

Abstract
We investigated the global patterns of abundance, diversity, and community structure of members of the Aminicenantes (candidate phylum OP8). Our aim was to identify the putative ecological role(s) played by members of this poorly characterized bacterial lineages in various ecosystems. Analysis of near full-length 16S rRNA genes identified four classes and eight orders within the Aminicenantes. Within 3,134 datasets comprising ∼1.8 billion high throughput-generated partial 16S rRNA genes, 47,351 Aminicenantes-affiliated sequences were identified in 913 datasets. The Aminicenantes exhibited the highest relative abundance in hydrocarbon-impacted environments, followed by marine habitats (especially hydrothermal vents and coral-associated microbiome samples), and aquatic, non-marine habitats (especially in terrestrial springs and groundwater samples). While the overall abundance of the Aminicenantes was higher in low oxygen tension as well as non-saline and low salinity habitats, it was encountered in a wide range of oxygen tension, salinities, and temperatures. Analysis of the community structure of the Aminicenantes showed distinct patterns across various datasets that appear to be, mostly, driven by habitat variations rather than prevalent environmental parameters. We argue that the detection of the Aminicenantes across environmental extremes and the observed distinct community structure patterns reflect a high level of intraphylum metabolic diversity and adaptive capabilities that enable its survival and growth in a wide range of habitats and environmental conditions.
Introduction
During the last quarter century, culture-independent diversity surveys have been extensively utilized to investigate bacterial diversity in almost all accessible habitats on earth [1]–[5]. These surveys have collectively demonstrated that the scope of bacterial diversity is much broader than previously expected based on culture-based assessments [6], [7], with a large fraction of the 16S rRNA gene sequences encountered not belonging to known cultured bacterial phyla. The term candidate phylum (CP) was thus proposed to describe such lineages [2].
One of the most important challenges facing microbial ecologists is to elucidate the putative metabolic capabilities and ecological roles of these candidate phyla, as well as the underlying ecological factors controlling their observed patterns of abundance, diversity, and community structure on a global scale. Various environmental genomics approaches have been utilized to obtain genomic fragments and partial genome assemblies from these lineages. These include construction and screening of large insert (Fosmid and BAC) libraries [8]–[11], direct metagenomic surveys and subsequent implementation of novel binning approaches to reconstruct genomes from metagenomic sequence data [12]–[15], and single cell genomics [16]–[19]. Collectively, these efforts have yielded valuable insight regarding the genomic characteristics and putative metabolic capabilities of multiple novel candidate phyla. Further, in several incidents, these insights were successfully utilized as a stepping-stone for enrichment and isolation of some of these lineages [20]–[22].
Genomic approaches are extremely valuable for deciphering putative metabolic capabilities of uncultured bacterial lineages. However, information from genomic studies is derived from a single sampling event in a single environment, and often from a single cell within the sample [17], [19]. Extrapolation of such information to imply similar capabilities and genomic features to all members and lineages within an entire bacterial phylum is hence inappropriate. This is especially true since a single bacterial phylum could exhibit a bewildering array of metabolic capabilities.
A complementary approach that has previously been utilized on an ecosystem level [23]–[27], but rarely utilized in a global phylocentric context, relies on using in silico database mining approaches to examine patterns of distribution of members of a specific candidate phyla in 16S rRNA gene diversity surveys. This approach could clarify the patterns of abundance, diversity, and community structure of the targeted lineage. This phylocentric strategy could greatly benefit from the dramatic increase in the number and size of publicly available 16S rRNA gene datasets; brought about by utilizing next generation sequencing technologies in recent ambitious initiatives to catalogue 16S rRNA gene diversity on a global scale [28]–[30].
Here, we describe a comprehensive examination of the global distribution of members of the Aminicenantes (candidate phylum OP8) using in silico database mining approaches. Our aim was to understand the putative ecological role(s) played by members of this poorly characterized bacterial lineages in various ecosystems and to demonstrate the utility of in silico database mining approaches in extracting meaningful ecological patterns from high throughput 16S rRNA gene datasets. Candidate phylum OP8 was first identified in sediments from the Obsidian Pool in Yellowstone National Park [2]. Since then, it has subsequently been identified in a wide range of terrestrial and marine habitats [31]–[34]. A recent study has described two near candidate phylum OP8 genome assemblies from 38 partial single cell genomes obtained from deep sediments of a brackish lake (Sakinaw lake, British Columbia, Canada), and the name Aminicenantes was proposed for this candidate phylum to highlight the high proportion of genes encoding aminolytic enzymes identified in both assemblies [16]. Our results highlight the ubiquitous nature of the Aminicenantes, and identify various environmental conditions impacting its global abundance and distribution in various habitats. We argue that these observed patterns suggest that, collectively, members of the Aminicenantes exhibit a high level of intra-phylum metabolic and adaptive diversities, and are hence capable of survival, and growth in a wide range of environmental extremes.
Materials and Methods
1. A taxonomic outline of the Aminicenantes
While the candidate phylum Aminicenantes (CD-OP8) is recognized in several curated taxonomic outlines e.g. Greengenes [35] and SILVA [36], only a fairly low number of Aminicenantes sequences are deposited in these databases (109, and 12, in Greengenes and SILVA, respectively). The continuous deposition of new near full-length 16S rRNA gene sequences in GenBank database repository, coupled to the sporadic updates of curated taxonomic schemes, raises the prospect that additional Aminicenantes 16S rRNA sequences putatively representing novel high rank (class/ order) lineages have been deposited in GenBank but have yet to be included in taxonomic schemes. Therefore, as a preliminary step, we aimed to identify and classify all GenBank-deposited Aminicenantes 16S rRNA gene sequences and produce an updated and comprehensive taxonomic outline of this phylum. To this end, we queried GenBank NR database using BlastN [37], to identify the closest relatives of each of the 109 Aminicenantes sequences currently recognized in Greengenes and SILVA databases. The 500 closest relatives of each sequence were downloaded; and duplicates, sequences shorter than 800 bp, and chimeric sequences, identified using Galaxy [38], were removed. The remaining sequences (n = 2955) were aligned to a collection of reference sequences representing all Aminicenantes sequences, as well as sequences from a collection of 17 phyla, and 8 candidate phyla using ClustalX [39]. The phylogenetic positions of putative Aminicenantes sequences were evaluated using Distance, Parsimony, Maximum likelihood, and Bayesian approaches as previously described [40]. Sequences were deemed representative of a new class/order within the Aminicenantes if two or more distinct sequences remained reproducibly monophyletic and formed a bootstrap-supported independent clade upon varying the composition and size of the data set used for phylogenetic analysis [41].
2. Identification of Aminicenantes members in next Generation 16S rRNA gene datasets
Publicly available datasets generated using high throughput sequencing technologies (Pyrosequencing and Illumina) were downloaded from MG-RAST [42], VAMPS (http://vamps.mbl.edu/index.php), and GenBank SRA[43] (through the mirror web interface of DNA Databank of Japan http://www.ddbj.nig.ac.jp) in December 2012. Preliminary analysis indicated the absence of the Aminicenantes in human and metazoan microbiome samples and hence these datasets were excluded from further analysis (with the notable exception of rumen samples which were included). In total, 3,141 datasets from 110 different studies with 1,820,857,401 distinct 16S rRNA sequences were included in the analysis (Table S1 in File S1). All datasets were quality screened to filter all the sequences with lengths less than 50 base pairs, sequences with ambiguous nucleotides, and sequences with hompolymer stretches more than 8 bps. Sequences were classified using classify.seqs commands package in MOTHUR v.1.29.0 [44], using Silva alignment and Greengenes classification scheme. Sequences were identified as members of the Aminicenantes using a cutoff of 70% confidence threshold, as well as by confirmation of such assignment by sporadic manual insertion of putative Aminicenantes sequences into reference phylogenetic trees as described above. The subphylum level affiliation of all high throughput Aminicenantes sequences identified were determined using the updated taxonomic scheme produced in this study using near full length 16S rRNA gene sequences as described above. All analyses were conducted on a the HPC Cowboy super computer, a 252 compute nodes with dual six core CPUs and 32 GB RAMs server, 2 fat nodes with 256 GB RAM, GPU cards and 120 TB very fast disk storage at the OSU High Performance Computing Center at Oklahoma State University.
3. Classification of next-generation datasets according to habitat type and prevalent environmental conditions
All datasets included in this study were classified according to two different classification schemes: habitat type as well as prevalent environmental conditions. These classifications were used to determine the ecological prevalence and distribution patterns of various members of the Aminicenantes. Habitat-based classification scheme involved binning all 3,141 datasets into five major habitat types: Marine, aquatic non-marine, soil, hydrocarbon-impacted, and rumen/other (dust, animal-associated habitats and air). Due to the heterogeneity of geochemical and environmental conditions observed in marine, aquatic non-marine, and soil habitats, these three habitats were further sub-classified into multiple sub-habitat types, determined through the analysis of the projects’ available metadata (Table 1). For classification of datasets according to prevalent environmental conditions, three different classification schemes using temperature, oxygen tension, and salinity were utilized (Table 2). Classification based on prevalent pH conditions was not feasible due to the frequent absence of accurate pH metadata in a large proportion of the datasets, as well as the exceedingly low number of datasets that appear to originate from environments with preeminently low (e.g. <3) or high (e.g. >9) pH. A detailed description of all habitats examined in this study is presented in Table S1 in File S1.
Table 1. Classification and overall patterns of Aminicenantes relative abundance in various habitats and sub-habitats1.
| Dataset type | Total datasets | Datasets with Aminicenantes (%) | Average Aminicenantes abundance (%) | Maximum relative abundance |
| Total datasets | 3,141 | 918 (29.22%) | 0.20%2 | 10.20% |
| Total 16S rRNA sequences | 1,820,857,401 | 47,315 | 0.0026% | |
| Marine datasets | 1,154 | 248 (21.50%) | 0.28% | 5.28% |
| Deep marine sediments | 32 | 30 | 0.50% | 2.89% |
| Coral associated microbiome | 19 | 10 | 0.89% | 4.67% |
| Pelagic | 390 | 40 | 0.20% | 2.46% |
| Hydrothermal vents | 101 | 60 | 0.23% | 5.28% |
| Coastal | 612 | 107 | 0.20% | 1.87% |
| Aquatic non-marine datasets | 1,665 | 645 (38.74%) | 0.15% | 10.20% |
| Spring and ground water | 25 | 10 | 2.80% | 10.20% |
| Temperate freshwater | 1569 | 587 | 0.11% | 2.50% |
| Salt marshes | 71 | 48 | 0.03% | 0.67% |
| Soil datasets | 276 | 14 (5.072%) | 0.07% | 0.80% |
| Agriculture | 28 | 2 | 0.03% | 0.06% |
| Grassland | 140 | 10 | 0.00% | 0.00% |
| Heavy metal/hydrocarbon contaminated | 8 | 1 | 0.00% | 0.01% |
| Arid and Semi-arid | 46 | 0 | 0% | 0% |
| Permafrost | 54 | 1 | 0.01% | 0.01% |
| Hydrocarbon-impacted datasets | 14 | 10 (71.43%) | 0.32% | 0.95% |
| Herbivorous gut and other datasets3 | 32 | 1 (3.125%) | 0.02% | 0.02% |
A detailed description of every dataset is provided as supplementary material (Table S1 in File S1).
Average abundance values in datasets where Aminicenantes sequences were identified.
26 Datasets were designated “other”; these datasets originated from dust, air and animal associated habitat. See Supplementary Table S1 in File S1 for details.
Table 2. Patterns of Aminicenantes relative abundance in datasets classified by prevalent environmental conditions1.
| Dataset type | Total datasets | Datasets with Aminicenantes (%) | Average Aminicenantes abundance (%)2 | Maximum relative abundance |
| Oxygen Tension | ||||
| Oxic | 2,787 | 735(26.4%) | 0.10% | 2.50% |
| Hypoxic | 101 | 35 (34.65%) | 0.17% | 2.90% |
| Anoxic | 253 | 148 (58.5%) | 0.46% | 10.20% |
| Temperature3 | ||||
| Low | 317 | 4 (1.26%) | 0.004% | 0.01% |
| Temperate | 2657 | 807 (30.372%) | 0.19% | 10.20% |
| Medium | 53 | 48 (90.56%) | 0.02% | 0.06% |
| Elevated | 11 | 6 (54.55%) | 0.06% | 0.20% |
| Extremely elevated | 103 | 53 (51.46%) | 0.24% | 5.28% |
| Salinity4 | ||||
| Non-Saline | 1,863 | 575 (30.86%) | 0.16% | 10.20% |
| Low Salinity | 1,179 | 274 (23.24%) | 0.26% | 5.30% |
| Moderate salinity | 77 | 51 (66.23%) | 0.02% | 0.20% |
| Hypersaline | 22 | 18 (81.81%) | 0.07% | 0.68% |
A detailed description of every dataset is provided as supplementary material (Supplementary Table S1 in File S1).
Average abundance values in datasets where Aminicenantes sequences were identified.
Temperature classifications: Low: Arctic, Antarctic, subarctic, and permafrost marine and terrestrial conducive to the growth of psychrophilic microorganisms; temperate: Habitats in temperate ecosystems e.g. lakes, soils in continental settings; Medium: Habitats with temperatures around 37°C e.g. rumen; Elevated: habitats with temperatures conducive to the growth of thermophiles (50–80°C) e.g. Alberta oil sands tailings pond; Extremely elevated: habitats conducive to the growth of hyperthermophiles (>80°C degrees) e.g. Hydrothermal vents.
Salinity classifications: Non-saline: Environments with <1% salinity; Low salinity: Marine environments, and environments with comparable salinities; Moderate salinities: Environments with salinities around 5–15% e.g. Alberta oil sands tailings pond and Huabei Oilfield in China; Hypersaline: Environments with >15% salinity.
4. Deciphering ecological preferences and patterns of distribution of the Aminicenantes
The distribution and preferences of Aminicenantes were identified by correlating Aminicenantes relative abundance (% of sequences affiliated with Aminicenantes in the dataset), diversity, and community structure to its distribution in various habitats and sub-habitats, as well as across various environmental conditions.
Rarefaction curves were used to compare diversities of Aminicenantes community in different datasets as previously described [45]. We chose rarefaction curve analysis since it provides a sample size unbiased estimate of diversity and is hence useful in comparing datasets with wide variations in the numbers of sequences examined. In brief, rarefaction curves were constructed for all datasets with more than 50 sequences belonging to the Aminicenantes. Rarefaction curve plots were used to rank the datasets in order of diversity. Datasets with intersecting rarefaction curves were given the same rank. The datasets were ranked from one (least diverse) to 198 (most diverse) and subsequently binned into diversity categories as follows; “very low” (ranks 1–40), “low” (41–80), “medium” (81–120), “high” (121–160), and “very high” (161–198) categories. The ranks were then used to correlate Aminicenantes diversity to specific environmental factors using Spearman rank correlation and the significance of these correlations were tested in R [46].
The community structure profiles i.e. the proportion of various Aminicenantes lineages in various datasets were examined to reveal overall patterns of community structure in different habitats and under different environmental conditions. In addition, to zoom in on the patterns of Aminicenantes community structure in datasets where Aminicenantes represents a significant fraction of the overall bacterial community, the community structures in datasets with more than 50 Aminicenantes sequences (n = 198) were compared using principal-component analysis (PCA) and biplots were constructed using the R statistical package. In this analysis, the relative position of datasets is indicative of the level of their similarity, the directions of the class/subclass arrows are indicative of their respective maximal abundances, and the lengths of the arrows are proportional to the differential abundances of such lineages.
Results
1. A revised taxonomic outline of the Aminicenantes
A total of 142 near-full length 16S rRNA Aminicenantes gene sequences were identified in GenBank NR database (Table S2 in File S1). Detailed phylogenetic analysis grouped the Aminicenantes sequences into four candidate classes: OP8-1, OP8-2, OP8-3 and OP8-unclassified. Candidate class OP8-1 has the largest number of near full-length Aminicenantes sequences and is comprised of five distinct orders (OP8-1_HMMV, OP8-1_SHA-124, OP8-1_OPB95, OP8-1_unclassified, and OP8-1_YNP) (Figure 1, Table S2 in File S1). In contrast, classes OP8-2, OP8-3, and OP8-unclassified have a lower number of near full-length sequences and are not further sub-classified into candidate orders. This revision of Aminicenantes phylogeny hence increased the number of recognized near full-length 16S rRNA gene sequences by 30.3%, and added one candidate class (OP8-3) and one candidate order (OP8-1_YNP) to the Greengenes taxonomic outline, the most detailed Aminicenantes classification scheme in curated databases.
Figure 1. An updated taxonomic outline of the Aminicenantes.
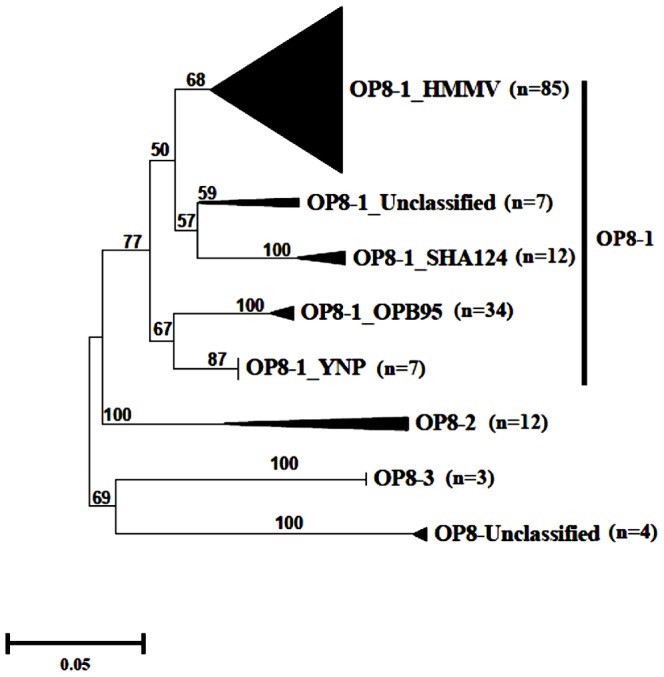
The Distance NJ tree was constructed using Jukes-Cantor corrections in MEGA5 [64]. Bootstrap values (in percent) are based on 1000 replicates and are shown for branches with more than 50% bootstrap support. Numbers in parentheses represent the number of sequences in each OP8 sub-phylum.
2. Identification of members of Aminicenantes in next generation 16S rRNA gene datasets
We used pyrosequencing- and Illumina-generated 16S rRNA gene datasets available in three publicly available gene repositories (VAMPS, GenBank, and MG-RAST) [42], [43] to identify the patterns of relative abundance, diversity, and community structure of members of the Aminicenantes. Within 3,141 datasets comprising ∼1.8 billion 16S rRNA gene sequences, 47,315 (0.0026%) from 918 (29.2%) different datasets were affiliated with the Aminicenantes (Table 1, Table S1 in File S1).
3. Patterns of Aminicenantes abundance
Overall relative abundance of Aminicenantes varied widely between various datasets, and ranged between 0 and 10.2% (encountered in MG-RAST dataset number 4455892, obtained from groundwater heavily contaminated by arsenic in the Ganges-Brahmaputra Delta region of Bangladesh, [47] (Table 1, Figure S1 in File S1). Although Aminicenantes has been identified in a substantial fraction (29.2%) of examined datasets, it invariantly constituted a minor fraction of the bacterial community identified, and rarely exceeded 5% in all datasets (Table 1, Figure S1 in File S1).
Based on incidence of occurrence (i.e. percentages of datasets in which sequences affiliated with the Aminicenantes were identified), and relative abundance of Aminicenantes in various datasets (Table 1, Figure S1 in File S1), members of the Aminicenantes appear to be most abundant in hydrocarbon-impacted habitats, being identified in 71.4% of the datasets (10/14), with an average abundance of 0.321%. The Aminicenantes was also frequently identified in marine (21.5% of datasets) and aquatic non-marine (38.74% of datasets) habitats, with average relative abundances of 0.275%, 0.146%, respectively (Table 1, Figure S1 in File S1). On the other hand, the Aminicenantes were rarely identified in soils and rumen habitats (Table 1, Figure S1 in File S1).
Aminicenantes abundance also demonstrated distinct patterns in relation to oxygen tension, temperature, and salinity (Table 2, Figure S2 in File S1). The Aminicenantes were most abundant in anaerobic habitats (58% of datasets, average 0.46%) e.g. Mai Po mangrove marshes in Hong Kong, heavy metal contaminated ground water in Bangladesh [47], active hydrothermal vent sediments from the Mid-Atlantic Ridge [48], anoxic sulfide and sulfur-rich terrestrial spring in southwestern Oklahoma (Zodletone spring) [40], and anoxic sediments from the Guaymas [3] and Cariaco Basins [49] (Table S1 in File S1). However, the Aminicenantes were also identified in much lower abundance in few oxic habitats e.g. water and sediments from coastal and open ocean sites surveyed from South Atlantic to the Caribbean seabed (Table S1 in File S1), coastal water of western English channel [50], and soils and sediments of hypersaline lake, La Sal del Rey’s in southern Texas, USA [51] (Table S1 in File S1). Temperature profile of Aminicenantes abundance indicated an extremely rare occurrence in low temperature terrestrial and marine habitats (e.g. in datasets from the Canadian, Alaskan and European tundra and arctic soils, as well as the Amundsen sea [50], [52]), (Table S1 in File S1), and a slightly higher preference (based on incidence of occurrence) to habitats with temperate, medium, elevated, and extremely elevated temperatures (Table 2, Figure S2 in File S1). Salinity wise, Aminicenantes was present at all levels of salinities, with slightly higher relative abundances in non-saline, and low salinity habitats (Table 2, Figure S2 in File S1).
4. Patterns of Aminicenantes community structure
Examination of patterns of Aminicenantes community composition across habitats revealed several distinct patterns. For example, order OP8-1_HMMV appears to be prevalent in marine environments, where it represented 53.5% of the total Aminicenantes sequences identified in marine datasets (Figure 2a). Class OP8-1_unclassified appeared to be the prominent lineage in aquatic non-marine environments, where it represented 77% of the total number of Aminicenantes sequences (Figure 2a). Order OP8-2 was the prevalent Aminicenantes lineage in hydrocarbon-impacted environments where it represented 66% of the total number of sequences. Although extremely rare in the rumen, the Aminicenantes sequences identified in a single dataset from this habitat belonged to order OP8-1_OPB95. PCA analysis conducted on datasets with more than 50 Aminicenantes sequences (n = 198, Figure 2b) confirmed such patterns where most of the environments from marine origins clustered along the OP8-1_HMMV species arrow (circles in Figure 2b), most of the environments from aquatic non-marine origins clustered along the OP8-1_unclassified species arrow (stars in Figure 2b), and the majority of the hydrocarbon-impacted environments clustered in the direction of the OP8-2 species arrow (diamonds in Figure 2b).
Figure 2. Aminicenantes relative abundance and community in various habitats.

(A) Relative abundance of Aminicenantes-affiliated sequences in marine, aquatic non-marine, soil, hydrocarbon-impacted, and rumen/other habitats. (B) PCA biplot of the community structure of Aminicenantes in datasets belonging to marine (•), aquatic non-marine (★), soil (n), hydrocarbon-impacted (u), and rumen ( ) with >50 Aminicenantes sequences. The biplot was generated in R using the prcomp and biplot functions in library labdsv. The first 2 axes explained 73% of the variance. There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (C) Relative abundance of Aminicenantes-affiliated sequences in various marine subhabitats. (D) PCA biplot of the community structure of Aminicenantes in marine datasets classified as coastal (blue), pelagic (green), hydrothermal vent (red), coral-associated (black), and deep sediment (yellow). There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (E) Relative abundance of Aminicenantes-affiliated sequences in environments originating from aquatic non-marine habitats. (F) PCA biplot of the community structure of Aminicenantes in aquatic non-marine datasets classified as freshwater (black), spring and groundwater (red), and salt marshes (blue). There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (G) Relative abundance of Aminicenantes-affiliated sequences in environments originating from soil habitats. Since only one soil dataset contained >50 Aminicenantes sequence, a PCA soil biplot is not feasible.
) with >50 Aminicenantes sequences. The biplot was generated in R using the prcomp and biplot functions in library labdsv. The first 2 axes explained 73% of the variance. There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (C) Relative abundance of Aminicenantes-affiliated sequences in various marine subhabitats. (D) PCA biplot of the community structure of Aminicenantes in marine datasets classified as coastal (blue), pelagic (green), hydrothermal vent (red), coral-associated (black), and deep sediment (yellow). There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (E) Relative abundance of Aminicenantes-affiliated sequences in environments originating from aquatic non-marine habitats. (F) PCA biplot of the community structure of Aminicenantes in aquatic non-marine datasets classified as freshwater (black), spring and groundwater (red), and salt marshes (blue). There are two sets of axis scales on the biplot; the ones on the right and top correspond to the axis scores for samples, and the bottom and left axes correspond to the loadings of the variables (in this case, OP8 subphyla). (G) Relative abundance of Aminicenantes-affiliated sequences in environments originating from soil habitats. Since only one soil dataset contained >50 Aminicenantes sequence, a PCA soil biplot is not feasible.
Sub-classification of habitats (Figures 2c-h) further revealed additional patterns at the sub-habitat level, especially in marine, soil, and aquatic non-marine habitats systems. Within marine environments, the prevalence of OP8-1_HMMV was more pronounced in coral-associated, pelagic, and deep marine datasets (Figure 2c). Indeed, in marine datasets with >50 Aminicenantes sequences, OP8-1_HMMV represents the majority (more than 80%) of the total Aminicenantes sequences in all coral-associated and pelagic datasets, as well as in the majority (10 out of 13) of deep sediment datasets. OP8-1_HMMV also represented the majority of Aminicenantes sequences in a few of the coastal (three out of 15) and hydrothermal (one out of six) datasets. Accordingly, those samples clustered together along the OP8-1_HMMV species arrow in the PCA biplot (red circles representing one vent sample, green circles representing five pelagic samples, yellow circles representing ten deep sediment samples, black circles representing five coral-associated samples, and blue circles representing three coastal samples in Figure 2d). In the remaining marine samples, the majority of Aminicenantes datasets has a mixed community of OP8-1_HMMV and other lineages, and so had an intermediary position between species arrows in the PCA biplot. In rare cases, some datasets did not contain any OP8-1_HMMV sequences. For example, all Aminicenantes-affiliated sequences from three hydrothermal vent samples belonged to the newly proposed candidate class OP8-3, and hence clustered in the direction of OP8-3 species arrow in the PCA biplot (red circles in Figure 2d).
Within aquatic non-marine habitats, the overall majority of Aminicenantes sequences belonged to subclass OP8-1_unclassified (Figure 2e). The majority (85.1% of datasets originating from the two non-saline aquatic non-marine sub-habitats (temperate freshwater lakes, and spring and groundwater samples) showed >70% of Aminicenantes-affiliated sequences belonging to the order OP8-1_unclassified and were hence clustered along the OP8-1_unclassified arrow in the PCA biplot (black and red stars, Figure 2f). However, two notable exceptions to this pattern were observed: 1. In several datasets, a mixed community of OP8-1_ unclassified with other lineages was observed (e.g. 11 freshwater lake samples had a mixed community of OP8-1_unclassified (53.6±2.4%), OP8-1_OPB95 (34.2±3.2%), and OP8-1_SHA-124 (11.3±1.9%), and one sample from a sinkhole had a mixed Aminicenantes community of OP8-1_OPB95 (43.7%), OP8-1_unclassified (32.1%), and OP8-2 (22.4%). 2. In few datasets, OP8-1_unclassified order was absent e.g. sewage samples with high abundance (> 90%) of OP8-1_OPB95 (Figure 2f).
While the Aminicenantes class OP8-1_unclassified was the prevalent lineage in the majority of aquatic non-marine habitats originating from temperate freshwater lakes, as well as spring and groundwater datasets; a distinct community structure was observed in aquatic non-marine habitats with low to moderate salinity (Figure 2e). Within these habitats, e.g. three samples from the Amazon-Guianas estuaries, and a salt marsh samples from Cabo Rojo, PR, the majority of Aminicenantes-affiliated sequences belonged to order OP8-1_HMMV (83.7±7.98%). Accordingly, those samples clustered along the HMMV species arrow in various PCA plot (Figure 2f).
Finally, a relatively small number of Aminicenantes-affiliated sequences were present in soil samples. Those were mainly affiliated with orders OP8-1_unclassified, OP8-1_OPB95, and OP8-1_SHA-124. Some unique patterns were observed at the sub-habitat level e.g. the prevalence of OP8-1_unclassified order in samples from permafrost soils (Figure 2g). However, it is important to note that the Aminicenantes exhibited an extremely rare distribution in all soil datasets examined, being only identified in 14 out of 276 datasets, with an extremely low average relative abundance (0.07%). Therefore, the significance of the observed patterns, given their extreme rarity, and doubtful ecological role in soil habitats, is questionable.
We also studied the effect of environmental conditions (O2 tension, temperature, and salinity) on Aminicenantes community structure in various datasets. When environments were classified based on their salinity, we observed a shift in the prevalence of various Aminicenantes lineages, with order OP8-1_unclassified representing the majority of Aminicenantes-affiliated sequences in non-saline habitats, as opposed to order HMMV in low and moderate salinity environments, and class OP8-2 in hypersaline environments (Figure 3a). We also observed an effect of temperature on the pattern of Aminicenantes community structure changes, where order OP8-1_unclassified and class OP8_2 dominated in low temperature and psychrophilic habitats, as opposed to orders OP8-1_OPB95 and HMMV in thermophilic and hyperthermophilic habitats (Figure 3b). However, the uneven number of samples belonging to each category (Table 3) could possibly skew these results. Finally, no remarkable effect of O2 tension on Aminicenantes community structure was observed (Figure 3c).
Figure 3. Relative abundance of Aminicenantes-affiliated sequences in different environments sub-classified according to (A) temperature, (B) oxygen tension, and (C) salinity.

Table 3. Diversity rankings of all datasets classified according to habitat and prevalent environmental conditions.
| Habitat/ Environmental parameter | Average diversity rank±SD | Number of samples belonging to this diversity rank | ||||
| Very low | Low | Medium | High | Very high | ||
| Marine | 115±68.5 | 11 | 1 | 6 | 10 | 12 |
| Pelagic | 89.9±63.3 | 1 | 0 | 2 | 1 | 0 |
| Coastal | 169.1±39.6 | 0 | 1 | 0 | 1 | 7 |
| Coral | 115.4±57.3 | 1 | 0 | 1 | 3 | 0 |
| Deep_sed | 106.4±65.9 | 5 | 0 | 3 | 4 | 5 |
| Hyd_vent | 34.3±59.1 | 4 | 0 | 0 | 1 | 0 |
| Non-marine | 96±52.8 | 26 | 38 | 34 | 31 | 21 |
| Freshwater | 93.7±51.6 | 26 | 38 | 34 | 30 | 18 |
| Spring/GW | 187.5±8.8 | 0 | 0 | 0 | 0 | 3 |
| Salt marsh | 148 | 0 | 0 | 0 | 1 | 0 |
| Hydrocarbon-Impacted Soil | 96.5±83.4 | 3 | 0 | 0 | 0 | 4 |
| Soil | 174.5 | 0 | 0 | 0 | 0 | 1 |
| Salinity | ||||||
| Non-saline | 95.8±52.4 | 29 | 38 | 34 | 29 | 24 |
| Low-salinity | 115.8±69.6 | 11 | 0 | 6 | 10 | 14 |
| Hypersaline | 115.3±47.4 | 0 | 1 | 0 | 2 | 0 |
| Temperature | ||||||
| Temperate | 102.6±56.1 | 33 | 39 | 40 | 40 | 38 |
| Elevated | 24.3±47 | 7 | 0 | 0 | 1 | 0 |
| O2 tension | ||||||
| Anoxic | 88.3±74.5 | 12 | 1 | 1 | 6 | 7 |
| Hypoxic | 84.6±44.1 | 26 | 36 | 37 | 27 | 5 |
| Oxic | 157.4±49.5 | 2 | 2 | 2 | 7 | 26 |
5. Patterns of Aminicenantes diversity
One hundred and ninety-eight datasets with more than 50 Aminicenantes-affiliated sequences were included in the diversity analysis. Due to the underrepresentation of hydrocarbon-impacted sites and soils, comparison of diversities was restricted to the marine and aqueous non-marine habitats and their subcategories. Within all habitats, the levels of diversity varied widely, but marine habitats showed higher diversity than freshwater habitats (Student t-test p-value = 0.037), with most of the marine environments (72%) showing medium to very high Aminicenantes diversity (Table 3). Within marine habitats, a higher average diversity rank was observed in coastal samples, and a lower average diversity was observed in hydrothermal vent samples. Indeed, coastal samples Aminicenantes diversities were significantly higher than those in all other marine environments (p-value ranging from 0.0004 to 0.041). Hydrothermal vent samples Aminicenantes diversities were significantly lower than those in coastal, and deep marine sediment samples (p-values 0.0004, and 0.04, respectively).
Aminicenantes diversity within aquatic non-marine environments varied, with high diversities observed in spring/groundwater samples and the single sample from a salt marsh. Significantly lower diversities were observed in samples from freshwater temperate environments (p-value = 0.002).
We also correlated diversity rankings to environmental conditions including temperature, salinity, and oxygen tension (Table 3). Interestingly, while no clear correlation was identified between temperature, or salinity and diversity levels of Aminicenantes at OTU0.03, a positive highly significant correlation existed between the dataset diversity rank and the environment’s oxygen tension (Spearman rank correlation coefficient = 0.4, p-value = 5.3E-9).
Discussion
In this study, we utilized in silico database mining approaches to provide an updated and expanded taxonomic outline of the candidate phylum Aminicenantes using near full-length 16S rRNA gene sequences, as well as to examine the global patterns of Aminicenantes distribution using high throughput (Pyrosequencing and Illumina) generated 16S rRNA gene datasets. We report that: 1. Members of the Aminicenantes are present in a substantial fraction (918 out of 3,141) of high throughput-generated datasets examined, where they represent a minor/rare fraction of the community, with very few exceptions. 2. Members of the Aminicenantes are ubiquitous, being encountered in all different types of habitats and across all spectra of environmental parameters (temperature, salinity, and oxygen tension) examined. 3. Distinct differences exist between the relative abundance of the Aminicenantes across different habitats and environmental conditions. 4. Members of the Aminicenantes exhibit a distinct community structure patterns across various datasets, and these patterns appear to be, mostly, driven by habitat variations rather than prevalent environmental parameters.
Utilizing high throughput-generated datasets of partial 16S rRNA gene sequences in dedicated sequence repositories (VAMPS, MG-RAST, and GenBank SRA) for analyzing patterns of prokaryotic diversity represents an extremely valuable, yet largely overlooked, resource. Next generation sequencing datasets are often deposited with a single accession number per dataset, often with inadequate metadata, and, unlike Sanger-generated sequences, these datasets are not readily amendable to online search queries. Nevertheless, when properly exploited, these datasets represent an excellent resource for testing specific ecological hypothesis. Examining Aminicenantes diversity in 3,141 distinct datasets, comprising a total of ∼1.8 billion partial sequences clearly demonstrates the presence of members of this candidate phylum in a large number (29.2% of datasets examined) of habitats. However, the Aminicenantes always represented a minor fraction of the overall community and often exhibited an extremely rare distribution: The relative abundance of the Aminicenantes was less than 0.01% of the total community in 70.1% of datasets examined, 0.01–0.1% in 16.1% of datasets examined, 0.1–1% in 12.9% of datasets, and more than 1% in only 0.9% of datasets examined (Table S1 in File S1). The reason for the occurrence, survival, and retention of various lineages as members of the rare biosphere (e.g. less than 0.1%) in various environments is an issue that has previously been thoroughly debated [6], [53], [54]. Possible reasons explaining this phenomenon vary and range between filling very specialized niches, acting as a backup system that readily responds to seasonal variations encountered in various ecosystem, exhibiting extremely slow growth or dormancy, introduction to the ecosystem through recent immigration of these rare phylotypes to the sampling site, or introduction to the dataset through contamination during sampling, DNA extraction, or amplification. Indeed, several of these explanations are plausible to elucidate the role of extremely rare members of the Aminicenantes in their respective ecosystems. Regardless, it is reasonable to assume that the detection of the Aminicenantes above a certain empirical threshold (e.g. 1%, equivalent to 105 cells/gram or ml in a community with a cell count of 107) reflects its successful colonization and propagation in a specific habitat, and suggests its importance in fulfilling vital ecosystem services that justifies its retention in that habitat. Therefore, examination of the few datasets in which the Aminicenantes are present in relatively higher abundances could offer a window on what factors are conducive for Aminicenantes survival and propagation in-situ. Datasets with more than 1% Aminicenantes relative abundance (Table S1, Figure S1 in File S1) (0.9% of the total number of datasets) were not restricted to one habitat type or one environmental condition, but occurred within the majority of the five habitats examined, and across a wide range of environmental conditions. Therefore, it is improbable that a single, specific, environmental condition e.g. hypersalinity or extreme temperature represents the only scenario for eliciting a competitive niche for the Aminicenantes. Rather, we argue that conditions at which Aminicenantes propagates appear to be induced by other types of natural or anthropogenic stressors, which effectively preclude a large fraction of the population, opening the window for Aminicenantes to propagate. This is apparent from the fact that many of the datasets with >1% Aminicenantes relative abundance came from environments with variable types of environmental stressors e.g. high levels of hydrocarbons (e.g. Alberta oil sands tailing ponds, Petroleum reservoirs in Huabei, China, and north slope oil facility) [55]–[57], or high levels of metal (arsenic) contamination in Araihazar, Bangladesh [47].
Overall relative abundance of the Aminicenantes appeared to vary widely across various habitats, as well as across specific environmental conditions. The Aminicenantes appear to be most abundant in hydrocarbon-impacted environments, being encountered in 71.4% of the datasets (10/14), with an average abundance of 0.321%. The association of specific lineages and phylotypes with hydrocarbon-impacted environments regardless of its origin (natural or anthropogenic), or chemical composition (natural gas, petroleum, enrichments on a single substrate) has previously been noted ([58], [59]. This prevalence in hydrocarbon-impacted settings is in agreement with the notion that success and propagation of members of the Aminicenantes in a specific environment is contingent on the occurrence of specific environmental stressors (hydrocarbon contamination and possibly associated anaerobiasis and high sulfide levels in such habitats) that partially alleviates competition, allowing for successful propagation of members of the Aminicenantes. The Aminicenantes were also identified in a considerable fraction of marine and aquatic non-marine habitats (Table 1, Table S1 in File S1). However, the complexity and variability of geochemical parameters encountered in these heterogeneous ecosystems prevents us from deciphering what exact environmental characteristics, or combination thereof, within these habitats favored Aminicenantes propagation. Correlating Aminicenantes abundance to environmental conditions (temperature, salinity, and oxygen tension) revealed that while members of the Aminicenantes could be encountered in a wide range of environmental conditions, it appears to exhibit significantly higher abundances in anoxic (compared to oxic and microoxic) habitats and a significantly lower abundance in low temperature (compared to temperate and elevated temperature) habitats. The relatively higher abundance of the Aminicenantes in anoxic environments suggests a prevalent anaerobic/facultative mode of metabolism within the Aminicenantes. Indeed, the majority of studies where the Aminicenantes represented more than 1% the total bacterial community originated from seemingly anaerobic habitats (e.g. arsenic contaminated ground water from Bangladesh, Guayamas methane seeps, and hypoliminion sites in Lake Mendota).
Analysis of the Aminicenantes community structure was conducted by: 1. Utilizing the classification of all (47,351) next generation sequences identified to examine the Aminicenantes community structure in various types of habitats and across various environmental conditions, and 2. PCA analysis of the Aminicenantes community structure in datasets where they exhibited relatively higher abundances (n>50). Overall, it appears that factors impacting Aminicenantes community structure are mostly habitat-driven (i.e. similar community structure observed in similar habitats), rather than driven by prevalent environmental conditions (temperature, salinity, oxygen tension) within an ecosystem. For example, class OP8-3 was exclusively identified in hydrothermal vent habitats; order OP8-1_ HMMV represented the majority of Aminicenantes sequences encountered in coral associated, pelagic, and deep marine habitats; OP8-1_unclassified represented the majority of sequences in aqueous non-marine habitats; and OP8-2 represented the majority of sequence in hydrocarbon-impacted habitats. The role of prevalent environmental condition in shaping the Aminicenantes microbial community is less certain, mostly due to the inadequate representation of special categories e.g. normal (body) temperature, elevated temperature, and hypersaline environments. However, one notable exception in which an environmental parameter appears to play a clear role in shaping the Aminicenantes microbial community is the distinct prevalence of order OP8-1_ HMMV in multiple low salinity datasets regardless of their habitat.
Finally, it is interesting to note that Aminicenantes sequences were identified across all ranges of salinity and temperatures including those conducive to the growth of obligate halophiles and hyperthermophiles, respectively. Collectively, the detection of Aminicenantes-affiliated sequences across environmental extremes, coupled to their observed ubiquitous distribution on a global scale and the distinct patterns of community structure exhibited argues for a high level of intraphylum metabolic and adaptive diversity within the Aminicenantes. Therefore it is probable that Aminicenantes cells in nature exhibit multiple distinct metabolic capabilities, wide array of survival weapons, and various adaptive strategies. This, in turn, highlights the importance of obtaining multiple genomic assemblies that adequately represents the broad phylogenetic diversity of this phylum, as well as its wide environmental distribution to truly gauge the pangenomic diversity within the Aminicenantes. The recently acquired genomic information from single cell-based efforts from Sakinaw Lake represents admirable effort to investigate this understudied and yet-uncultured lineage. However, information from such assemblies should not be extrapolated to describe all members of the Aminicenantes. Indeed, the discovery of novel capabilities within well-establish lineages e.g. phototrophy amongst Acidobacteria [60], methane oxidation amongst the Verrucomicrobia [61], [62], anaerobic oxidation of ammonia amongst the Planctomycetes [63] highlights the importance of continued efforts to decipher and expand genomic diversity within various bacterial phyla.
Supporting Information
Contains the files: Figure S1 Aminicenantes relative abundance in different habitat types. Figure S2 Aminicenantes relative abundance in response to various geochemical conditions. Table S1 Summary of all high throughput-generated datasets analyzed in this study. Table S2 List of all near full-length 16S rRNA sequences belonging to the Aminicenantes and their class/order level phylogenetic affiliations to Aminicenantes.
(DOC)
Acknowledgments
We would like to thank Dana Brunson at the OSU high performing computer center for technical assistance.
Funding Statement
This work was supported by the National Science Foundation Microbial Observatories Program (grant EF0801858). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Dojka MA, Hugenholtz P, Haack SK, Pace NR (1998) Microbial diversity in a hydrocarbon- and chlorinated-solvent-contaminated aquifer undergoing intrinsic bioremediation. Appl Environ Microbiol 64: 3869–3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hugenholtz P, Pitulle C, Hershberger KL, Pace NR (1998) Novel division level bacterial diversity in a Yellowstone hot spring. J Bacteriol 180: 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Teske A, Hinrichs KU, Edgcomb V, de Vera Gomez A, Kysela D, et al. (2002) Microbial diversity of hydrothermal sediments in the Guaymas Basin: evidence for anaerobic methanotrophic communities. Appl Environ Microbiol 68: 1994–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Roesch LF, Fulthorpe RR, Riva A, Casella G, Hadwin AK, et al. (2007) Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J 1: 283–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y, Sheng HF, He Y, Wu JY, Jiang YX, et al. (2012) Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl Environ Microbiol 78: 8264–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, et al. (2006) Microbial diversity in the deep sea and the underexplored "rare biosphere". Proc Natl Acad Sci U S A 103: 12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hugenholtz P (2002) Exploring prokaryotic diversity in the genomic era. Genome Biol 3: REVIEWS0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vergin KL, Urbach E, Stein JL, DeLong EF, Lanoil BD, et al. (1998) Screening of a fosmid library of marine environmental genomic DNA fragments reveals four clones related to members of the order Planctomycetales. Appl Environ Microbiol 64: 3075–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Treusch AH, Kletzin A, Raddatz G, Ochsenreiter T, Quaiser A, et al. (2004) Characterization of large-insert DNA libraries from soil for environmental genomic studies of Archaea. Environ Microbiol 6: 970–980. [DOI] [PubMed] [Google Scholar]
- 10. Elshahed MS, Najar FZ, Aycock M, Qu C, Roe BA, et al. (2005) Metagenomic analysis of the microbial community at Zodletone Spring (Oklahoma): insights into the genome of a member of the novel candidate division OD1. Appl Environ Microbiol 71: 7598–7602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kielak AM, van Veen JA, Kowalchuk GA (2010) Comparative analysis of acidobacterial genomic fragments from terrestrial and aquatic metagenomic libraries, with emphasis on acidobacteria subdivision 6. Appl Environ Microbiol 76: 6769–6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Narasingarao P, Podell S, Ugalde JA, Brochier-Armanet C, Emerson JB, et al. (2012) De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J 6: 81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, et al. (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. [DOI] [PubMed] [Google Scholar]
- 14. Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, et al. (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665. [DOI] [PubMed] [Google Scholar]
- 15. Pelletier E, Kreimeyer A, Bocs S, Rouy Z, Gyapay G, et al. (2008) "Candidatus Cloacamonas acidaminovorans": genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol 190: 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- 17. Youssef NH, Blainey PC, Quake SR, Elshahed MS (2011) Partial genome assembly for a candidate division OP11 single cell from an anoxic spring (Zodletone Spring, Oklahoma). Appl Environ Microbiol 77: 7804–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Campbell JH, O'Donoghue P, Campbell AG, Schwientek P, Sczyrba A, et al. (2013) UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci U S A 110: 5540–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McLean JS, Lombardo MJ, Badger JH, Edlund A, Novotny M, et al. (2013) Candidate phylum TM6 genome recovered from a hospital sink biofilm provides genomic insights into this uncultivated phylum. Proc Natl Acad Sci U S A 110: E2390–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tourna M, Stieglmeier M, Spang A, Konneke M, Schintlmeister A, et al. (2011) Nitrososphaera viennensis, an ammonia oxidizing archaeon from soil. Proc Natl Acad Sci U S A 108: 8420–8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Konneke M, Bernhard AE, de la TorreJR, Walker CB, Waterbury JB, et al. (2005) Isolation of an autotrophic ammonia-oxidizing marine archaeon. Nature 437: 543–546. [DOI] [PubMed] [Google Scholar]
- 22. Girguis PR, Cozen AE, DeLong EF (2005) Growth and population dynamics of anaerobic methane-oxidizing archaea and sulfate-reducing bacteria in a continuous-flow bioreactor. Appl Environ Microbiol 71: 3725–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bates ST, Clemente JC, Flores GE, Walters WA, Parfrey LW, et al. (2013) Global biogeography of highly diverse protistan communities in soil. ISME J 7: 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Freitas S, Hatosy S, Fuhrman JA, Huse SM, Welch DB, et al. (2012) Global distribution and diversity of marine Verrucomicrobia. ISME J 6: 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergmann GT, Bates ST, Eilers KG, Lauber CL, Caporaso JG, et al. (2011) The under-recognized dominance of Verrucomicrobia in soil bacterial communities. Soil Biol Biochem 43: 1450–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones RT, Robeson MS, Lauber CL, Hamady M, Knight R, et al. (2009) A comprehensive survey of soil acidobacterial diversity using pyrosequencing and clone library analyses. ISME J 3: 442–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Buckley DH, Huangyutitham V, Nelson TA, Rumberger A, Thies JE (2006) Diversity of Planctomycetes in soil in relation to soil history and environmental heterogeneity. Appl Environ Microbiol 72: 4522–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gilbert JA, Meyer F, Jansson J, Gordon J, Pace N, et al. (2010) The Earth Microbiome Project: Meeting report of the "1 EMP meeting on sample selection and acquisition" at Argonne National Laboratory October 6 2010. Stand Genomic Sci 3: 249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huse SM, Ye Y, Zhou Y, Fodor AA (2012) A core human microbiome as viewed through 16S rRNA sequence clusters. PLoS One 7: e34242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Knight R, Jansson J, Field D, Fierer N, Desai N, et al. (2012) Unlocking the potential of metagenomics through replicated experimental design. Nat Biotechnol 30: 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chouari R, Le Paslier D, Daegelen P, Ginestet P, Weissenbach J, et al. (2005) Novel predominant archaeal and bacterial groups revealed by molecular analysis of an anaerobic sludge digester. Environ Microbiol 7: 1104–1115. [DOI] [PubMed] [Google Scholar]
- 32. Joynt J, Bischoff M, Turco R, Konopka A, Nakatsu CH (2006) Microbial community analysis of soils contaminated with lead, chromium and petroleum hydrocarbons. Microb Ecol 51: 209–219. [DOI] [PubMed] [Google Scholar]
- 33. Losekann T, Knittel K, Nadalig T, Fuchs B, Niemann H, et al. (2007) Diversity and abundance of aerobic and anaerobic methane oxidizers at the Haakon Mosby Mud Volcano, Barents Sea. Appl Environ Microbiol 73: 3348–3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dhillon A, Teske A, Dillon J, Stahl DA, Sogin ML (2003) Molecular characterization of sulfate-reducing bacteria in the Guaymas Basin. Appl Environ Microbiol 69: 2765–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, et al. (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6: 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41: D590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, et al. (2008) NCBI BLAST: a better web interface. Nucleic Acids Res 36: W5–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goecks J, Nekrutenko A, Taylor J, Galaxy T (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: R86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 40. Youssef N, Steidley BL, Elshahed MS (2012) Novel high-rank phylogenetic lineages within a sulfur spring (Zodletone Spring, Oklahoma), revealed using a combined pyrosequencing-sanger approach. Appl Environ Microbiol 78: 2677–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dalevi D, Hugenholtz P, Blackall LL (2001) A multiple-outgroup approach to resolving division-level phylogenetic relationships using 16S rDNA data. Int J Syst Evol Microbiol 51: 385–391. [DOI] [PubMed] [Google Scholar]
- 42. Meyer F, Paarmann D, D'Souza M, Olson R, Glass EM, et al. (2008) The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9: 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, et al. (2013) GenBank. Nucleic Acids Res 41: D36–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75: 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Youssef NH, Elshahed MS (2009) Diversity rankings among bacterial lineages in soil. ISME J 3: 305–313. [DOI] [PubMed] [Google Scholar]
- 46.Team RDC (2011) R: A Language and Environment for Statistical Computing. Reference Index. Vienna, Austria: R Foundation for Statistical Computing.
- 47. Legg TM, Zheng Y, Simone B, Radloff KA, Mladenov N, et al. (2012) Carbon, metals, and grain size correlate with bacterial community structure in sediments of a high arsenic aquifer. Front Microbiol 3: 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Flores GE, Campbell JH, Kirshtein JD, Meneghin J, Podar M, et al. (2011) Microbial community structure of hydrothermal deposits from geochemically different vent fields along the Mid-Atlantic Ridge. Environ Microbiol 13: 2158–2171. [DOI] [PubMed] [Google Scholar]
- 49. Madrid VM, Taylor GT, Scranton MI, Chistoserdov AY (2001) Phylogenetic diversity of bacterial and archaeal communities in the anoxic zone of the Cariaco Basin. Appl Environ Microbiol 67: 1663–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zinger L, Amaral-Zettler LA, Fuhrman JA, Horner-Devine MC, Huse SM, et al. (2011) Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS One 6: e24570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hollister EB, Engledow AS, Hammett AJ, Provin TL, Wilkinson HH, et al. (2010) Shifts in microbial community structure along an ecological gradient of hypersaline soils and sediments. ISME J 4: 829–838. [DOI] [PubMed] [Google Scholar]
- 52. Chu H, Fierer N, Lauber CL, Caporaso JG, Knight R, et al. (2010) Soil bacterial diversity in the Arctic is not fundamentally different from that found in other biomes. Environ Microbiol 12: 2998–3006. [DOI] [PubMed] [Google Scholar]
- 53. Elshahed MS, Youssef NH, Spain AM, Sheik C, Najar FZ, et al. (2008) Novelty and uniqueness patterns of rare members of the soil biosphere. Appl Environ Microbiol 74: 5422–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pedros-Alio C (2012) The rare bacterial biosphere. Ann Rev Mar Sci 4: 449–466. [DOI] [PubMed] [Google Scholar]
- 55. Stevenson BS, Drilling HS, Lawson PA, Duncan KE, Parisi VA, et al. (2011) Microbial communities in bulk fluids and biofilms of an oil facility have similar composition but different structure. Environ Microbiol 13: 1078–1090. [DOI] [PubMed] [Google Scholar]
- 56. Saidi-Mehrabad A, He Z, Tamas I, Sharp CE, Brady AL, et al. (2013) Methanotrophic bacteria in oilsands tailings ponds of northern Alberta. ISME J 7: 908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li H, Yang SZ, Mu BZ, Rong ZF, Zhang J (2006) Molecular analysis of the bacterial community in a continental high-temperature and water-flooded petroleum reservoir. FEMS Microbiol Lett 257: 92–98. [DOI] [PubMed] [Google Scholar]
- 58. Elshahed MS, Senko JM, Najar FZ, Kenton SM, Roe BA, et al. (2003) Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring. Appl Environ Microbiol 69: 5609–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davis JP, Struchtemeyer CG, Elshahed MS (2012) Bacterial communities associated with production facilities of two newly drilled thermogenic natural gas wells in the Barnett Shale (Texas, USA). Microb Ecol 64: 942–954. [DOI] [PubMed] [Google Scholar]
- 60. Bryant DA, Costas AM, Maresca JA, Chew AG, Klatt CG, et al. (2007) Candidatus Chloracidobacterium thermophilum: an aerobic phototrophic Acidobacterium. Science 317: 523–526. [DOI] [PubMed] [Google Scholar]
- 61. Pol A, Heijmans K, Harhangi HR, Tedesco D, Jetten MS, et al. (2007) Methanotrophy below pH 1 by a new Verrucomicrobia species. Nature 450: 874–878. [DOI] [PubMed] [Google Scholar]
- 62. Dunfield PF, Yuryev A, Senin P, Smirnova AV, Stott MB, et al. (2007) Methane oxidation by an extremely acidophilic bacterium of the phylum Verrucomicrobia. Nature 450: 879–882. [DOI] [PubMed] [Google Scholar]
- 63. Strous M, Fuerst JA, Kramer EH, Logemann S, Muyzer G, et al. (1999) Missing lithotroph identified as new planctomycete. Nature 400: 446–449. [DOI] [PubMed] [Google Scholar]
- 64. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Contains the files: Figure S1 Aminicenantes relative abundance in different habitat types. Figure S2 Aminicenantes relative abundance in response to various geochemical conditions. Table S1 Summary of all high throughput-generated datasets analyzed in this study. Table S2 List of all near full-length 16S rRNA sequences belonging to the Aminicenantes and their class/order level phylogenetic affiliations to Aminicenantes.
(DOC)