

Abstract
The androgen receptor (AR) is a master regulator transcription factor in normal and cancerous prostate cells. Canonical AR activation requires binding of androgen ligand to the AR ligand binding domain, translocation to the nucleus, and transcriptional activation of AR target genes. This regulatory axis is targeted for systemic therapy of advanced prostate cancer. However, a new paradigm for AR activation in castration-resistant prostate cancer (CRPC) has emerged wherein alternative splicing of AR mRNA promotes synthesis of constitutively active AR variants that lack the AR ligand binding domain (LBD). Recent work has indicated that structural alteration of the AR gene locus represents a key mechanism by which alterations in AR mRNA splicing arise. In this review, we examine the role of truncated AR variants (ARVs) and their corresponding genomic origins in models of prostate cancer progression, as well as the challenges they pose to the current standard of prostate cancer therapies targeting the AR ligand binding domain. Since ARVs lack the COOH-terminal LBD, the genesis of these AR gene rearrangements and their resulting ARVs provides strong rationale for the pursuit of new avenues of therapeutic intervention targeted at the AR NH2-terminal domain. We further suggest that genomic events leading to ARV expression could act as novel biomarkers of disease progression that may guide the optimal use of current and next-generation AR-targeted therapy.
Keywords: Androgen Receptor, alternative splicing, castration resistance, genomic rearrangement, prostate cancer
INTRODUCTION
The AR is a 110 kDa protein with a modular domain organization found in members of the steroid hormone receptor superfamily [1, 2]. The NH2-terminal domain (NTD), also referred to as transcriptional activation function (AF)-1, is a potent transcriptional activation domain in isolation and is responsible for the majority of AR transcriptional activity through the recruitment of diverse co-regulatory proteins. The central domain of the AR is the DNA binding domain (DBD), which is comprised of two zinc-finger motifs. The first zinc finger is responsible for making direct contact with the DNA major groove of an androgen response element (ARE) half-site, while the second zinc finger mediates dimerization with a second AR molecule bound to an adjacent ARE half-site [3]. The DBD is followed by a short, flexible hinge region which contains the bipartite nuclear localization signal. The COOH-terminal domain (CTD) of the AR houses both the ligand binding domain (LBD) and a secondary transcriptional activation domain termed AF-2.
The primary role of the AR is to sense and respond to circulating androgens, the most abundant of which are testosterone and dihydrotestosterone (DHT) [4, 5]. In the absence of ligand stimulation, AR is cytoplasmic, bound in a chaperone complex of heat shock proteins and high molecular weight immunophilins, which maintains AR protein in an inactive conformation with a high affinity for ligand binding [6]. Following ligand binding, the AR undergoes a conformational change, causing dissociation of a subset of chaperone proteins and exposing the nuclear localization signal in the hinge region. Upon translocation of the AR/DHT complex to the nucleus, the AR DBD engages with genomic AREs [7], mediating chromosomal looping and structural reorganization of the genome [8–10]. In order for productive gene transcription to occur, the AR is reliant on interactions with a wide variety of transcriptional co-regulators, of which nearly 200 have been identified to date [11]. These transcriptional co-regulators form large complexes that result in recruitment of the basal transcriptional machinery and a finely-tuned level of androgen-responsive gene transcription [12, 13]. In healthy prostate tissue, these androgen-responsive genes are important for normal prostate architecture, homeostasis, and physiological function. In prostate cancer (PCa), these target genes support ongoing proliferation and survival of tumor cells.
Structure and Function of the AR COOH-Terminal Domain
The CTD of the AR is the best understood functional domain by virtue of its structural homology and regulatory similarities with other steroid receptors [14]. The AR gene locus, located at Xq11-12, is approximately 180 kilobases in length and consists of eight coding exons separated by intronic segments of varying length. Exon 1 codes for the entire AR NTD, or approximately 60% of the total protein, while exons 2 and 3 code for the two zinc finger domains of the AR DBD. Exons 4–8 are located in close proximity to one another in the AR gene locus, and code for the hinge region and CTD/LBD of the AR (Fig. 1). Importantly, all AR-targeted therapeutics currently approved for clinical use modulate AR activity by exerting action on this domain [15]. The CTD contains 11 α-helices that form the binding pocket of the AR LBD, while a twelfth helix forms a “kickstand” which locks into place upon androgen binding [16–18]. This upswing of helix 12 stabilizes AR binding to DHT and forms the AF-2 protein interaction interface [19, 20]. AF-2 has been shown to exert transcriptional activity in the presence of bound agonist by binding to nuclear receptor (NR)-box motifs in coactivators, such as SRC-2 [19], and is also capable of mediating intramolecular interaction with FxxLF or WxxLF motifs in the AR NTD [21]. Furthermore, the CTD contains a ligand-regulated nuclear export signal which is dominant over the AR nuclear localization signal but is inhibited by ligand binding [22].
Figure 1. Androgen Receptor Functional Domains.
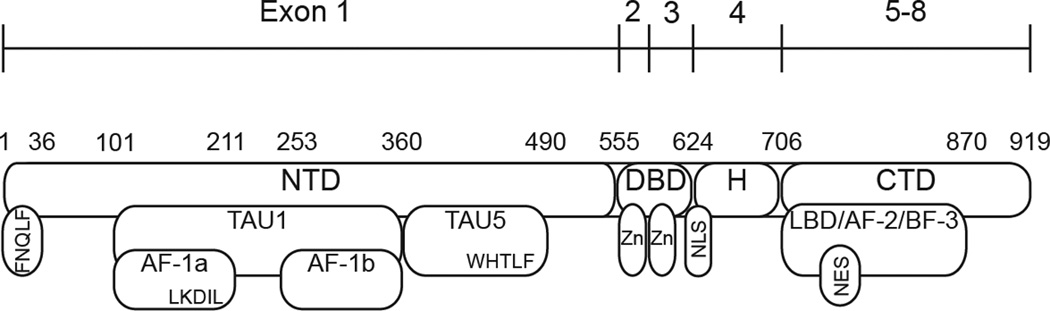
The AR possesses a modular domain organization common to members of the steroid hormone receptor family of nuclear receptor transcription factors. The amino-terminal domain (NTD) harbors transcriptional activation unit (TAU)-1 and TAU-5. Transcriptional activation function (AF)-1a and AF-1b are subdomains of TAU-1. The DNA binding domain (DBD) is comprised of two zinc finger motifs (Zn), and a flexible hinge (H) region containing the AR nuclear localization signal (NLS). The COOH-terminal domain (CTD) harbors the ligand binding domain (LBD), a ligand-regulated nuclear export signal (NES), and the protein interaction domains AF-2 and binding function (BF)-3.
Recent work has provided evidence for a second protein interaction domain within the CTD, termed binding function (BF)-3, which communicates allosterically with AF-2 [23, 24]. Interestingly, a host of mutations identified in both PCa and androgen insensitivity syndrome map to BF-3, supporting the concept that this domain may play an important role in allosteric regulation of AR function [25]. Another recent study provided evidence that FKBP52, a co-chaperone protein critical for maintaining AR in a conformation competent for ligand binding, may interact with AR through the BF-3 domain [26]. Importantly, these critical AF-2/BF-3 mediated functions are amenable to targeting with small molecules [23, 26–28] which could potentially lead to new avenues of AR-targeted therapy.
Therapeutic Targeting of the COOH-Terminal Domain in Prostate Cancer
PCa is the most frequently diagnosed male cancer and second leading cause of cancer deaths [29]. For tumors that are relapsed, locally advanced, and/or metastatic, the current standard of care is androgen deprivation therapy (ADT), achieved by suppression of AR signaling through the use of AR antagonists such as bicalutamide or flutamide, or by preventing production of testosterone by the testes using gonadotropin releasing hormone agonists such as leuprolide [15]. ADT initially provides a robust therapeutic benefit by blocking tumor cell proliferation and inducing apoptosis, resulting in clinical regression. Invariably, however, AR signaling is eventually reactivated via diverse mechanisms including AR amplification and/or AR protein overexpression, gain of function AR mutations [30, 31], enhanced uptake and conversion of adrenal androgens, or de novo androgen synthesis by tumor cells [32]. These mechanisms have been reviewed in detail elsewhere [33–35]. These molecular events mark a transition from the initial androgen-dependent PCa to a lethal castration recurrent phenotype, also referred to as “castration resistant PCa” (CRPC). Enzalutamide (formerly MDV3100) and abiraterone acetate, two next-generation AR-targeted therapeutics, were developed to address these mechanisms of disease progression [36, 37]. In Phase III trials, abiraterone and MDV3100 increased overall survival CRPC patients by 3.9 and 4.8 months, respectively [38, 39]. Clinical trials have demonstrated that both drugs provide significant therapeutic benefit to a high proportion of patients [38–41], but a subset of patients continue to experience disease progression, either through acquired resistance or through de novo insensitivity prior to treatment. Currently, it is of major interest to identify mechanisms that may drive these types of resistance to next-generation AR-targeted therapies. Importantly, unlike other steroid hormone receptors in which AF-2 is the dominant transactivation domain, the AR CTD plays a primarily regulatory role, and the AR NTD is responsible for the majority of AR transactivation [19]. Therefore, recent work describing the discovery and characterization of constitutively active, pathogenic AR splice variants which lack the CTD regulatory domain have generated significant interest, as these species may be capable of restoring AR signaling in PCa tissues following ADT through a mechanism of constitutive AR NTD transcriptional activity.
CONSTITUTIVELY ACTIVE AR SPLICE VARIANTS PROMOTE CASTRATION RESISTANCE IN PROSTATE TUMORS
Splice variants of the AR have been recognized for over two decades in the context of loss-of-function splicing alterations in androgen insensitivity syndrome, which is a topic that has been reviewed in detail elsewhere [42]. The first gain-of-function AR splice variant (ARV) was identified in 22Rv1 cells due to the presence of a smaller, 75–80 kDa AR immunoreactive species on western blot that was initially thought to be a proteolytic degradation fragment of full length AR [43]. This AR subspecies was shown to lack a ligand binding domain and was constitutively active in both the presence and absence of androgen. Similarly, a subsequently identified Q640Stop mutation resulted in premature truncation and constitutive AR signaling in bone metastases from a patient who relapsed following ADT with luprorelin and flutamide [44]. It was initially postulated that the smaller band observed in the 22Rv1 cell line resulted from calpain-mediated cleavage of full length AR at a consensus calpain recognition site in the AR hinge region [45]. However, later work demonstrated that RNA interference (RNAi) targeted against AR exon 7 (Fig. 1) had no effect on expression levels of the smaller species, despite robust ablation of full length AR. Conversely, RNAi targeted against AR exon 1 led to ablation of both the full length and the truncated species [46]. These data strongly suggested that the truncated ARV was not a product of full length AR mRNA or protein, but instead derived from an alternate mRNA species. The ability to differentially target full-length vs. truncated ARV species with discrete RNAi reagents further revealed that the constitutive activity of the truncated ARV was the driving force behind the androgen independent proliferation of 22Rv1 cells.
Since their initial identification, nearly a dozen different ARV mRNA species have been identified in PCa cell lines, xenografts, and clinical samples [42]. ARVs arise as a result of the incorporation of alternative, or cryptic, exons coded for in the AR gene locus [46–49], or through an exon skipping mechanism in which non-contiguous AR exons are spliced together [50]. Characterization of these novel ARV mRNAs has revealed multiple alternative, or “cryptic” exons in the AR locus, most of which flank AR exon 3. For example, alternative exon 2b (also termed cryptic exon 4, or “CE4”) is located upstream of exon 3, whereas many others are within AR intron 3 (CE1, CE2, CE3, CE5, and exon 3'). The products of these splicing aberrations generally incorporate canonical AR exons 1–3, which code for the AR NTD as well as the DBD. These three exons appear to form the minimum requirement for a transcriptionally active ARV [51]. However, ARVs differ in their utilization of exons 4–8, with most ARVs incorporating one of the seven currently identified cryptic exons coded for by the AR locus [42]. Problematically, multiple naming systems have been proposed to refer to the various ARVs. For example, the ARV encoded by contiguously spliced exons 1, 2, 3, and 2b has been alternately named AR-V4 [48], AR5 [47], and ARV6 [49]. Therefore, for the purposes of this review, we will refer to the variants by their exon composition, e.g. AR 1/2/3/2b, to alleviate confusion. To date, only three ARV transcripts have been mechanistically investigated in cell lines, xenografts, or clinical samples: AR 1/2/3/CE3, AR 1/2/3/2b, and AR 1/2/3/4/8.
The most studied and currently best characterized ARV is coded for by AR exons 1/2/3/CE3, alternatively termed AR-V7 and AR3 [47, 48]. AR 1/2/3/CE3 has been shown to be expressed at the mRNA and protein level in normal and cancerous prostate tissue, multiple commonly used PCa cell lines, and human tumor xenografts. Expression of this isoform was also demonstrated to be increased in locally recurrent and metastatic castration resistant PCa tissue compared to prostatectomy specimens from hormone naïve men [47]. A separate study found that 1/2/3/CE3 mRNA expression levels in prostatectomy specimens could predict the likelihood of biochemical recurrence after surgery [48]. Biochemically, 1/2/3/CE3 was shown to function as a constitutively active transcription factor independent of androgen ligand [47]. However, the exact transcriptional program mediated by this variant may differ slightly from full-length AR. In one study, transient transfection of LNCaP cells with an AR 1/2/3/CE3 expression vector was shown to effect a strikingly similar transcriptional program compared with ligand-activated full length AR [48]. On the other hand, a second study using targeted siRNA knockdown of endogenous full length AR versus 1/2/3/CE3 demonstrated that the CE3 isoform activated Akt expression, whereas full-length AR did not [48]. More recently, Hu and colleagues [52] have reported a unique role for AR 1/2/3/CE3 in the activation of M-phase specific cell cycle genes. For example, whereas full-length AR target genes appeared to be largely associated with pathways important for biosynthesis and metabolism, the 1/2/3/CE3 variant was able to activate transcription of pro-mitotic cell cycle regulators such as UBE2C, CDCA5, ZWINT, TPX2, and CDC25C. Taken together, these data strongly support a role for the AR 1/2/3/CE3 splice variant as a constitutively active AR isoform with significant clinical implications for biology and treatment of castration resistant PCa tumors.
The AR 1/2/2b and AR 1/2/3/2b mRNA variants were initially identified by 5' RACE experiments in the 22Rv1 cell line [46]. Specific knockdown of these ARVs using an exon 2b-targeted siRNA resulted in an expected reduction in the truncated 75–80 kDa AR species, suggesting that one or both of the 1/2/2b and 1/2/3/2b mRNAs were translated. However, our laboratory recently developed an antibody specific to the COOH-terminal extension encoded by AR exon 2b, which revealed that only the AR 1/2/3/2b variant is productively translated to functional protein in 22Rv1 cells [51]. Importantly, siRNA knockdown of AR 1/2/3/2b significantly reduced the ability of 22Rv1 cells to proliferate in the absence of androgen, but had no effect on androgen dependent proliferation, supporting a role for this ARV as a driver of castration resistance in 22Rv1 cells [46].
Finally, AR 1/2/3/4/8 was shown to arise through the skipping of exons 5–7 in the mRNA transcript [50]. This exon skipping event places exon 8 out-of-frame, resulting in formation of a premature translation termination codon in exon 8. This variant, originally named ARv567es, is expressed at the mRNA level in a wide range of normal and cancerous prostate tissues, although protein expression has not yet been confirmed using variant-specific antibodies. Sun and colleagues also demonstrated that AR 1/2/3/4/8 mRNA is expressed endogenously in the LuCaP 86.2 and 136 xenografts, and inferred that this protein was expressed endogenously due to its molecular weight. Interestingly, upon castration, mRNA levels of AR 1/2/3/4/8 increased in these xenografts compared with intact hosts. When expressed ectopically in LNCaP cells, AR 1/2/3/4/8 displayed constitutive transcriptional activity and could interact directly with full length AR, resulting in enhanced ligand dependent and -independent activity of the full length receptor in these cells. A later study by Hu and colleagues demonstrated that a novel ninth exon, located downstream of AR exon 8, was incorporated into the mRNA transcript of this AR 1/2/3/4/8 variant in VCaP cells [52]. However, because incorporation of exon 9 does not affect the premature translation stop codon in exon 8, this exon simply alters the 3’ untranslated region of this mRNA. Therefore, AR 1/2/3/4/8/9 mRNA codes for exactly the same protein as AR 1/2/3/4/8 mRNA, and thus it is not surprising that this variant displayed constitutive, ligand independent activity in promoter-reporter assays. Interestingly, in this study, the strength of AR 1/2/3/4/8/9 transcriptional activity appeared to depend on which cell line it was tested in, with higher activity apparent in PC-3 vs. LNCaP PCa cell lines.
Although all active gain of function ARVs identified to date consist of the AR NTD and DBD, they harbor unique COOH-terminal extensions encoded by the various exons that can be spliced into the 3’ mRNA termini of AR variant transcripts. This has been proposed to bear significant implications for ARV biochemistry because the bipartite nuclear localization signal (NLS), RKx10RKLKK, spans the exon 3–4 junction in wild type AR [53]. Therefore, since most of the ARV mRNAs identified to date do not harbor exon 4, the bipartite NLS would be disrupted in these variants. However, recent work has demonstrated that the AR NTD/DBD core displays a high level of constitutive nuclear localization in the absence of ligand that is independent of both HSP90 and the nuclear import adapter protein importin-β, resulting in transcriptional activation of endogenous AR targets [51]. Moreover, this study demonstrated that differences in ARV transcriptional activity that have been observed are promoter-dependent phenomena as opposed to arising from differential rates of nuclear access.
GENOMIC REARRANGEMENTS PROMOTE DISEASE PROGRESSION AT MULTIPLE STAGES OF PROSTATE CANCER DEVELOPMENT
Gene Rearrangements Prior to ADT: From PIN to PCa and Beyond
Beginning with the discovery of recurrent Ets-family gene rearrangements in 2005 [54], it has become increasingly clear that structural alterations are frequent events in the PCa genome and underpin many aspects of tumor biology and disease progression. These events include the highly prevalent TMPRSS2-Ets family of gene fusions [54] as well as fusions involving Raf family members [55]. A number of other rearrangements have also been identified in primary prostate tumors, which may represent novel mechanisms for driving tumor invasiveness, proliferation/survival, and anchorage-independent growth in PCa [56, 57]. These gene rearrangements have been reviewed in detail elsewhere [58]. More recently, chromosomal alterations involving the PTEN locus have been shown to cooperate with allelic loss to drive PCa progression [59]. Interestingly, this mechanism of PTEN inactivation, as well as the rearrangements reported by Pflueger et al. [57] was highly correlated with underlying ERG rearrangement, supporting a role for Ets family rearrangements as a genome-destabilizing event early in prostate tumorigenesis. This may also explain the observation that patients with fusion-positive PCa experience more aggressive and lethal disease compared with fusion-negative cases [60, 61], though a number of studies have reported that TMPRSS2:Ets rearrangement may alternatively correlate with low Gleason grade [62], favorable prognosis [63], or may not be predictive of disease outcome at all [64]. Regardless, clinical samples from men with metastatic castration-recurrent PCa exhibit a wide range of mutations, deletions, and rearrangements as determined by exome sequencing [65]. Overall, these genomic rearrangements have been shown to be associated with and predictive of PCa genesis and/or progression, such that molecular subtyping of based on these criteria may result in improvements in patient management and/or clinical trial designs [58].
Rearrangements of the AR Locus: a New Paradigm for ARV Expression and Activity Following ADT?
These recent whole genome studies have also supported the long-held fundamental concept that the AR signaling axis is a critical master regulator in PCa. Foremost, this axis has been shown to be the most frequently-altered pathway in hormone-naïve PCa, and 100% of castration-resistant PCa metastases display genomic and/or mRNA expression alterations in this pathway, most frequently in the AR gene itself [66]. The observation that AR exon 2b is incorporated into the 1/2/3/2b transcript downstream of exon 3 in 22Rv1 cells, despite the fact that 2b is located 5' of exon 3 in the normal reference genome [46, 48] (Fig. 2), raised an intriguing question: what is the molecular basis for this splicing pattern? One clue came from the observation that the full-length AR in 22Rv1 cells is slightly larger due to an extra zinc finger in the DBD encoded by tandem duplication of AR exon 3. Interestingly, in addition to these unanticipated splicing patterns, it was demonstrated that the 22Rv1 cell line exhibits significantly increased mRNA expression of the AR 1/2/3/CE3 variant [67]. In the same study, the androgen-dependent CWR22Pc cell line, which was derived from the same original CWR22 xenograft model as 22Rv1, was found by quantitative RT-PCR analysis to express extremely low but detectable transcript expression of these ARVs. These observations suggested that the observed splicing patterns may not be true “alternative splicing” events in 22Rv1 cells, but may instead be due to an underlying alteration in AR gene structure. Indeed, interrogation of AR gene structure demonstrated that the region harboring exon 2b, 3, and CE1-3 was present in the genome at two-fold higher copy number in castration-recurrent 22Rv1 cells, but not CWR22Pc, suggesting the presence of a tandem duplication [67]. More detailed analysis confirmed that a ~35kb segment, comprised of exon 3 and its flanking cryptic exons, was involved in a tandem duplication event within 22Rv1 cells (Fig. 2). Importantly, long term culture of the lineage-related CWR22Pc cell line in the absence of androgen resulted in the outgrowth of a castration resistant population of cells that harbored the exact same break fusion junction and repair signature as 22Rv1, and displayed increased expression of truncated ARVs mRNAs and proteins including 1/2/3/2b and 1/2/3/CE3. These data indicate that a subset of cells within the original CWR22 tumor harbor this rearrangement and are driven by constitutive, ligand-independent ARV activity prior to androgen deprivation. In this cell line, ADT simply results in selective outgrowth of these ARV-driven cells harboring the 35kb tandem duplication. Importantly, complex patterns of AR gene copy imbalance were also observed in metastatic CRPC samples, but not in hormone-naïve primary tumors [67], suggesting that generation of constitutively active ARV through genomic rearrangements may be a recurring theme in human disease progression.
Figure 2. AR Genomic Alterations and Altered Splicing Patterns Leading to ARV Expression.
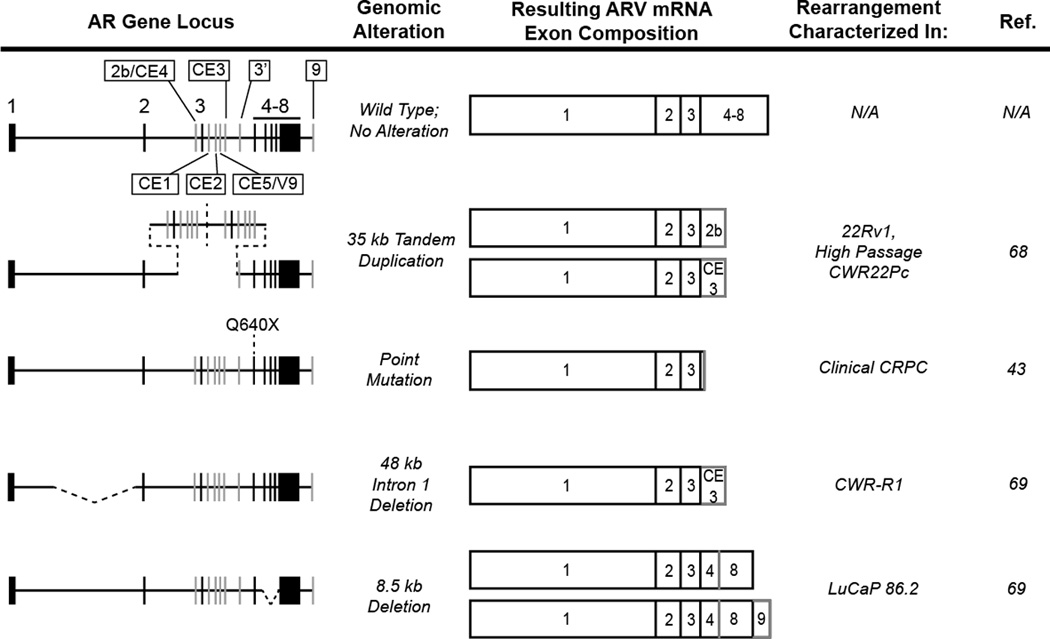
The 180 kb androgen receptor gene locus harbors eight canonical exons (black vertical hashes) that code for the wild type AR mRNA and protein (black boxes). Seven alternative, or cryptic, exons have also been identified (gray vertical hashes) that can be incorporated into the AR transcript upon activation of alternative splicing pathways (gray borders/boxes). Four discrete AR gene rearrangements or mutations, depicted as dashed black lines, have been shown to disrupt AR splicing and favor the expression of AR variants in PCa cell lines and xenografts.
Interestingly, ARV have also been described in the mouse PCa cell line Myc-CaP [68], in which the AR is amplified through genomic copy number gain. Mouse AR (mAR)-V2 was shown to result from splicing of exons 1–3 together with a novel cryptic exon located ~250 kb downstream of the AR gene locus. Perhaps even more compelling, a second ARV termed mAR-V4 was generated by splicing of exons 1–4 and a novel cryptic exon located nearly 1 Mb upstream of the AR transcriptional start site. Whereas mAR-V2 showed little activity in functional assays, mAR-V4 was constitutively active and localized to the nucleus, similar to ARVs identified in human cell lines and tissues. Though the molecular basis for splicing of mAR-V4 was not addressed in this study, it is likely contingent upon the known amplification of the AR gene in Myc-CaP cells. Following this rearrangement of the AR gene, the V4 cryptic exon could be situated downstream of the AR open reading frame, thus accounting for the incorporation of the V4 exon at the 3’ terminus of the transcript.
Further investigation of genomic copy number imbalance in additional models of CRPC progression has confirmed AR gene rearrangements as an important mechanism involved in the generation of constitutively active ARVs [69]. Multiplex ligation dependent probe assays (MLPA) were employed to query the copy number of AR exons in a variety of PCa cell lines and tissues. Interestingly, LuCaP 86.2 cells displayed reduced copy number of AR exons 5–7, which was shown subsequently to result from an 8.5kb intragenic deletion of this genomic segment (Fig. 2). Clearly, deletion of AR exons 5–7 provides an attractive mechanistic explanation for synthesis of the AR 1/2/3/4/8 variant in this xenograft model [50]. Interestingly, deletion of exons 5–7 prevents synthesis of full-length AR, indicating that this CRPC tumor would no longer be driven by androgen/AR signaling, but rather depends exclusively on the AR 1/2/3/4/8 variant for ongoing growth and survival.
An additional model of CRPC that has been shown to express high levels of truncated ARVs is the CWR-R1 cell line. To determine the basis for the splicing alterations in this model, Illumina paired-end massively parallel sequencing was employed to determine the sequence and structure of the AR locus [69]. This approach detected copy number loss spanning a 48 kb region of AR intron 1 (Fig. 2), which was supported by MLPA data querying copy number throughout AR intronic sequences. Interestingly, this deletion was initially observed only within a subpopulation of CWR-R1 cells. However, long term culture of CWR-R1 cells in androgen-depleted growth medium resulted in the outgrowth of the deletion-positive population. Importantly, the outgrowth of this subpopulation was accompanied by a corresponding increase in the protein expression of the constitutively active AR 1/2/3/CE3 variant. This finding supports the possibility that within at least some prostate tumors, subpopulations of ARV-driven cells with underlying rearrangements in the AR gene may exist prior to administration of AR-targeted therapies and, by virtue of constitutively active ARV expression, be able to overcome any drug in the current arsenal of AR-based therapies to repopulate the tumor. Based on the finding that ARV expression is an important feature of CRPC progression [47, 48, 50, 70, 71] and these recent data demonstrating AR gene rearrangements as a mechanism for altered AR splicing [67, 69], it is possible that AR gene rearrangements may represent a new class of genomic markers with predictive and/or prognostic value in CRPC.
STRUCTURE AND FUNCTION OF THE AR NTD
The role of ARVs in clinical PCa and castration resistance highlights the need for a greater understanding of NTD structure and function to aid in the design of AR-targeted therapeutics that do not require an intact AR LBD. The principal role of the NTD is to serve as a docking site for AR transcriptional co-regulators [72], and it is well established that the NTD is the predominant transcriptional activation domain of the AR [2, 19, 73]. This stands in contrast to other steroid hormone receptors, in which the CTD harbors the primary transcriptional activation domain [19]. The NTD is divided into two primary transcriptional activation units (TAUs) termed TAU-1 and TAU-5, which have been shown to have distinct roles in AR-mediated transcription [2, 74] (Fig. 1). The TAU-5 domain maps to amino acids 361–490 of the AR NTD, and has been shown to promote AR activity specifically under conditions of low/no androgens [74, 75]. Deletion of TAU-5 causes near-complete loss of AR function in the absence of DHT in both androgen dependent LNCaP [74] and castration resistant C4-2 cells [75]. Further work mapped TAU-5 activity to a conserved Trp-His-Thr-Leu-Phe (WHTLF) motif, and deletion or mutation of the hydrophobic W/L/F residues to alanine significantly inhibited androgen independent AR activity [76]. Interestingly, however, a recurring W435L mutation found in metastatic PCa tissue from patients relapsing after ADT was shown to increase ligand-dependent AR transcriptional activity, possibly through stabilization of an N/C intramolecular interaction between this domain and the AF-2 region [31].
The other major domain within the NTD, TAU-1, maps to amino acids 101–360 and houses two smaller subdomains, known as activation function (AF)-1a (amino acids 101–211) and AF-1b (amino acids 252–360). Deletion of either of the AF-1 sub domains causes complete loss of AR transcriptional activity in both androgen dependent LNCaP cells as well as castration resistant C4-2 cells, whereas deletion of the internal spacer region between AF-1a and -1b actually enhances AR transcription [75]. The transcriptional activity of TAU1 has traditionally been ascribed to an LxxLL-like motif, LKDIL, located within AF-1a [77]. Deletion or mutation of this sequence causes significant loss of AR activity, similar to deletion of the entire AF-1a fragment [72]. Intriguingly, the LKDIL motif overlaps an Lx7LL motif described by Zhu and colleagues [78], which is critical for mediating interaction with the transcriptional co-repressor NCoR and its binding partner TAB2. When TAB2 was phosphorylated by MEKK1, the NCoR/TAB2 complex was released, resulting in AR de-repression. However, no transcriptional co-activators have yet been identified that specifically bind to the LKDIL motif following co-repressor dissociation [79]. Furthermore, attributing TAU-1 activity exclusively to LKDIL does not account for the transcriptional loss observed upon deletion of AF-1b, suggesting that other elements within the TAU-1 domain may be important to the activity of this region [75].
Therapeutic Targeting of the AR NTD
The AR represents a nearly ideal drug target, in that pharmacologic targeting of the AR signaling axis produces profound results on prostate tumor biology with relatively minimal toxic side effects. While therapies that require an intact AR LBD have proven effective in treating PCa, mounting evidence suggests that the CTD may be ultimately dispensable for AR function in the context of CRPC. Observations of CTD-truncated ARVs, which function as potent, constitutively active transcription factors independent of the CTD, suggest that the NTD itself could be an alternative target for inhibition of AR transcriptional activity. Despite significant progress in defining the structural and functional composition of the AR NTD with the goal of therapeutic targeting, one principal challenge is the inherent flexibility and lack of tertiary structure throughout the NTD [80]. These structural characteristics are likely to be of fundamental importance to the transcriptional activity of the AR, but they have also proven to be a major obstacle to crystallographic analysis of AR structure and subsequent intelligent drug design. Nonetheless, two promising classes of drugs have recently been identified that seem to interact specifically with the NTD to mediate its inhibition. EPI-001 is a chlorinated bisphenol A diglycidyl ether (BADGE) identified in a high-throughput screen for compounds that could inhibit AR NTD activity. EPI-001 was demonstrated to function by preventing binding of the CBP/p300 histone acetyltransferase to the AR NTD, thereby preventing AR activity at target gene enhancers and preventing outgrowth of castration recurrent tumors in a xenograft model [81]. More recently, a similar drug screen for compounds isolated from the marine sponge Niphates digitalis identified a class of drugs termed Niphatenones [82], which were shown by click chemistry to covalently bind to an unknown portion of the AR NTD. This binding was shown to rely on a glycerol ether substructure and an extended saturated alkyl chain flanking the central ketone of Niphatenone B, which was shown to mediate growth inhibition in AR-expressing LNCaP cells but not AR-negative PC3 cells. These data suggest that the effect is AR specific, though no studies were performed to rule out effects on other steroid hormone receptors. Importantly, these compounds have not yet been tested against cell or xenograft models bearing truncated ARV, a critical experiment that will likely determine their impact and usefulness in the long-term maintenance of PCa. Taken together, however, the recent identification of these AR NTD inhibitors provides strong proof of principle that the NTD remains a vital and viable therapeutic target, and may be a key to containing the progression of late-stage PCa.
SUMMARY AND FUTURE DIRECTIONS
The discovery and characterization of ARVs has indicated that tumors driven by these species may represent a clinically relevant molecular subtype of CRPC that may require different therapeutic intervention than tumors only harboring full length AR. Whereas Ets family gene rearrangements have been proposed as a specific biomarker of PCa, high levels of ARV expression may serve as a marker of true androgen independence: ARV-positive tumors are highly unlikely to respond to any currently available antiandrogens or ADT strategies. Even the potent next-generation therapeutics enzalutamide and abiraterone fail in a significant fraction of patients, and it is tempting to hypothesize that these patients might progress due to AR gene rearrangements and/or expression of ARVs that lack the domain targeted by these new drugs. Used in conjunction with the classification schema suggested by Rubin and colleagues [58], ARV status could serve as an additional biomarker to inform the optimal use of current and next-generation ADT, as well as non-AR based therapies, in the treatment of PCa.
ACKNOWLEDGEMENTS
Studies in our laboratory have been funded by the Prostate Cancer Foundation, American Cancer Society Research Scholar Grant (RSG-12-031-01 to S.M.D.), and a Department of Defense Prostate Cancer Research Program New Investigator Award (W81XWH-10-1-0353 to S.M.D.). S.M.D. is a Masonic Scholar of the Masonic Cancer Center, University of Minnesota.
LIST OF ABBREVIATIONS
- ADT
androgen depletion therapy
- AF
transcriptional activation function
- AR
androgen receptor
- ARV
androgen receptor splice variants
- BF
binding function
- CRPC
castration resistant prostate cancer
- CTD
COOH-terminal domain
- DBD
DNA binding domain
- LBD
ligand binding domain
- NTD
NH2-terminal domain
- PCa
prostate cancer
- RNAi
RNA interference
- TAU
transcriptional activation unit
REFERENCES
- 1.Heinlein CA, Chang C. Androgen receptor in prostate cancer. Endocr Rev. 2004;25:276–308. doi: 10.1210/er.2002-0032. [DOI] [PubMed] [Google Scholar]
- 2.Claessens F, Denayer S, Van Tilborgh N, et al. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008;6:e008. doi: 10.1621/nrs.06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shaffer PL, Jivan A, Dollins DE, et al. Structural basis of androgen receptor binding to selective androgen response elements. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4758–4763. doi: 10.1073/pnas.0401123101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson JD. The role of 5alpha-reduction in steroid hormone physiology. Reprod Fertil Dev. 2001;13:673–678. doi: 10.1071/rd01074. [DOI] [PubMed] [Google Scholar]
- 5.Tindall DJ, Rittmaster RS. The rationale for inhibiting 5alpha-reductase isoenzymes in the prevention and treatment of prostate cancer. J Urol. 2008;179:1235–1242. doi: 10.1016/j.juro.2007.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- 7.Shang Y, Myers M, Brown M. Formation of the androgen receptor transcription complex. Mol Cell. 2002;9:601–610. doi: 10.1016/s1097-2765(02)00471-9. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Carroll JS, Brown M. Spatial and Temporal Recruitment of Androgen Receptor and Its Coactivators Involves Chromosomal Looping and Polymerase Tracking. Molecular Cell. 2005;19:631–642. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 9.Makkonen H, Kauhanen M, Paakinaho V, et al. Long-range activation of FKBP51 transcription by the androgen receptor via distal intronic enhancers. Nucleic Acids Research. 2009;37:4135–4148. doi: 10.1093/nar/gkp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu D, Zhang C, Shen Y, Nephew KP, Wang Q. Androgen receptor-driven chromatin looping in prostate cancer. Trends in Endocrinology & Metabolism. 2011;22:474–480. doi: 10.1016/j.tem.2011.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agoulnik IU, Weigel NL. Coactivator selective regulation of androgen receptor activity. Steroids. 2009;74:669–674. doi: 10.1016/j.steroids.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naar AM, Lemon BD, Tjian R. Transcriptional coactivator complexes. Annu Rev Biochem. 2001;70:475–501. doi: 10.1146/annurev.biochem.70.1.475. [DOI] [PubMed] [Google Scholar]
- 13.Heemers HV, Tindall DJ. Androgen receptor (AR) coregulators: a diversity of functions converging on and regulating the AR transcriptional complex. Endocr Rev. 2007;28:778–808. doi: 10.1210/er.2007-0019. [DOI] [PubMed] [Google Scholar]
- 14.Pereira de Jesus-Tran K, Cote PL, Cantin L, et al. Comparison of crystal structures of human androgen receptor ligand-binding domain complexed with various agonists reveals molecular determinants responsible for binding affinity. Protein Sci. 2006;15:987–999. doi: 10.1110/ps.051905906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Knudsen KE, Scher HI. Starving the Addiction: New Opportunities for Durable Suppression of AR Signaling in Prostate Cancer. Clin Cancer Res. 2009;15:4792–4798. doi: 10.1158/1078-0432.CCR-08-2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matias PM, Donner P, Coelho R, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–26171. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 17.Sack JS, Kish KF, Wang C, et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Nat Acad Sci. 2001;98:4904–4909. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jasuja R, Ulloor J, Yengo CM, et al. Kinetic and Thermodynamic Characterization of Dihydrotestosterone-Induced Conformational Perturbations in Androgen Receptor Ligand-Binding Domain. Mol Endocrinol. 2009;23:1231–1241. doi: 10.1210/me.2008-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He B, Gampe RT, Kole AJ, et al. Structural Basis for Androgen Receptor Interdomain and Coactivator Interactions Suggests a Transition in Nuclear Receptor Activation Function Dominance. Molecular Cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 20.Watanabe C, Watanabe H, Tanaka S. An interpretation of positional displacement of the helix12 in nuclear receptors: Preexistent swing-up motion triggered by ligand binding. Biochimica et Biophysica Acta - Proteins & Proteomics. 2010;1804:1832–1840. doi: 10.1016/j.bbapap.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 21.Dubbink HJ, Hersmus R, Verma CS, et al. Distinct Recognition Modes of FXXLF and LXXLL Motifs by the Androgen Receptor. Mol Endocrinol. 2004;18:2132–2150. doi: 10.1210/me.2003-0375. [DOI] [PubMed] [Google Scholar]
- 22.Saporita AJ, Zhang Q, Navai N, et al. Identification and characterization of a ligand-regulated nuclear export signal in androgen receptor. J Biol Chem. 2003;278:41998–42005. doi: 10.1074/jbc.M302460200. [DOI] [PubMed] [Google Scholar]
- 23.Estebanez-Perpina E, Arnold LA, Nguyen P, et al. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Nat Acad Sci. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grosdidier S, Carbo LR, Buzon V, et al. Allosteric Conversation in the Androgen Receptor Ligand-Binding Domain Surfaces. Mol Endocrinol. 2012;26:1078–1090. doi: 10.1210/me.2011-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buzon V, Carbo LR, Estruch SB, et al. A conserved surface on the ligand binding domain of nuclear receptors for allosteric control. Molecular and Cellular Endocrinology. 2012;348:394–402. doi: 10.1016/j.mce.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 26.De Leon JT, Iwai A, Feau C, et al. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proc Nat Acad Sci. 2011;108:11878–11883. doi: 10.1073/pnas.1105160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joseph JD, Wittmann BM, Dwyer MA, et al. Inhibition of prostate cancer cell growth by second-site androgen receptor antagonists. Proc Nat Acad Sci. 2009;106:12178–12183. doi: 10.1073/pnas.0900185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axerio-Cilies P, Lack NA, Nayana MRS, et al. Inhibitors of Androgen Receptor Activation Function-2 (AF2) Site Identified through Virtual Screening. J Med Chem. 2011;54:6197–6205. doi: 10.1021/jm200532b. [DOI] [PubMed] [Google Scholar]
- 29.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: A Cancer Journal for Clinicians. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 30.O'Mahony OA, Steinkamp MP, Albertelli MA, et al. Profiling Human Androgen Receptor Mutations Reveals Treatment Effects in a Mouse Model of Prostate Cancer. Mol Cancer Res. 2008;6:1691–1701. doi: 10.1158/1541-7786.MCR-08-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steinkamp MP, O'Mahony OA, Brogley M, et al. Treatment-Dependent Androgen Receptor Mutations in Prostate Cancer Exploit Multiple Mechanisms to Evade Therapy. Cancer Res. 2009;69:4434–4442. doi: 10.1158/0008-5472.CAN-08-3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Locke JA, Guns ES, Lubik AA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–6415. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 33.Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nat Rev Clin Oncol. 2011;8:12–23. doi: 10.1038/nrclinonc.2010.136. [DOI] [PubMed] [Google Scholar]
- 34.Lamont KR, Tindall DJ. Minireview: Alternative Activation Pathways for the Androgen Receptor in Prostate Cancer. Mol Endocrinol. 2011;25:897–907. doi: 10.1210/me.2010-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nadiminty N, Gao AC. Mechanisms of persistent activation of the androgen receptor in CRPC: recent advances and future perspectives. World J Urol. 2012;30:287–295. doi: 10.1007/s00345-011-0771-3. [DOI] [PubMed] [Google Scholar]
- 36.Tran C, Ouk S, Clegg NJ, et al. Development of a Second-Generation Antiandrogen for Treatment of Advanced Prostate Cancer. Science. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Attard G, Reid AHM, Olmos D, de Bono JS. Antitumor Activity with CYP17 Blockade Indicates That Castration-Resistant Prostate Cancer Frequently Remains Hormone Driven. Cancer Res. 2009;69:4937–4940. doi: 10.1158/0008-5472.CAN-08-4531. [DOI] [PubMed] [Google Scholar]
- 38.de Bono JS, Logothetis CJ, Molina A, et al. Abiraterone and Increased Survival in Metastatic Prostate Cancer. New Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Scher HI, Fizazi K, Saad F, et al. Increased Survival with Enzalutamide in Prostate Cancer after Chemotherapy. New Engl J Med. 2012;367:1187–1197. doi: 10.1056/NEJMoa1207506. [DOI] [PubMed] [Google Scholar]
- 40.Payton S. Prostate cancer: MDV3100 has antitumor activity in castration-resistant disease. Nat Rev Urol. 2010;7:300. doi: 10.1038/nrurol.2010.69. [DOI] [PubMed] [Google Scholar]
- 41.Scher HI, Beer TM, Higano CS, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1/2 study. The Lancet. 2010;375:1437–1446. doi: 10.1016/S0140-6736(10)60172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dehm SM, Tindall DJ. Alternatively spliced androgen receptor variants. Endocrine-Related Cancer. 2011;18:R183–R196. doi: 10.1530/ERC-11-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tepper CG, Boucher DL, Ryan PE, et al. Characterization of a novel androgen receptor mutation in a relapsed CWR22 prostate cancer xenograft and cell line. Cancer Res. 2002;62:6606–6614. [PubMed] [Google Scholar]
- 44.Céraline J, Cruchant MD, Erdmann E, et al. Constitutive activation of the androgen receptor by a point mutation in the hinge region: A new mechanism for androgen-independent growth in prostate cancer. Int J Cancer. 2004;108:152–157. doi: 10.1002/ijc.11404. [DOI] [PubMed] [Google Scholar]
- 45.Libertini SJ, Tepper CG, Rodriguez V, et al. Evidence for Calpain-Mediated Androgen Receptor Cleavage as a Mechanism for Androgen Independence. Cancer Res. 2007;67:9001–9005. doi: 10.1158/0008-5472.CAN-07-1072. [DOI] [PubMed] [Google Scholar]
- 46.Dehm SM, Schmidt LJ, Heemers HV, et al. Splicing of a Novel Androgen Receptor Exon Generates a Constitutively Active Androgen Receptor that Mediates Prostate Cancer Therapy Resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guo Z, Yang X, Sun F, et al. A Novel Androgen Receptor Splice Variant Is Up-regulated during Prostate Cancer Progression and Promotes Androgen Depletion–Resistant Growth. Cancer Res. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu R, Dunn TA, Wei S, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marcias G, Erdmann E, Lapouge G, et al. Identification of novel truncated androgen receptor (AR) mutants including unreported pre-mRNA splicing variants in the 22Rv1 hormone-refractory prostate cancer (PCa) cell line. Hum Mutat. 2010;31:74–80. doi: 10.1002/humu.21138. [DOI] [PubMed] [Google Scholar]
- 50.Sun S, Sprenger CCT, Vessella RL, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–2730. doi: 10.1172/JCI41824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chan SC, Li Y, Dehm SM. Androgen receptor splice variants activate androgen receptor target genes and support aberrant prostate cancer cell growth independent of canonical androgen receptor nuclear localization signal. J Biol Chem. 2012;287:19736–19749. doi: 10.1074/jbc.M112.352930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu R, Lu C, Mostaghel EA, et al. Distinct transcriptional programs mediated by the ligand-dependent full-length androgen receptor and its splice variants in castration-resistant prostate cancer. Cancer Res. 2012;72:3457–3462. doi: 10.1158/0008-5472.CAN-11-3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhou ZX, Sar M, Simental JA, et al. A ligand-dependent bipartite nuclear targeting signal in the human androgen receptor. Requirement for the DNA-binding domain and modulation by NH2-terminal and carboxyl-terminal sequences. Journal of Biological Chemistry. 1994;269:13115–13123. [PubMed] [Google Scholar]
- 54.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent Fusion of TMPRSS2 and ETS Transcription Factor Genes in Prostate Cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 55.Palanisamy N, Ateeq B, Kalyana-Sundaram S, et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med. 2010;16:793–798. doi: 10.1038/nm.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Berger MF, Lawrence MS, Demichelis F, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–220. doi: 10.1038/nature09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pflueger D, Terry Sp, Sboner A, et al. Discovery of non-ETS gene fusions in human prostate cancer using next-generation RNA sequencing. Genome Res. 2011;21:56–67. doi: 10.1101/gr.110684.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rubin MA, Maher CA, Chinnaiyan AM. Common Gene Rearrangements in Prostate Cancer. J Clin Oncol. 2011;29:3659–3668. doi: 10.1200/JCO.2011.35.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reid AHM, Attard G, Brewer D, et al. Novel, gross chromosomal alterations involving PTEN cooperate with allelic loss in prostate cancer. Mod Pathol. 2012;25:902–910. doi: 10.1038/modpathol.2011.207. [DOI] [PubMed] [Google Scholar]
- 60.Perner S, Demichelis F, Beroukhim R, et al. TMPRSS2:ERG Fusion-Associated Deletions Provide Insight into the Heterogeneity of Prostate Cancer. Cancer Res. 2006;66:8337–8341. doi: 10.1158/0008-5472.CAN-06-1482. [DOI] [PubMed] [Google Scholar]
- 61.Attard G, Clark J, Ambroisine L, et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene. 2008;27:253–263. doi: 10.1038/sj.onc.1210640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fine SW, Gopalan A, Leversha MA, et al. TMPRSS2-ERG gene fusion is associated with low Gleason scores and not with high-grade morphological features. Mod Pathol. 2010;23:1325–1333. doi: 10.1038/modpathol.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saramaki OR, Harjula AE, Martikainen PM, et al. TMPRSS2:ERG Fusion Identifies a Subgroup of Prostate Cancers with a Favorable Prognosis. Clin Cancer Res. 2008;14:3395–3400. doi: 10.1158/1078-0432.CCR-07-2051. [DOI] [PubMed] [Google Scholar]
- 64.Gopalan A, Leversha MA, Satagopan JM, et al. TMPRSS2-ERG Gene Fusion Is Not Associated with Outcome in Patients Treated by Prostatectomy. Cancer Res. 2009;69:1400–1406. doi: 10.1158/0008-5472.CAN-08-2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grasso CS, Wu Y-M, Robinson DR, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Taylor BS, Schultz N, Hieronymus H, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li Y, Alsagabi M, Fan D, et al. Intragenic Rearrangement and Altered RNA Splicing of the Androgen Receptor in a Cell-Based Model of Prostate Cancer Progression. Cancer Res. 2011;71:2108–2117. doi: 10.1158/0008-5472.CAN-10-1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watson PA, Chen YF, Balbas MD, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Nat Acad Sci. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li Y, Hwang TH, Oseth LA, et al. AR intragenic deletions linked to androgen receptor splice variant expression and activity in models of prostate cancer progression. Oncogene. 2012;31:4759–4767. doi: 10.1038/onc.2011.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hornberg E, Ylitalo EB, Crnalic S, et al. Expression of Androgen Receptor Splice Variants in Prostate Cancer Bone Metastases is Associated with Castration-Resistance and Short Survival. PLoS ONE. 2011;6:e19059. doi: 10.1371/journal.pone.0019059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang X, Morrissey C, Sun S, et al. Androgen Receptor Variants Occur Frequently in Castration Resistant Prostate Cancer Metastases. PLoS ONE. 2011;6:e27970. doi: 10.1371/journal.pone.0027970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alen P, Claessens F, Verhoeven G, et al. The Androgen Receptor Amino-Terminal Domain Plays a Key Role in p160 Coactivator-Stimulated Gene Transcription. Mol Cell Biol. 1999;19:6085–6097. doi: 10.1128/mcb.19.9.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bevan CL, Hoare S, Claessens F, et al. The AF1 and AF2 Domains of the Androgen Receptor Interact with Distinct Regions of SRC1. Mol Cell Biol. 1999;19:8383–8392. doi: 10.1128/mcb.19.12.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Christiaens V, Bevan CL, Callewaert L, et al. Characterization of the Two Coactivator-interacting Surfaces of the Androgen Receptor and Their Relative Role in Transcriptional Control*. J Biol Chem. 2002;277:49230–49237. doi: 10.1074/jbc.M209322200. [DOI] [PubMed] [Google Scholar]
- 75.Dehm SM, Tindall DJ. Ligand-independent Androgen Receptor Activity Is Activation Function-2-independent and Resistant to Antiandrogens in Androgen Refractory Prostate Cancer Cells. J Biol Chem. 2006;281:27882–27893. doi: 10.1074/jbc.M605002200. [DOI] [PubMed] [Google Scholar]
- 76.Dehm SM, Regan KM, Schmidt LJ, Tindall DJ. Selective role of an NH2-terminal WxxLF motif for aberrant androgen receptor activation in androgen depletion independent prostate cancer cells. Cancer Res. 2007;67:10067–10077. doi: 10.1158/0008-5472.CAN-07-1267. [DOI] [PubMed] [Google Scholar]
- 77.Chamberlain NL, Whitacre DC, Miesfeld RL. Delineation of two distinct type 1 activation functions in the androgen receptor amino-terminal domain. J Biol Chem. 1996;271:26772–26778. doi: 10.1074/jbc.271.43.26772. [DOI] [PubMed] [Google Scholar]
- 78.Zhu P, Baek SH, Bourk EM, et al. Macrophage/Cancer Cell Interactions Mediate Hormone Resistance by a Nuclear Receptor Derepression Pathway. Cell. 2006;124:615–629. doi: 10.1016/j.cell.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 79.Callewaert L, Van Tilborgh N, Claessens F. Interplay between Two Hormone-Independent Activation Domains in the Androgen Receptor. Cancer Res. 2006;66:543–553. doi: 10.1158/0008-5472.CAN-05-2389. [DOI] [PubMed] [Google Scholar]
- 80.Hilser VJ, Thompson EB. Structural Dynamics, Intrinsic Disorder, and Allostery in Nuclear Receptors as Transcription Factors. J Biol Chem. 2011;286:39675–39682. doi: 10.1074/jbc.R111.278929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Andersen RJ, Mawji NR, Wang J, et al. Regression of castrate-recurrent prostate cancer by a small-molecule inhibitor of the amino-terminus domain of the androgen receptor. Cancer Cell. 2010;17:535–546. doi: 10.1016/j.ccr.2010.04.027. [DOI] [PubMed] [Google Scholar]
- 82.Meimetis LG, Williams DE, Mawji NR, et al. Niphatenones, Glycerol Ethers from the Sponge Niphates digitalis Block Androgen Receptor Transcriptional Activity in Prostate Cancer Cells: Structure Elucidation, Synthesis, and Biological Activity. J Med Chem. 2012;55:503–514. doi: 10.1021/jm2014056. [DOI] [PubMed] [Google Scholar]