

Short abstract
A computational method for whole-genome prediction of microRNA target genes is presented. Application of this method to the Drosophila melanogaster, Drosophila pseudoobscura and Anopheles gambiae genomes identifies several hundred target genes potentially regulated by one or more known microRNAs.
Abstract
Background
The recent discoveries of microRNA (miRNA) genes and characterization of the first few target genes regulated by miRNAs in Caenorhabditis elegans and Drosophila melanogaster have set the stage for elucidation of a novel network of regulatory control. We present a computational method for whole-genome prediction of miRNA target genes. The method is validated using known examples. For each miRNA, target genes are selected on the basis of three properties: sequence complementarity using a position-weighted local alignment algorithm, free energies of RNA-RNA duplexes, and conservation of target sites in related genomes. Application to the D. melanogaster, Drosophila pseudoobscura and Anopheles gambiae genomes identifies several hundred target genes potentially regulated by one or more known miRNAs.
Results
These potential targets are rich in genes that are expressed at specific developmental stages and that are involved in cell fate specification, morphogenesis and the coordination of developmental processes, as well as genes that are active in the mature nervous system. High-ranking target genes are enriched in transcription factors two-fold and include genes already known to be under translational regulation. Our results reaffirm the thesis that miRNAs have an important role in establishing the complex spatial and temporal patterns of gene activity necessary for the orderly progression of development and suggest additional roles in the function of the mature organism. In addition the results point the way to directed experiments to determine miRNA functions.
Conclusions
The emerging combinatorics of miRNA target sites in the 3' untranslated regions of messenger RNAs are reminiscent of transcriptional regulation in promoter regions of DNA, with both one-to-many and many-to-one relationships between regulator and target. Typically, more than one miRNA regulates one message, indicative of cooperative translational control. Conversely, one miRNA may have several target genes, reflecting target multiplicity. As a guide to focused experiments, we provide detailed online information about likely target genes and binding sites in their untranslated regions, organized by miRNA or by gene and ranked by likelihood of match. The target prediction algorithm is freely available and can be applied to whole genome sequences using identified miRNA sequences.
Introduction
MicroRNAs (miRNAs) are a novel class of gene products that repress mRNA translation or mediate mRNA degradation in a sequence-specific manner in animals and plants [1-4]. To date, several hundred different miRNAs have been identified from various organisms and their sequences are archived and accessible at the RFAM miRNA registry website [5,6]. Currently, this database contains 21 miRNAs from Arabidopsis thaliana, 48 from Caenorhabditis briggsae, 106 from C. elegans, 73 from D. melanogaster, 122 from Mus musculus, and 130 from Homo sapiens.
With few exceptions, the target genes and the mechanism of target suppression are currently unknown because reliable experimental methods for comprehensively identifying the miRNA targets have yet to be developed. Founding members of the miRNA family, lin-4 and let-7 in C. elegans, have a central role as key regulators of developmental timing through cell fate decisions [7,8]. Because these miRNA genes are also conserved in other animals and mammals [9,10], it is not surprising to find that homologous genes, which were initially identified by genetic interaction, also possess conserved miRNA binding sites [11]. In insects, the bantam miRNA has been found to regulate cell proliferation and cell death by targeting the apoptosis gene hid (wrinkled) [12]. D. melanogaster miR-14 has been implicated in fat metabolism and stress resistance as well as cell death, however the precise target genes of this miRNA have not been identified [13]. The identification of animal miRNA targets is computationally difficult because animal miRNAs are relatively short and are only partially complementary to their mRNA targets, possibly because of additional interactions involving RNA binding proteins. As a result, it is challenging to define an algorithm and thresholds to predict reliably such target sites.
In contrast to animal miRNAs, some plant miRNA targets are more readily identified because of near-perfect complementarity to their target sequence [14]. Many of these plant mRNA targets encode transcription factors that regulate morphogenesis [15-19]. As a consequence of near-perfect complementarity, plant miRNAs predominantly act as small interfering RNAs (siRNAs) guiding destruction of their mRNA target, though some have also been found to behave like animal miRNAs. In addition, up to this point, plant miRNA target sites are predominantly found within the protein-coding segment of the target mRNAs [14], while animal miRNAs appear to primarily target the 3' untranslated region (UTR) [4,12,20-25].
The miRNA and siRNA pathways overlap at several points. Both siRNAs and miRNAs are processed from double-stranded RNA precursors requiring dsRNA-specific RNase III enzymes [26-30]. By an unknown molecular mechanism, the excised small RNAs become associated with Argonaute member proteins to form an RNA-induced silencing complex (RISC) that is able to target near-perfect complementary RNAs for degradation or for the control of translation [31-34]. In animal systems, it was shown that the introduction of a certain number of mismatches at centrally located positions allows for a switch from targeted mRNA degradation to translational repression [35,36]. In general, however, mutations in siRNAs typically abolish gene silencing without switching to translational repression [37]. Intriguingly though, siRNA-guided cleavage activity can be detected with sometimes distantly-related complementary sequences when siRNA specificity is evaluated at a genome-wide level [38].
About 10% of the miRNAs identified in invertebrates are also conserved in mammals, indicating that the regulatory function of these genes is likely to be conserved cross-species. Since miRNA-containing species have been separated by hundreds of millions of years of evolution, it is striking that many 22 nucleotide miRNAs do not exhibit stronger sequence divergence. This absence of sequence-evolution in many miRNAs suggests that these miRNAs have more than one target site and that evolution by compensatory base-pair changes has become extremely unlikely. Therefore, a miRNA may regulate few or many genes depending on its apparent birth date. It is also conceivable that additional evolutionary constraints, such as the presence of certain protein-binding sites within the miRNA-targeted mRNAs, are conferring specificity to these small RNA regulated processes.
In order to address the question of miRNA target identification in animals, we have developed a computational method to detect miRNA targets. This approach ranks the likelihood of each gene to be a miRNA target and conversely for each miRNA to target a gene (Figure 1). The target prediction method relies on the maintenance of evolutionary relationships between miRNAs and their targets, using three completely sequenced insect genomes (D. melanogaster, D. pseudoobscura and A. gambiae). We identify distinct networks of miRNA-mediated gene regulation such as the control of cell fate, morphogenesis and nervous system function, which appear to be preferentially targeted by miRNAs [39].
Figure 1.
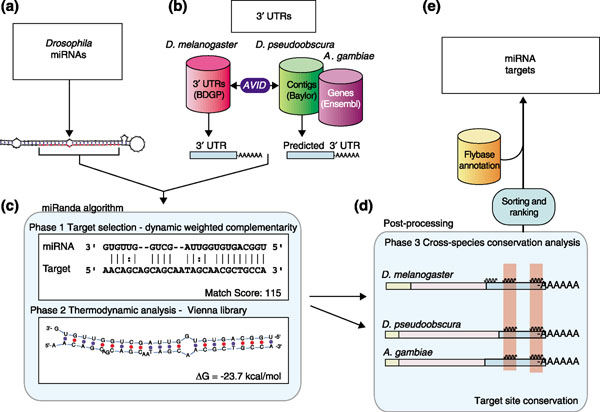
Algorithm and analysis pipeline. Source data consisting of (a) miRNAs and (b) 3' UTRs are processed initially by (c) the miRanda algorithm, which searches for complementarity matches between miRNAs and 3' UTRs using dynamic programming alignment (Phase 1) and thermodynamic calculation (Phase 2). (d) All results are then post-processed by first filtering out results not consistently conserved according to target sequence similarity with D. pseudoobscura and A. gambiae (Phase 3), then by sorting and ranking all remaining results. (e) Finally, all miRNA target gene predictions are annotated using data from FlyBase and stored for further analysis.
Target prediction
Every gene in D. melanogaster is a potential target for one or more of the characterized D. melanogaster miRNAs [40-43]. Reliable identification of miRNA targets is qualitatively different from standard sequence similarity analysis and requires new methods. In traditional sequence analysis, one tries to assess the likelihood of a hypothesis, for example, whether similarity between two sequences is due to common ancestry and continuity of functional constraints or a chance occurrence. Here, however, we aim to assess the likelihood of an actual physical interaction between two molecular species with phenotypic consequences, and have to assume that the interacting molecular species are present in the cell at the same time and at sufficient effective concentrations to facilitate the interaction. One of these species is a mature miRNA, the other a full length and translation-competent mRNA, or, in this study, its 3' UTR. The analysis is complicated by the fact that the sequence of the miRNA is small (approximately 22 nucleotide) and that the interaction may be substantially affected by a protein complex, that is, the interaction is probably not simple hybridization by optimal base pairing. To address these difficulties, at least in part, we have developed a three-phase method (miRanda) for target site identification from sequence information (Figure 1). The three phases are as follows: sequence-matching to assess first whether two sequences are complementary and possibly bind; free energy calculation (thermodynamics) to estimate the energetics of this physical interaction; and evolutionary conservation as an informational filter. We have validated this method using experimentally verified target genes (and target sites) from the literature [4,20-25] and against a randomized background model (see Materials and methods).
Sequence match
Using each of the 73 available D. melanogaster miRNAs as probes, we scan the 3' UTRs of 9,805 D. melanogaster genes for possible complementarity matches using a dynamic programming algorithm. For the remaining genes of D. melanogaster, accurate 3' UTR sequences were not available. The algorithm takes into account G-U wobble pairs, allows moderate insertions and deletions and, importantly, uses a weighting scheme that rewards complementarity at the 5' end of the miRNA, as observed in known miRNA:target-mRNA duplexes. In addition, we have applied position-specific empirically defined rules (see Materials and methods). The result is a score (S) for each detected complementarity match between a miRNA and a potential target gene.
Free energy calculation
For each match, the free energy (ΔG) of optimal strand-strand interaction between miRNA and UTR is calculated using the Vienna package [44]. We cannot, however, take into account the energy of interaction with possible protein components, as these details are at present largely unknown [2].
Evolutionary conservation
Given imperfect rules for sequence pairing and energy estimation, the conservation of predicted miRNA-target pairs in closely-related species is an important additional criterion for this analysis. Given the surprisingly high level of sequence conservation of miRNAs across phyla [9,45], we assume that the set of miRNAs in D. melanogaster is shared identically with D. pseudoobscura and A. gambiae. We only consider a miRNA target pair to be conserved across species if the following three criteria are met: a specific miRNA independently matches orthologous UTRs in both species; sequences of detected target sites in both species exhibit more than a specified threshold of nucleotide identity (ID) with each other; and the positions of both target sites are equivalent according to a cross-species UTR alignment [46] (see Materials and methods).
For this three-phase assessment of miRNA-target matches, we use cut-off values that provide a balance between false positives and false negatives, by inspection of known targets (see Materials and methods). The thresholds for sequence conservation (≥80% identity for D. pseudoobscura and ≥60% identity for A. gambiae) were chosen after extensive analysis of alignments between D. melanogaster, D. pseudoobscura and A. gambiae 3' UTR sequences. To maximize predictive power, we have kept the number of adjustable parameters and cutoffs as small as possible (see Materials and methods).
We take into account potential many-to-one relationships between miRNAs and their target genes using an additive scoring scheme. This system allocates a score to a miRNA (or multiple miRNAs) and target gene by summing over all scores for all conserved target sites detected for that pair (see Materials and methods). All predictions are then sorted and ranked according to this scheme. This means that miRNA target predictions with high scores or multiple detected sites, or both, are ranked preferentially. Finally, for each miRNA, the ten highest scoring target genes are selected for further analysis. These represent our highest quality target predictions for each miRNA, and, as such, are suitable for experimental validation, although lower-ranked predictions are also available (see Additional data files).
Validation using known miRNA targets
Application of our target prediction method to experimentally verified targets serves as initial validation. However, as the method was developed using known target sites as a guide [4,12,20-25], independent validation and refinement of the method will depend on future experiments.
The method correctly identifies nine of the ten currently characterized target genes (Table 1) for the miRNAs lin-4 and let-7 in C. elegans and bantam in D. melanogaster. At this threshold level, the details (position and base pairing as reported by others) of most target sites are largely reproduced, together with interesting alternative target sites on the known target genes. This comparison is only partially conclusive, as not all reported target sites on known target genes have been individually verified by experiment. The missed duplex between the lin-4 miRNA and its reported target gene (lin-14) (Table 1) contains an unusually long loop structure in the target sequence, which cannot easily be detected without adversely affecting the rate of false positive detection. In conclusion, we not only detect the majority of known miRNA targets, but the rankings obtained from our additive scoring scheme for these targets are also consistently high (Table 1). For example, the two target genes of the let-7 miRNA (hbl-1 and lin-41) are detected as the number 1 and number 2 ranked genes hit respectively, from a scan against 1,014 C. elegans 3' UTR sequences.
Table 1.
Validation of prediction method on experimentally verified miRNA targets
| MiRNA | Organism | Target gene (3' UTR) | Number of experimental sites | Number of predicted sites | Rank | Number of predicted sites with conservation | Matches experimental to predicted | Matches experimental to predicted (%) |
| lin-4 | cel/cbr | lin-14 (Abnormal cell-lineage protein 14) | 7 | 1 | 0 | 0 | 0% | |
| lin-4 | cel/cbr | lin-28 | 1 | 1 | 4/1,014 | 1 | 1 | 100% |
| lin-4 | cel/cbr | lin-41a lin41b | 1 | 1 | 5/1,014 | N/A | 1† | 100%† |
| let-7 | cel/cbr | lin-14 (Abnormal cell-lineage protein 14) | 2 | 6 | 9/1,014 | 2 | 2 | 100% |
| let-7 | cel/cbr | lin-28 | 1 | 1 | 12/1,014 | 1 | 1 | 100% |
| let-7 | cel/cbr | lin-41a lin41b | 2 | 6 | 2/1,014 | N/A | 2† | 100%† |
| let-7 | cel/cbr | daf-12 | 3 | 10 | 7/1,014 | 1 | 1 | 33% |
| let-7 | cel/cbr | hbl-1 (hunchback-related protein) | 8 | 14 | 1/1,014 | 8 | 5 | 63% |
| bantam | dme/dps | hid (Head involution defective (wrinkled)) | 2 | 2 | 1/11,318 | 2 | 2 | 100% |
| miR-13 | dme/dps | CG10222 | 1 | 1 | 4/11,318 | 1 | 1 | 100% |
Using intermediate thresholds (S: 80; ΔG: -14 kcal/mol), for each known miRNA and target gene pair (in either C. elegans or D. melanogaster), we list the number of known experimental target sites, the number of sites detected here, both raw and conserved in C. briggsae or D. pseudoobscura; and, the number and percentage of known sites that correspond to computationally detected conserved sites, with larger values indicating more successful (retrospective) prediction († and 'N/A' indicate that no 3' UTR was available to scan against in C. briggsae, hence no conservation analysis was possible, results assume conservation). cel/cbr: C. elegans/C. briggsae; dme/dps: D. melanogaster/D. pseudoobscura.
We also perform control calculations by running multiple trials using randomized miRNA sequences (see Materials and methods). A simple estimate of the rate of false positive results from applying the same fixed match, energy and conservation thresholds to target sites using actual and randomized miRNA sequences and assuming that all above-threshold target sites using random sequences are not biologically meaningful. The estimated rate of false positives is (R-/R+), where R- is the total number of above-threshold hits for randomized miRNA sequences and R+ is the total number of above-threshold hits for the actual miRNAs. The overall estimate is a false positive rate of 35% (Table 2). Interestingly, target genes for actual miRNAs with two or more conserved sites occur 11 times more frequently than for randomized miRNAs, representing a much lower false positive rate of 9% (Table 2). This apparent increase in reliability for predictions with multiple sites per target gene may be related to cooperativity of miRNA-target interactions.
Table 2.
Whole genome comparison of real versus randomized miRNAs against the complete genomes of D. melanogaster and D. pseudoobscura
| Total hits | Total conserved hits | 1 site | ≥ 2 sites | |
| 73 D. melanogaster miRNAs (A) | 6,864 | 589 | 556 | 33 |
| 73 Random miRNAs (B) | 5,152 | 204 | 201 | 3 |
| Standard deviation (100 experiments) | ± 132 | ± 43 | ± 40 | ± 3 |
| Ratio (A/B) | 2.9 | 2.8 | 11.0 | |
| Estimated false positives (%) | 35% | 36% | 9% |
Detected conserved hits (especially those with multiple detected sites in the 3' UTR) are significantly over-represented (2.8× and 11× as many cases, on average, respectively) in analyses with actual miRNAs compared to randomly shuffled miRNAs. The thresholds used for this analysis were S: 100; ΔG: -19 kcal/mol; ID: 70%.
Finally, we test whether specific functional classes of miRNA target genes are highly over-represented among the predicted miRNAs compared to random expectation (Table 3). For each GO molecular function class [47] we count the number of conserved targets detected for actual miRNAs and for the sequence-randomized miRNAs. Statistical significance of over-represented classes is measured by a Z-score (see Materials and methods). These results indicate that some functional classes (for example, translational regulators and apoptosis regulators) are significantly enhanced among miRNA target gene predictions.
Table 3.
Functional analysis of actual versus random miRNAs
| GO molecular function class | ||||||||||||
| Transcription regulator | Apoptosis regulator | Cell adhesion molecule | Binding | Enzyme | Transporter | Signal transducer | Translation regulator | Motor | Enzyme regulator | Structural molecule | Chaperone | |
| Actual miRNAs | 113 | 6 | 10 | 146 | 128 | 35 | 66 | 4 | 6 | 9 | 6 | 1 |
| Random miRNAs (average) | 43 | 1 | 2 | 98 | 88 | 22 | 41 | 2 | 4 | 9 | 7 | 1 |
| Standard deviation | ± 7.5 | ± 0.9 | ± 1.5 | ± 13.3 | ± 13.7 | ± 4.9 | ± 9.7 | ± 1.3 | ± 2.2 | ± 3.9 | ± 2.6 | ± 1.2 |
| Z-score | 9.3 | 5.9 | 5.0 | 3.6 | 2.9 | 2.7 | 2.6 | 1.4 | 1.0 | 0.01 | -0.3 | -0.3 |
Integers are number of detected conserved cases in each class. The standard deviation is for 50 experiments. The Z-scores for seven functional classes indicate over-represention for actual miRNA target genes. The thresholds used are S: 100; ΔG: -19 kcal/mol; ID: 70%.
Results
Potential miRNA targets
We report 535 potential target genes for the 73 known D. melanogaster miRNAs in decreasing order of match score for sites in detected 3' UTR targets. All results are available online [39]. All of these targets have passed the filters for free energy estimates, and conservation of target site sequence and position between D. melanogaster and D. pseudoobscura (see Materials and methods).
Of these predicted target genes in D. melanogaster, 264 have some functional annotation [48,49], while 231 have more than one predicted target interaction site. Results obtained from our random model suggest that 3' UTRs with more than one predicted target site for a given miRNA are more reliable than those with a single site. For this reason, the most promising candidates for target validation experiments may be the 231 target genes with multiple miRNA target sites. In general, cooperative binding of miRNAs to a single target gene can involve multiple hits by one or several distinct miRNAs. Specific examples are: The eye pigmentation gene brown (bw) is hit by the miRNAs bantam (three sites) and miR-314 (two sites); the apoptosis gene hid/wrinkled (W) is hit by bantam (two sites), miR-309 and miR-286; and the eye development gene seven-up (svp) is hit by miR-33 (two sites), miR-124, miR-277 and miR-312.
Given the strong conservation of miRNA sequences between D. melanogaster and A. gambiae [42], we searched for a subset of the above targets also conserved in the 3' UTRs of A. gambiae genes using the same procedure (Figure 1). In this way, we find 150 potential targets in corresponding A. gambiae genes. Of these, some 40% exhibit target site conservation of more than 60% identity to the corresponding D. melanogaster site. Notable examples are Scr (miR-10), netrin-B (miR-184, miR-284), sticks and stones (miR-282; two hits), and VACht (miR-9) (notation is: target gene (miRNA)).
Given the availability of an essentially complete set of protein coding genes and (perhaps) nearly all miRNAs in D. melanogaster, one can identify both biological processes and molecular functions predicted to be preferentially targeted by miRNAs. Processes we find to be over-represented by detected miRNA targets include: transcriptional control, translational control, cell-adhesion, enzyme regulation and apoptosis regulation (Figure 2). Separate groups of miRNAs appear to be specific to particular functional classes of target genes: for example, a group of seven miRNAs, miR-281, miR-311, miR-79, miR-92, miR-305, miR-131 and miR-31a, enriched two to four times in target genes in larval development; a group of five, bantam, miR-286, miR-309, miR-14 and miR-306, enriched three to six times in targets implicated in death or cell death; and the group bantam, miR-286 and the miR-2/miR-13 family, enriched five to six times in genes involved in regulation of apoptosis. A group of nine miRNAs is also two to three times enriched in genes involved in pattern specification. Overall, target genes annotated as transcription factors are detected twice as frequently as expected by chance (21% of annotated identified target genes, compared to 9.5% of all annotated FlyBase [50] genes). Translation factors are increased four times over expectation (miR-318, miR-304, miR-276b). This may represent a more general control mechanism for miRNA regulation of translation, in addition to the specific control of translation of individual target genes.
Figure 2.
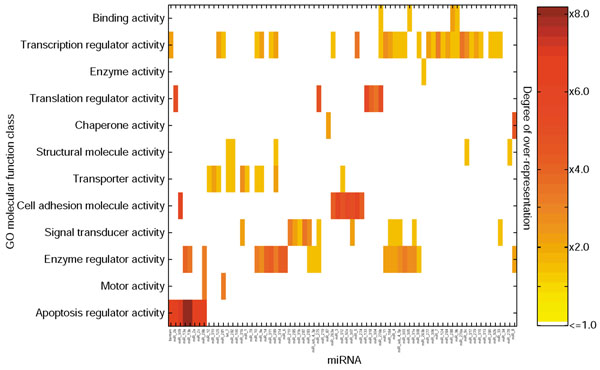
Functional map of miRNAs and their target genes. Left axis: selected over-represented FlyBase [49] derived GO [87] classifications from the 'molecular function' hierarchy. Bottom axis: ordered list of the 73 miRNAs. Each cell in the matrix is color-coded according to the degree of over-representation (right axis) for a miRNA hitting a specific functional class. For example, a bright red box indicates that a given miRNA hits six to eight times more targets in a particular class then one would expect by chance. The matrix is built by two-dimensional hierarchical clustering after normalization for classes that are over-represented in FlyBase annotations as a whole.
Investigating possible connections between genomic location and function, we analyzed 12 clusters of miRNAs in the D. melanogaster genome the members of which are potentially co-expressed, for example, let-7, miR-125 and miR-100 [40-43]. Contrary to expectation, we did not find any obvious links between genomic location and predicted target gene. One possible exception is the link between the position of three of the five copies of the miR-2 family in the intron of the gene spitz (involved in growth) and one of its top target genes, reaper (involved in cell death).
The well-characterized miRNA let-7 (in C. elegans) has two annotated top ten targets: tamo and lar. The gene tamo is thought to be required for the nuclear import of the NF-κB homolog Dorsal and recent work has connected it to the expression of a small RNA regulated by ecdysone [51,52]. We do not predict hunchback as a target of let-7 in D. melanogaster, given the thresholds and parameters used, although some below-threshold hits do appear to be conserved in D. pseudoobscura. Instead, we predict miR-12 and miR-184 to have a stronger effect on hunchback.
Many genes involved in the maternal genetic system, determining germ cell fate and anterior-posterior polarity of the egg, are well known to be translationally regulated. We clearly predict the following subset of these genes to be miRNA targets: germ cell less, bicoid, hunchback, caudal, staufen, arrest (bruno-1) and bruno-2. In addition, although the genes oskar and nanos are not top-ten ranked predictions, oskar has two conserved target sites (miR-3, miR-6), ranked below the top-ten hits for these miRNAs, and nanos has five strongly predicted target sites for miR-9c (three) and miR-263b (two), but below the 80% conservation threshold. Taken together, these data may indicate that this system is at least partially under miRNA regulation. We detail below three more biological processes predicted to be subject to miRNA regulation.
Hox genes and body axis specification
A multi-tiered hierarchy of transcription factors establishes the morphological segmentation and diversification of the anterior-posterior body axis of the Drosophila embryo [53]. The Hox genes (lab, pb, Dfd, Scr, Antp, Ubx, abd-A and Abd-B) play a key role in diversification by switching the fates of embryonic segments between alternative developmental pathways [54]. The genes are organized in two separate clusters on chromosome 3R, the Antennapedia (lab, pb, Dfd, Scr, Antp) and Bithorax (Ubx, abd-A, Abd-B) complexes. Both the genes and their relative order within the complexes are conserved in vertebrates [55].
Our predictions indicate that five of the eight Hox genes are regulated by miRNAs (Table 4). The 3' UTR of Scr is a potential target of miR-10, which is located within the Antennapedia complex between Dfd and Scr (and similarly, near the homologs of Dfd (hox4) in the Hox gene clusters of A. gambiae, Tribolium castaneum, zebrafish, pufferfish, mouse and human [56]). Scr is also a strong target for bantam, the miRNA associated with the apoptosis gene hid [12], and for miR-125, the putative Drosophila homolog of the miRNA lin-4 in C. elegans. Another strong target match for miR-125 is ftz, which is involved in the regulation of Hox genes and lies within the Hox cluster between Scr and Antp. All three of the Bithorax complex genes are likely to be regulated by multiple miRNAs. Interestingly, abd-A and Abd-B are both targeted by miR-iab-4-3-p, which is located within the complex between abd-A and Abd-B.
Table 4.
Potential miRNA targets of Hox cluster genes and their regulators
| Gene name | Genes identifier | MiRNA |
| Abd-A | CG10325* | miR-263a |
| Abd-B | CG11648 | miR-3,miR-5, miR-306 |
| Antp | CG1028* | miR-304 |
| Ftz | CG2047 | miR-125 |
| Hth | CG17117 | miR-276a, miR-265b, miR-279, miR-287† |
| Pc | CG32443 | miR-100, miR-313 |
| Scr | CG1030 | bantam, miR-10†, miR-125, miR-315†, miR-275 |
| Trr | CG3848* | miR-124 |
| Trx | CG8651 | miR-283, miR-307 |
| Ubx | CG10388 | miR-280, miR-315, miR-316*, miR-317* |
*Target gene based on top 20 hits of each miRNA; all others are based on top ten hits. †Target site also conserved in A. gambiae.
Aside from the Hox genes themselves, several other regulators of Hox gene function also appear to be miRNA targets. These include members of the trithorax activator (trx, trr) and the Polycomb (Pc) repressor groups, which control the spatial patterns of Hox gene expression by maintaining chromatin structure [57], and homothorax (hth), which is required for the nuclear translocation of the Hox cofactor extradenticle [58].
Ecdysone signaling and developmental timing
Ecdysone signaling triggers and coordinates many of the developmental transitions in the life cycle of Drosophila. Ecdysone pulses occur during embryonic, the three larval instar, prepupal, pupal and adult stages and regulate numerous physiological processes including morphogenetic cell shape changes, differentiation and death [59-62]. The regulation of these diverse processes by ecdysone is achieved through a complex genetic hierarchy. At the top of the hierarchy is the ecdysone receptor (EcR), a member of the nuclear hormone receptor family; it regulates the expression of different sets of transcription factors, including the zinc finger proteins of the Broad-Complex and many other nuclear hormone receptors, which in turn control key regulators of the different physiological processes.
Our predictions show many potential miRNA targets at several levels of the ecdysone cascade. These include EcR and several of the downstream transcription factors and co-factors (for example, br, E71, E74, E93, crol, fkh) [63-66]. Interestingly, broad (br), whose expression is exquisitely timed and differentially controlled in different tissues, has seven alternate splice forms with five different 3' UTRs. All five 3' UTRs contain high-ranking predicted targets for miRNA regulation (Table 5). The fact that three miRNAs control different splice forms in varying combinations supports the analogy made to transcriptional regulation for describing the combinatorial mechanisms to achieve specificity and redundancy in targeting genes.
Table 5.
Potential miRNA targets of ecdysone induction
| Gene name | Gene identifier | MiRNA |
| Amon | CG6438 | miR-2a |
| Aop | CG3166 | miR-7§ |
| Bon | CG5206 | miR-iab-4-5p |
| Br‡ | CG11491-RA | miR-14 |
| CG11491-RB&RC | miR-9, miR-14, miR-210 | |
| CG11491-RD | let-7, miR-9 | |
| CG11491-RE&RG | miR-9, miR-210 | |
| CG11491-RF | miR-316 | |
| Crol | CG14938 | miR-210§, miR-79 |
| Cyp314a1 | CG13478 | miR-308 |
| Eh | CG5400 | miR-279 |
| EcR | CG1765 | miR-14§ |
| Eip71CD | CG7266 | miR-34 |
| Eip74EF | CG32180 | miR-306 |
| Eip93F | CG18389 | miR-14, miR-286 |
| Fkh | CG10002 | miR-281 |
| Hr38 | CG1864 | miR-308 |
| Hr46 | CG33183* | miR-1,miR-9a, miR-9c, miR-11§, miR-124, miR-318 |
| Hr96 | CG11783† | miR-92a§ |
| Msn | CG16973† | |
| Pcs | CG7761 | miR-308 |
| Rab6 | CG6601 | miR-317 |
| Rpr | CG4319 | miR-13a, miR-13b, miR-2a, miR-2b, miR-2c |
| Rep2 | CG1975 | miR-210 |
| Slpr | CG2272† | miR-3 |
| W | CG5123 | bantam§ |
| Woc | CG5965 | miR-100 |
*,†Target gene based on the top 20 and top 30 hits of each miRNA, respectively; all others based on top ten hits. ‡The gene br has seven splice variants RA to RG, five of which have unique UTRs. § Target site also conserved in A. gambiae.
In addition to the core transcription factors of the ecdysone cascade, several of its effector pathways are likely to be directly targeted by miRNAs. These include genes in morphogenetic/stress signaling (aop, msn, slpr, hep), biogenesis (rab6) and the cell death pathway (hid, rpr, parcas, Rep2) [67-69]. They also include several miRNAs target genes that are involved in the biosynthesis of ecdysone (woc, CypP450s) and of other hormones triggering developmental transitions (amon/ETH, Eh) [70,71]. Despite their synchronous expression with ecdysone pulses in late larvae and pre-pupae [9], let-7 and miR-125 are not prominently targeting the core factors of the ecdysone cascade.
Development and function of the nervous system
We predict a large number of miRNA target genes that are involved in cell fate decisions in the developing nervous system. In particular, we predict several miRNA target genes within components of the Notch pathway, which regulates the early decision between the neuronal and the ectodermal fate [72,73]. These include Notch ligands and factors regulating their stability (Ser, neur), as well as factors that bind to Notch (dx) or modify its sensitivity to ligands (sca, gp150, fng). They also include genes of the E(spl) complex (CG8328, CG8346) and of the Brd complex (Tom) [74]. Genes in these two complexes are known to share motifs for translational regulation in their 3' UTR (Bearded- and K-box), some of which have previously been predicted to be miRNA target sites [75]. In addition, our predicted miRNA target genes include factors involved in the asymmetric cell division of neuroblasts (insc, par 6) and transcription factors regulating different aspects of neuronal differentiation (Dr, jim, Lyra, nerfin, SoxN, svp, unc4).
The establishment of neural connectivity is a complex morphogenetic process comprising the growth and guidance of axons and dendrites, and the formation of synapses. Many miRNAs target these processes at several different levels (Figure 3; Table 6). These targets include a remarkably large number of secreted and transmembrane factors known to mediate axon guidance decisions (netrin A and B, Slit; Drl, Dscam, Eph, PTPs, Robos, Semas; FasI, beatIV) [76-78] (Table 6). All these factors are conserved and have similar axon guidance functions in vertebrates. Interestingly, netrin1 and robo are also predicted miRNA target genes in vertebrates (our unpublished observations), suggesting that translational control is an important conserved aspect of the regulation of axon guidance factors.
Figure 3.
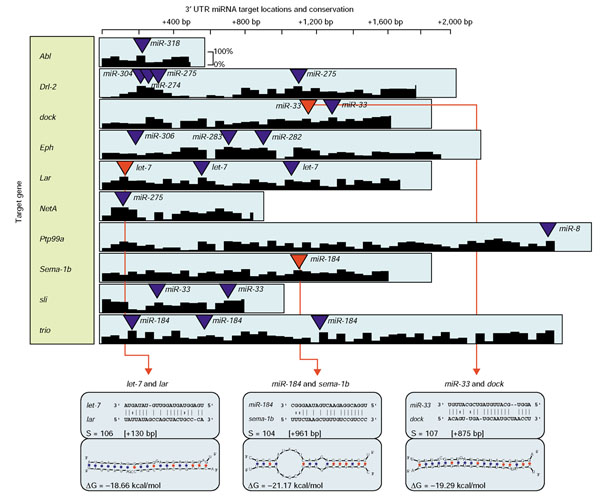
Representation of 3' UTRs for potential miRNA target genes involved in axon guidance. Each individual conserved hit between a miRNA and a target gene is marked by an annotated triangle on a conservation plot (D. melanogaster versus D. pseudoobscura) for that UTR. Red triangles indicate target site locations that are illustrated in more detail (alignment and secondary structure) below. Multiple target sites on a 3' UTR for one or more miRNAs are not uncommon and reflect cooperative regulation of transcription.
Table 6.
Potential miRNA targets of the axon guidance pathway
| Gene name | Gene identifier | MiRNA |
| Abl | CG4032* | miR-318 |
| Beat-Ia | CG4846† | miR-263b |
| Dock | CG3727 | miR-33 |
| Drl-2 | CG3915 | miR-274, miR-275‡ miR-304 |
| Eph | CG1511 | miR-282, miR-283, miR-306 |
| Lar | CG10443 | let-7‡ |
| NetA | CG18657* | miR-275‡, miR-288, miR-14 |
| NetB | CG10521† | miR-31b |
| Ptp99A | CG2005 | miR-8‡ |
| Robo | CG13521* | miR-282‡, miR-284‡, miR-307, miR-133‡ |
| Sema-1a | CG18405* | miR-iab-4-5p |
| Sema-1b | CG6446* | miR-184 |
| Sema-2a | CG4700* | miR-4‡ |
| sli | CG8355 | miR-33‡ |
| Spen | CG18497† | let-7‡ |
| Trio | CG18214 | miR-184‡ |
*,†Target gene based on the top 20 and top 30 hits of each miRNA, respectively; all others based on top 10 hits. ‡Target site also conserved in A. gambiae.
In addition to these cell surface factors, miRNAs target the cellular machinery that effects cell shape change and adhesion, including regulators and components of the cytoskeleton (for example, dock, trio, RhoGAPs, Rho1, Abl, tricornered, wasp, Sop2, nesprin, Khc-73, gamma-tubulin) and of the cell junctions (for example, crumbs, dlt, mbc, skiff).
Many of the developmental factors are re-employed in the mature nervous system to control synaptic function by effecting morphogenetic changes in synapse size, shape or strength. Additional miRNA target genes that are active in the mature nervous system include neurotransmitter receptors, ion channels and pumps (clumsy, DopR, nAChr; Hk, Shaker), as well as factors involved in neurotransmitter transport and synaptic release (SerT, vAChT, Eaat1; Cirl, Rab3, Sap47, unc13) [79].
Why would translational regulation by miRNAs feature so prominently in the development and function of the nervous system? The distances between the nucleus and dendrites/axon projections are relatively large, making nuclear regulation difficult. Furthermore, differential gene activity between and even within compartments (for example, between different portions of a growth cone or different branches of a dendritic tree) is crucial for neuronal function. Therefore, translational control near the site of action is a more efficient means of modulating gene activity than transcriptional control. For axon guidance, it has been shown that the relative abundance of adhesion molecules and chemotropic receptors on the surface of the growth cone is post-transcriptionally regulated in response to external cues presented by intermediate targets. Regulation by miRNAs would thus provide an excellent additional mechanism of post-transcriptional control.
Discussion
The precise rules and energetics for pairing between a miRNA and its mRNA target sites, with probable involvement of a protein complex, are not known and cannot easily be deduced from the few experimentally proven examples. Therefore, any computational methods for the identification of potential miRNA target sites are at risk of having a substantial rate of false positives and false negatives. Based on analysis of the known examples, we have biased our method toward stronger matches at the 5' end of the miRNA, and used energy calculation plus conservation of target site sequence to provide our current best estimate of biologically functional matches. Overall, we find that conservation is a crucial filter and reduces the rate of prediction error.
Our results suggest that miRNAs target the control of gene activity at multiple levels, specifically transcription, translation and protein degradation, in other words, that miRNAs act as meta-regulators of expression control. Among biological processes, we find that the most prominent targets include signal transduction and transcription control in cell fating and developmental timing decisions, as well as morphogenetic processes such as axon guidance. These processes share the need for the precise definition of boundaries of gene activity in space and time. Our findings therefore support and expand earlier work on the role of miRNAs in developmental processes [42,80]. In addition, we predict that miRNAs also play an important role in controlling gene activity in the mature nervous system.
As miRNA and mRNA have to be present simultaneously at minimum levels in the same cellular compartment for a biologically meaningful interaction, more precise expression data as a function, for example, of developmental stage [42], will be extremely useful and will be incorporated in future versions of target prediction methods. Similarly, further work will include the analysis of potential target sites in coding regions and 5' UTRs, as well as conservation and adaptation of target sites in many species.
This genome-wide scan for potential miRNA target genes gives us a first glimpse of the complexity of the emerging network of regulatory interactions involving small RNAs (see Additional data). Both multiplicity (one miRNA targets several genes) and cooperativity (one gene is targeted by several distinct miRNAs) appear to be general features for many miRNAs, as already apparent with the discovery of the targets for lin-4 and let-7. The analogy of these many-to-one and one-to-many relationships to those of transcription factors and promoter regions is tempting and elucidation of the network of regulation by miRNAs will make a major contribution to cellular systems biology. In the meantime, we would not be surprised if experiments focusing on target candidates filtered in this way have a high rate of success and help to unravel the biology of regulation by miRNA-mRNA interaction.
Materials and methods
MiRNA sequences
An initial set of D. melanogaster miRNA sequences was built using the RFAM miRNA database [45]. Mature miRNA sequences were placed in a FASTA formatted sequence file. In total, the final file contained 73 unique miRNA sequences. All sequences used for this analysis are available from RFAM and as supplementary material [39].
3' UTR sequences
Sequences for D. melanogaster 3' UTRs were obtained from the Berkeley Drosophila Genome Project (BDGP). In total, 3' UTR sequences were available for 14,287 transcripts, representing 9,805 individual D. melanogaster genes. A corresponding set of D. pseudoobscura 3' UTR sequences was then built from the March 2003 first freeze of the D. pseudoobscura genome project at Baylor College of Medicine. Each D. melanogaster 3' UTR was mapped to D. pseudoobscura contigs by searching both the actual D. melanogaster 3' UTR sequence, using NCBI BLASTn [81], and the peptide sequence of each gene, using NCBI tBLASTn [81], against D. pseudoobscura contigs [81]. Results from these two scans were then used to identify candidate 2,000 bp regions of D. pseudoobscura contigs, within which we believe an orthologous D. pseudoobscura 3' UTR is present. The AVID [46] alignment tool was used to align the real D. melanogaster 3' UTR and a candidate D. pseudoobscura region. Finally, this alignment was used to trim each candidate region, leaving the predicted D. pseudoobscura 3' UTR. In total 12,416 transcripts and 8,282 genes from D. pseudobscura were mapped to orthologous D. melanogaster UTRs in this fashion. The Ensembl database [47] Application Programming Interface was used to construct A. gambiae predicted 3' UTRs by taking 2,000 bp downstream from the last exon of each transcript. Orthology mappings between A. gambiae and D. melanogaster UTRs were then obtained by searching all Ensembl A. gambiae peptides against all D. melanogaster peptides using BLASTp. In total 9,823 A. gambiae genes were mapped to D. melanogaster genes in this manner.
MiRanda scanning algorithm
The miRanda algorithm is similar to the Smith-Waterman algorithm [82], however, instead of building alignments based on matching nucleotides (A-A or U-U, for example), it scores based on the complementarity of nucleotides (A=U or G≡C). The scoring matrix used for this analysis also allows G=U 'wobble' pairs, which are important for the accurate detection of RNA:RNA duplexes [44]. Complementarity parameters at individual alignment positions are: +5 for G≡C, +5 for A=U, +2 for G=U and -3 for all other nucleotide pairs. The algorithm uses affine penalties (linear in the length of a gap after an initial opening penalty) for gap-opening (-8) and gap-extension (-2). In addition, following observation of known target sites, complementarity scores (positive and negative values) at the first eleven positions are multiplied by a scaling factor (here set at 2.0), so as to reflect the observed 5'-3' asymmetry. Finally, the following four empirical rules are applied, with positions counted starting at the 5' end of the miRNA: no mismatches at positions 2 to 4; fewer than five mismatches between positions 3-12; at least one mismatch between positions 9 and L-5 (where L is total alignment length); and fewer than two mismatches in the last five positions of the alignment. With these parameters, the dynamic programming algorithm optimizes the complementarity score between a miRNA sequence and an mRNA sequence (typically a 3' UTR), summed over all aligned positions, and finds all non-overlapping hybridization alignments in decreasing order of complementarity score down to some cutoff value (default value 80). The detection of sub-optimal alignments follows heuristics previously used in sequence alignment [81,83].
In order to estimate the thermodynamic properties of a predicted duplex, the algorithm uses folding routines from the Vienna 1.3 RNA secondary structure programming library (RNAlib) [44]. The expanded thermodynamic parameters used [83] are more computationally intensive than the initial scan, but allow potential hybridization sites to be scored according their respective folding energies. The miRNA sequence and 3' UTR sequence from a hybridization alignment are joined into a single sequence with an eight base sequence linker containing artificial 'X' bases that cannot base pair. This strand-linker-strand configuration assumes the phase space entropy of strand-strand association is constant for all miRNA-target matches [44,83]. The minimum energy of this structure with the last matching base pair (from initial sequence alignment) constrained is then calculated using RNAlib.
Conservation of target sites
All miRNA sequences are scanned against the 3' UTR datasets of D. melanogaster, D. pseudoobscura and A. gambiae. The thresholds used for hit detection are: initial Smith-Waterman hybridization alignments must have S ≥ 80, and the minimum energy of the duplex structure ΔG ≤ -14 kcal/mol. Each hit between a miRNA and a UTR sequence is then scored according to the total energy and total score of all hits between those two sequences. Hits are deemed to be conserved in D. pseudobscura or A. gambiae if a target site equivalent to that detected in a D. melanogaster UTR can be found in the orthologous D pseudoobscura or A. gambiae UTR at the same position in the UTR alignments. Our definition of equivalence between target sites is that their sequences are more than 80% identical for D. pseudoobscura and 60% identical for A. gambiae. All results from the scan are then ranked and sorted according to total score of conserved target sites detected. For each miRNA, the ten highest ranked genes are selected as its candidate target genes in this way. Multiple miRNAs binding the same site on a target gene are resolved using a greedy algorithm that assigns the highest scoring and lowest free energy miRNA target duplex to each potential site so that different miRNA target sites cannot overlap.
Validation
For the initial validation, 3' UTR sequences for C. elegans and C. briggsae were obtained, if possible, from UTRdb [84]. If unavailable, UTR sequences were estimated by taking 2,000 bp of flanking nucleotide sequence downstream of the last exon of the gene in question using the Ensembl database [48].
Randomized test
Control sequences for the randomized experiment were constructed by assembling 100 sets of 73 miRNAs each generated by random shuffling of each D. melanogaster miRNA. Each of these sets of 73 randomized miRNAs was independently searched against all D. melanogaster and D. pseudoobscura 3' UTRs as in the reference experiment. Results and counts were then averaged over all 100 random sets, and were compared with the results of the actual miRNA scan. For the functional analysis, GO classes for known D. melanogaster genes were obtained from FlyBase and conserved hits for the real and random miRNAs for each class are counted. The Z-scores are generated from the actual miRNA counts, averaged random miRNA counts and their standard deviations.
Note that recent work by Stark et al. [85] and also by Rajewski and Socci [86] addresses similar issues to those described in this work.
Additional data files
The following files are available with this article and at [39]: a supplementary target functional plot (GO biological process; Additional data file 1); a supplementary target:miRNA network graph (Additional data file 2); an Excel table of the top 10 miRNA target predictions by miRNA (Additional data file 3); and an Excel table of the top 10 miRNA target predictions by gene (Additional data file 4). Additional data files of top 20, 30, and 40 both by microRNA and by gene and the source code of the core miRanda algorithm are available at [39].
Supplementary Material
A supplementary target functional plot
A supplementary target:miRNA network graph
An Excel table of the top 10 miRNA target predictions by miRNA
An Excel table of the top 10 miRNA target predictions by gene
Acknowledgments
Acknowledgements
We thank Gary Bader, Boris Reva, Carl Thummel and Mihaela Zavolan for interesting discussions and also Rebecca Ward, Ulrich Unnerstall and Kerstin Dose for support and assistance.
A previous version of this manuscript was made available before peer review at http://genomebiology.com/2003/4/11/P8
Contributor Information
Chris Sander, Email: sanderc@mskcc.org.
Debora S Marks, Email: Debbie@maple.bioc.columbia.edu.
References
- Pasquinelli AE. MicroRNAs: deviants no longer. Trends Genet. 2002;18:171–173. doi: 10.1016/S0168-9525(01)02624-5. [DOI] [PubMed] [Google Scholar]
- Ambros V. MicroRNA pathways in flies and worms: growth, death, fat, stress, and timing. Cell. 2003;113:673–676. doi: 10.1016/s0092-8674(03)00428-8. [DOI] [PubMed] [Google Scholar]
- Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336–338. doi: 10.1126/science.1085242. [DOI] [PubMed] [Google Scholar]
- Lin SY, Johnson SM, Abraham M, Vella MC, Pasquinelli A, Gamberi C, Gottlieb E, Slack FJ. The C. elegans hunchback Homolog, hbl-1, controls temporal patterning and is a probable MicroRNA target. Dev Cell. 2003;4:639–650. doi: 10.1016/s1534-5807(03)00124-2. [DOI] [PubMed] [Google Scholar]
- The miRNA registry http://www.sanger.ac.uk/Software/Rfam/mirna/
- Ambros V, Bartel B, Bartel DP, Burge CB, Carrington JC, Chen X, Dreyfuss G, Eddy SR, Griffiths-Jones S, Marshall M, et al. A uniform system for microRNA annotation. RNA. 2003;9:277–279. doi: 10.1261/rna.2183803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack F, Ruvkun G. Temporal pattern formation by heterochronic genes. Annu Rev Genet. 1997;31:611–634. doi: 10.1146/annurev.genet.31.1.611. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Ruvkun G. Control of developmental timing by microRNAs and their targets. Annu Rev Cell Dev Biol. 2002;18:495–513. doi: 10.1146/annurev.cellbio.18.012502.105832. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, Hayward DC, Ball EE, Degnan B, Muller P, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Yalcin A, Meyer J, Lendeckel W, Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr Biol. 2002;12:735–739. doi: 10.1016/S0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- Moss EG, Tang L. Conservation of the heterochronic regulator Lin-28, its developmental expression and microRNA complementary sites. Dev Biol. 2003;258:432–442. doi: 10.1016/S0012-1606(03)00126-X. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Hipfner DR, Stark A, Russell RB, Cohen SM. bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell. 2003;113:25–36. doi: 10.1016/s0092-8674(03)00231-9. [DOI] [PubMed] [Google Scholar]
- Xu P, Vernooy SY, Guo M, Hay BA. The Drosophila microRNA Mir-14 suppresses cell death and is required for normal fat metabolism. Curr Biol. 2003;13:790–795. doi: 10.1016/S0960-9822(03)00250-1. [DOI] [PubMed] [Google Scholar]
- Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP. Prediction of plant microRNA targets. Cell. 2002;110:513–520. doi: 10.1016/s0092-8674(02)00863-2. [DOI] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC. Cleavage of scarecrow-like mRNA targets directed by a class of Arabidopsis miRNA. Science. 2002;297:2053–2056. doi: 10.1126/science.1076311. [DOI] [PubMed] [Google Scholar]
- Palatnik JF, Allen E, Wu X, Schommer C, Schwab R, Carrington JC, Weigel D. Control of leaf morphogenesis by microRNAs. Nature. 2003;425:257–263. doi: 10.1038/nature01958. [DOI] [PubMed] [Google Scholar]
- Chen X. A microRNA as a translational repressor of APETALA2 in Arabidopsis flower development. Science. 11 September 2003 (10.1126/science.1088060) [DOI] [PMC free article] [PubMed]
- Tang G, Reinhart BJ, Bartel DP, Zamore PD. A biochemical framework for RNA silencing in plants. Genes Dev. 2003;17:49–63. doi: 10.1101/gad.1048103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Kasschau KD, Carrington JC. Negative feedback regulation of dicer-like1 in Arabidopsis by microRNA-guided mRNA degradation. Curr Biol. 2003;13:784–789. doi: 10.1016/S0960-9822(03)00281-1. [DOI] [PubMed] [Google Scholar]
- Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75:843–854. doi: 10.1016/0092-8674(93)90529-y. [DOI] [PubMed] [Google Scholar]
- Wightman B, Ha I, Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- Moss EG, Lee RC, Ambros V. The cold shock domain protein LIN-28 controls developmental timing in C. elegans and is regulated by the lin-4 RNA. Cell. 1997;88:637–646. doi: 10.1016/S0092-8674(00)81906-6. [DOI] [PubMed] [Google Scholar]
- Slack FJ, Basson M, Liu Z, Ambros V, Horvitz HR, Ruvkun G. The lin-41 RBCC gene acts in the C. elegans heterochronic pathway between the let-7 regulatory RNA and the LIN-29 transcription factor. Mol Cell. 2000;5:659–669. doi: 10.1016/s1097-2765(00)80245-2. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvie AE, Horvitz HR, Ruvkun G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, Rougvie AE. The Caenorhabditis elegans hunchback-like Gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev Cell. 2003;4:625–637. doi: 10.1016/s1534-5807(03)00127-8. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Caudy AA, Hammond SM, Hannon GJ. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature. 2001;409:363–366. doi: 10.1038/35053110. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, McLachlan J, Pasquinelli AE, Balint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol. 2002;12:1484–1495. doi: 10.1016/S0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Radmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lee Y, Jeon K, Lee JT, Kim S, Kim VN. MicroRNA maturation: stepwise processing and subcellular localization. EMBO J. 2002;21:4663–4670. doi: 10.1093/emboj/cdf476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SM, Bernstein E, Beach D, Hannon GJ. An RNA-directed nuclease mediates post-transcriptional gene silencing in Drosophila cells. Nature. 2000;404:293–296. doi: 10.1038/35005107. [DOI] [PubMed] [Google Scholar]
- Martinez LA, Naguibneva I, Lehrmann H, Vervisch A, Tchenio T, Lozano G, Harel-Bellan A. Synthetic small inhibiting RNAs: efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc Natl Acad Sci USA. 2002;99:14849–14854. doi: 10.1073/pnas.222406899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvagner G, Zamore PD. A microRNA in a multiple-turnover RNAi enzyme complex. Science. 2002;297:2056–2060. doi: 10.1126/science.1073827. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Yi R, Cullen BR. MicroRNAs and small interfering RNAs can inhibit mRNA expression by similar mechanisms. Proc Natl Acad Sci USA. 2003;100:9779–9784. doi: 10.1073/pnas.1630797100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs. Genes Dev. 2003;17:438–442. doi: 10.1101/gad.1064703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena S, Jonsson ZO, Dutta A. Small RNAs with imperfect match to endogenous mRNA repress translation: implications for off-target activity of siRNA in mammalian cells. J Biol Chem. 2003;278:44312–44319. doi: 10.1074/jbc.M307089200. [DOI] [PubMed] [Google Scholar]
- Elbashir SM, Martinez J, Patkaniowska A, Lendeckel W, Tuschl T. Functional anatomy of siRNAs for mediating efficient RNAi in Drosophila melanogaster embryo lysate. EMBO J. 2001;20:6877–6888. doi: 10.1093/emboj/20.23.6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol. 2003;21:635–637. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- Supplementary Material for: 'MicroRNA targets in Drosophila http://www.microrna.org/
- Lagos-Quintana M, Rauhut R, Lendeckel W, Tuschl T. Identification of novel genes coding for small expressed RNAs. Science. 2001;294:853–858. doi: 10.1126/science.1064921. [DOI] [PubMed] [Google Scholar]
- Sempere LF, Sokol NS, Dubrovsky EB, Berger EM, Ambros V. Temporal regulation of microRNA expression in Drosophila melanogaster mediated by hormonal signals and broad-Complex gene activity. Dev Biol. 2003;259:9–18. doi: 10.1016/S0012-1606(03)00208-2. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T. The small RNA profile during Drosophila melanogaster development. Dev Cell. 2003;5:337–350. doi: 10.1016/s1534-5807(03)00228-4. [DOI] [PubMed] [Google Scholar]
- Lai EC, Tomancak P, Williams RW, Rubin GM. Computational identification of Drosophila microRNA genes. Genome Biol. 2003;4:R42. doi: 10.1186/gb-2003-4-7-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuchty S, Fontana W, Hofacker IL, Schuster P. Complete suboptimal folding of RNA and the stability of secondary structures. Biopolymers. 1999;49:145–165. doi: 10.1002/(SICI)1097-0282(199902)49:2<145::AID-BIP4>3.3.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Griffiths-Jones S, Bateman A, Marshall M, Khanna A, Eddy SR. Rfam: an RNA family database. Nucleic Acids Res. 2003;31:439–441. doi: 10.1093/nar/gkg006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray N, Dubchak I, Pachter L. AVID: A global alignment program. Genome Res. 2003;13:97–102. doi: 10.1101/gr.789803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T, et al. The Ensembl genome database project. Nucleic Acids Res. 2002;30:38–41. doi: 10.1093/nar/30.1.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FlyBase Consortium The FlyBase database of the Drosophila genome projects and community literature. Nucleic Acids Res. 2002;30:106–108. doi: 10.1093/nar/30.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart WM, Crosby M, Matthews B, Rindone WP, Chillemi J, Russo Twombly S, Emmert D, Ashburner M, Drysdale RA, Whitfield E, et al. FlyBase: a Drosophila database. The FlyBase consortium. Nucleic Acids Res. 1997;25:63–66. doi: 10.1093/nar/25.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakhina S, Steward R. Several small RNAs, one precursor and two genes - a complex locus essential for viability. A Dros Res Conf. 2003;44:403A. [Google Scholar]
- Minakhina S, Yang J, Steward R. Tamo selectively modulates nuclear import in Drosophila. Genes Cells. 2003;8:299–310. doi: 10.1046/j.1365-2443.2002.00634.x. [DOI] [PubMed] [Google Scholar]
- Akam M. Hox genes: from master genes to micromanagers. Curr Biol. 1998;8:R676–678. doi: 10.1016/s0960-9822(98)70433-6. [DOI] [PubMed] [Google Scholar]
- McGinnis W, Krumlauf R. Homeobox genes and axial patterning. Cell. 1992;68:283–302. doi: 10.1016/0092-8674(92)90471-n. [DOI] [PubMed] [Google Scholar]
- Mann RS. Why are Hox genes clustered? Bioessays. 1997;19:661–664. doi: 10.1002/bies.950190804. [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M, Rauhut R, Meyer J, Borkhardt A, Tuschl T. New microRNAs from mouse and human. RNA. 2003;9:175–179. doi: 10.1261/rna.2146903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon JA, Tamkun JW. Programming off and on states in chromatin: mechanisms of Polycomb and trithorax group complexes. Curr Opin Genet Dev. 2002;12:210–218. doi: 10.1016/S0959-437X(02)00288-5. [DOI] [PubMed] [Google Scholar]
- Rieckhof GE, Casares F, Ryoo HD, Abu-Shaar M, Mann RS. Nuclear translocation of extradenticle requires homothorax, which encodes an extradenticle-related homeodomain protein. Cell. 1997;91:171–183. doi: 10.1016/s0092-8674(00)80400-6. [DOI] [PubMed] [Google Scholar]
- Riddiford M. Hormones and Drosophila Development. In: Martinez Arias M, Bate A, editor. In The Development of Drosophila melanogaster. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1993. pp. 899–940. [Google Scholar]
- Baehrecke EH. Steroid regulation of programmed cell death during Drosophila development. Cell Death Differ. 2000;7:1057–1062. doi: 10.1038/sj.cdd.4400753. [DOI] [PubMed] [Google Scholar]
- Thummel CS. Molecular mechanisms of developmental timing in C. elegans and Drosophila. Dev Cell. 2001;1:453–465. doi: 10.1016/s1534-5807(01)00060-0. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Chory J. Steroid signaling in plants and insects - common themes, different pathways. Genes Dev. 2002;16:3113–3129. doi: 10.1101/gad.1042102. [DOI] [PubMed] [Google Scholar]
- Simon AF, Shih C, Mack A, Benzer S. Steroid control of longevity in Drosophila melanogaster. Science. 2003;299:1407–1410. doi: 10.1126/science.1080539. [DOI] [PubMed] [Google Scholar]
- Broadus J, McCabe JR, Endrizzi B, Thummel CS, Woodard CT. The Drosophila beta FTZ-F1 orphan nuclear receptor provides competence for stage-specific responses to the steroid hormone ecdysone. Mol Cell. 1999;3:143–149. doi: 10.1016/s1097-2765(00)80305-6. [DOI] [PubMed] [Google Scholar]
- D'Avino PP, Thummel CS. crooked legs encodes a family of zinc finger proteins required for leg morphogenesis and ecdysone-regulated gene expression during Drosophila metamorphosis. Development. 1998;125:1733–1745. doi: 10.1242/dev.125.9.1733. [DOI] [PubMed] [Google Scholar]
- Beckstead R, Ortiz JA, Sanchez C, Prokopenko SN, Chambon P, Losson R, Bellen HJ. Bonus, a Drosophila homolog of TIF1 proteins, interacts with nuclear receptors and can inhibit betaFTZ-F1-dependent transcription. Mol Cell. 2001;7:753–765. doi: 10.1016/S1097-2765(01)00220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne JC, Kondylis V, Rabouille C. Ecdysone triggers the expression of Golgi genes in Drosophila imaginal discs via broad-complex. Dev Biol. 2002;245:172–186. doi: 10.1006/dbio.2002.0632. [DOI] [PubMed] [Google Scholar]
- Jiang C, Lamblin AF, Steller H, Thummel CS. A steroid-triggered transcriptional hierarchy controls salivary gland cell death during Drosophila metamorphosis. Mol Cell. 2000;5:445–455. doi: 10.1016/s1097-2765(00)80439-6. [DOI] [PubMed] [Google Scholar]
- Jiang C, Baehrecke EH, Thummel CS. Steroid regulated programmed cell death during Drosophila metamorphosis. Development. 1997;124:4673–4683. doi: 10.1242/dev.124.22.4673. [DOI] [PubMed] [Google Scholar]
- Wismar J, Habtemichael N, Warren JT, Dai JD, Gilbert LI, Gateff E. The mutation without children(rgl) causes ecdysteroid deficiency in third-instar larvae of Drosophila melanogaster. Dev Biol. 2000;226:1–17. doi: 10.1006/dbio.2000.9811. [DOI] [PubMed] [Google Scholar]
- Rayburn LY, Gooding HC, Choksi SP, Maloney D, Kidd AR, 3rd, Siekhaus DE, Bender M. amontillado, the Drosophila homolog of the prohormone processing protease PC2, is required during embryogenesis and early larval development. Genetics. 2003;163:227–237. doi: 10.1093/genetics/163.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadesch T. Notch signaling: a dance of proteins changing partners. Exp Cell Res. 2000;260:1–8. doi: 10.1006/excr.2000.4921. [DOI] [PubMed] [Google Scholar]
- Baker KD, Warren JT, Thummel CS, Gilbert LI, Mangelsdorf DJ. Transcriptional activation of the Drosophila ecdysone receptor by insect and plant ecdysteroids. Insect Biochem Mol Biol. 2000;30:1037–1043. doi: 10.1016/S0965-1748(00)00075-8. [DOI] [PubMed] [Google Scholar]
- Lai EC, Burks C, Posakony JW. The K box, a conserved 3' UTR sequence motif, negatively regulates accumulation of enhancer of split complex transcripts. Development. 1998;125:4077–4088. doi: 10.1242/dev.125.20.4077. [DOI] [PubMed] [Google Scholar]
- Lai EC, Rubin GM. Neuralized is essential for a subset of Notch pathway-dependent cell fate decisions during Drosophila eye development. Proc Natl Acad Sci USA. 2001;98:5637–5642. doi: 10.1073/pnas.101135498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm A, Tessier-Lavigne M. Conservation and divergence of axon guidance mechanisms. Curr Opin Neurobiol. 1999;9:603–615. doi: 10.1016/S0959-4388(99)00021-5. [DOI] [PubMed] [Google Scholar]
- Mueller BK. Growth cone guidance: first steps towards a deeper understanding. Annu Rev Neurosci. 1999;22:351–388. doi: 10.1146/annurev.neuro.22.1.351. [DOI] [PubMed] [Google Scholar]
- Giniger E. How do Rho family GTPases direct axon growth and guidance? A proposal relating signaling pathways to growth cone mechanics. Differentiation. 2002;70:385–396. doi: 10.1046/j.1432-0436.2002.700801.x. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Ganetzky B. Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron. 2000;26:35–43. doi: 10.1016/s0896-6273(00)81135-6. [DOI] [PubMed] [Google Scholar]
- Ambros V, Moss EG. Heterochronic genes and the temporal control of C. elegans development. Trends Genet. 1994;10:123–127. doi: 10.1016/0168-9525(94)90213-5. [DOI] [PubMed] [Google Scholar]
- Waterman MS, Eggert M. A new algorithm for best subsequence alignments with application to tRNA-rRNA comparisons. J Mol Biol. 1987;197:723–728. doi: 10.1016/0022-2836(87)90478-5. [DOI] [PubMed] [Google Scholar]
- Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- Schneider R, Sander C. The HSSP database of protein structure-sequence alignments. Nucleic Acids Res. 1996;24:201–205. doi: 10.1093/nar/24.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesole G, Liuni S, Grillo G, Licciulli F, Mignone F, Gissi C, Saccone C. UTRdb and UTRsite: specialized databases of sequences and functional elements of 5' and 3' untranslated regions of eukaryotic mRNAs. Update 2002. Nucleic Acids Res. 2002;30:335–340. doi: 10.1093/nar/30.1.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Brennecke J, Russell RB, Cohen SM. Identification of Drosophila microRNA targets. PLoS Biology. October 13 2003 (DOI: 10.1371/journal.pbio.0000060) [DOI] [PMC free article] [PubMed]
- Rajewsky N, Socci ND. Computational identification of microRNA targets. Dev Biol. [DOI] [PubMed]
- Ashburner M, Lewis S. On ontologies for biologists: the Gene Ontology - untangling the web. Novartis Found Symp. 2002;247:66–80. doi: 10.1002/0470857897.ch6. [discussion 80-63, 84-90, 244-252] [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A supplementary target functional plot
A supplementary target:miRNA network graph
An Excel table of the top 10 miRNA target predictions by miRNA
An Excel table of the top 10 miRNA target predictions by gene