosphate pathway, and purine metabolism), and pathways relevant to cellular process (adherens junction, focal adhesion, tight junction, lysosome, and cell cycle relevant processes) and information processing (aminoacyl-tRNA biosynthesis, proteasome, spliceosome, ubiquitin-mediated proteolysis, and ECM—receptor interaction). Additionally, it has been noted that expression levels of many CS-affected genes are mediated through the binding motifs located on their control regions, called GREs. We thus examined the presence of this binding site on the promoter of genes in each of the enriched pathways to assess the potential effect of GRE on common functions. Following the approach in our previous study,30 genes shown in bold are those whose promoters contain GRE motifs located on conserved regions identified from the corresponding sets of orthologous promoters (details in Supplemental Material 2). The average ratio between genes containing GRE motifs vs. genes without GRE motifs in liver gene batteries is 0.35 where 7 out of 23 gene batteries have ratios ≥0.5. The average ratio in muscle is 0.47 where there are 10 out of 19 gene batteries with ratios ≥0.5. Also, we find that many CS-responsive genes in signaling cascades and information processing pathways (eg, MAPK signaling, Wnt signaling, aminoacyl-tRNA biosynthesis, and proteasome) are more likely to be directly regulated by the CSs and GR complex in both tissues. Some other functions, eg insulin signaling, PPAR signaling, focal adhesion, and cell cycle relevant processes, contain a greater number of genes directly regulated by the CS complex in muscle (Table 2).
Potential transcriptional regulators of CS-responsive common functions
During chronic infusion, MPL concentrations reach and remain at a stable steady state after six hours.35 The drug binds to cytosolic GRs and then rapidly translocates into the nucleus to alter the expression of target-genes. As GRs are greatly diminished in response to CSs,22,23,27,28 mRNA levels of CS target-genes should quickly return to the baseline. However, as observed in Table 2 many gene data sets exhibit a long-term response before returning to baseline or even reach a new steady state without returning. In addition to a number of possibilities (eg, multiple GR isoforms, multiple GREs with different affinities to the drug—receptor complex, multiple receptors that can mediate the effect of CSs),35,40 we hypothesize that these effects can be caused by the regulation of secondary bio-signals where TFs are the most likely candidates. After activation by CSs, they in turn further modulate the expression of their target-genes as a continuous cascade of events that were initiated by the drug.
Consequently, with the hypothesis that common functions activated across multiple tissues are more likely to be important as CS-responsive, the gene batteries selected in Table 2 were further analyzed to predict putative transcriptional regulators that are potential secondary bio-signals relevant to the regulation of transcriptional responses. For 18 common functions expressed across multiple patterns, we found 23 critical gene batteries in liver responses and 19 in muscle responses. Using the context-specific CRM search technique, a set of TFs associated with TFBSs in CRMs that are statistically significantly present on the corresponding promoter set was extracted for each gene battery (see Materials and Methods). Frequent TFs present in more than 20% of all gene batteries in each tissue are shown in bold (Tables 3 and 4). Specifically, we have 22 frequent TFs relevant to the regulation of transcriptional responses in liver and 20 in muscle. Among them, 17 relevant TFs are common including CLOX, CREB, CTCF, E2FF, EGRF, ETSF, FKHD, HOMF, HOXF, NKXH, NR2F, OCT1, RXRF, SORY, SP1F, STAT, and ZBPF. Almost all TF families consist of TF members that are recognized as differentially expressed genes in one or both tissues (see Supplemental Material 2). This finding highlights the possibility that secondary bio-signals are involved in the regulatory complexities of expression changes for CS-affected genes.
Table 3.
Functions and transcriptional regulators of gene batteries in liver.
| NO. | FUNCTIONS | PATTERNS | TRANSCRIPTION FACTORS* |
|---|---|---|---|
| 1 | Adherens junction | 7 | ZBPF, ETSF, SP1F, EBOX, EGRF, NKXH, ZF5F, GATA, IRXF, E2FF, EVI1, HAND, STAF, AP2F, PAX5 |
| 2 | Aminoacyl-tRNA biosynthesis | 5 | MYBL, P53F, GATA, NKXH, YY1F, CAAT, E2FF, EBOX, ETSF, HESF, HNF1, HOMF, PARF, SP1F |
| 3 | ECM-receptor interaction | 7 | CTCF, MYOD, ZF5F, E2FF, CDEF, AHRR, HESF, EGRF, NRF1, PAX5, GLIF, MAZF, EKLF, SP1F, ETSF |
| 4 | Focal adhesion | 7 | NKXH, ETSF, RUSH, TBPF, SORY, PARF, HOXC, ZBPF, MYT1, SIXF, MZF1, CLOX, CREB, DMRT, HOXF |
| 5 | Insulin signaling pathway | 6 | CAAT, CLOX, EBOX, EVI1, NEUR, NR2F, RXRF |
| 6 | Lysosome | 11 | FKHD, SNAP, ZBPF, CLOX, HEAT, HOMF, PAX3, OCT1, BRNF, E2FF, MYT1, PRDF |
| 7 | MAPK signaling pathway | 3 | HOMF, LHXF, HOXF, TBPF, FKHD, HESF, MYT1, AP4R, NKXH, E2FF, ETSF, RXRF, SORY, STAT, ZBPF |
| 8 | Neurotrophin signaling pathway | 5 | CREB, EGRF, ETSF, HOXF, ZBPF, CTCF |
| 9 | Cell cycle relevant-processes | 5 | NRF1, EGRF, EKLF, E2FF, HESF, CREB, NR2F, SP1F, GLIF, MAZF, MYBL, SRFF, CTCF, PAX5, RXRF |
| 10 | Oxidative phosphorylation | 6 | ETSF, NKXH, OCT1, RUSH, SRFF |
| 10a | Oxidative phosphorylation | 11 | IRFF, ABDB, FKHD, HOXF, CLOX, CREB, CTCF, E2FF, LHXF, NKXH, NR2F, SORY |
| 10b | Oxidative phosphorylation | 8 | ETSF, NFKB, SP1F, STAT, ZBPF |
| 11 | Pentose phosphate pathway | 6 | FKHD, BRNF, GREF, CLOX, RXRF, CAAT, HOXF, SIXF, ZFHX, BRN5, E2FF, NR2F, SP1F |
| 12 | PPAR signaling pathway | 12 | PARF, NFKB, HOXF, P53F, CREB, ETSF, LEFF, NR2F, GKLF, STAT, SP1F, CEBP, NKXH, CLOX, MOKF |
| 13 | Proteasome | 5 | CAAT, ETSF, CLOX, MYBL, NFAT, STAT |
| 14 | Purine metabolism | 5 | ETSF, EKLF, CAAT, SP1F, NFKB, NRSF, SORY, BRNF, CART, CLOX, GREF |
| 15 | Spliceosome | 3 | HOXF, ABDB, ETSF, CDXF, EVI1, GATA, HOMF, SORY |
| 15a | Spliceosome | 5 | HOMF, SORY, BRNF, HOXF, OCT1, PDX1, CLOX, FKHD, HBOX, MYBL, NKXH, NR2F |
| 16 | Tight junction | 3 | NRF1, HESF, SP1F, NKXH, LEFF, MYT1, EGRF, CAAT, EKLF, ETSF, FKHD, GATA |
| 16a | Tight junction | 7 | ZFHX, ETSF, EBOX, STAT, OCT1, CREB, EGRF, HNF1, NRSF, PARF, RXRF, ZF5F, ZFTR |
| 17 | Ubiquitin mediated proteolysis | 3 | ABDB, OCT1, HOXH, NFAT, BRNF, CREB, SP1F, STAT, DLXF, CLOX, E2FF, HAND, HNF6, HOMF, XBBF |
| 17a | Ubiquitin mediated proteolysis | 5 | EGRF, ETSF, ZBPF, HESF, NRF1, E2FF, PAX9, SP1F, DMRT, CTCF, HNF1, EBOX, FKHD, MAZF, ZF5F |
| 18 | Wnt signaling pathway | 3 | NKXH, AP4R, HOMF, MYT1, SORY, DLXF, TBPF, HOXF, E2FF, OCT1, ATBF, LHXF, BRNF, GKLF, FKHD |
Notes:
TFs associated with overrepresented cis-regulatory modules. if the number of associated TFs is ≥15, only the first 15 most frequently-associated TFs in statistical significant CRMs are reported. TFs shown in bold type are those present commonly more than 20% across all gene batteries.
Table 4.
Functions and transcriptional regulators of gene batteries in muscle.
| NO. | FUNCTIONS | PATTERNS | TRANSCRIPTION FACTORS* |
|---|---|---|---|
| 1 | Adherens junction | 2 | E2FF, NR2F, SP1F, RXRF, NKXH, HOXF, ETSF, EVI1, BRNF, FKHD, MYBL, SORY, OCT1, CTCF, EGRF |
| 2 | Aminoacyl-tRNA biosynthesis | 1 | HOXF, ETSF, NKX6, OCT1, CREB, RUSH, STAT, HOMF, P53F |
| 3 | ECM-receptor interaction | 2 | ETSF, RXRF, SP1F, NR2F, CTCF, E2FF, GKLF, GLIF, NEUR, SORY, SRFF |
| 4 | Focal adhesion | 2 | EGRF, SP1F, ETSF, RXRF, ZBPF, CTCF |
| 4a | Focal adhesion | 3 | NR2F, EGRF, OCT1, ETSF, MYOD, NOLF, SORY, RXRF, GLIF, MZF1, EKLF, EREF, EVI1, HOMF, NKXH |
| 5 | Insulin signaling pathway | 2 | EGRF, ZBPF, AHRR, E2FF, EBOX, MAZF, SP1F, CTCF, GLIF, HESF, HOXF, SNAP |
| 6 | Lysosome | 1 | EBOX, ETSF, EGRF, GRHL, ABDB, AHRR, CREB, CTCF, HOMF, MAZF, NR2F |
| 7 | MAPK signaling pathway | 2 | E2FF, NKXH, EGRF, OCT1, HOXF, CTCF, FKHD, HOMF, NKX6, PAX6, ZBPF |
| 8 | Neurotrophin signaling pathway | 2 | ZBPF, HESF, SP1F, PLAG, EGRF, ETSF, E2FF, EKLF, MAZF, GLIF |
| 9 | Cell cycle relevant-processes | 2 | HOMF, HOXF, FKHD, ETSF, NKXH, CREB, LHXF, E2FF, ZF5F, HEAT, STAT, EKLF, CTCF, CLOX, ZBPF |
| 10 | Oxidative phosphorylation | 5 | PAX5, BRNF, NR2F, ETSF, HNF1, FKHD, IRFF, AP1F, SORY, HNF6, HOXF, FAST, CREB, MYBL, OVOL |
| 11 | Pentose phosphate pathway | 2 | NR2F, TBPF, CLOX, GATA, HOXF, PAX2, TALE |
| 12 | PPAR signaling pathway | 1 | CLOX, ETSF, EVI1, GATA, NKXH, NR2F, STAT, ZBPF |
| 13 | Proteasome | 1 | HOXF, ETSF, HOMF, E2FF, STAT, NKXH, CLOX, EVI1, CAAT |
| 14 | Purine metabolism | 1 | RXRF, ETSF, HOXF, STAT, NKXH, PAX6, PAX8, SORY, TBPF, EREF, AP1R, CAAT, CREB, FKHD, NR2F |
| 15 | Spliceosome | 1 | CREB, ETSF, HEAT, EVI1, E2FF, HNF6, HOXF, ABDB, CLOX, DMRT, IRFF, MYT1, NFKB |
| 16 | Tight junction | 2 | PAX5, OCT1, HOXC, SP1F, NR2F, TBPF, NKXH |
| 17 | Ubiquitin mediated proteolysis | 1 | HOXF, BRNF, FKHD, TBPF, NKXH, LHXF, SORY, HOMF, NKX6, PARF, OCT1, CDXF, GATA, YY1F, ETSF |
| 18 | Wnt signaling pathway | 2 | ETSF, CREB, HOMF, HOXF, EGRF, NKXH |
Notes:
TFs associated with overrepresented cis-regulatory modules. If the number of associated TFs is ≥15, only the first 15 most frequently-associated TFs in statistical significant CRMs are reported. TFs shown in bold type are those present commonly more than 20% across all gene batteries.
Putative functional regulatory networks
As biological processes do not happen in isolation, we defined a hypothetical quantity, called the “regulatory closeness,” which putatively reflects the similarity of transcriptional regulatory mechanisms between two gene batteries to characterize the relationship among expression, function, and transcriptional regulation (see Materials and Methods). We graphed these relationships in a so-called functional regulatory network with nodes representing gene batteries and edges high transcriptional regulatory similarities (RC ≥0.8 in this case). Figure 3 exhibits corresponding functional regulatory networks in liver (top) and in muscle (bottom). Gene batteries having no relationship of high RC with any other gene battery are not displayed in the graph. The edges present on both graphs are displayed in “red” color. As a result, we have 19 gene batteries with 25 edges for the functional regulatory network in liver and 19 gene batteries with 38 edges in muscle. The networks show that gene batteries with similar expression patterns are better connected to each other than batteries with different expression patterns. This is the main reason why muscle that has the same number of gene batteries as liver contains more connections. Moreover, under the same condition and for the same function, involved genes can exhibit different expression patterns in liver than muscle. Also, even in the same tissue, different sets of genes with the same function can have different expression patterns (eg, gene battery “tight junction” in liver). Additionally, gene expression patterns in liver are more diverse than those in muscle, which may result in the potential capability of sharing common regulatory mechanisms among gene batteries, which is higher in muscle. These results provide an overview landscape of tissue-specific expression and regulation under chronic MPL infusion through critically identified transcriptional responses and CS-affected functions as well as their transcriptional regulatory relationships.
Figure 3.

Tissue-specific regulation represented by putative functional regulatory networks (top: liver; bottom: muscle). Each node represents a gene battery which is a set of coexpressed genes sharing a common function (pathway). Edges characterize the regulatory closeness between two gene batteries if the ratio of sharing common transcriptional regulators is greater than 0.8; ‘red’ edges express high regulatory closeness that occurs in both liver and muscle. Although these gene batteries are involved in similar functions under chronic corticosteroid administration, their expression patterns and transcriptional regulatory relationships are specific in liver vs. in muscle.
Discussion
Although CSs affect gene expression in multiple tissues, the array of genes that are regulated by these steroids is diverse, highly tissue specific, and depends on their functions in the tissues. Liver is an organ that plays a critical role in performing and regulating important physiological processes such as energy metabolism, detoxification, inflammation, and immune responses against pathogens. Muscle plays a key role in maintaining systemic energy homeostasis and accounts for 80% of insulin-directed glucose disposal. In addition, both liver and muscle are important sites for immune reactions caused by different immune cell types. Therefore, we explored tissue-specific CS-responsive gene expression profiles, their functional significance, and the effect of secondary bio-signals in the form of TFs to gain better insight into CS pharmacogenomics.
In this study, 2,285 differentially expressed genes in liver during chronic MPL infusion were parsed into 12 distinct temporal clusters. Functional analysis of the pathways in which these clustered genes are involved suggests that each cluster is enriched with specific biological functions. For example, most of the genes from cluster 6 are present in pathways involved in performing or regulating energy metabolism (oxidative phosphorylation, pentose phosphate pathway, and starch and sucrose metabolism). Similarly, genes from cluster 7 are primarily involved in pathways regulating immune response and inflammation (complement and coagulation cascades, leukocyte transendothelial migration, antigen processing and presentation) or associated processes (cell adhesion, ECM—receptor interaction, focal adhesion). It is interesting to see that the expression of genes in cluster 6 is up-regulated over time with continuous MPL infusion reaching a plateau. In contrast, expression of genes in cluster 7 is continuously down-regulated, ultimately reaching a plateau. These observations go well with the fact that CSs repress inflammation and immune response cascades that lead to their therapeutic applications while steroid effects on metabolic processes lead to their adverse effects. The CSs, being potent anti-inflammatory agents, not only repress inflammatory signaling but also affect immune cell trafficking. This is confirmed by the continuous down-regulation of genes involved in leukocyte migration and associated processes including cell adhesion. Another interesting observation is the development of tolerance, which is a commonly observed phenomenon in chronic CS treatment because of the down-regulation in the expression of GRs that mediate their genomic effects. For example, in liver gene expression, clusters 1, 2, 3, 4, and 5 show clear tolerance, which may occur because of the down-regulation in receptor expression following CS dosing.
In muscle responses during CS infusion, 2,291 differentially expressed genes were parsed into seven distinct temporal clusters. Similar to liver, genes present in pathways regulating immune response (B-cell receptor signaling pathway, Fc gamma R-mediated phagocytosis, chemokine signaling pathway) and related processes (ECM—receptor interaction, focal adhesion) are down-regulated in muscle during MPL infusion (clusters 2 and 3), although the specific immune regulating pathways are different between these two tissues. In the case of energy metabolism in muscle, genes involved in oxidative phosphorylation show a robust up-regulation followed by the development of tolerance, genes involved in fatty acid metabolism show a transient down-regulation followed by a robust up-regulation (cluster 1), and genes involved in glycolysis, pyruvate metabolism, and pentose phosphate pathway show exactly the opposite expression profile with initial transient up-regulation followed by robust down-regulation (cluster 2). This observation is consistent with the fact that glucocorticoids direct the muscle to use free fatty acids as the primary energy source, producing NADPH which is used for the production of ATPs through oxidative phosphorylation. Furthermore, the observation that genes involved in glycolysis and pyruvate metabolism are down-regulated confirms that GC decrease the dependence of muscle on glucose, which helps in the maintenance of proper plasma glucose concentrations. It is interesting to see that, even though the genes involved in oxidative phosphorylation are up-regulated both in liver and muscle at some point in time after MPL dosing, the expression profiles are very different between the two tissues with the liver showing three different sets of expression patterns and muscle showing the development of tolerance by 10 hours after the start of MPL infusion. Furthermore, expression of genes involved in ubiquitin-mediated proteolysis falls under cluster 1, where the genes show a robust up-regulation in muscle suggesting that the proteasomal machinery is activated by MPL. This is one of the important causes of clinical side-effects of CS, which is muscle wasting caused by the breakdown of muscle proteins to provide the amino acid carbon for gluconeogenesis in liver.
Apart from the above-mentioned processes that are affected by CSs in both liver and muscle, CSs have tissue-specific effects on certain processes and pathways. For example, they cause up-regulation in the expression of ribosomal genes in liver, but have no effects on muscle ribosomal gene expression. In liver, clusters 1 and 5 consist of ribosomal genes that show an up-regulation of expression with MPL infusion with the peak expression occurring at 18 and 10 hours after the start of drug infusion and the development of tolerance thereafter. CSs are known to cause hypertrophy in liver, and up-regulation of expression ribosomal genes is a part of the process of increasing the cell mass, which is not observed in muscle. Similarly, genes involved in the metabolism of drugs and retinol show initial down-regulation followed by a rebound in expression in liver after the drug infusion, while the expression of these genes is unaffected in muscle. Xenobiotic and retinol metabolism primarily occur in liver, and the muscle has no role in these processes; hence, the expression of these genes is not affected in muscle. However, the expression of genes present in pathways involved in adipocytokine signaling, calcium signaling, ErbB signaling, GnRH signaling, and phosphatidylinositol signaling are all affected by CS dosing in muscle, but the drug has no effects on these genes in liver, at least in part because of the higher relevance for these signaling pathways in muscle compared to liver.
Although the plasma concentrations of MPL attained steady state and remained almost constant from six hours after the start of the infusion, the GR mRNA expression and the receptor density are diminished because of the negative feedback regulation. This will cause a drastic reduction in the activated drug receptor complex, which is the primary driving force for the changes in gene expression. Hence, if the gene expression changes in these animals are caused only by the activated drug–receptor complex in the nucleus, then the expression profile for all the genes would go to a steady-state expression close to the baseline because of the marked tolerance caused by the drastic reduction in receptor density. However, from the expression profiles in both tissues, there are many genes that show long-term responses. It is well known that CS can regulate the expression of many TFs, and hence, we hypothesized that many of the long-term responses in the gene expression profiles are caused by other TFs that are affected and differentially expressed by CS. As shown in Tables 3 and 4, we were able to computationally identify the important TFs that could be involved in CS-responsive transcriptional profiles. Furthermore, from this analysis we could see that CSs regulate many of these TFs even at genomic levels (see Supplemental Material 2). For example, important regulators of inflammation and immune response (eg, nuclear factor kappa-B (NF-κB), interferon regulatory factor (IRF)) are found to be involved in pathogenesis of inflammatory disorders when CSs are used as therapeutic drugs. In this analysis, both are identified as potential TFs that are relevant to the CS-responsive transcription profiles, and their members are also down-regulated at the genomic level by CS in both liver and muscle. Similarly, many other TFs including STAT, CREB, and RXRF, which play important roles in energy metabolism and many signaling pathways, are also identified as potential TFs with distinct expression profiles during CS dosing. These results suggest that many TFs act as secondary bio-signals in controlling CS-responsive transcriptional behaviors, which in turn regulate the processes that are involved in both the therapeutic effects (caused by anti-inflammatory effects and immune suppression) and adverse effects (impact on metabolic processes).
Although similar pathways are affected by CS in both tissues, the genes and their expression patterns can be very different depending on significance of that pathway in each tissue. For example, as shown in Figure 4, the insulin signaling pathway is affected by MPL in both liver and muscle, but the individual genes that are CS-responsive and the expression patterns are very different between the two tissues. Among these, there is a tissue-specific coherent set of genes that are significantly coexpressed and regulate a certain set of different/similar downstream functions dependent on the tissue. Insulin signaling plays an important role in regulating many aspects of energy metabolism (protein synthesis and lipid and carbohydrate metabolism) and cell cycle processes (proliferation, differentiation, and apoptosis). Components of insulin signaling pathway that are involved in protein synthesis are responsive to CS only in liver, whereas genes involved in anti-lipolysis, proliferation, and differentiation are responsive to CS only in muscle. However, genes involved in glycogenesis, glycolysis, and lipogenesis are CS-responsive in both liver and muscle, although the expression patterns are very different between the two tissues. The mTOR and EIF4EBP1 genes are continuously up-regulated only in liver with MPL infusion. They play an important role in activating liver protein synthesis, which is required to support the increase in liver mass caused by CS. As this phenomenon is not observed in muscle, these proteins may not be relevant and hence are not CS-responsive. Similarly, muscle is the major consumer of lipids as the energy source with high CS concentrations, and hence PRKAR1A, the gene involved in anti-lipolysis, is down-regulated only in muscle and is not affected in liver. It is also important to remember that TFs in the form of secondary bio-signals play an important role in regulating tissue-specific expression. As shown in Figure 4, all TFs involved in insulin signaling except for EBOX are different between the two tissues and hence result in regulating different genes involved in the same pathway and also cause differences in the expression profiles. This observation reinforces the tissue-specificity of transcriptional responses during MPL infusion (see more in Supplemental Material 3).
Figure 4.
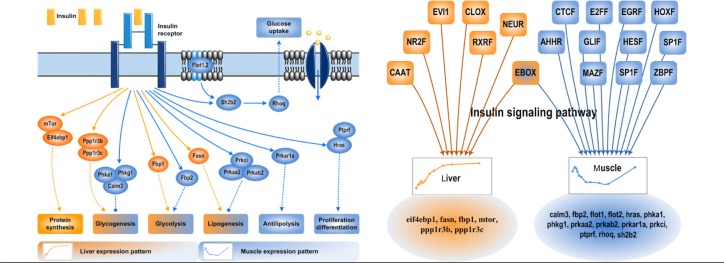
Tissue-specific expression within individual functions—a case of insulin signaling pathway. The left subfigure shows an abstract of the insulin signaling pathway where expressed genes in liver and muscle and their corresponding downstream affected functions are included. The right panel displays the tissue-specific regulation of genes within the same functional category; putative TFs are those significantly overrepresented on the promoters of corresponding genes.
Supplementary Files
Supplementary file 1.Critical dynamic transcriptional responses in liver under chronic corticosteroid administration.
Supplementary file 2.Regulation analysis in liver and in muscle.
Supplementary file 3.Heat map of regulatory closeness between selected gene batteries, and illustration of tissue-specific expression within individual functions.
Footnotes
ACADEMIC EDITOR: James Willey, Editor in Chief
FUNDING: TTN and IPA gratefully acknowledge the financial support from NIH grant GM082974. WJJ, RRA, DCD, and SS appreciate the financial support from NIH grant GM24211.
COMPETING INTERESTS: Author(s) disclose no potential conflicts of interest.
Author Contributions
IPA conceived and designed the experiments. TTN and SS analyzed the data. TTN and IPA wrote the first draft of the manuscript. TTN, IPA, RRA, WJJ, SS, and DCD contributed to the writing of the manuscript. TTN, IPA, RRA, WJJ, SS, and DCD agreed with the manuscript results and conclusions. TTN, IPA, RRA, WJJ, SS, and DCD jointly developed the structure and arguments for the paper. TTN, IPA, RRA, WJJ, SS, and DCD made critical revisions and approved the final version. All authors reviewed and approved the final manuscript.
DISCLOSURES AND ETHICS
As a requirement of publication the authors have provided signed confirmation of their compliance with ethical and legal obligations including but not limited to compliance with ICMJE authorship and competing interests guidelines, that the article is neither under consideration for publication nor published elsewhere, of their compliance with legal and ethical guidelines concerning human and animal research participants (if applicable), and that permission has been obtained for reproduction of any copyrighted material. This article was subject to blind, independent, expert peer review. The reviewers reported no competing interests.
REFERENCES
- 1.Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids – new mechanisms for old drugs. N Engl J Med. 2005;353(16):1711–23. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 2.Barnes PJ. Corticosteroid effects on cell signalling. Eur Respir J. 2006;27(2):413–26. doi: 10.1183/09031936.06.00125404. [DOI] [PubMed] [Google Scholar]
- 3.Baxter JD. Advances in glucocorticoid therapy. Adv Intern Med. 2000;45:317–49. [PubMed] [Google Scholar]
- 4.Bialas MC, Routledge PA. Adverse effects of corticosteroids. Adverse Drug React Toxicol Rev. 1998;17(4):227–35. [PubMed] [Google Scholar]
- 5.Frauman AG. An overview of the adverse reactions to adrenal corticosteroids. Adverse Drug React Toxicol Rev. 1996;15(4):203–6. [PubMed] [Google Scholar]
- 6.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96(1):23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 7.Locsey L, Asztalos L, Kincses Z, Gyorfi F, Berczi C. Dyslipidaemia and hyperlipidaemia following renal transplantation. Int Urol Nephrol. 1996;28(3):419–30. doi: 10.1007/BF02550506. [DOI] [PubMed] [Google Scholar]
- 8.Almon RR, Dubois DC, Jin JY, Jusko WJ. Pharmacogenomic responses of rat liver to methylprednisolone: an approach to mining a rich microarray time series. AAPS J. 2005;7(1):E156–94. doi: 10.1208/aapsj070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almon RR, DuBois DC, Piel WH, Jusko WJ. The genomic response of skeletal muscle to methylprednisolone using microarrays: tailoring data mining to the structure of the pharmacogenomic time series. Pharmacogenomics. 2004;5(5):525–52. doi: 10.1517/14622416.5.5.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dezso Z, Nikolsky Y, Sviridov E, et al. A comprehensive functional analysis of tissue specificity of human gene expression. BMC Biol. 2008;6:49. doi: 10.1186/1741-7007-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reverter A, Ingham A, Dalrymple BP. Mining tissue specificity, gene connectivity and disease association to reveal a set of genes that modify the action of disease causing genes. BioData Min. 2008;1(1):8. doi: 10.1186/1756-0381-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greco D, Somervuo P, Di Lieto A, et al. Physiology, pathology and relatedness of human tissues from gene expression meta-analysis. PLoS One. 2008;3(4):e1880. doi: 10.1371/journal.pone.0001880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lage K, Hansen NT, Karlberg EO, et al. A large-scale analysis of tissue-specific pathology and gene expression of human disease genes and complexes. Proc Natl Acad Sci USA. 2008;105(52):20870–5. doi: 10.1073/pnas.0810772105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagaraj SH, Ingham A, Reverter A. The interplay between evolution, regulation and tissue specificity in the Human Hereditary Diseasome. BMC Genomics. 2010;11(suppl 4):S23. doi: 10.1186/1471-2164-11-S4-S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Yu X, Zack DJ, Zhu H, Qian J. TiGER: a database for tissue-specific gene expression and regulation. BMC Bioinformatics. 2008;9:271. doi: 10.1186/1471-2105-9-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang X, Ye Y, Wang G, Huang H, Yu D, Liang S. VeryGene: linking tissue-specific genes to diseases, drugs, and beyond for knowledge discovery. Physiol Genomics. 2011;43(8):457–60. doi: 10.1152/physiolgenomics.00178.2010. [DOI] [PubMed] [Google Scholar]
- 17.Petretto E, Mangion J, Dickens NJ, et al. Heritability and tissue specificity of expression quantitative trait loci. PLoS Genet. 2006;2(10):e172. doi: 10.1371/journal.pgen.0020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Su AI, Li WH. Gene expression evolves faster in narrowly than in broadly expressed mammalian genes. Mol Biol Evol. 2005;22(10):2113–8. doi: 10.1093/molbev/msi206. [DOI] [PubMed] [Google Scholar]
- 19.Winter EE, Goodstadt L, Ponting CP. Elevated rates of protein secretion, evolution, and disease among tissue-specific genes. Genome Res. 2004;14(1):54–61. doi: 10.1101/gr.1924004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature. 2001;414(6865):799–806. doi: 10.1038/414799a. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen TT, Nowakowski RS, Androulakis IP. Unsupervised selection of highly coexpressed and noncoexpressed genes using a consensus clustering approach. OMICS. 2009;13(3):219–37. doi: 10.1089/omi.2008.0074. [DOI] [PubMed] [Google Scholar]
- 22.Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Pharmacodynamics and pharmacogenomics of methylprednisolone during 7-day infusions in rats. J Pharmacol Exp Ther. 2002;300(1):245–56. doi: 10.1124/jpet.300.1.245. [DOI] [PubMed] [Google Scholar]
- 23.Sun YN, DuBois DC, Almon RR, Jusko WJ. Fourth-generation model for corticosteroid pharmacodynamics: a model for methylprednisolone effects on receptor/gene-mediated glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm. 1998;26(3):289–317. doi: 10.1023/a:1023233409550. [DOI] [PubMed] [Google Scholar]
- 24.Dong Y, Poellinger L, Gustafsson JA, Okret S. Regulation of glucocorticoid receptor expression: evidence for transcriptional and posttranslational mechanisms. Mol Endocrinol. 1988;2(12):1256–64. doi: 10.1210/mend-2-12-1256. [DOI] [PubMed] [Google Scholar]
- 25.Oakley RH, Cidlowski JA. Homologous down regulation of the glucocorticoid receptor: the molecular machinery. Crit Rev Eukaryot Gene Expr. 1993;3(2):63–88. [PubMed] [Google Scholar]
- 26.Vedeckis WV, Ali M, Allen HR. Regulation of glucocorticoid receptor protein and mRNA levels. Cancer Res. 1989;49(8 Suppl):2295s–302s. [PubMed] [Google Scholar]
- 27.Almon RR, DuBois DC, Brandenburg EH, et al. Pharmacodynamics and pharmacogenomics of diverse receptor-mediated effects of methylprednisolone in rats using microarray analysis. J Pharmacokinet Pharmacodyn. 2002;29(2):103–29. doi: 10.1023/a:1019762323576. [DOI] [PubMed] [Google Scholar]
- 28.Sun YN, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Dose-dependence and repeated-dose studies for receptor/gene-mediated pharmacodynamics of methylprednisolone on glucocorticoid receptor down-regulation and tyrosine aminotransferase induction in rat liver. J Pharmacokinet Biopharm. 1998;26(6):619–48. doi: 10.1023/a:1020746822634. [DOI] [PubMed] [Google Scholar]
- 29.Ramakrishnan R, DuBois DC, Almon RR, Pyszczynski NA, Jusko WJ. Fifth-generation model for corticosteroid pharmacodynamics: application to steady-state receptor down-regulation and enzyme induction patterns during seven-day continuous infusion of methylprednisolone in rats. J Pharmacokinet Pharmacodyn. 2002;29(1):1–24. doi: 10.1023/a:1015765201129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen TT, Almon RR, Dubois DC, Jusko WJ, Androulakis IP. Comparative analysis of acute and chronic corticosteroid pharmacogenomic effects in rat liver: transcriptional dynamics and regulatory structures. BMC Bioinformatics. 2011;11:515. doi: 10.1186/1471-2105-11-515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tong W, Cao X, Harris S, et al. ArrayTrack – supporting toxicogenomic research at the US Food and Drug Administration National Center for Toxicological Research. Environ Health Perspect. 2003;111(15):1819–26. doi: 10.1289/ehp.6497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Genomatix. http://www.genomatix.de.
- 33.Nguyen TT, Foteinou PT, Calvano SE, Lowry SF, Androulakis IP. Computational identification of transcriptional regulators in human endotoxemia. PLoS One. 2011;6(5):e18889. doi: 10.1371/journal.pone.0018889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cartharius K, Frech K, Grote K, et al. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21(13):2933–42. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 35.Almon RR, DuBois DC, Jusko WJ. A microarray analysis of the temporal response of liver to methylprednisolone: a comparative analysis of two dosing regimens. Endocrinology. 2007;148(5):2209–25. doi: 10.1210/en.2006-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pavlidis P. Using ANOVA for gene selection from microarray studies of the nervous system. Methods. 2003;31(4):282–9. doi: 10.1016/s1046-2023(03)00157-9. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen TT, Almon RR, DuBois DC, Jusko WJ, Androulakis IP. Importance of replication in analyzing time-series gene expression data: corticosteroid dynamics and circadian patterns in rat liver. BMC Bioinformatics. 2010;11:279. doi: 10.1186/1471-2105-11-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelander S, Larsson E, Kristiansson E, et al. Predictive screening for regulators of conserved functional gene modules (gene batteries) in mammals. BMC Genomics. 2005;6:68. doi: 10.1186/1471-2164-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Britten RJ, Davidson EH. Gene regulation for higher cells: a theory. Science. 1969;165(891):349–57. doi: 10.1126/science.165.3891.349. [DOI] [PubMed] [Google Scholar]
- 40.Almon RR, DuBois DC, Yao Z, Hoffman EP, Ghimbovschi S, Jusko WJ. Microarray analysis of the temporal response of skeletal muscle to methylprednisolone: comparative analysis of two dosing regimens. Physiol Genomics. 2007;30(3):282–99. doi: 10.1152/physiolgenomics.00242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1.Critical dynamic transcriptional responses in liver under chronic corticosteroid administration.
Supplementary file 2.Regulation analysis in liver and in muscle.
Supplementary file 3.Heat map of regulatory closeness between selected gene batteries, and illustration of tissue-specific expression within individual functions.