

Abstract
The complete stereostructures of xestoproxamines A–C, from the Bahamian sponge Neopetrosia proxima, were assigned from spectroscopic analysis, including MS, 2D NMR, and integrated degradation CD-analysis. Two new CD application protocols are described for defining absolute configuration: one for allylic methyl groups in branched chains and a second for the heterocyclic core bis-piperidine with specific applicability to other members of this class alkaloids – known for their stereo-heterogeneity – and tertiary cyclic amines in general.
Polycyclic amines comprise a family of biologically active macrocycles reported from sponges of the genera Haliclona, Xestospongia, Petrosia, Neopetrosia and Reniera, among others1. Their structures contain two or more nitrogen atoms woven into networks of fused heterocyclic and carbocyclic rings, mainly based on piperidine or cyclohexane. From the standpoint of structural characterization, polycyclic amines present difficulties in their purification due to their amphiphilic physical properties, while structure elucidation is hampered by both limited proton chemical shift dispersion presented in their 1H NMR spectra and stereochemical heterogeneity. In fact, the structures of several alkaloids in this class have succumbed only to X-ray crystallography. We report here three new bis-piperidine alkaloids, xestoproxamines A–C (1–3) from Neopetrosia proxima2 and assignment of their complete absolute stereostructures by an integrated approach based on NMR and circular dichroism (CD).3 A notable advancement is chiroptical analysis by CD, following double quaternization of the bis-piperidine, to give N, N′-bis-bromophenacyl derivatives which display characteristic split Cotton effects that reliably reflect the absolute configuration of the bis-piperidine core.
The molecular formula for 1 was established as C30H52N2 based on HRESIMS data (m/z 441.4207, [M+H]+). The presence of two 1,2-disubstituted carbon-carbon double bonds was supported by two coupled pairs of CH=CH spin systems in the 1H-1H COSY spectrum (Table 1, δ 5.49 m; 5.37 m; 5.31, m, and 5.28, m) and four CH sp2 carbons in the 13C DEPT spectrum (δ 131.74, d; 131.69, d; 130.1, d, and 129.1, d). NOESY correlations observed between allylic methylene groups (H16b/H19b and H26b/H29b) confirmed the Z-configuration of both double bonds. Because all sp2 carbons were accounted for and no C=N bonds were present, the remaining N atoms must be sp3 hybridized; therefore, 1 is a di-tertiary amine related to the known alkaloids halicyclamine A (4),4 B (5),5 haliclonacyclamines A (6), B (7),6 arenosclerin A (8) and haliclonacyclamine E (9).7 The remaining four degrees of unsaturation were completed by four rings. Interpretation of 13C, DEPT, and HSQC data showed six N-substituted CH2 groups (δ 58.5, 56.9, 56.4, 55.3, 48.9, and 47.4) that were linked to the remaining ring and chain elements (substructures a–c, Figure 1), including two substituted piperidine rings, from additional 2D NMR evidence (DQF-COSY, TOCSY, gHSQC, and gHMBC), in particular, HMBC cross-peaks at of H3/C9, H4/C9, and H10/C3. Contiguous spin systems identified by TOCSY cross-peaks of H13b/H16b and H11b/H14b secured two long hydrocarbon linking chains between the CH2-N and CH-N groups. Substructure c was joined to a by HMBC cross-peaks from H2 to both C29 and C30. The final ring was closed by connecting substructure b to c through HMBC correlations from H22 to C24.
Table 1.
1H (600 MHz) and 13C NMR (125 MHz) for 1 (CD3OD)
| No. | δC, mult.a | δH, mult (J in Hz, ax/eq) | DQF-COSY | HMBCb |
|---|---|---|---|---|
| 1 | 55.3, CH2 | 3.32, dd (12.0, ax)e | 2 | 2, 3, 11, 30 |
| 3.18, m (eq) | 2, 3, 5, 11, 30 | |||
| 2 | 29.7, CH | 2.29, m (ax) | 1, 3, 30 | 3, 29, 30 |
| 3 | 36.0, CH | 1.91, m (ax) | 2, 4, 4 | 4, 5, 8, 9, 10, 30 |
| 4 | 25.2, CH2 | 1.80, m (eq) | 3 | 3, 5, 9, |
| 1.69, m (ax) | 2, 3 | |||
| 5 | 47.4, CH2 | 3.48, brd (13.2, eq) | 4 | 1, 3, 4, 11 |
| 3.19, m (ax) | 1, 3, 4 | |||
| 6 | 58.5, CH2 | 3.21, brd (12.0, eq) | 7 | 7, 8, 9, 10, 20, 21 |
| 2.84, dd (12.0, 12.0, ax) | 7, 8, 9, 10 | |||
| 7 | 34.3, CH | 1.90, m (ax) | 6, 8, 20 | 6, 8, 9,10, 20 |
| 8 | 27.5, CH2 | 1.84, m (eq) | 7, 9 | 3,6, 7 |
| 1.06, ddd (12.0, 12.0, 12.0, ax) | 6, 7, 20 | |||
| 9 | 42.1, CH | 1.74, m (ax) | 8, 10 | 2, 3, 4, 7, 8 |
| 10 | 56.9, CH2 | 3.69, m (eq) | 9 | 3, 6, 8, 9, 21 |
| 2.84, dd (12.0, 12.0 ax) | 6, 8, 9 | |||
| 11 | 48.9c, CH2 | 3.32, m | 12 | 1, 5, 12, 13 |
| 3.02, ddd (12.8, 12.8, 3.7) | 5, 12, 13 | |||
| 12 | 25.2, CH2 | 1.79, m | 11, 13 | 11, 13, 14 |
| 1.66, m | 11, 14 | |||
| 13 | 21.7, CH2 | 1.79, m | 12, 14 | 11, 12, 14, 15 |
| 1.65, m | 11,12, 14 | |||
| 14 | 27.3, CH2 | 1.52, m | 13, 15 | 12, 13, 15, 16 |
| 1.44, m | 12, 13, 15, 16 | |||
| 15 | 29.0, CH2 | 1.43, m | 14, 16 | 14, 16, 17 |
| 1.25, m | 14, 16, 17 | |||
| 16 | 29.4, CH2 | 2.25, m | 15, 17 | 15, 17, 18 |
| 2.00, m | 14, 17, 18 | |||
| 17 | 131.69d, CH | 5.49, m | 16, 18 | 15, 16, 18, 19 |
| 18 | 129.1, CH | 5.31, m | 17, 19 | 16, 19, 20, |
| 19 | 23.4, CH2 | 2.50, m | 18, 20 | |
| 2.12, brd (15.4) | 7, 17, 18, 20 | |||
| 20 | 31.5, CH2 | 1.74, m | 7 | 6, 7, 8, 18, 19 |
| 1.27, m | 6, 7, 8 | |||
| 21 | 56.4, CH2 | 3.20, m | 22 | 6, 22, 23 |
| 22 | 22.6, CH2 | 1.81, m | 21, 23 | 21,23, 24, 25 |
| 23 | 28.1, CH2 | 1.62, m | 22, 24 | 21, 22, 24, 25 |
| 1.42, m | 21, 22, 24 | |||
| 24 | 27.0, CH2 | 1.42, m | 23, 25 | 22, 23, 25, 26 |
| 25 | 29.0, CH2 | 1.53, m | 24, 26 | 23, 24, 26, 27 |
| 1.45, m | 23, 24, 26, 27 | |||
| 26 | 27.1, CH2 | 2.35, m | 25, 27 | 28 |
| 1.91, m | 24, 25, 27, 28 | |||
| 27 | 131.74d, CH | 5.37, m | 26, 28 | 25, 26, 28, 29 |
| 28 | 130.1, CH | 5.28, m | 27, 29 | 26, 27, 29, 30 |
| 29 | 21.5, CH2 | 2.24, m | 28, 30 | 28, 30 |
| 1.82, m | 27, 28, 30 | |||
| 30 | 31.9, CH2 | 1.72, m | 29 | 1, 3, 28, 29 |
| 1.57, m | 1, 3, 28, 29 |
Determined from DEPT and HSQC.
b HMBC correlations, optimized for 8 Hz, are from proton(s) stated to the indicated carbon.
Observed by HSQC.
May be interchanged.
Coupling constant assigned by 1-D TOCSY (irradiation of H5eq).
Figure 1.


Substructures a–c of xestoproxamine A (1). Points of attachment of linking chains are indicated by •.
The relative configuration of the conjoined bis-piperidine rings in 1 (designated rings A and B, Figure 2) could be established from J coupling constants and NOESY data. The relative orientation of H2ax and H3ax was established by a 1D-TOCSY experiment; irradiation of H5eq (δ 3.48, brd) showed H1ax (dd, J = 12.0, 12.0 Hz) revealed two large couplings to H1eq (δ 3.18, m) and to H2ax (δ 2.29, m) respectively. In addition, H3ax was vicinally coupled by two large scalar couplings (J = 11.0 Hz) to H2ax and H4ax. Additional large couplings (J = 12.0 Hz and 12.0 Hz, respectively) from H7ax and H9ax to H8ax (δ 1.06, ddd, J = 12.0, 12.0, 12.0 Hz) showed the three protons to be axially disposed. The relative configuration of the remaining stereocenters in 1 was assigned by interpretation of NOESY correlations. The dihedral angle of ~90 °C between H3 and H9 was supported by lack of vicinal coupling (3J~ 0 Hz). Conformational constraints placed on the rings by NOE and J data, in particular, mutual correlations between the pairs H2ax and H8ax and H10eq and H3ax, lead to the depicted relative configuration and a conformer that places the averaged planes of ring A and B orthogonal to each other (Figure 2).
Figure 2.
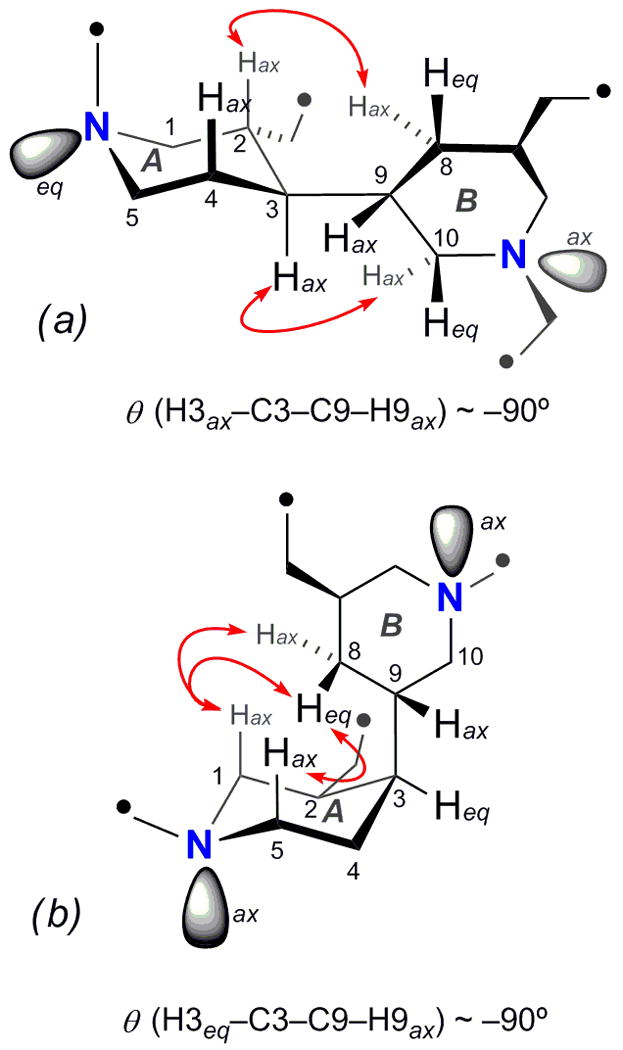
Selected NOESY correlations between the piperidine rings of (a) xestoproxamine A (1) and (b) xestoproxamine C (3). Points of attachment of linking chains are indicated by •. Note, ring ‘A’ has undergone ring-flip in 3 with respect to 1.
Xestoproxamine B (2) showed a molecular formula of C30H54N2, which has one degree of unsaturation less than xestoproxamine A (1). The 1H NMR signals for 2 (Table 2) were similar to those of 1 except for the presence of two vinyl protons (δ 5.48, m and 5.33, m) indicating only one C=C double bond. 2D NMR data confirmed both 1 and 2 share the same bis-piperidine ring system, including relative configuration (JHH and NOESY). The double bond was positioned between C17/C18 by COSY and TOCSY correlations to H19. Therefore xestoproxamine B (2) is a dihydro derivative of xestoproxamine A (1).
Table 2.
1H (600 MHz) and 13C NMR (125 MHz) for xestoproxamine B (2) (CD3OD)
| No. | δC, mult.a | δH, mult (J in Hz, ax/eq) | COSY | HMBCb |
|---|---|---|---|---|
| 1 | 55.7, CH2 | 3.23, dd (12.0, 12.0, ax) | 3 | 2, 3, 5, 11, 30 |
| 3.16, m (eq) | 2, 3, 5, 11, 30 | |||
| 2 | 29.9, CH | 2.24, m (ax) | 1, 3, 30 | 1, 3, 29, 30 |
| 3 | 36.1, CH | 1.87, m (ax) | 2, 4 | 1, 2, 4, 5 8, 10, 29 30 |
| 4 | 25.3, CH2 | 1.81, m (eq) | 3, 5 | 2, 3, 9 11 |
| 1.68, m (ax) | 2, 3, 9 | |||
| 5 | 47.5, CH2 | 3.45, brd (13.7, eq) | 4 | 1, 3, 4 |
| 3.17, m (ax) | 1, 3, 4 | |||
| 6 | 57.4, CH2 | 3.23, brd (12.0, eq) | 7 | 7, 8, 10, 20, 21 |
| 2.85, dd (12.0, 12.0, ax) | 7, 8, 10, 21 | |||
| 7 | 34.5, CH | 1.91, m (ax) | 6, 8, 20 | 6, 8, 19, 20 |
| 8 | 27.9, CH2 | 1.87, m (eq) | 7, 9 | 3, 6, 7, 9, 10, 20 |
| 1.08, ddd (12.0, 12.0, 12.0, ax) | 3, 6, 7, 9, 10, 20 | |||
| 9 | 42.1, CH | 1.78, m (ax) | 8, 10 | 4, 7, |
| 10 | 56.7, CH2 | 3.53, m (eq) | 9 | 6, 8 9, 21 |
| 2.98, dd (12.0, 12.0, ax) | 6, 8, 9 | |||
| 11 | 49.3, CH2 | 3.30, m | 12 | 1, 5, 12, 13 |
| 3.02, m | 1. 5, 12, 13 | |||
| 12 | 21.7, CH2 | 1.77, m | 11, 13 | 11, 13, 14 |
| 1.65, m | 11, 13 | |||
| 13 | 25.4, CH2 | 1.68, m | 12, 14 | 11, 12, 14, 15 |
| 1.60, m | 11, 12, 15 | |||
| 14 | 27.5, CH2 | 1.44, m | 13, 15 | 12, 13, 15 |
| 15 | 28.9, CH2 | 1.44, m | 14, 16 | 13, 16, 17 |
| 1.26, m | 13, 16, 17 | |||
| 16 | 29.5, CH2 | 2.22, m | 15, 17 | 14, 15, 17, 18 |
| 2.00, m | 14, 15, 17, 18 | |||
| 17 | 131.7, CH | 5.48, m | 16, 18 | 16, 18, 19 |
| 18 | 129.2, CH | 5.33, m | 17, 19 | 17, 18, 19 |
| 19 | 23.5, CH2 | 2.45, m | 18, 20 | |
| 2.15, m | 17, 20 | |||
| 20 | 31.7, CH2 | 1.75, m | 19, 7 | 6, 7, 18, |
| 1.28, m | 6, 7, 8, 18, 19 | |||
| 21 | 56.1, CH2 | 3.30, m | 22 | 6, 10, 22, 23 |
| 3.07, m | 6, 10, 22, 23 | |||
| 22 | 20.0, CH2 | 1.77, m | 21, 23 | 21, 23 |
| 23 | 26.2, CH2 | 1.50, m | 22, 24 | 22 |
| 1.37, m | 21, 22 | |||
| 24–28 | 29.0-26.4, CH2 | 1.53-1.25, m | ||
| 29 | 21.8, CH2 | 1.25, m | 30 | |
| 1.21, m | ||||
| 30 | 32.1, CH2 | 1.69, m | 2, 29 | 1, 2, 3 |
| 1.44, m | 1, 2, 3 |
Determined from DEPT and HSQC.
HMBC correlations, optimized for 8 Hz, are from proton(s) stated to the indicated carbon.
Observed by HSQC.
May be interchanged.
The third new bis-piperidine alkaloid, xestoproxamine C (3) C31H56N2, showed a characteristic signal for a secondary methyl branch in the 1H NMR spectrum (Table 3, δ 0.87, d, J = 6.7 Hz) that was absent in 1 and 2. 2D NMR and NOESY data indicated the configuration of 3 in the core bis-piperidine heterocycle differed from 1 and 2 but corresponded to the relative configuration found in haliclonacyclamine A (6)5. While ring B remained in the same conformation as 1 and 2, ring A in 3 had undergone ring flip to the opposite chair conformation, apparently without inversion at the ring A sp3 nitrogen (NOESY, Figure 2), a consequence of the inverted C2 configuration, yet in the new torsional arrangement a dihedral angle of ~90° for H3-C3-C9-H9 (3JH3–H9 ~0 Hz) was retained.
Table 3.
1H (600 MHz) and 13C NMR (125 MHz) NMR Data for xestoproxamine C (3) (CDCl3)
| No. | δC, mult.a | δH, mult (J in Hz, ax/eq) | DQF-COSY | HMBCb |
|---|---|---|---|---|
| 1 | 53.1, CH2 | 2.93, dd (12.0, 12.0, ax) | 2 | |
| 2.55, brd (12.0, eq) | 2, 5 | |||
| 2 | 40.1, CH | 2.03, m (ax) | 1, 3, 30 | |
| 3 | 34.0, CH | 1.96, m (eq) | 2, 4 | 1, 2, 4, 8, 9, 10 |
| 4 | 34.7, CH2 | 2.12, m (ax) | 3, 5 | 2 |
| 1.80, m (eq) | ||||
| 5 | 46.7, CH2 | 3.14, dd (12.0, 12.0, ax) | 4 | |
| 2.80, m (eq) | ||||
| 6 | 60.0, CH2 | 2.76, m (eq) | 7 | |
| 1.84, dd (12.0, 12.0, ax) | ||||
| 7 | 37.4, CH | 1.60, m (ax) | 6, 8, 20 | |
| 8 | 37.9, CH2 | 2.43, m (eq) | 7, 9 | 6, 7, 9, 10, 20 |
| 1.00, ddd (12.0, 12.0, 12.0, ax) | ||||
| 9 | 44.9, CH | 1.98, m (ax) | 8, 10 | 8 |
| 10 | 59.9, CH2 | 2.98, brd (9.8, eq) | 9 | |
| 2.17, m (ax) | 6, 8 | |||
| 11 | 56.5, CH2 | 2.91, m | 12 | 1, 5, 12, 13 |
| 12 | 22.0, CH2 | 1.65, m | 11, 13 | 11, 13 |
| 1.56, m | 11, 13 | |||
| 13 | 26.8, CH2 | 1.40, m | 12 | 11, 12 |
| 14 | 26.8–27.7 | 1.50-1.20, m | ||
| 15 | 27.3, CH2 | 1.37, m | ||
| 16 | 26.6, CH2 | 2.03, m | 15, 17 | 15, 17, 18 |
| 1.96, m | 15, 17, 18 | |||
| 17 | 129.5, CH | 5.44, m | 16, 18 | 16 |
| 18 | 129.5, CH | 5.41, m | 17, 19 | 16 |
| 19 | 24.8, CH2 | 2.37, m | 18, 20 | 17, 18, 20 |
| 1.81, m | 7, 17, 18, 20 | |||
| 20 | 34.1, CH2 | 1.51, m | 7, 19 | 8, 18, 19 |
| 0.93, m | 6, 7, 8, 18, 19 | |||
| 21 | 55.0, CH2 | 2.70, m | 22 | 10 |
| 2.48, m | 6, 10 | |||
| 22 | 27.9, CH2 | 1.58, m | 21, 23 | 21, 23, 24 |
| 1.48, m | 21, 24, 31 | |||
| 23 | 30.5, CH | 1.51, m | 22, 24, 31 | |
| 24 | 35.9, CH2 | 1.39, m | 23, 25 | 22, 23, 31 |
| 1.09, m | ||||
| 25–30 | 26.8–27.7 | 1.50-1.20, m | ||
| 31 | 21.2, CH3 | 0.87, d (6.7) | 23 | 22, 23, 24 |
Assigned from HSQC/HMBC.
b HMBC correlations, optimized for 8 Hz, are from proton(s) stated to the indicated carbon.
Compound 3 failed to produce suitable X-ray quality crystals under a variety of conditions. Stereoanalysis by spectroscopic means would require separate assignments of the two stereoelements in 3; the C23 stereocenter and the bis-piperidine core. We devised the following degradative approach for assignment of the lone C23 stereocenter exo to the heterocyclic core. Hofmann elimination of suitably quaternized nitrogen atoms in 3 would provide a terminal olefin (δ21), which could then be subjected to cross metathesis with a styrenyl derivative to insert a chromophore next to the secondary Me branch (first sphere of asymmetry), rendering the molecular amenable to analysis by CD. Due to the scarcity of 3, the method was first piloted with more abundant xestoproxamine A (1) (Scheme 1). Catalytic hydrogenation of 1 (Pd-C, MeOH, H2), purification, and conversion to the free base 10 by HPLC with buffered solvent (Lux-cellulose; CH3CN/i-PrOH/Et2NH), and immediate exhaustive alkylation with CH3I cleanly provided the bis-methiodide salt 11. The iodide counter ion was exchanged with hydroxide by treatment of 11 with either Ag2O or strong anion exchange resin (Amberlite-IRA 400, HO− form). Two possible major products were anticipated from cleavage of C21-N and cleavage of either C5-N or C11-N, based on the Hofmann rule that leads to the least substituted alkenes. In the event, Hofmann elimination (10 min, 300 W, 140°C) of the neat quaternary ammonium hydroxide double salt afforded only one major alkene product, 12. The structure of 12 was confirmed by 2D NMR (COSY, TOCSY, HSQC, and HMBC), which showed piperidine ring B was intact: exo cleavage of the C5-N bond was supported by the multiplicity of H4 (δ 5.55, dt, J = 17.0, 10.3 Hz). In contrast, ring A had undergone endo cleavage of the C21-N bond, and the vinyl methine signal exhibited the more complex pattern for vinyl proton H22 associated with a terminal allyl group (δ 5.80 (dddd, J = 17.0, 10.3, 6.8, 6.8 Hz, 1H). Alkene 12 was subjected to cross metathesis with excess 2-methoxy-6-vinylnaphthalene 138 (~10 eq) in the presence of Grubbs’ second generation catalyst9 which selectively engaged only the less hindered vinyl group derived from ring B to give the conjugated naphthalene 14. The UV spectrum of 14 in CH3CN showed characteristic λmax (246 and 292 nm) for the conjugated naphthalene chromophore, however – as expected – this styrenyl system was CD silent due to the remoteness of the chromophore from the distal sphere of asymmetry near C2 (xestoproxamine numbering, Figure 1).
Scheme 1.

Hofmann degradation of xestoproxamine A (1) and cross metathesis with 6-MeO-2-vinylnaphthalene (13).
In order to assign the observed Cotton effect to the configuration of the C23 center in 3, an appropriate methyl-branched model was required. Because the Cotton effect anticipated for the degradation/cross metathesis product of 3 would arise from asymmetric perturbation of the vinyl-conjugated 6-methoxynaphthalene, the model could consist of a chromophore adjacent to an allylic methyl branch of known configuration, appended to a short chain. Preparation of the model was achieved as depicted in Scheme 2. The α-branched propionamide 15, obtained by diastereoselective Myers alkylation of the enolate of (R, R)-(−)-N-propionylpseudoephedrine10 (16) (LDA, LiCl),11 with 1-iodosilyloxy ether 17 was selectively reduced (LiBH3N(CH2)4, rt) to the alcohol 1812 (%ee >94% by modified Mosher method).13 Swern oxidation of 18 to the corresponding aldehyde, followed by Wittig olefination (Ph3PC=CH2) gave terminal olefin 19. Finally, cross metathesis of 19 (Grubbs’ II, MeO-NpV) provided model alkene (S)-20.
Scheme 2.
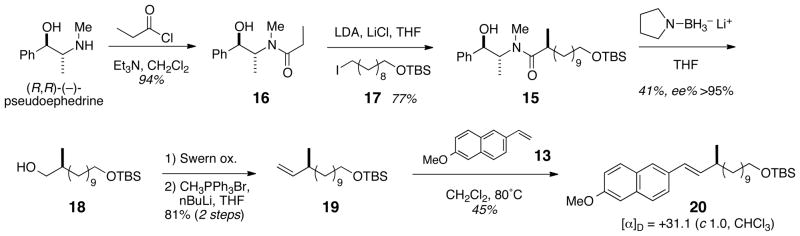
Synthesis of model compound 20.
Xestoproxamine C (3) was converted to 21, via the intermediates 22–24 (Scheme 3), by a similar sequence of reactions used to convert 1 to 14. The CD spectra (Figure 3 and Table 4) of synthetic (S)-20 [λ 256 nm (δε +3.9)] and 21 [λ 255 nm (δε +3.8)] were of the same sign and magnitude, therefore, 21 and xestoproxamine C (3) have the 23S configuration.
Scheme 3.
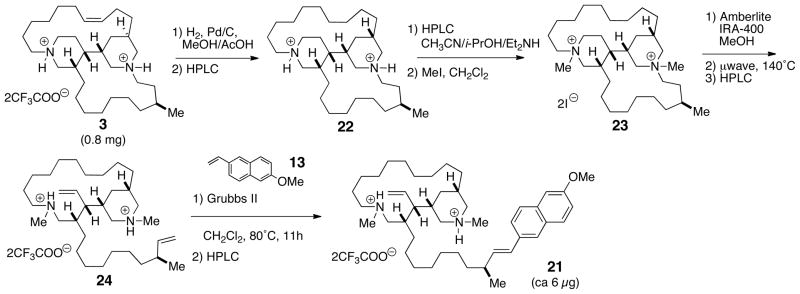
Hofmann degradation of xestoproxamine C (3) and cross metathesis with 6-MeO-2-vinylnaphthalene (13).
Figure 3.

CD spectra (CH3CN, 23°C) of (a) 20, (b) 21, (c) 14.
Table 4.
CD Data (23 °C) for Vinyl-naphthalene and bis-p-Bromophenacyl Derivatives.
No simple spectroscopic method was available for defining the absolute configuration of the bis-piperidine ring system in 1–3. Since this is an outstanding problem of configurational analysis in this class of alkaloids, we turned to refinement of a general method for assignment of cyclic tertiary amines involving derivatization and CD first published by Nakanishi and coworkers.14 Nakanishi’s assignment of the absolute configuration of quinuclidinols14 – bicyclic tertiary amines – followed simultaneous quaternization of the tertiary nitrogen and esterification of the pendant secondary OH group with excess p-phenylbenzylchloride and interpretation of the resultant split CD spectrum arising from exciton coupling of the two arene chromophores. Although the xestoproxamines differ considerably from the quinuclidinols the same principle could be applied: quaternization of both N atoms with a suitable chromophore and interpretation of exciton coupling CD (ECCD) would be informative of the absolute configuration of the heterocyclic bis-piperidine core. However, three possible problems were anticipated. The distance between chromophores attached to the N atoms in 1–3 is considerably larger than those in derivatized quinuclidinols with an expectedly weaker ECCD. This could be compensated for by substitution of the p-phenylbenzyl group with a more polarized chromophore: a phenacyl group. It was anticipated that possible differences in conformations of the natural products and quaternized derivatives and the introduction of two new stereocenters at N would complicate non-empirical interpretation of ECCD effects. On the other hand, the stereochemical outcome of N-quaternization should be predictable. The alkylating reagent should approach both N atoms in 1–3 along a trajectory in line with the lone pair without changing the N-configuration. In any case, the resultant configuration and conformation of the derivatives would be revealed by CD spectra and could be interpreted as ‘fingerprint’ spectra that relate the handedness of the heterocyclic core. This would confer an important advantage to chiroptical analysis of antipodal bis-piperidines by CD over comparisons using [α]D. Optical rotations of alkanamines are typically weak in magnitude and comparisons of specific rotation alone are notoriously unreliable due to changes in sign resulting from even slight structural variants (unsaturation). By comparing a fingerprint Cotton effect with that of a bis-piperidine phenacyl derivative of known absolute configuration, the configuration of any member of the series could be assigned.
To ensure a reasonably strong CD signal in the final product, an N-(4-bromophenacyl) chromophore (λmax265 nm, ε ~12,000) was introduced by exhaustive quaternization of the alkaloids. The bis-TFA salt of tetrahydroxestoproxamine (10) was converted to the free base (KOH/MeOH) followed by alkylation with p-bromophenacylbromide (toluene, 90 °C) to give 25 after HPLC purification (Scheme 4). The CD spectrum (Table 4) of 25 showed a characteristic negative split Cotton effect (λ 273 nm, δε −6.0; 253 (+2.6)) arising from exciton coupling of equatorial and axial N-p-bromophenacyl chromophores disposed with a negative helicity (see Figure 4). The relative conformations of the conjoined bis-piperidine ring system in 25 and 1 maintain the same conformations as starting material (analysis of J and NOESY data), however, the exact orientation of the transition dipole vectors that align along the average axes of the N-p-Br-phenacyl groups is highly dependent upon the conjoining C3-C9 bond which is subject to subtle torsional angle changes from non-bonding effects of the side chains and not easily predictable. Instead, we chose to make empirical chiroptical comparisons with p-Br-phenacyl derivatives with a compound of known configuration.
Scheme 4.
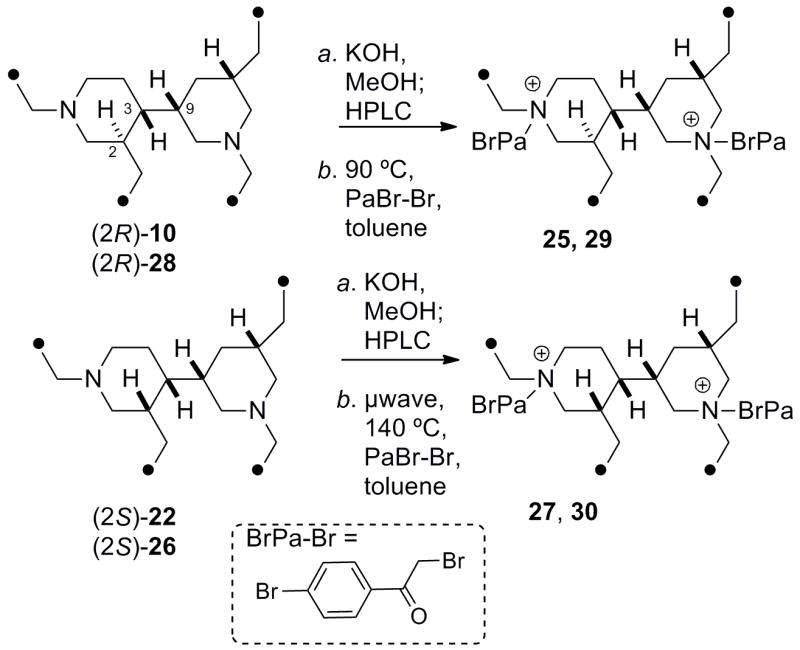
Preparation of N, N′-p-Br-phenacyl derivatives of bis-piperidine alkaloids.
Figure 4.

CD spectra (MeOH, 23°C) of (a) 25, (b) 27, (c) 29, (d) 30.
The relative configuration of (−)-‘perhaliclonacyclamine’ (26),15 the hydrogenation product of (+)-tetradehydrohaliclonacyclamine A (31) obtained from an Indonesian sponge, Halichondria sp., differs from the tetrahydroxestoproxamine A (10) in the relative configuration at C2 and the presence of two additional CH2 groups in the lower linking chain.16 A sample of (−)-26,17 was alkylated in the same manner (see above) to give derivative 27. The CD spectrum of 27 (λ 273 nm (δε −6.1); 254, (+3.2); Figure 4, Table 4) is almost identical to that of 25. Therefore, despite differences in relative configuration in the two piperidine rings in 25 and 27, both conform to the same chromophore alignments (a negative N–(C=O)–(C=O)–N’ angle) – imposed largely by the constraint of the C3-C9 torsional angle – and CD reveals they share the same absolute stereostructure, with the exception of the epimeric C2 position. A sample of haliclonacyclamine E (9), isolated from a Brazilian sample of Arenosclera braziliensis,18 was hydrogenated (H2, Pd-C) to the tetrahydro derivative (28), and exhaustively alkylated (p-bromo-phenacylbromide, 90 °C) as before to the bis-p-Br-phenacylated compound 29 that showed a CD spectrum (λ 273 nm, δε −6.0; 255 (+3.1), Table 4) almost identical with those of 25 and 27. Finally, the CD spectrum of the bis-p-bromophenylacyl derivative (30, λ 273 nm, δε ––7.1; 253 (+3.5)), obtained from tetrahydroxestoproxamine C (22) was identical to those of 25, 27 and 30.19 Therefore the absolute configuration of the heterocyclic cores in xestoproxamine A (1), haliclonacyclamine E (9), (−)-perhaliclonacyclamine (26) and xestoproxamine C (3) are the same, with the exception of C2 as noted above. Since the absolute configuration of (−)-26 was established earlier by X-ray crystallography,15 the stereostructures of 1–3 and 9 are now completely assigned. Bis-piperidine 8 co-occurs with 97 and it is likely both of these natural products share the same heterocyclic core configuration.
A summary of the configurational analysis of 1–3 and the CIP descriptors for the core stereocenters of related bis-piperidine alkaloids is given in Table 5, along with a comparison of reported [α]D values. No clear trend can be seen except that the compounds have high stereochemical heterogeneity that may reflect their geographic origins. The specific rotations vary in sign and magnitude with a strong dependence upon the presence and position of unsaturation in the top and bottom linking chains and possibly the chain lengths. Haliclonacyclamines A (6) and (7), from Haliclona sp. from the Great Barrier Reef, differ in the linking chains but the cores are antipodal to those of (−)-26 and (+)-31 from Halichondria sp. collected in Indonesia. We note that the recently reported (−)-neopetrosiamine (32)20 from the sponge Neopetrosia proxima,2 collected in the Caribbean sea south of the Bahamas, is very similar to 1–3, but the configuration was not defined.
Table 5.
Specific rotations and Configurational Assignment of bis-Piperidine Alkaloids.a
| cmpd. | [α]D | conc. g/100 mL, solvent | linker chain Cn | absolute confign.a | ||
|---|---|---|---|---|---|---|
|
| ||||||
| top | bottom | |||||
| 1b | +4.4 | c 2.0, | CH3OH | C10 | C10 | 2R,3S,7S,9S |
| 2b | +2.7 | c 2.4, | CH3OH | C10 | C10 | 2R,3S,7S,9S |
| 3b | −18.5 | c 0.67, | CHCl3 | C10 | C10 | 2S,3S,7S,9S,23S |
| 4c | −7.3 | c, 0.73, | CH2Cl2 | C8 | C12 | 2R*,3S*,9R* h |
| 5d | −143.5 | c, 0.65, | ? | C10 | C8 | 2R*,3S*,7S* h |
| 6e | −3.4 | c 1.21, | CH2Cl2 | C10 | C12 | 2R,3R,7R,9R |
| 7e | +3.4 | c 0.55, | CH2Cl2 | C10 | C12 | 2R,3R,7R,9R |
| 8f | −3 | c 0.015, | MeOH | C10 | C12 | 2R,3S,7S,9S b |
| 9f | +14 | c 0.02, | MeOH | C10 | C12 | 2R,3S,7S,9S b |
| 26g | −20.9 | c 0.205, | CHCl3 | C10 | C12 | 2S,3S,7S,9S |
| 31g | +19.4 | c 0.515, | CHCl3 | C10 | C12 | 2S,3S,7S,9S |
| 32i | −10 | c 1.0, | CHCl3 | C10 | C10 | 2R*,3R*,7R*,9R*h |
Bis-piperidine alkaloids have shown modest activity against various cancer cell lines. Compounds 1–3 were assayed in vitro against human colon tumor cells (HCT-116) and showed IC50 values of 21.2, 6.3, and 5.4 μM respectively.
In conclusion, three new bis-piperidine alkaloids, xestoproxamines A–C (1–3) are reported and their complete structures assigned by integrated MS, NMR, and chiroptical analysis. We assigned the absolute configuration of the remote methyl group in xestoproxamine C (3) by CD following a Hoffman degradation/cross metathesis protocol, a sensitive technique that can be extended to other natural products containing acyclic allylic methyl branches. Finally, we have shown that ECCD of N, N′-di-p-bromophenacyl bis-piperidine alkaloids, obtained by quaternization of the tertiary amines, reliably reflects the absolute configurations of the heterocyclic cores in 1–3, independent of the relative configuration at C2. This observation was exploited to show the two groups of structures, 1, 2, 8, 9, and 3, 26, have the same absolute configuration in their core heterocyclic rings and should be applicable to other bis-piperidine alkaloids in this class.
Experimental Section
General Experimental Procedures
General procedures are described elsewhere.21 Yields were determined gravimetrically except for those of masses of less than ~100 μg which are estimated from UV extinction coefficients or by quantitative solvent 13C satellite (QSCS)22 analysis of cryomicroprobe-measured 1H NMR spectra. HPLC was carried out using either a Gilson Model 302 pump equipped with tandem detectors – UV-visible (ISCO Model UA-5, λ 254 nm) and refractive index (Waters R401) – or a Rainin HPXL dual-pump with split flow (7:1) between two detectors – a Jasco CD-2095 UV-CD and an ESA Model 301 evaporative light scattering detector (ELSD).
Animal Material
The sponge Neopetrosia proxima2 was collected in June 2008 at Stirrup Cay, the Bahamas (25° 49.511N 77° 53.924′ W) at a depth of 8.5 m and identified by Sven Zea (Universidad Nacional de Colombia, InveMar). The surface of the tissue was dark brown and free of epiphytic zoanthids. A voucher sample of the sponge (08-13-073) is stored at UC San Diego.
Extraction and Isolation
A sample of N. proxima (252.6 g wet wt.) was extracted with MeOH (3 × 2.5 L, 25 °C, overnight) then CH2Cl2/MeOH (2 × 2.5 L, 25 °C, overnight). The resulting extract was partitioned between hexanes (3 × 500 mL) and 9:1 MeOH/H2O (1 L). The aqueous MeOH layer was removed and the H2O content adjusted to 1:1 MeOH/H2O before extraction with CH2Cl2 (3 × 1 L). The aqueous MeOH layer was concentrated under reduced pressure and then applied directly onto a reversed-phase (C18) silica column (~ 400 g) successively eluting with 1:9, 3:7, 1:1, 9:1 CH3CN/H2O + 0.2% TFA, and i-PrOH + 0.2% TFA to give five fractions. A portion (305 mg) of the third fraction (1.22 g) was subjected to preparative HPLC (reversed-phase, C18, 10 mL.min−1, gradient elution with 3:7 to 2:5 CH3CN/H2O + 0.3% TFA over 40 min.) to give 12 fractions. The third preparative HPLC fraction (27.2 mg) was subjected to semi-preparative HPLC (reversed-phase Synergi-HydroRP, 2.5 mL min−1, mobile phase: 2:2:6:0.05 CH3CN/i-PrOH/H2O/TFA) to give xestoproxamine A (1, 14.0 mg). A portion (4 mg) of the fourth preparative HPLC fraction (24.0 mg) was subjected to semi-preparative HPLC over the same column (2.5 mL.min−1, 22.5:22.5:55:0.5 CH3CN/i-PrOH/H2O/TFA) to give xestoproxamine B (2, 2.9 mg). The entire sixth HPLC fraction was subjected to HPLC (Phenomenex Lux-cellulose, 1.5 mL.min−1, mobile phase: 9:1:0.02 CH3CN/i-PrOH/Et2NH) to give xestoproxamine C (3, 1.5 mg).
Xestoproxamine A (1)
Colorless glass; [α]D24 +4.4 (c 2.0, MeOH); FTIR (ATR): ν 1676, 1437, 1203, 1133, 841, 801, 723 cm−1; 1H and 13C NMR, see Table 1. HRESIMS m/z 441.4207 [M+H]+ (calcd for C30H53N2, 441.4203)
Xestoproxamine B (2)
Colorless glass; [α]D24 +2.7 (c 2.4, MeOH); FTIR (ATR): ν 2934, 2862, 1674, 1463, 1199, 1131, 833, 799, 721 cm−1; 1H and 13C NMR, see Table 2; HRESIMS m/z 443.4362 [M+H]+ (calcd for C30H55N2, 443.4358)
Xestoproxamine C (3)
Colorless solid; [α]D24 −18.5 (c 0.67, CHCl3); FTIR (ATR): ν 2933, 2862, 1673, 1470, 1199, 1128, 829, 797, 721 cm−1; 1H and 13C NMR, see Table 3; HRESIMS m/z 457.4513 [M+H]+ (calcd for C31H57N2, 457.4516)
Tetrahydroxestoproxamine A Free Base (10)
A solution of xestoproxamine A (1) TFA salt (1.0 mg) and Pd/C (10%, 0.2 mg) in MeOH + 2% AcOH (0.5 mL) was vigorously stirred under 1 atmosphere of H2 overnight. The mixture was passed through a membrane filter (0.45 μ). The solvent was removed from the filtrate under reduced pressure, and the residue subjected to HPLC (Phenomenex Lux-cellulose, 1.5 mL.min−1, mobile phase: 9:1:0.02 CH3CN/i-PrOH/Et2NH) to give tetrahydroxestoproxamine A free base (10, 0.55 mg) which was used immediately after characterization. Colorless solid; [α]D23 −12.1 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 2.90 (dt, 1H, J = 12.0, 5.5 Hz), 2.80 (dd, 1H, J = 12.5, 4.0 Hz), 2.73 (brd, 1H, J = 10.0 Hz), 2.70-2.61 (m, 5H), 2.54 (brp, 1H, J = 5.5 Hz), 2.41 (brt, 1H, J = 9.8 Hz), 2.24 (t, 1H, 11 Hz), 2.04 (d, 1H, J = 12.5 Hz), 1.87 (t, 1H, J = 11 Hz), 1.82 (m, 1H), 1.64-1.21 (m, 40H), 0.97 (m, 1H), 0.72 (q, 1H, J = 12 Hz); HRESIMS m/z 445.4518 [M+H]+ (calcd for C30H57N2, 445.4516)
Tetrahydroxestoproxamine A bis-Methiodide Salt (11)
Free base 10 (0.55 mg) was dissolved in CH2Cl2 (0.8 mL), excess CH3I (0.2 mL) was added, and the mixture stirred overnight in the dark. Volatiles were removed under a stream of N2 to give the bis-methiodide salt (11, 0.9 mg) as an off-white solid. 1H NMR (500 MHz, CD3OD) δ 3.84 (d, J = 10.5 Hz, 1H), 3.54-3.49 (m, 3H), 3.41-3.15 (m, 8H) 3.17 (s, 3H), 3.12 (s, 3H), 2.20 – 1.22 (m, 44H), 1.19 (q, J = 12.5 Hz, 1H); 13C (125 MHz, CD3OD) δ 69.4 (CH2), 66.5 (CH2), 64.6 (CH2), 63.6 (CH2), 60.4 (CH2), 57.9 (CH2), 53.3 (CH3), 47.6 (CH3), 37.0 (CH), 35.2 (CH), 32.2 (CH), 32.1 (CH2), 31.8 (CH2), 31.7 (CH), 29.2 (CH2), 29.1(CH2), 29.0 (CH2), 28.8 (CH2), 28.6 (2 × CH2), 28.3 (CH2), 28.2 (CH2), 28.1 (CH2), 27.4 (CH2), 27.3 (CH2), 26.8 (CH2), 26.6 (CH2), 25.7 (CH2), 25.3 (CH2), 22.3 (CH2), 20.7 (CH2), 20.2 (CH2); HRESIMS m/z 237.2453 [M]2+ (calcd for C32H62N2, 474.4908)
Hofmann degradation of Tetrahydroxestoproxamine A bis-Methiodide salt
A solution of the bis-methiodide (11, 0.9 mg) was eluted with MeOH through a short column of strong anion exchange resin (Amberlite IRA-400, HO− form, prepared immediately before from Cl− form by, elution with aqueous 1N NaOH, followed by washing with distilled H2O until the eluate was neutral, then MeOH). The eluate was concentrated under reduced pressure to remove the volatiles, and solvent-free methohydroxide was irradiated (microwave, 300 W, 140 °C, 10 min). The crude product was taken up in MeOH and passed through a reversed-phase silica (C18) cartridge (200 mg) by elution with MeOH + 0.1 % TFA. The solvent was removed under a stream of N2, and the residue subjected to HPLC (reversed-phase, C18 Phenomenex Luna, 5μ, 10 × 250 mm, gradient: 40–100% CH3CN/H2O +0.1 % TFA over 40 min) to give a single major elimination product 12 (0.45 mg).
12: 1H NMR (600 MHz, CD3OD, representative signals) δ 5.80 (dddd, J = 17.0, 10.3, 6.8, 6.8 Hz, 1H), 5.55 (dt, J = 17.0, 10.3 Hz, 1H), 5.31 (dd, J = 10.3, 1.4 Hz, 1H), 5.18 (dd, J = 17.0, 1.4 Hz), 4.98 (dq, J = 17.0, 1.9 HZ, 1H), 4.92 (1H, under solvent), 3.44 (brd, J = 12.5 Hz, 1H), 3.40 (brd, J = 12.5 Hz, 1H), 3.20 (td, J = 12.5, 4.7 Hz, 1H), 3.10 (td, J = 12.5, 5.2 Hz, 1H), 2.92 (s, 3H), 2.85 (s, 3H), 2.54 (t, J = 12 Hz, 1H), 2.53 (t, J = 12 Hz, 1H), 2.18 (t, J = 10.4 Hz, 1H), 2.05 (q, 7.3 Hz, 2H), 0.88 (q, J = 12 Hz, 1H); HRESIMS m/z 473.4828 [M+H]+ (calcd for C32H61N2, 473.4829)
Cross Metathesis of 12 with 2-Methoxy-6-vinylnaphthalene (13)
To a solution of alkene 12 (450 μg, 0.642 μmol) and 6-methoxy-2-vinylnaphthalene8 (13, 1.2 mg, 6.42 μmol) in CH2Cl2 (0.5 mL) was added Grubbs’ 2nd generation catalyst9 (226 μg, 0.265 μmol) in CH2Cl2 (1 mL) in three portions over a period of 11 h at 80 °C. The solvent was evaporated under a stream of N2. The crude reaction mixture was passed through a reversed-phase C18 cartridge by elution with MeOH + 0.1% TFA, then subjected to HPLC (Luna, C18, 250 × 10 mm, 40–100% CH3CN/H2O + 0.1 % TFA over 40 min) to give the 6-methoxy-2-naphthyl-ethenyl derivative 14 (240 μg). UV (CH3CN) λmax 246, 292 nm; CD: See Table 4; 1H NMR (600 MHz, CD3OD, representative signals) δ 7.685 (d, J = 9.0 Hz, 1H), 7.681 (d, J = 8.6 Hz, 1H), 7.60 (brs, 1H), 7.55 (dd, J = 8.6, 2.0 Hz, 1H), 7.18 (d, J = 2.0 Hz, 1H), 7.09 (dd, J = 9.0, 2.0 Hz, 1H), 6.51 (d, J = 16.0 Hz, 1H), 6.32 (dt, J = 16.0, 7.0 Hz, 1H), 5.55 (dt, J = 17.0, 10.3 Hz, 1H), 5.31 (dd, J = 10.3, 1.4 Hz, 1H), 5.18 (dd, J = 17.0, 1.4 Hz), 3.90 (s, 3H), 2.91 (s, 3H), 2.83 (s, 3H), 2.50 (t, J = 12 Hz, 1H), 2.49 (t, J = 12 Hz, 1H), 2.26 (q, J = 7.0 Hz, 2H), 0.86 (q, J = 12 Hz, 1H); HRESIMS m/z 629.5403 [M]+ (calcd for C43H69N2O, 629.5404)
Degradation of Xestoproxamine C
Dihydroxestoproxamine C TFA salt (22)
A solution of xestoproxamine C (3) (0.8 mg) and Pd/C (10%, 0.2 mg) in MeOH + 2% AcOH (0.5 mL) was vigorously stirred overnight under H2 (1 atm). The solution was passed through a membrane filter (0.45 m) and the volatiles removed under a stream of N2. The residue was purified by HPLC (reversed-phase, Phenomenex, Synergi-HydroRP, 4 μ, 10 × 250 mm; 3 mL.min−1; mobile phase: 25:25:50:0.5 CH3CN/i-PrOH/H2O/TFA) to give dihydroxestoproxamine C (22). Colorless glass; 1H NMR (500 MHz, CD3OD, representative signals) δ 3.68 (brd, J = 11.2 Hz, 1H), 3.48 (td, J = 13.0, 4.0 Hz, 1H), 2.91 (t, J = 12.0 Hz, 1H), 2.84 (t, J = 12.0 Hz, 1H), 2.33 (brd, J = 12.0 Hz, 1H), 0.95 (d, J = 7.0 Hz, 1H); 13C NMR (125 MHz, CD3OD) δ 58.0 (CH2), 57.5 (CH2), 56.6 (CH2), 54.9 (CH2), 53.1 (CH2), 43.3 (CH), 40.3 (CH), 37.1 (CH), 35.74 (CH2), 35.67 (CH2), 33.7 (CH2), 33.6 (CH2), 33.3 (CH), 32.9 (CH2), 31.5 (CH), 28.8 (CH2), 25.56 (CH2), 28.53 (CH2), 27.8 (3 x CH2), 27.7 (CH2), 26.8 (CH2), 26.6 (CH2), 26.3 (CH2), 26.2 (CH2), 26.0 (CH2), 25.6 (CH2), 21.9 (CH2), 21.0 (CH3); HRESIMS m/z 460.4748 [M+H]+ (calcd for C31H60N2, 460.4751)
Tetrahydroxestoproxamine C bis-Methiodide Salt (23)
The TFA salt 22 was subjected to HPLC (chiral column, Lux-cellulose, 1.5 mL.min−1., mobile phase: 9:1:0.02 CH3CN/i-PrOH/DEA) to give the free base (0.5 mg). Immediately after removal of the volatiles, the free base (0.5 mg) was dissolved in CH2Cl2 (0.8 mL), treated with excess CH3I (0.2 mL), and the mixture stirred overnight in the dark. The solvent was evaporated under a stream of N2 to give the methiodide (23, 0.8 mg) as an off-white solid. 1H NMR (500 MHz, CD3OD, representative signals) δ 3.87 (brd, J = 11.4 Hz, 1H), 3.77 (td, J = 13.8, 3.0 Hz, 1H), 3.21 (s, 3H), 3.12 (s, 3H), 3.04 (t, J = 12.0 Hz, 1H), 1.03 (d, J = 7.0 Hz, 3H); 13C (125 MHz, CD3OD) δ 69.5 (CH2), 69.0 (CH2), 67.4 (CH2), 65.3 (CH2), 61.7 (CH2), 57.2 (CH2) [obscured by solvent] (CH3), 45.9 (CH3), 39.3 (CH), 36.4 (CH2), 36.2 (CH), 35.2 (CH2), 34.0 (CH2), 33.3 (CH), 33.1 (CH), 32.6 (CH2), 31.2 (CH), 30.2 (CH2), 29.5 (CH2), 29.3 (CH2), 28.7 (CH2), 28.4 (CH2), 28.2 (CH2), 27.94 (CH2), 27.86 (2 × CH2), 27.5 (CH2), 26.9 (CH2), 26.1 (CH2), 25.3 (CH2), 25.2 (CH2), 22.1 (CH3), 22.0 (CH2); HRESIMS m/z 244.2531 [M]2+ (calcd for C33H64N2, 488.5064).
Hofmann Degradation of Tetrahydroxestoproxamine C bis-Methiodide Salt (23)
A solution of the methiodide 23 in MeOH was passed through a short column of strong anion exchange resin (Amberlite IRA-400 (Cl−), converted to the HO− form with 1N NaOH). After removal of solvent under reduced pressure, the methohydroxide was transferred to a 10 mL microwave vessel, dried, and irradiated (μ wave, 300W, 140 °C over 7.5 min, then an additional 4.5 min). The crude product obtained was taken up in MeOH passed through a reversed-phase silica cartridge (C18, 200 mg) eluting with MeOH + 0.1 % TFA. Solvent was removed under a stream of N2, and the residue purified by HPLC (reversed-phase, C18 Phenomenex Luna, 5 μ, 10 × 250 mm, gradient: 40–100% CH3CN/H2O +0.1 % TFA over 40 min) to give the double-elimination product 24 (~140 μg).
24: 1H NMR (600 MHz, CD3OD, representative signals) δ 5.67 (ddd, J = 17.2, 10.2, 7.8 Hz, 1H), 5.62 (dt, J = 17.2, 10.0 Hz, 1H), 5.35 (d, J = 10.0 Hz, 1H), 5.29 (d, J = 17.2 Hz, 1H), 4.94 (brd, J = 17.2 Hz, 1H), 2.93 (s, 3H), 2.86 (s, 3H), 2.58 (t, J = 12.2 Hz, 1H), 2.50 (t, J = 12.2 Hz, 1H), 0.98 (d, J = 7 Hz, 3H); HRESIMS m/z 487.4983 [M+H]+ (calcd for C33H63N2, 487.4886).
Cross Metathesis of 24 with 2-Methoxy-6-vinylnaphthalene (13)
Grubbs’ 2nd generation catalyst (95 μg, 0.112 μmol) in CH2Cl2 (0.8 mL) was added in three portions over a period of 11 h to alkene 24 (140 μg, 0.199 μmol) and 2-methoxy-6-vinylnaphthalene8 (0.8 mg, 4.20 μmol) and the mixture stirred vigorously in a sealed vial at 80 °C. The solvent was removed under a stream of N2 and the residue passed through a reversed-phase silica cartridge (C18, 200 mg) eluting eluting with MeOH + 0.1% TFA, and finally purified by HPLC (reversed-phase, C18 Phenomenex Luna, 5 μ, 10 × 250 mm, gradient: 40–100% CH3CN/H2O +0.1 % TFA over 40 min) to give the 6-methoxy-2-naphthyl-ethenyl derivative 21 (6 μg). UV (CH3CN) λ 246, 292 nm; CD (CH3CN) λ 256 (δε +3.9); 1H NMR (600 MHz, CD3OD, representative signals) δ 7.69 (d, J = 8.6 Hz, 2H), 7.62 (brs, 1H), 7.55 (dd, J = 8.6, 2.0 Hz, 1H), 7.19 (brs, J = 2.0 Hz, 1H), 7.10 (dd, J = 9.0, 2.0 Hz, 1H), 6.48 (d, J = 16.2 Hz, 1H), 6.16 (dd, J = 16.2, 8.0 Hz, 1H), 5.58 (dt, J = 17.2, 10.0 Hz, 1H), 5.32 (d, J = 10.0 Hz, 1H), 5.27 (d, J = 17.2 Hz, 1H), 3.90 (s, 3H), 2.89 (s, 3H), 2.81 (s, 3H), 1.12 (d, J = 7 Hz, 3H); HRESIMS m/z 643.5563 [M]+, calcd 643.5561 for C44H71N2O.
Preparation of p-bromophenacyl derivatives
bis-p-Bromophenacyl Tetrahydroxestoproxamine A (25)
The acetate salt of tetrahydroxestoproxamine A (0.6 mg, 1.06 μmol) was converted to the free base 10 by addition of KOH (238 μg, 4.25 μmol) in MeOH (0.5 mL). After a few minutes the solvent was evaporated under a stream of N2 then on high vacuum. Toluene (0.5 mL) and p-bromophenacyl bromide (5.9 mg, 21.2 μmol) were added and the mixture was stirred at 90°C for 12 h. The solvent was evaporated under a stream of N2, and the mixture was subjected to HPLC (reversed-phase Phenomenx, Luna, C18, 10 × 250 mm; 2 mL.min−1 40–100 % CH3CN/H2O + 0.1% TFA over 40 min) to give the bis-p-bromophenacyl TFA salt 25 (0.7 mg). UV (MeOH) λmax 265 (log ε 4.38) nm; CD (MeOH) λ 254 (δε +2.6), 276 (δε −6.0) nm; 1H NMR (500 MHz, CD3OD, representative signals) δ 7.99 (d, J = 8.8 Hz, 2H), 7.94 (d, J = 8.8 Hz, 2H), 7.78 (d, J = 8.8 Hz, 2H), 7.77 (d, J = 8.8 Hz, 2H), 5.32 (d, J = 18.2 Hz, 1H), 5.17 (d, J = 18.2 Hz, 1H), 5.17 (d, J = 18.2 Hz, 1H), 5.14 (s, 2H), 4.18 (brd, J = 11.3 Hz, 1H), 4.11 (brd, J = 12.7 Hz, 1H); 3.99 (m, 1H), 3.98 (m, 1H), 3.81 (m, 2H), 3.77 (m, 1H), 3.58 (td, J = 13.1, 3.3 Hz, 1H), 3.56 (m, 1H), 3.54 (t, J = 12.8 Hz, 1H), 3.31 (under solvent), 3.24 (t, J = 12.8 Hz, 1H), 1.21 (q, J = 12.6 Hz, 1H); HRESIMS m/z 838.3632 [M]2+ (calcd for C46H68Br2N2O2, 838.3636).
bis-p-Bromophenacyl Perhaliclonacyclamine (27)
(−)-perhaliclonacyclamine15 (26, 150 μg, 0.317 μmol) was stirred with KOH (71 μg, 1.27 μmol) in MeOH (0.5 mL). After a few minutes the solvent was evaporated under a stream of N2 then on high vacuum. Toluene (0.5 mL) and p-bromophenacylbromide (1.8 mg, 6.34 μmol) were added and the mixture was stirred at 90 °C for 12 h. The solvent was evaporated under a stream of N2, and the mixture was subjected to HPLC (reversed-phase Phenomenex, Luna, C18, 10 × 250 mm; 2 mL.min−1.; mobile phase/gradient: 40–100 % CH3CN/H2O + 0.1% TFA over 40 min) to give the bis-p-bromophenacyl TFA salt 27 (ca 44 μg). UV (MeOH) λ 265 nm; CD (MeOH) λ 254 (δε +3.2), 272 (−6.1) nm; HRESIMS m/z 866.3946 [M]2+ (calcd for C48H72Br2N2O2, 866.3950).
bis-p-Bromophenacyl Perhydrohaliclonacyclamine E (29)
A solution of (+)-haliclonacyclamine E7 (9, TFA salt, 0.8 mg, 1.15 μmol) in MeOH/AcOH (49:1, 0.25 mL) was stirred with Pd/C (10%, 0.1 mg) at room temperature under H2 (1 atm) for 48 h. The mixture was filtered through a nylon syringe filter (0.45 μm) and the solvent removed from the filtrate under a stream of N2. The residue was subjected to HPLC (Phenomenex, Synergi Hydro-RP, 4μ, 10 × 250 mm; 2.5 mLmin−1, 25:25:50:0.5 CH3CN/i-PrOH/H2O/TFA) to give perhydrohaliclonacyclamine E TFA salt (28, 0.3 mg). The TFA salt was subjected to HPLC (Phenomenex Lux-cellulose, 1.5 mL.min−1, 9:1:0.02 CH3CN/i-PrOH/Et2NH) and the solvent removed under a stream of N2 and further dried under high vacuum. Toluene (0.5 mL) and p-bromophenacylbromide (1.2 mg, 4.28 μmol) were added and the mixture was stirred at 90 °C for 12 h. The solvent was removed under a stream of N2, and the mixture was subjected to HPLC (reversed-phase, Phenomenx, Luna, C18, 10 × 250 mm; 2 mL.min−1, 40–100 % CH3CN/H2O + 0.1% TFA over 40 min) to give the bis-p-bromophenacyl TFA salt 29 (ca 7.0 μg). UV (MeOH) λ 265 nm (log ε 4.38); CD (MeOH) λ 254 (δε +2.6), 272 (−6.0); HRESIMS m/z 866.3940 [M]2+ (calcd for C48H72Br2N2O2, 866.3950).
bis-p-bromophenacyl dihydroxestoproxamine C (30)
Xestoproxamine C free base (22, 0.4 mg, 0.876 mmol) was stirred with Pd/C (10%, 0.1 mg) in 0.25 mL MeOH/AcOH (49:1) at room temperature under H2 (1 atm) for 48 h. The mixture was filtered through a membrane filter (0.45 m) and the solvent was evaporated under a stream of N2 then by high vacuum. The dihydroxestoproxamine C acetate salt was subjected to HPLC (Phenomenex Lux-cellulose, 1.5 mL.min−1, mobile phase: 9:1:0.02 CH3CN/i-PrOH/Et2NH) and the solvent concentrated under a stream of N2 then dried under reduced pressure. Dihydroxestoproxamine C free base was stirred with p-bromophenacylbromide (3.0 mg, 10.8 mmol) in toluene at 80 °C for 2 h in a microwave reactor (300W). The solvent was removed under a stream of N2 and the mixture subjected to RPHPLC (column: Phenomenx, Luna, C18(2), 10 × 250 mm; 2 mL.min−1; 40–100 % CH3CN/H2O + 0.1% TFA over 40 min) to give the bis-p-bromophenacyl TFA salt derivative 30 (137 μg). UV (MeOH) λmax 265 nm; CD (MeOH) λ 254 (δε +3.5), 276 (δε −7.1) nm; HRESIMS m/z 852.3792 [M]2+ (calcd for C47H70Br2N2O2, 852.3799).
Synthesis of Model Compound 20
Amide (15)
n-BuLi (2.21 M in hexanes, 3.0 mL, 6.64 mmol) was added to a stirred solution of anhydrous lithium chloride (0.84 g, 19.8 mmol) and diisopropylamine (0.94 mL, 6.64 mmol) in THF (20 mL) at −78 °C. The mixture was kept at −78 °C for 20 min, then placed in an ice bath for 30 min, then cooled back to −78 °C. Amide 16 (0.70 g, 3.16 mmol, prepared by acylation of pseudoephedrine with propionyl chloride) was added over 10 min, and the solution slowly warmed to room temperature over 1.5 h, and stirred an additional 15 min, then cooled to −40 °C. The 1-O-TBS-9-iododecane 17 (1.75 g, 4.39 mmol) was added dropwise and the mixture allowed to warm to room temperature overnight with stirring. The mixture was poured into saturated NH4Cl solution (75 mL), and the organic layer separated. The aqueous layer was extracted with EtOAc (4 × 50 mL) and the combined organic layers were dried (MgSO4), the solvent removed under reduced pressure and the crude product purified by flash chromatography (SiO2, 1:3 EtOAc/hexanes) to give the amide 15 (1.20 g, 77% yield) [α]D22 −36.7 (c 3.1, CHCl3); FTIR (ATR): ν 3376, 2926, 2854, 1620, 1464, 1408, 1254, 1099, 1052, 835, 775, 701 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.38-7.23 (m, 5H), 4.61 (t, J = 7.2 Hz, 1H, major), 4.59 (m, 1H, minor), 4.39 (brs, 1H, major), 4.08 (p, J = 8.0 Hz, 1H, minor), 3.59 (t, J = 6.4 Hz, 2H, major), 3.58 (t, J = 6.4 Hz, 2H, minor), 2.91 (s, 3H, minor), 2.84 (s, 3H, major), 2.78 (t, J = 6.4 Hz, 1H, minor), 2.58 (sex, J = 7.2 Hz, 1H, major), 2.29 (brs, 1H, minor), 1.71 (brs, 1H, major), 1.60-1.46 (m), 1.31-1.19 (m), 1.14 (d, J = 6.8 Hz, 3H, major), 1.08 (d, J = 6.8 Hz, 3H, major), 1.02 (d, J = 6.8 Hz, 3H, minor), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (75 MHz, CDCl3): δ 179.3 (C), 142.6 (C), 128.3 (CH), 127.5 (CH), 126.3 (CH), 76.5 (CH), 63.3 (CH2), 36.6 (CH), 34.0 (CH2), 32.9 (CH2), 29.9-29.4 (5 × CH2), 27.4 (CH2), 26.0 (3 × CH3), 25.8 (CH2), 18.4 (C), 17.3 (CH3), 14.5 (CH3), −5.3 (2 × CH3); HRESIMS m/z 514.3685 [M+Na]+ (calcd for C29H53NO3SiNa, 514.3687)
Alcohol (18)
BH3•THF complex (1 M in THF, 3.0 mL, 3 mmol) was added to pyrrolidine (246 mL, 3 mmol) at 0 °C, and the solution was warmed to room temperature and stirred for 45 min. The solution was cooled back to 0 °C and treated, dropwise, with n-BuLi (2.21 M in hexanes, 1.36 mL, 3 mmol) with stirring for an additional 30 min. The amide 15 (500 mg, 1 mmol) in THF (10 mL) was added and stirred at room temperature overnight. 3M HCl (5 mL) was added to quench the excess hydride and the layers were separated. Ether (5 mL) was added to the aqueous layer, and the mixture cooled to 0 °C before being made basic (pH 9–10) by addition of 2 N NaOH. The aqueous mixture was extracted with ether (3 × 5 mL) and the combined organic extracts washed with 1:1 brine/1 N NaOH (2 × 10 mL), dried (Na2SO4), the solvent evaporated, and subjected to flash chromatography (SiO2, 12.5% EtOAc/hexanes) to give the alcohol 18 (135 mg, 41% yield) [α]D23 −5.5 (c 2.1, CHCl3); FTIR (ATR): ν 3340, 2926, 2855, 1464, 1254, 1102, 1041, 835, 775 cm−1; 1H NMR (400 MHz, CDCl3): δ 3.59 (t, J = 6.4 Hz, 2H), 3.50 (dd, J = 10.0, 5.6 Hz, 1H), 3.41 (dd, J = 10.0, 6.8 Hz, 1H), 1.59 (m, 1H), 1.50 (p, J = 6.8 Hz, 2H), 1.41-1.26 (m, 15H), 1.09 (m, 1H) 0.91 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 68.4 (CH2), 63.3 (CH2), 35.7 (CH), 33.2 (CH2), 32.9 (CH2), 29.9-29.4 (5 × CH2), 27.0 (CH2), 26.0 (3 × CH3), 25.8 (CH2), 18.4 (C), 16.6 (CH3), −5.3 (2 × CH3); HRESIMS m/z 353.2849 [M+Na]+ (calcd for C19H42O2SiNa, 353.2846)
Aldehyde (19a)
DMSO (64 mL, 0.91 mmol) was added dropwise to a stirred solution of oxalyl chloride (51 mL, 0.60 mmol) in CH2Cl2 (3 mL) at −78°C, and stirred for 10 min. A solution of the alcohol 18 (100 mg, 0.302 mmol) in CH2Cl2 was added, and the mixture stirred for 30 min at −50°C. Et3N (168 mL, 1.21 mmol) was added and the mixture brought to −30°C followed by stirring an additional 30 min. Saturated NH4Cl solution (5 mL) was added, and the layers were separated. The aqueous layer was extracted with CH2Cl2 (2 × 3 mL) and the combined organic extracts were dried (MgSO4), concentrated and the residue purified by flash chromatography (SiO2, 3% EtOAc/hexanes) to give the aldehyde 19a (91 mg, 92% yield) [α]D24 +12.8 (c 3.4, CHCl3); FTIR (ATR): ν 2926, 2854, 1730, 1463, 1254, 1099, 835, 774 cm−1; 1H NMR (400 MHz, CDCl3): δ 9.61 (d, J = 2.4 Hz, 1H), 3.59 (t, J = 6.4 Hz, 2H), 2.32 (sd, J = 7.2, 1.6 Hz, 1H), 1.69 (m, 1H), 1.50 (p, J = 7.2 Hz, 2H) 1.25 (m, 15H), 1.08 (d, J = 7.2 Hz, 3H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 205.5 (CH), 63.3 (CH2), 46.3 (CH), 32.9 (CH2), 30.5 (CH2), 29.6-29.4 (5 × CH2), 26.9 (CH2), 26.0 (3 × CH3), 25.8 (CH2), 18.4 (C), 13.3 (CH3), −5.3 (2 × CH3); HRESIMS m/z 351.2691 [M+Na]+ (calcd for C19H40O2SiNa, 351.2690)
Olefin (19)
n-BuLi (1.2 M in hexanes, 143 uL, 0.171 mmol) was added to a slurry of MePPh3Br (65 mg, 0.183 mmol) in THF (0.5 mL) at 0 °C, then the mixture stirred at room temperature for 5 min. The aldehyde 19a (40 mg, 0.122 mmol) in THF (0.5 mL) was added and stirring continued for 2 h at room temperature. Saturated NH4Cl solution (1 mL) was added followed by ether (2 mL) and the layers were separated. The aqueous layer extracted with ether (2 × 2 mL) and the combined organic extracts dried (MgSO4), concentrated, and the residue purified by flash chromatography (SiO2, hexanes) to give the chiral olefin 19 (35 mg, 88% yield) [α]D23 +6.5 (c 2.8, CHCl3) FTIR (ATR): ν 2925, 2854, 1463, 1254, 1099, 994, 909, 834, 773 cm−1; 1H NMR (400 MHz, CDCl3): δ 5.69 (ddd, J = 17.2, 10.0, 7.2 Hz, 1H), 4.94 (ddd, J = 17.2, 2.0, 1.2 Hz, 1H), 4.89 (ddd, J = 10.0, 2.4, 1.2 Hz, 1H), 3.51 (t, J = 6.8 Hz, 2H), 2.10 (m, 1H), 1.50 (p, J = 7.2 Hz, 1H), 1.25 (m, 16H), 0.97 (d, J = 6.8 Hz, 3H), 0.89 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3): δ 145.1 (CH), 112.2 (CH2), 63.3 (CH2), 37.8 (CH), 36.7 (CH2), 32.9 (CH2), 29.8-29.4 (5 × CH2), 27.2 (CH2), 26.0 (3 × CH3), 25.8 (CH2), 20.2 (CH3), 18.4 (C), −5.3 (2 × CH3); HRESIMS m/z 327.3081 [M+H]+ (calcd for C20H43O2SiNa, 327.3078)
Alkene (20)
A mixture of alkene 19 (2 mg, 6.12 mmol) and 2-methoxy-6-vinylnaphthalene (11.3 mg, 61.2 mmol) was treated with Grubbs’ 2nd generation catalyst (1 mg, 1.22 mmol) in CH2Cl2 (3 mL) at 70 °C in a sealed vial for 12 h. Additional catalyst (1 mg, 1.22 mmol) was added and the mixture stirred an additional 6 h at 80 °C. The solvent was removed under a stream of N2, and the mixture passed through a reversed-phase (C18) silica cartridge (CH3CN then MeOH), followed by HPLC (reversed-phase, C8, 85% CH3CN/H2O) to give alkene 20 (1.3 mg, 42%) [α]D22 +31.1 (c 1.0, CHCl3); UV (CH3CN) 246 nm (log ε 4.62), 292 (4.20); CD (CH3CN) λ 255 nm (δε +3.8). FTIR (ATR): ν 2925, 2853, 1256, 1100, 1035, 837, 777 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.68 (d, J = 9.0 Hz, 1H), 7.66 (d, J = 9.0, 1H), 7.62 (brs, 1H), 7.55 (dd, J = 9.0, 2.0 Hz, 1H), 7.11 (dd, J = 9.0. 2.0 Hz, 1H), 7.10 (brs, 1H), 6.46 (d, J = 16 Hz, 1H), 6.16 (dd, J = 16.0, 8.5 Hz, 1H), 3.91 (s, 3H), 3.59 (t, J = 7.0 Hz, 2H), 2.32 (sep, J = 6.5 Hz, 1H), 1.50 (m, 2H), 1.38 (m, 2H), 1.27 (m, 14H), 1.10 (d, J = 6.5 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H); 13C NMR (125 MHz, CDCl3): δ 157.4 (C), 136.6 (CH), 133.7 (C), 133.4 (C), 129.3 (CH), 129.1 (C), 128.0 (CH), 126.9 (CH), 125.1 (CH), 124.2 (CH), 118.8 (CH), 105.8 (CH), 63.3 (CH2), 55.3 (CH3), 37.4 (CH), 37.2 (CH2), 32.9 (CH2), 29.8-29.4 (5 × CH2), 27.4 (CH2), 26.0 (3 × CH3), 25.8 (CH2), 20.8 (CH3), 18.4 (C), −5.3 (2 × CH3); HRESIMS m/z 482.3573 [M]+ (calcd for C31H50O2Si, 482.3575)
Supplementary Material
Acknowledgments
We thank S. Zea (INVEMAR, Universidad Nacional de Colombia) for identification of the sponge, J. R. Pawlik (University of North Carolina, Wilmington), and the captain and crew of the RV Seward Johnson for logistical support during collecting expeditions, D. Dalisay for assistance with biological assays. The NSF Biological Oceanography Program (OCE-0095724, 0550468 to J.R.P). is acknowledged for ship time, and NSF (CRIF, CHE0741968) for funds to acquire the 500 MHz NMR spectrometer. This work was supported by grants from NIH (CA122256 and AI039987 to T. F. M.), and a Ruth L. Kirschstein National Research Service Award NIH/NCI (T32 CA009523 to B. I. M).
Footnotes
Dedicated to Dr. Koji Nakanishi of Columbia University for his pioneering work on bioactive natural products.
Supporting Information Available: 1H, 13C NMR and 2D NMR spectra of 1–3, 13C NMR spectra of 10, 11, 22, and 23, 1H NMR spectra of 12, 14, 24, 25, and 30, and 1H and 13C NMR of all new synthetic intermediates (15, 18, 19a, 19, and 20). This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.a) Andersen RJ, Van Soest RWM, Kong F. In: Alkaloids: Chemical and Biological Perspectives. Pelletier SW, editor. Vol. 10. Pergamon; New York: 1996. pp. 301–355. [Google Scholar]; b) Berlinck RGS. Top Heterocycl Chem. 2007;10:211–238. [Google Scholar]
- 2.Neopetrosia proxima and Xestoproxima proxima are synonymous; the former name now replaces the latter. Campos M, Mothes B, Eckert R, Van Soest RWM. Zootaxa. 2005;963:1–22.We retain the names ‘xestoproxamines’, based on the latter genus name, to comply with precedent disclosure. See Ref. 3.
- 3.Presented in part at the 13th International Symposium on Marine Natural Products; Phuket, Thailand. 17–22 October, 2010. [Google Scholar]
- 4.Jaspars M, Pasupathy V, Crews P. J Org Chem. 1994;59:3253–3255. [Google Scholar]
- 5.Harrison B, Talapatra S, Lobkovsky E, Clardy J, Crews P. Tetrahedron Lett. 1996;37:9151–9154. [Google Scholar]
- 6.a) Charan RD, Garson MJ, Brereton IM, Willis AC, Hooper JNA. Tetrahedron. 1996;52:9111–9120. [Google Scholar]; b) Clark RJ, Field KL, Charan RD, Garson MJ, Brereton IM, Willis AC. Tetrahedron. 1998;54:8811–8826. [Google Scholar]; c) Mudianta IW, Garson MJ, Bernhardt PV. Aust J Chem. 2009;62:667–670. [Google Scholar]
- 7.Torres YR, Berlinck RGS, Magalhães A, Schefer AB, Ferreira AG, Hajdu E, Muricy G. J Nat Prod. 2000;63:1098–1105. doi: 10.1021/np9905618. [DOI] [PubMed] [Google Scholar]
- 8.2-Methoxy-6-vinylnaphthalene was synthesized from 2-bromo-6-methoxynaphthalene. Lindh J, Savmarker J, Nilsson P, Sjoberg PJR, Larhed M. Chem Eur J. 2009;15:4630–4636. doi: 10.1002/chem.200802744.
- 9.Scholl M, Ding S, Lee CW, Grubbs RH. Org Lett. 1999;1:953–956. doi: 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- 10.Myers AG, Yang BH, Chen H, Gleason JL. J Am Chem Soc. 1994;116:9361–9362. [Google Scholar]
- 11.Prepared from 1,10-decanediol. Tully SE, Cravatt BF. J Am Chem Soc. 2010;132:3264–3265. doi: 10.1021/ja1000505.
- 12.Fisher GB, Fuller JC, Harrison J, Alvarez SG, Burkhardt ER, Goralski CT, Singaram B. J Org Chem. 1994;59:6378–6385. [Google Scholar]
- 13.Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092–4096. [Google Scholar]
- 14.Zhao N, Kumar N, Neuenschwander K, Nakanishi K, Berova N. J Am Chem Soc. 1995;117:1844–1845. [Google Scholar]
- 15.The absolute configuration of (−)-perhaliclonacyclamine was established from X-ray single crystal structure determination of the parent compound, (+)-tetradehydro-haliclonacyclamine A, obtained with Cu-Kα radiation, and interpretation of anomalous scattering (Bijvoet method). Mudianta IW, Katavic PL, Lambert LK, Hayes PY, Banwell MG, Munro MHG, Bernhardt PV, Garson MJ. Tetrahedron. 2010;66:2752–2760.
- 16.It’s worth recalling that it is not possible, strictly, to assign absolute configuration to chiral compounds by comparison of signs of [α]D, alone, except for enantiomers, even those as similar as 1–3 and 26. Two counter example are prescient and sufficient to illustrate this often ignored tenet in stereochemistry. The paired compounds (−)-perhydrohaliclonacyclamine (26) and the parent (+)-tetradehydrohaliclonacyclamine A (31), and (−)-haliclonacyclamine A (6) ([α]D = −3.4) and (+)-haliclonacyclamine B (7) ([α]D = +3.4), differ only in position of a single double bond in the lower linking chain, yet the pairs show opposite signs of specific rotation. Also, the absolute configuration of (−)- 26 ([α]D = −20.9])15 was shown to be antipodal to that of (−)- 6 despite the same sign of [α]D. Lastly, the signs of [α]D of alkaloid salts and their parent free bases may also differ.
- 17.We are grateful to Professor Mary Garson, University of Queensland, for the generous gift of 26.
- 18.We are grateful to Professor Roberto Berlinck, University of São Paulo, for the generous gift of 9.
- 19.The chair-chair conformations and N-configurations adopted by the bis-piperidine ring systems in the pairs 25 and 29, and 27 and 30 are preserved with respect to their starting materials (see Supporting Information for assignment of conformation of 25 using NOESY data). Although the Br-phenacyl substitutents are disposed differently – equatorial/axial on rings A and B of 25 and 29, and axial/axial for 27 and 30 – examination of molecular models show the C-N bond vectors subtend similar angles (θ ~90°) in all four derivatives. This explains why the diastereomeric variants give the same CD spectra because helicity and the consequent exciton coupling is dictated largely by the orthogonality of the piperidine ring planes. It is likely that the reactive conformers of each bis-piperidine ring requires an equatorial lone pair and, consequently, a ring flip for the B ring in all four starting materials and an A ring flip in 22 than 26 (see Figure 2). Reaction of 22, with both lone pairs on N in the axial orientation, was particularly sluggish and required more forcing conditions (microwave reactor, 140 °C) compared to 10, 26, and 28 (90 °C), possibly a consequence of torsional constraints imposed on 22 for the A ring flip (two less CH2 groups in the ‘northern’ linker chain) compared to the more flexible 26.
- 20.Wei X, Nieves K, Rodríguez AD. Bioorg Med Chem Lett. 2010;20:5905–5908. doi: 10.1016/j.bmcl.2010.07.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dalisay DS, Rogers EW, Edison A, Molinski TF. J Nat Prod. 2009;72:732–738. doi: 10.1021/np8007649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dalisay DS, Molinski TF. J Nat Prod. 2009;72:739–744. doi: 10.1021/np900009b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.