

Abstract
Cancer stem cells likely survive chemotherapy or radiotherapy by acquiring mutations that inactivate the endogenous apoptotic machinery or by cycling slowly. Thus, knowledge about the mechanisms linking the activation of an alternative cell death modality and the cell cycle machinery could have a transformative impact on the development of new cancer therapies, but the mechanisms remain completely unknown. We investigated the regulation of alternative cell death in Drosophila larval brain neural stem cells (neuroblasts) in which apoptosis is normally repressed. From a screen, we identified two novel loss-of-function alleles of the Cdc20/fizzy (fzy) gene that lead to premature brain neuroblast loss without perturbing cell proliferation in other diploid cell types. Fzy is an evolutionarily conserved regulator of anaphase promoting complex/cyclosome (APC/C). Neuroblasts carrying the novel fzy allele or exhibiting reduced APC/C function display hallmarks of necrosis. By contrast, neuroblasts overexpressing the non-degradable form of canonical APC/C substrates required for cell cycle progression undergo mitotic catastrophe. These data strongly suggest that Fzy can elicit a novel pro-survival function of APC/C by suppressing necrosis. Neuroblasts experiencing catastrophic cellular stress, or overexpressing p53, lose Fzy expression and undergo necrosis. Co-expression of fzy suppresses the death of these neuroblasts. Consequently, attenuation of the Fzy-dependent survival mechanism functions downstream of catastrophic cellular stress and p53 to eliminate neuroblasts by necrosis. Strategies that target the Fzy-dependent survival mechanism might lead to the discovery of new treatments or complement the pre-existing therapies to eliminate apoptosis-resistant cancer stem cells by necrosis.
Keywords: Cdc20/Fizzy, Drosophila, Brain, Catastrophic cellular stress, Necrosis, Neuroblast
INTRODUCTION
Conventional chemotherapy and radiotherapy induce catastrophic cellular stress in cancer cells and eliminate them by apoptosis through the activation of the p53-dependent checkpoint mechanism (Deckbar et al., 2011; Galluzzi et al., 2011). However, these treatments often fail to eradicate cancer stem cells, possibly because these cells acquire mutations that inactivate the apoptotic machinery or simply undergo cell division infrequently. Nevertheless, cancer stem cells maintain their dependence on the cell cycle machinery to expand the tumor mass. As such, strategies that couple activation of an alternative cell death modality to the cell cycle machinery can potentially improve the targeting efficiency against cancer stem cells.
Programmed cell death plays a pivotal role during the maintenance of homeostasis and constitutes the molecular basis for various chemotherapies and radiotherapies for cancers (Fuchs and Steller, 2011; Galluzzi et al., 2011). Necrosis frequently serves as the main alternative cell death modality to eliminate apoptosis-deficient cells under certain physiological conditions (Vandenabeele et al., 2010; Han et al., 2011). Recent studies have indicated that necrosis can be activated in a regulated manner. The receptor interacting protein (RIP) family kinases play a central role in activating necrosis in vertebrates (Hitomi et al., 2008; Cho et al., 2009; He et al., 2009; Zhang et al., 2009). Unlike vertebrates, however, the Drosophila genome does not appear to encode a functional ortholog of the RIP kinase (Igaki et al., 2011). Thus, studies of the regulation of necrosis in flies will likely provide novel insight into the mechanisms that elicit necrosis independently of the RIP kinases.
Approximately 100 brain neuroblasts are born from the procephalic neurogenic region during Drosophila embryogenesis. These neuroblasts undergo asymmetric divisions to generate the differentiated cell types necessary for the function of larval brains, and all but five neuroblasts enter transient mitotic quiescence at the end of embryogenesis (Datta, 1995). These quiescent brain neuroblasts progressively re-enter the cell cycle in the first to the early third larval instar, and all 100 brain neuroblasts are actively dividing in the late third larval instar (Datta, 1995; Lee et al., 2006). Brain neuroblasts appear incapable of undergoing apoptosis during larval development as the Polycomb proteins actively silence the apoptotic genes (Bello et al., 2007; Siegrist et al., 2010). Consistently, overexpression of p53 reduces the total number of larval brain neuroblasts in the absence of caspase activation (Ouyang et al., 2011). Thus, fly larval brain neuroblasts provide a unique system for discovering genes that elicit an alternative cell death modality in a stem cell type that is apoptosis deficient.
Cdc20/Fzy elicits the function of APC/C in a myriad of biological responses, including cell cycle progression, by regulating the specificity of substrate recognition and recruitment (Yu, 2007; Pesin and Orr-Weaver, 2008; Eguren et al., 2011; McLean et al., 2011; Pines, 2011). Residues located in a channel between the first and the seventh β-propellers or at the center of the top side of the WD40 repeat domain of Cdc20/Fzy mediate direct interaction with the D-box or the KEN box in the mitotic substrates of APC/C (Chao et al., 2012). Whether these residues also mediate substrate recognition and recruitment in other APC/C-regulated responses remains unknown. Insight linking the specificity of substrate recognition by the WD40 repeat domain of Cdc20/Fzy to the activation of necrosis might provide novel strategies for targeting apoptosis-resistant cancer stem cells.
We describe a novel mechanism by which Cdc20/Fzy maintains the survival of neuroblasts in larval brains by suppressing necrosis independently of its function in promoting cell proliferation. We identified two novel fzy mutant alleles that specifically lead to brain neuroblast loss without perturbing the proliferation of other diploid cell types. Neuroblast loss in these fzy mutant brains occurred through necrosis rather than through premature differentiation, apoptosis or autophagy. Fzy suppresses necrosis in neuroblasts partly by antagonizing Aif and JNK signaling, because removing the function of either signaling mechanism significantly prolonged the survival of fzy mutant neuroblasts. Neuroblasts experiencing catastrophic cellular stress, including protein mis-folding response or the activation of DNA damage responses, lose Fzy expression and undergo necrosis. Similarly, neuroblasts overexpressing p53 also lose Fzy expression and die by necrosis. Most importantly, overexpression of fzy suppresses the necrosis of neuroblasts overexpressing p53 or experiencing catastrophic cellular stress, indicating that the attenuation of Fzy expression directly contributes to the death of these neuroblasts. We propose that the Fzy-dependent survival mechanism provides a novel link between the regulation of necrosis by p53 and the cell cycle machinery to regulate the viability of apoptosis-deficient neuroblasts during larval brain neurogenesis.
RESULTS
Fzy maintains neuroblasts independently of its role in promoting cell proliferation
In order to identify genes that regulate an alternative cell death modality in the absence of apoptosis, we screened a collection of EMS-induced mutants for a premature neuroblast loss phenotype in late third instar larval brains. We identified two novel fzy alleles, fzy5032 and fzyAC-10, that result in premature loss of brain neuroblasts (Fig. 1A). Although the number of neuroblasts plateaued at 100 in wild-type brains at 96 hours after hatching, the number of neuroblasts in fzy mutant brains briefly plateaued at ∼40 at 48 hours after hatching and then declined sharply towards the end of larval development. We next determined the kinetics of neuroblast loss following the removal of fzy function. All wild-type clones contained neuroblasts, but 35% of fzy5032 clones and 55% of fzy4 [a strong fzy loss-of-function allele (Dawson et al., 1993)] clones did not contain neuroblasts (Fig. 1B). Together, these data indicate that Fzy functions cell autonomously to maintain neuroblasts. The fzy5032 and fzyAC-10 mutations result in single amino acid substitutions that most likely perturb a unique function of the Fzy protein (Fig. 1D). Consistently, restoring fzy function completely rescued the neuroblast loss phenotype in these fzy mutant brains (Fig. 1C). For the rest of our study we used the fzy5032 allele because the amino acid residue altered by this mutation is conserved and fzy5032 larvae displayed a similar level of fzy transcript and a similar expression pattern of Fzy protein as control larvae (supplementary material Fig. S1A-C).
Fig. 1.

The fzy5032 mutation perturbs the function of fzy in maintaining neuroblasts but not in promoting cell proliferation. (A) fzy5032 and fzyAC-10 larval brains showed progressive neuroblast loss. Larvae of the indicated genotype were aged for 24, 48, 72 or 96 hours after hatching, and brains were stained for the neuroblast marker Deadpan (Dpn). The average number of neuroblasts per brain lobe is shown (n=10 brain lobes per genotype). (B) Neuroblasts were rapidly lost in fzy mutant clones. Larvae with GFP-marked mosaic clones derived from single neuroblasts of the indicated genotypes were aged for 24 hours after clone induction, and brains were stained for GFP, to identify the clones, and Dpn. Greater than 90% of wild-type clones were derived from neuroblasts and contained a single neuroblast per clone. Less than 10% of wild-type clones were derived from intermediate neural progenitor cells and did not contain neuroblasts. The percentage of the clones lacking identifiable neuroblasts is shown. (C) Restoring fzy function rescued the neuroblast loss phenotype in fzy5032 and fzyAC-10 larval brains. Larvae of the indicated genotype were aged for 72 hours after hatching, and brains were stained for Dpn. fzytDa indicates the transgene containing the entire fzy genomic locus. The average number of neuroblasts per brain lobe is shown. (D) The molecular lesion induced by the fzy5032 or fzyAC-10 mutation. (E-H) fzy5032 pharate adults have poorly developed brains. (E,G) Control and fzy5032 pharate adults at 120 hours after white puparium formation. Scale bar: 1 mm. (F,H) Paraffin sections of control and fzy5032 pupal brains aged for 96 hours after white puparium formation, stained with Hematoxylin and Eosin. Scale bar: 200 μm. (I-N) Overexpression of the fzy5032 transgene restored proliferation in fzy4 neuroepithelial cells. (I-M) Larvae containing GFP-marked mosaic clones (outlined in light gray) derived from single neuroepithelial cells of the indicated genotypes were aged for 72 hours after clone induction, and brains were stained for the indicated markers. Prospero (Pros) serves as a marker for immature neurons. Scale bar: 10 μm. (N) The percentage of clones containing more than two cells versus clones containing two cells or fewer is shown. (O-T) Overexpression of the fzy5032 transgene prolonged proliferation in fzy4 neuroblasts. (O-S) Larvae containing GFP-marked mosaic clones (outlined in light gray) derived from single neuroblasts (light gray arrow) of the indicated genotypes were aged for 72 hours after clone induction, and brains were stained for the indicated markers. Scale bar: 10 μm. (T) The percentage of clones containing more than two cells versus clones containing two cells or fewer is shown. (U) A summary model of the analyses of the fzy5032 allele. In this and all subsequent figures error bars indicate ± s.d.; *P<0.001 for the marked genotype in comparison with the control genotype in the same bar graph as determined by Student’s t-test; n.s. indicates that the difference is not statistically significant.
We hypothesize that the fzy5032 mutation uniquely perturbs the maintenance of larval brain neuroblasts because fzy5032 flies possess all adult-specific appendages but exhibit poor motor function probably because of a dramatically reduced neuropil (Fig. 1E-H). Consistently, wild-type or fzy5032 neuroepithelial cells in the larval optic lobe proliferated to generate a large number of progeny, but fzy4 neuroepithelial cells failed to proliferate (Fig. 1I-K,N). Importantly, overexpression of the wild-type Fzy transgenic protein or the Fzy5032 transgenic protein encoded by the fzy5032 mutation restored cell proliferation in fzy4 optic lobe neuroepithelial cells (Fig. 1L-N). Similarly, overexpression of the mouse wild-type Cdc20 transgenic protein or the Cdc20mut transgenic protein that carries an amino acid substitution homologous to Fzy5032 also restored proliferation in fzy4 optic lobe neuroepithelial cells (Fig. 1N; data not presented). Thus, the Fzy5032 or Cdc20mut mutant protein can functionally substitute endogenous Fzy in optic lobe neuroepithelial cells. By contrast, overexpression of the Fzy5032 or Cdc20mut transgenic protein can prolong proliferation in fzy4 brain neuroblasts, but cannot suppress the neuroblast loss phenotype (Fig. 1O-T). Thus, the Fzy5032 or Cdc20mut mutant protein cannot functionally substitute endogenous Fzy to maintain brain neuroblasts. Together, these data led us to conclude that the fzy5032 mutation uniquely abolishes the function of fzy in maintaining larval brain neuroblasts.
Because Cdc20/Fzy is also a component of the mitotic checkpoint complex that regulates proper chromosome segregation during mitosis (Jia et al., 2013), we wanted to know whether defects in chromosome segregation could contribute to the neuroblast loss phenotype in fzy5032 brains. Mutations in the roughdeal (rod) and mitotic 15 [mit(1)15] genes lead to nondisjunction and aneuploidy, whereas mutations in the abnormal spindle (asp) gene lead to prolonged metaphase arrest (Starr et al., 1998; Basto et al., 2000). Consistent with previously reported observations, neuroblasts in rod or mit(1)15 mutant brains exhibited chromosomal nondisjunction, but these brains did not display a premature neuroblast loss phenotype (supplementary material Fig. S1D-F). Furthermore, despite exhibiting increased metaphase figures, asp mutant brains also did not display a neuroblast loss phenotype (supplementary material Fig. S1F). These data indicate that defects in chromosome segregation are not sufficient to induce a neuroblast loss phenotype. Importantly, we never detected defects in chromosome segregation in mitotic neuroblasts in fzy5032 brains (data not presented). Thus, the neuroblast loss phenotype in fzy5032 brains is unlikely to be due to peculiar neuroblast-specific chromosome-segregation defects.
Neuroblasts in fzy5032 brains undergo necrosis
We hypothesized that the neuroblast loss phenotype in fzy5032 brains is due to premature cell death, for the following reasons. fzy5032 neuroblasts never expressed the differentiation markers including Pros and Elav (Fig. 1P,Q; supplementary material Fig. S2A,B), and fzy5032 brains contain many ‘ghost neuroblasts’ that lacked the expression of all known neuroblast markers and detectable nuclear DNA (supplementary material Fig. S2A,B; data not presented). We ruled out that premature apoptosis leads to neuroblast loss in fzy5032 brains because fzy5032 neuroblasts did not show the activation of caspases (data not presented). Consistently, removing the function of genes crucial for the activation of apoptosis also did not suppress neuroblast loss in fzy5032 brains (Fig. 2A). We also ruled out that premature autophagy contributes to neuroblast loss in fzy5032 brains because removing the function of the genes required for the activation of autophagy did not prevent neuroblast loss in fzy5032 brains (Fig. 2A). Thus, we conclude that the neuroblast loss phenotype in fzy5032 brains is most likely due to the activation of an alternative cell death modality.
Fig. 2.

Neuroblasts in fzy5032 brains undergo necrosis. (A) Removing the apoptotic or autophagic genes did not prevent neuroblast loss in fzy5032 brains. The average number of neuroblasts per brain lobe from larvae aged for 72 hours after hatching is shown. (B,C) Neuroblasts in fzy5032 brains exhibited morphological hallmarks of necrosis. Transmission electron micrograph of control and fzy5032 brains from larvae aged for 96 hours after hatching is shown. Higher magnifications of the areas marked by yellow boxes in B and C are shown below. Black arrowheads indicate mitochondria. Scale bars: 500 nm. (D-F) Neuroblasts in fzy5032 brains exhibited mitochondrial aggregates. Brains from control and fzy5032 larvae were stained for the mitochondria marker ATP5a. Scale bar: 10 μm. (F) The percentage of neuroblasts with evenly interspersed mitochondria (green) versus aggregated mitochondria (blue). (G-I) Neuroblasts in fzy5032 brains exhibited increased cytoplasmic ROS production. Brains from control and fzy5032 larvae carrying a PCNA::3XEmGFP transgene were stained with DHE. Scale bar: 20 μm. (I) The percentage of neuroblasts with high levels of ROS (green) versus low levels of ROS (blue). (J-L) Neuroblasts in fzy5032 brains exhibited increased cytoplasmic calcium ions. Brains from control and fzy5032 larvae were stained for GFP, which detects the expression of a genetically encoded calcium sensor in vivo (Wor >GCaMP3.0). Scale bar: 20 μm. (L) The percentage of neuroblasts with low cytoplasmic calcium (green) or high cytoplasmic calcium (blue) is shown. (M-O) Neuroblasts in fzy5032 brains exhibited accumulation of ubiquitin conjugates. Brains from control and fzy5032 larvae were stained with FK2, which recognizes ubiquitin-conjugated to proteins (Lee et al., 2008). Scale bar: 10 μm. (O) The percentage of neuroblasts with (blue) or without (green) ubiquitin conjugates is shown. Here and in all subsequent figures, yellow arrows indicate neuroblasts not expressing necrosis markers; white arrows indicate neuroblasts expressing necrosis markers. The yellow dotted line indicates the boundary between the optic lobe and the brain.
To investigate how fzy5032 neuroblasts die, we first characterized their morphological changes by transmission electron microscopy. Neuroblasts in control brains always contained single nuclei and cytoplasm with distinct organelles including mitochondria, ribosomes and endoplasmic reticulum (Fig. 2B). By contrast, neuroblasts in fzy5032 brains often did not have identifiable nuclei, and their cytoplasm appeared largely devoid of organelles and vacuoles (Fig. 2C). In these neuroblasts, mitochondria were swollen and ruptured, and remnants of the endoplasmic reticulum clumped with remaining mitochondria into electron dense aggregates. Consistently, we detected mitochondrial aggregates in 25% of the remaining fzy5032 neuroblasts, whereas mitochondria were evenly interspersed in the cytoplasm of control neuroblasts (Fig. 2D-F). Furthermore, we detected elevated levels of reactive oxygen species (ROS) and calcium ions in fzy5032 neuroblasts compared with control neuroblasts (Fig. 2G-L). In addition, almost 40% of fzy5032 neuroblasts contained ubiquitin-conjugated protein aggregates, which were virtually nonexistent in control neuroblasts (Fig. 2M-O). This combination of morphological features and molecular marker expression closely resembles those displayed by cells undergoing necrosis (Vandenabeele et al., 2010; Zhivotovsky and Orrenius, 2011; Galluzzi et al., 2012a). Thus, we conclude that an aberrant onset of necrosis leads to neuroblast loss in fzy5032 brains.
Because Fzy is essential for cell cycle progression, we tested whether neuroblast loss in fzy null mutant clones is primarily due to an aberrant onset of necrosis. Unexpectedly, we detected the expression of necrosis markers indicative of mitochondrial aggregates, ubiquitin aggregates and increased cytosolic calcium ions in neuroblasts in fzy4 clones far less frequently than in identically staged fzy5032 clones (supplementary material Fig. S2C-E). These data suggest that loss of cell cycle function is dominant in contributing to the neuroblast loss phenotype in fzy null mutant clones.
Fzy suppresses necrosis in neuroblasts through a novel APC/C-dependent mechanism
Fzy elicits the function of APC/C by regulating the specificity of substrate recognition and recruitment (Yu, 2007; Pesin and Orr-Weaver, 2008; Eguren et al., 2011; McLean et al., 2011; Pines, 2011). Thus, we tested whether Fzy suppresses necrosis in neuroblasts through an APC/C-dependent mechanism. Larval brains mutant for genes encoding either the scaffolding, catalytic or substrate binding subunit of APC/C all showed a drastic reduction in neuroblasts (supplementary material Fig. S3A-H). Furthermore, neuroblasts in these mutant larval brains also displayed mitochondrial aggregates, increased cytosolic ROS and increased cytoplasmic calcium ions (supplementary material Fig. S3I-Z). These data indicate that neuroblasts mutant for APC/C also undergo necrosis. Thus, Fzy likely suppresses necrosis in neuroblasts through an APC/C-dependent mechanism.
We next tested whether aberrant accumulation of known APC/C substrates required for cell cycle progression might contribute to the neuroblast loss phenotype in fzy5032 brains. Identical to wild-type neuroblasts, fzy5032 neuroblasts maintained single nuclei and the cell diameters were 10-12 μm (Fig. 3A,B). By contrast, neuroblasts mis-expressing a non-degradable form of Cyclin A, Cyclin B or the fly homolog of securin, Pimples (Pim), displayed hallmarks of mitotic catastrophe including multinucleation and drastically enlarged cell diameter (Fig. 3E,F; data not presented). The severity of mitotic catastrophe directly correlated with the degree of neuroblast loss in these brains; none of these mutant brains displayed a neuroblast loss phenotype as strong as fzy5032 brains (Fig. 3F). Furthermore, we never detected aberrant accumulation of Cyclin A and Cyclin B in fzy5032 neuroblasts (data not presented). Thus, we conclude that it is unlikely that aberrant accumulation of APC/C substrates required for cell cycle progression contributes to the necrosis of neuroblasts in fzy5032 brain.
Fig. 3.

Aberrant accumulation of APC/C substrates required for cell cycle progression does not lead to the necrosis of neuroblasts in fzy5032 brains. (A-F) Overexpression of non-degradable known APC/C substrates required for cell cycle progression leads to mitotic catastrophe in neuroblasts. Brains from control and fzy5032 larvae were stained for Lamin (Lam) and with Hoechst. Worniu-Gal4 (Wor) allows neuroblast-specific overexpression of the UAS transgene. cycAΔ170 encodes non-degradable Cyclin A. The hs-cycBS transgene allows overexpression of non-degradable Cyclin B induced by a heat-shock promoter. pimdba encodes non-degradable Pim. Neuroblasts are outlined in yellow. Scale bar: 10 μm. (F) The average number of neuroblasts per brain lobe is shown. (G) Aberrant accumulation of toxic proteins or blocking proteasome function is not sufficient to induce neuroblast loss. TDP43Δnls encodes a mutant form of TDP43. VCPR152H encodes a mutant form of VCP. pros26B and prosβ encode dominant-negative forms of 26S proteasome components. The average number of neuroblasts per brain lobe is shown.
We also examined whether aberrant accumulation of toxic proteins is sufficient to induce neuroblast loss. Expression of Transactive response DNA-binding protein 43, lacking the nuclear localization signal (TDP-43ΔNLS), or Valosin containing protein, which carries a substitution of histidine for arginine 152 (VCPR152H) induces ubiquitin inclusion and the degeneration of fly ommatidia (Ritson et al., 2010). Ectopically expressing either transgenic protein also induced the accumulation of ubiquitin conjugates in neuroblasts but was insufficient to induce neuroblast loss (supplementary material Fig. S4A-C; Fig. 3G). Separately, we tested whether blocking ubiquitin-mediated protein degradation induced neuroblast loss by co-expressing dominant-negative forms of two 26S proteosome components. Despite the presence of ubiquitin conjugates, blocking the 26S proteosome function was also insufficient to induce neuroblast loss (supplementary material Fig. S4D; Fig. 3G). Thus, it is unlikely that aberrant accumulation of toxic proteins leads to the necrosis of neuroblasts in fzy5032 brains.
Fzy suppresses necrosis in neuroblasts by multiple downstream mechanisms
We tested whether the mechanisms that regulate necrosis in vertebrates might contribute to neuroblast loss in fzy5032 brains. Aif is a mitochondrial oxidoreductase protein, which, upon activation, can trigger necrosis by recruiting an endonuclease complex (Delavallée et al., 2011). aif (also known as AIF - FlyBase) is dispensable for the maintenance and the proliferation of neuroblasts because an aif null mutant neuroblast clone maintained a single neuroblast and consisted of many neuronal progeny (Fig. 4A,B,E). Importantly, an aif, fzy double mutant clone consistently contained more progeny than a fzy single mutant clone, albeit the frequency of neuroblast-containing clones appeared similar between these two genotypes (Fig. 4C-E). Thus, removing aif function prolongs the survival of fzy5032 neuroblasts, and aif acts downstream of fzy to regulate necrosis in neuroblasts.
Fig. 4.

Fzy maintains neuroblast survival by antagonizing Aif and JNK signaling. (A-E) Removing aif function prolonged the survival of fzy5032 neuroblasts. (A-D) Larvae containing GFP-marked mosaic clones (outlined in light gray) derived from single neuroblasts (light gray arrow) of the indicated genotypes were aged for 72 hours after clone induction, and brains were stained for the indicated markers. Scale bar: 10 μm. (E) The average number of cells per clone. (F) Aberrant activation of JNK signaling leads to neuroblast loss. Tak1 encodes wild-type Tak1; Tak1DN encodes a dominant-negative form of Tak1. P35 encodes an inhibitor of activated caspases. hepCA encodes a constitutively active form of Hep. ask1 encodes wild-type Ask1. The average number of neuroblasts per brain lobe is shown. (G-I) Neuroblasts in fzy5032 brains show activated JNK signaling. Brains from larvae of the indicated genotype were stained for phosphorylated JNK (p-JNK). Scale bar: 10 μm. (I) The percentage of neuroblasts with (blue) or without (green) p-JNK expression. (J-L) Removing the function of JNK signaling prolonged the survival of fzy5032 neuroblasts. (J,K) Larvae containing GFP-marked mosaic clones (outlined in light gray) derived from single neuroblasts (light gray arrow) of the indicated genotypes were aged for 72 hours after clone induction, and brains were stained for the indicated markers. Scale bar: 10 μm. (L) The average number of cells per clone.
The binding of TNF (Eiger in flies) to TNFR (Wengen in flies) triggers necrosis in vertebrates and caspase-independent cell death in flies through the activation of JNK signaling (Laster et al., 1988; Vercammen et al., 1998; Igaki et al., 2011; Kanda et al., 2011; Cabal-Hierro and Lazo, 2012; Galluzzi et al., 2012b). We tested whether overexpression of eiger in glial cells, which surround individual neuroblast lineages (Pereanu et al., 2005; Weng et al., 2010), can induce the death of larval brain neuroblasts, which express wengen (Wang et al., 2006; Berger et al., 2012). Surprisingly, overexpression of eiger failed to induce neuroblast loss in larval brains (data not presented). Conversely, removing the function of eiger or wengen also failed to suppress neuroblast loss in fzy5032 brains (data not presented). Thus, Fzy probably suppresses the necrosis of neuroblasts by antagonizing JNK signaling, which acts downstream of TNFR, or a TNF-independent mechanism.
We first tested whether aberrant activation of JNK signaling is sufficient to induce neuroblast loss. Overexpression of a JNK kinase kinase encoded by the Transforming growth factor beta-activated kinase (Tak1) gene but not the kinase-dead Tak1 encoded by Tak1KD, efficiently induced neuroblast loss (Fig. 4F). Similarly, overexpression of JNK kinase encoded by the hemipterous (hep) gene also efficiently induced neuroblast loss (Fig. 4F). However, expression of P35 failed to prevent Tak1-induced neuroblast loss (Fig. 4F). Thus, aberrant activation of JNK signaling is sufficient to induce caspase-independent death of larval brain neuroblasts. We next tested whether JNK signaling contributes to the necrosis of neuroblasts in fzy5032 brains. Indeed, neuroblasts in fzy5032 brains but not in control brains expressed puc-lacZ and phosphorylated JNK, two well established functional readouts of activated JNK signaling (Fig. 4G-I; data not presented). The basket (bsk) gene, which encodes the fly homolog of JNK, is dispensable for the maintenance and the proliferation of neuroblasts because a bsk mutant neuroblast clone maintained a single neuroblast and consisted of many neuronal progeny (Fig. 4A,J,L). Importantly, a bsk, fzy double mutant neuroblast clone reproducibly contained more progeny than a fzy mutant clone, albeit the frequency of neuroblast-containing clones appeared similar between these two genotypes (Fig. 4C,J-L). Thus, removing bsk function prolongs the survival of fzy5032 neuroblasts. We conclude that JNK signaling acts downstream of fzy to regulate necrosis in neuroblasts.
Catastrophic cellular stress triggers necrosis in neuroblasts by attenuating the Fzy-dependent survival mechanism
Cells experiencing catastrophic cellular stress frequently express the same necrosis markers as fzy5032 neuroblasts. Thus, we hypothesized that attenuation of the Fzy-dependent survival mechanism functions to eliminate larval brain neuroblasts experiencing irreparable cellular damage because apoptosis is actively repressed in these cells. We tested this hypothesis by inducing three different types of catastrophic cellular stress in larval brain neuroblasts. The chaperonin containing Tcp-1 (CCT) complex is responsible for the folding of major structural proteins including Tubulin (Hughes et al., 2008). Overexpression of the UAS-RNAi transgene efficiently reduced the function of CCT2 or CCT4, which encodes a distinct subunit of CCT, in neuroblasts, leading to the formation of a much thinner spindle apparatus during mitosis (supplementary material Fig. S5A-D). Knocking-down the function of CCT2 or CCT4 resulted in a severe neuroblast loss phenotype (Fig. 5A). Many of the remaining neuroblasts displayed the necrosis markers and lost Fzy expression despite maintaining the expression and the nuclear localization pattern of Dpn (Fig. 5B-H; supplementary material Fig. S5J-L,P-R,V-X,AA-CC). Thus, attenuation of Fzy expression correlates with the necrosis of neuroblasts induced by defects in protein folding. The telomere capping complex encoded by genes including caravaggio (cav) and suppressor of variegated 205 [Su(var)205] is essential for chromosome stability, as inappropriately capped telomeres are recognized as double-stranded DNA breaks, triggering DNA damage responses (Raffa et al., 2011). Neuroblasts in cav or Su(var)205 mutant brains but not wild-type brains showed robust expression of phosphorylated histone H2A variant (pH2Av), a DNA-damage response marker (supplementary material Fig. S5E-G,I). Importantly, cav or Su(var)205 mutant brains exhibited severe neuroblast loss (Fig. 5A). Many of the remaining neuroblasts in these mutant brains displayed the necrosis markers and lost Fzy expression while maintaining the expression and the nuclear localization pattern of Dpn (Fig. 5B-E,I,J; supplementary material Fig. S5M,N,S,T,Y,DD,EE). Hence, attenuation of Fzy expression also correlates with the necrosis of neuroblasts induced by uncapping of the telomeres. The flap endonuclease-1 (fen1) gene encodes a metallonuclease, which plays a pivotal role in the penultimate steps of Okazaki fragment maturation during DNA replication (Zheng et al., 2011). Neuroblasts in fen1 mutant brains exhibited robust pH2Av expression (supplementary material Fig. S5H-I). fen1 mutant brains displayed severe neuroblast loss (Fig. 5A). Many of the remaining neuroblasts in fen1 mutant brains also showed the necrosis markers and lost Fzy expression while maintaining the expression and the nuclear localization pattern of Dpn (Fig. 5B-E,K; supplementary material Fig. S5O,U,Z,FF). Therefore, attenuation of Fzy expression correlates with the necrosis of neuroblasts induced by defects in DNA replication. Taken together, the necrosis of neuroblasts experiencing catastrophic cellular stress consistently correlates with the attenuation of Fzy expression.
Fig. 5.

Catastrophic cellular stress induces necrosis in neuroblasts by attenuating the Fzy-dependent survival mechanism. (A) Catastrophic cellular stress led to a drastic reduction in neuroblasts. The average number of neuroblasts per brain lobe is shown. (B-D) Neuroblasts experiencing catastrophic cellular stress exhibited necrosis markers. Brains from larvae of the indicated genotype were stained with (B) ATP5a for mitochondrial morphology, (C) GFP for calcium content and (D) FK2 for ubiquitylated aggregates. The percentage of neuroblasts with (blue) or without (green) the expression of the indicated marker is shown. The percentage of neuroblasts with (blue) or without (green) the expression of the indicated marker is shown. (E-K) Neuroblasts experiencing catastrophic cellular stress lost Fzy expression. Brains from larvae of the indicated genotype were stained for Fzy. Scale bar: 10 μm. The percentage of neuroblasts with (green) or without (blue) Fzy expression is shown in E. (L-N) Overexpression of fzy suppressed the necrosis of neuroblasts (outlined in yellow in L and M) experiencing catastrophic cellular stress. Scale bar: 10 μm. The average number of neuroblasts per brain lobe is shown in N. (O) A summary cartoon showing how catastrophic cellular stress induces the necrosis of neuroblasts by attenuating Fzy expression.
We directly tested whether attenuating Fzy expression indeed contributes to the necrosis of neuroblasts experiencing catastrophic cellular stress. Consistent with this hypothesis, overexpression of fzy prevented the necrosis of CCT2, Su(var)205 or fen1 mutant neuroblasts (Fig. 5L-N). However, these surviving neuroblasts continued to proliferate and displayed phenotypes indicative of mitotic catastrophe including multinucleation and enlargement of cell diameter (Fig. 5M). These data indicate that attenuation of Fzy expression is indeed the major cause of the necrosis of neuroblasts experiencing catastrophic cellular stress (Fig. 5O).
Upregulation of p53 induces necrosis in neuroblasts by attenuating the Fzy-dependent survival mechanism
Catastrophic cellular stress often leads to cell death through the activation of the p53-dependent checkpoint mechanism, prompting us to test the role of p53 in the necrosis of neuroblasts experiencing catastrophic cellular stress. Consistently, we detected the expression of a p53 reporter transgene (p53RE-gfp) in many neuroblasts in Su(var)205 or fen1 mutant brains (Fig. 6A-D). Most importantly, removing p53 function suppressed the neuroblast loss phenotype in fen1 mutant brains to a similar extent as over-expression of fzy (Fig. 5N; Fig. 6E-G). Together, these data strongly suggest that p53 is required for the necrosis of neuroblasts experiencing catastrophic cellular stress.
Fig. 6.

Overexpression of p53 induces the necrosis of neuroblasts by attenuating the Fzy-dependent survival mechanism. (A-D) Neuroblasts experiencing catastrophic cellular stress expressed the p53 reporter transgene. Brains from larvae of the indicated genotype were stained for GFP, which indicates the expression of the p53-RE-gfp reporter transgene. Scale bar: 10 μm. (D) The percentage of neuroblasts with (green) or without (blue) GFP expression is shown. (E-G) Removing p53 function suppressed the necrosis of neuroblasts in fen1 mutant brains. Scale bar: 10 μm (E,F). (G) The average number of neuroblasts per brain lobe. (H) Overexpression of p53 led to neuroblast loss. The average number of neuroblasts per brain lobe is shown. (I-K) Neuroblasts overexpressing p53 were positive for necrosis markers. Brains from larvae of the indicated genotype were processed for staining with (I) ATP5a for mitochondrial morphology, (J) GFP for calcium content and (K) FK2 for ubiquitylated aggregates. The percentage of neuroblasts with (blue) or without (green) the expression of the indicated marker is shown. (L-N) Neuroblasts overexpressing p53 lost Fzy expression. Scale bar: 10 μm (L,M). The percentage of neuroblasts with (green) or without (blue) Fzy expression is shown in N. (O) Overexpression of fzy suppressed the necrosis of neuroblasts induced by overexpression of p53. The average number of neuroblasts per brain lobe is shown.
We next examined whether overexpression of p53 can induce necrosis in neuroblasts. Overexpressing p53 efficiently triggered neuroblast loss (Fig. 6H). Many of the remaining neuroblasts in larval brains overexpressing p53 expressed necrosis markers and lost Fzy expression while maintaining the expression and the nuclear localization pattern of Dpn (Fig. 6I-N; supplementary material Fig. S6A-H). To exclude the possibility that overexpression of p53 induces necrosis in neuroblasts by triggering cell cycle arrest, we tested whether knocking down the function of Cdk1 encoded by the cdc2 gene or overexpressing the wee1 gene can induce premature neuroblast loss. Reducing cdc2 function or increasing wee1 activity efficiently triggered G2 arrest in brain neuroblasts as indicated by the enlargement of neuroblasts and the reduction in mitosis (supplementary material Fig. S6I-L). However, neither approach was sufficient to induce neuroblast loss or loss of Fzy expression (supplementary material Fig. S6M-Q). These data strongly suggest that increased p53 transcriptional activity probably induces necrosis in neuroblasts by attenuating Fzy expression rather than triggering G2 cell cycle arrest.
We directly tested whether attenuating fzy expression contributes to the necrosis of neuroblasts induced by overexpression of p53. Consistent with this hypothesis, larval brains co-expressing fzy and p53 possessed significantly more neuroblasts than brains overexpressing p53 alone (Fig. 6O). Thus, fzy acts downstream of p53 to maintain the survival of neuroblasts. Taken together, we conclude that overexpression of p53 induces the necrosis of neuroblasts by attenuating the Fzy-dependent survival mechanism.
DISCUSSION
The inability to eradicate cancer stem cells undermines the effectiveness of current cancer therapies. The persistence of cancer stem cells directly contributes to cancer recurrence, threatening long-term patient survival. Cancer stem cells are able to survive catastrophic cellular damage induced by conventional chemotherapy and radiotherapy because these cells acquire mutations that inactivate checkpoint-induced apoptosis or undergo cell division infrequently. Thus, strategies that couple the activation of alternative cell death modality to the control of cell cycle might increase the likelihood of targeting cancer stem cells. One form of cell death that is tightly coupled to the cell cycle is mitotic catastrophe. However, the mechanisms by which mitotic catastrophe kills cells remain poorly defined. The other strategy is controlled activation of necrosis, which frequently serves as the main alternative cell death modality in cells where the activation of apoptosis is blocked. Despite recent advances in the regulation of necrosis, in-depth knowledge that will enable an efficient manipulation of this type of cell death remains limited in scope. Here, we report a novel Fzy-dependent cell-survival mechanism that likely functions as a ‘cell health checkpoint’ and appears to be poised to eliminate apoptosis-deficient neuroblasts experiencing catastrophic cellular stress through necrosis. Our data strongly suggest that Fzy interfaces with p53 in the regulation of necrosis. The two amino acid residues in the Fzy protein that function specifically to suppress necrosis in apoptosis-deficient neuroblasts might provide novel targeting sites to activate necrosis directly or indirectly by modulating the functional output of p53 (Fig. 7). Future investigation of the Fzy-dependent survival mechanism will provide crucial insight that could lead to the discovery of novel cancer therapies.
Fig. 7.
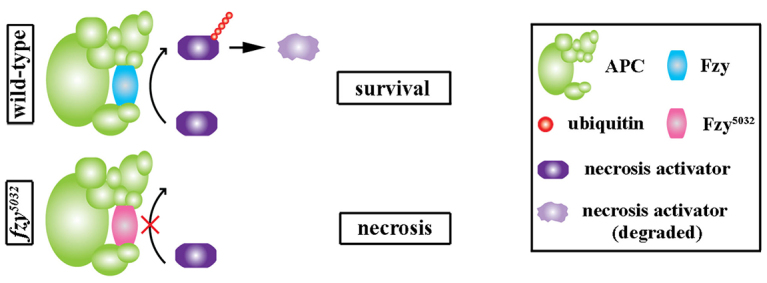
Attenuation of the Fzy-dependent survival mechanism links catastrophic cellular stress to the activation of necrosis in neuroblasts. (A) Fzy maintains neuroblast survival by targeting an unidentified necrosis activator for degradation through an APC/C-dependent mechanism. Wild-type Fzy (blue) recruits the presumed necrosis activator for degradation (purple). The Fzy5032 mutant protein is unable to recruit the necrosis activator to APC/C, leading to the necrosis of neuroblasts.
Activation of necrosis by a p53-dependent mechanism
Neuroblasts in fly larval brains provide an ideal model system for gaining mechanistic insight into how p53 triggers necrosis. The results from our study led us to conclude that overexpression of p53 induces necrosis in larval brain neuroblasts by attenuating the Fzy-dependent survival mechanism. Specifically, neuroblasts overexpressing p53 expressed the necrosis markers and lost Fzy, and co-expression of fzy suppressed the necrosis of neuroblasts induced by overexpression of p53 (Fig. 6). Thus, p53 might trigger necrosis in larval brain neuroblasts by destabilizing Fzy protein or repressing fzy gene transcription. We favor the second possibility for the following two reasons. First, p53 can directly repress the transcription of CDC20 in vertebrates (Kidokoro et al., 2008; Banerjee et al., 2009). Second, we identified two putative p53 binding sites in the regulatory element 5′ of the fzy transcription start site that is sufficient to drive a reporter transgene to express in neuroblasts (C.K. and C.-Y.L. unpublished observation). Additional future experiments will be necessary to confirm that p53 indeed transcriptionally represses the expression of fzy.
A previous study showed that overexpression of p53 can suppress the supernumerary neuroblast phenotype induced by loss of numb function in the absence of caspase activation (Ouyang et al., 2011). This study also showed that co-expression of Cyclin E can abolish the suppressive effect of the overexpression of p53 on the supernumerary neuroblast phenotype in numb mutant clones. Thus, the authors concluded that the inhibitory effect of p53 on ectopic neuroblast formation is largely mediated through the regulation of Cyclin E. However, co-expression of Cyclin E can only weakly suppress the necrosis of neuroblasts induced by overexpression of p53 in wild-type brains, at best (supplementary material Fig. S7A,B,E). Although we cannot exclude the possibility that the difference in the levels of transgene expression might contribute to this discrepancy, it is more likely that overexpression of Cyclin E can only suppress the effects of p53 on neuroblast formation in a numb mutant genetic background. Most importantly, overexpression of Cyclin E had absolutely no effects on the necrosis of neuroblasts in fzy5032 brains (supplementary material Fig. S7C-E). Thus, we conclude that defects in cell cycle progression do not contribute to the necrosis of neuroblasts in fzy5032 brains. This interpretation is completely consistent with other data from various experiments involving the manipulation of the cell cycle machinery described in this study.
Regulation of necrosis in Drosophila
Several studies have reported caspase-independent cell death in the developing fly retina and ovary, but whether the death of these cells indeed occurs through necrosis remains unclear (Bakhrat et al., 2010; Kanda et al., 2011; Yang et al., 2013). To our knowledge, the data presented in the current study provide the most direct evidence that necrosis indeed occurs in Drosophila, and that fly larval brain neuroblasts could provide a novel system for investigating the activation of necrosis in the absence of RIP kinases. We showed that two signaling mechanisms that have well-established roles in the activation of necrosis in vertebrates are essential for the necrosis of fzy5032 neuroblasts. Aif normally resides in mitochondria, and following its release, soluble Aif can translocate to the nucleus where it may activate DNA fragmentation and cell death (Delavallée et al., 2011). Although removal of aif function delayed necrosis in fzy5032 neuroblasts, overexpression of soluble Aif failed to induce neuroblast loss (Fig. 4A-E; data not presented). These data strongly suggest that additional downstream executioner proteins of necrosis must exist. Our data also indicate that aberrant activation of JNK signaling contributes to the necrosis of fzy5032 neuroblasts (Fig. 4F-L). We tested whether JNK signaling functions in parallel to Aif to induce the necrosis of neuroblasts. Unexpectedly, simultaneous removal of aif and bsk function failed to significantly prolong the survival of fzy5032 neuroblasts (data not presented). These results strongly suggest that JNK signaling and Aif function in the same pathway to elicit the necrosis of neuroblasts. Additional experiments in the future will be required to confirm this hypothesis. These results also indicate that additional mechanisms must exist to trigger the necrosis of neuroblasts in the absence of fzy function. Elucidating these yet unknown mechanisms is crucial to fully understand the regulation of necrosis in flies and might provide novel insight into the regulation of necrosis in vertebrates.
The developing fly retina has provided a sensitized genetic model system for investigating the complex gene network underlying the regulation of physiological cell death. For example, an earlier study showed that Tak1 or another JNKKK encoded by the apoptotic signal-regulating kinase 1 gene (ask1) regulates caspase-dependent cell death in the retina (Takatsu et al., 2000; Kuranaga et al., 2002). A recent study, however, showed that TNF signaling induces caspase-independent cell death in the retina by activating JNK signaling (Kanda et al., 2011). The results from these separate studies are consistent with the theory that JNK signaling functions at a nodal point and can elicit both apoptosis and non-apoptotic cell death in vertebrates (Galluzzi et al., 2012b). Intriguingly, although overexpression of Tak1 efficiently induces caspase-independent death of neuroblasts, overexpression of Ask1 did not have any effects on neuroblast survival (Fig. 4F). These data strongly suggest that only a specific JNK signaling cascade is sufficient to induce caspase-independent death of larval brain neuroblasts. We speculate that Fzy might suppress the necrosis of neuroblasts by antagonizing JNK signaling through the regulation of Tak1 or Hep. The mechanistic link between loss of Fzy expression and the activation of JNK signaling is key to understanding how catastrophic cellular stress leads to the activation of necrosis in neuroblasts.
fzy5032 neuroblasts as well as neuroblasts experiencing catastrophic cellular stress or overexpressing p53, showed an aberrant accumulation of ROS, cytosolic calcium ions and ubiquitin conjugates (Figs 2, 5, 6). Testing whether the ablation of ROS can suppress loss of fzy5032 neuroblasts is a crucial future experiment. Calcium-dependent signaling plays a vital role in regulating necrosis and can activate proteases such as calpain in vertebrates (Zhivotovsky and Orrenius, 2011). A recent study showed that an increase in calcium ions can trigger necrosis in the developing fly retina (Yang et al., 2013). As such, testing the contribution of increased calcium ions to the necrosis of fzy5032 neuroblasts will also be crucial in the immediate future. Finally, aberrant accumulation of ubiquitin conjugates has been observed in many neurodegenerative diseases (Irwin et al., 2013). Uniquely, fly neuroblasts exhibit a high tolerance to the accumulation of ubiquitin conjugates (Fig. 3G; supplementary material Fig. S4). Thus, understanding the role of ubiquitin conjugates in the Fzy-regulated cell survival mechanism will probably provide insight into novel therapies for cancer as well as neurodegeneration.
MATERIALS AND METHODS
Drosophila culture and genetics
All Drosophila stocks were maintained on yeast-glucose fly medium. Embryos were collected on apple juice agar supplemented with yeast paste. Larvae were transferred to cornmeal agar shortly after hatching. In all experiments, heterozygous larvae with balancers were used as controls, because none of these animals had any detectable phenotypes at any stage.
The fzy5032 mutant allele was isolated from an unbiased EMS mutagenesis screen. The UAS-fzy, UAS-fzy5032, UAS-cdc20 and UAS-cdc20mut flies were generated using Φc31-mediate transgenesis (Bischof et al., 2007).
The following mutations were used in this study: fzy1, fzy4 and fzyAC-10 (Nüsslein-Volhard and Wieschaus, 1980; Ashburner et al., 1990), fzytDa (Dawson et al., 1995), dreddB118 (Leulier et al., 2000), atg1 mutant alleles (unc-513 and unc-5125) (Toda et al., 2008), apc2 mutant alleles (mr3 and mr4) (Reed and Orr-Weaver, 1997), apc11 (lmg138) (Nagy et al., 2012), aif (aifKO) (Joza et al., 2008), bsk1 (Nüsslein-Volhard and Wieschaus, 1980), dronc (Nc51) (Chew et al., 2004), ark82 (Akdemir et al., 2006), apc5 (idaB4 and idaD14) (Vaskova et al., 2000), pink1B8 (Park et al., 2006), cav1 (Cenci et al., 2003), cav2248 (Li et al., 2011), Su(var)2054 and Su(var)2055 (Eissenberg et al., 1992), mir-8Δ3 (Karres et al., 2007), p535A-1-4 and p5311-1B-1 (Rong et al., 2002). The following transgenic fly lines were used in this study puc-lacZ (pucE69) (Ring and Martinez Arias, 1993), p53-RE-gfp (Brodsky et al., 2000), UAS-GCaMP3.0 (Tian et al., 2009), UAS-dTak1 and UAS-dTak1DN (Takatsu et al., 2000), UAS-hep (Boutros et al., 1998), UAS-hepCA (Adachi-Yamada et al., 1999), UAS-ask1 (Kuranaga et al., 2002), UAS-p35 (Hay et al., 1994), GUS-p53 and GUS-p53DN (Brodsky et al., 2000), UAS-wee (Price et al., 2002), UAS-tdp43Δnls and UAS-vcaR152H (Ritson et al., 2010), UAS-cycAΔ170, UAS-pim, UAS-pimdba-myc and UAS-pimkena-myc (Leismann et al., 2000; Leismann and Lehner, 2003), hs-cycBs (Su and O’Farrell, 1997), UAS-pros26β61; UAS-prosβ21 (Belote and Fortier, 2002). The following stocks were obtained from the Bloomington Drosophila Stock Center, apc10e01070, apc6 (cdc16MB09129), fen1EY12786, rodX-1, asp1 and mit(1)15, hs-flp, Elav-Gal4, Tub-Gal4, UAS-cyclin E, UAS-mCD8-GFP, FRT40A, Tub-Gal80, Df(3L)BSC283, Df(2R)Exel6065, Df(2R)Exel7157, Df(2R)BSC355, Df(3L)ED4341, Df(3R)Exel9012, Df(2L)Exel7038 and UAS-cdc2RNAi (28368). The following stocks were obtained from the Vienna Drosophila RNAi Center UAS-ND42RNAi (14444), UAS-ND75RNAi (52047), UAS-CCT2RNAi (41190) and UAS-CCT4RNAi (22154).
Immunofluorescence staining and imaging
Larval brains were processed for immunofluorescence staining following a standard protocol (Weng et al., 2012). The following primary antibodies were used: rat anti-Dpn (1:1000), rabbit anti-Mira (1:1000), mouse anti-Pros (1:100), chicken anti-GFP (1:2000, GFP-1020; Aves Labs), mouse anti-Fzy (1:20) and rabbit anti-Fzy (1:500, kindly provided by Dr J. Raff, University of Oxford, UK), mouse anti-Lamin (1:400, ADL67.10; DSHB), mouse anti-phospho-histone H3 (1:2000, 3H10; Upstate), mouse anti-CycB (1:50, F2F4; DSHB), mouse anti-Elav (1:100, 9F8A9; DSHB), mouse anti-ATP5A (1:500, MS507; Abcam), rabbit anti-phospho-SAPK/JNK (1:100, 9251; Cell Signaling), anti-conjugated ubiquitin (1:100, FK2; Enzo), anti-phospho-H2Av (1:100; Rockland). Species-specific fluorophere-conjugated secondary antibodies (Jackson ImmunoResearch, 703-545-155, 112-605-167; Life Technologies, A-11034, A-11035, A-11074, A31553, A-31556) were used at 1:500. We used Alexa-Fluor-488-phalloidin (1:100, A12379; Life Technologies) and Rhodamine-phalloidin (1:100, R415; Life Technologies). The following small molecules were used: Hoechst 33342 (2 μg/ml, C10340 COMPONENT G; Life Technologies). For dihydroethidium [DHE, a small molecule that indicates the presence of superoxide free radicals (Owusu-Ansah et al., 2008)] staining, brains were dissected in PBS, incubated in DHE (1:1000, 30 μM; a gift from Craig Byersdorfer, University of Michigan, USA) for 5 minutes at room temperature with rocking, washed in PBS for 20 minutes with rocking, and immediately mounted for imaging with PBS on glass slides. Samples were examined using a Leica TCS SP5 scanning confocal microscope system operated using LAS AF v.2.6.0.7266.
Identification of neuroblasts
Neuroblasts were identified based on the expression of Deadpan (Dpn).
Clonal analyses
All clones were induced by heat-shocking larvae at 37°C for 90 minutes at 24 hours after hatching. Following clone induction, larvae were aged at 25°C for 72 hours, and larval brains were processed for immunofluorescence staining. The mosaic clones were marked by GFP expression.
Quantitative PCR
RNA was isolated from four to six larvae per genotype aged for 96 hours after hatching using Trizol reagent (Gibco) following manufacturer’s suggested protocol. Primers used for qPCR: fzy1 forward 5′-TCCACAAAGAGTGGCTCACGC-3′, fzy1 reverse 5′-CAAATAGACGCAACTGCCCAAGG-3′; fzy2 forward 5′-GCTGGAGGCGTCTCTAAATGGA-3′, fzy2 reverse 5′-GGGCGTGGTGTTGGACTT-3′; rp49 forward 5′-ATCGGTTACGGATCGAACAA-3′, rp49 reverse 5′-GACAATCTCCTTGCGCTTCT-3′.
Histology
Larvae were allowed to pupate and develop at 25°C. Anterior pupal cases were removed from pharate adults to expose heads and whole pupae were fixed in 1% glutaraldehyde, 4% formaldehyde, acid and alcohol, overnight at 4°C. The samples were washed in 80% ethanol, dehydrated in ascending concentrations of ethanol, washed in xylenes, and embedded in paraffin. Sections (5 μm) were prepared and stained with Mayer’s Hematoxylin and Eosin. The sections were examined using a Leica DM5000 B upright microscope equipped with a QImaging Retiga 2000R camera. Images were acquired using QCapture Pro v.5.1.1.14 software (Media Cybernetics, Inc.).
Transmission electron microscopy
Larval brains were fixed in 2.5% glutaraldehyde in 0.1 M Sorensen’s buffer, pH 7.4, overnight at 4°C. After several rinses in buffer, they were post-fixed in 1% osmium tetroxide in the same buffer. They were then rinsed in double distilled water to remove phosphate and then en bloc stained with aqueous 3% uranyl acetate for 1 hour. They were dehydrated in ascending concentrations of ethanol, rinsed two times in propylene oxide, and embedded in epoxy resin. Ultra-thin sections (70 nm) were prepared and stained with uranyl acetate and lead citrate. The sections were examined using a Philips CM100 electron microscope at 60 kV. Images were recorded digitally using a Hamamatsu ORCA-HR digital camera system operated using AMT software (Advanced Microscopy Techniques Corp.). Images were processed using Adobe Photoshop CS3.
Statistical analysis
Data are presented as means ± s.d.; single asterisks in all figures indicate a P-value <0.001 for the marked genotype in comparison to the control genotype in the same graph, as determined by the Student’s t-test.
Supplementary Material
Acknowledgments
We thank Dr C. Doe in whose lab the fzy5032 mutant allele was isolated when C.-Y.L. was a postdoctoral fellow. We thank Drs J. Abrams, E. Baehrecke, U. Banerjee, I. Dawson, P. Deák, M. Dominguez, R. Kellum, C. Lehner, B. Lemaitre, M. Miura, T. Orr-Weaver, J. Penninger, C. Pfleger, J. Raff, R. Read, T. Schupbach, O. Shafer, P. Taylor, P. Todd, T. Tomoda and T. Xu for fly stocks and antibodies. We also thank the Bloomington Stock Center and Vienna Drosophila RNAi Center for fly stocks and Developmental Studies Hybridoma Bank (University of Iowa) for antibodies. We thank BestGene for generating transgenic fly lines. Molecular graphics and analyses were performed with the UCSF Chimera package. We thank members of the Lee lab for critical reading of the manuscript. We thank Dr J. Smith for advice with the structure of Cdc20. We thank Dr S. Meshinchi (Microscopy and Image Analysis Laboratory, University of Michigan) for assistance in transmission electron microscopy sample preparation and imaging.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
C.K., L.B. and C.-Y.L. designed the experiments. C.K., K.L.G., C.R.S., J.D. and C.E.G. performed the experiments. C.K., C.R.S., J.D. and C.-Y.L. analysed the data. C.K. and C.-Y.L. wrote the manuscript.
Funding
C.R.S wishes to thank Minas Gerais State Research Council for financial support. C.K. was initially supported by the National Institutes of Science (NIH) Medical Scientist Training Program (MSTP) Training Grant [T32-GM07863]. C.-Y.L was supported by University of Michigan start-up, the Burroughs Wellcome Fund Career Award in the Biomedical Sciences [1006160.01], a Sontag Foundation Distinguished Scientist Award and an NIH grant [R01-GM092818]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.104786/-/DC1
References
- Adachi-Yamada T., Fujimura-Kamada K., Nishida Y., Matsumoto K. (1999). Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400, 166–169 [DOI] [PubMed] [Google Scholar]
- Akdemir F., Farkas R., Chen P., Juhasz G., Medved’ová L., Sass M., Wang L., Wang X., Chittaranjan S., Gorski S. M., et al. (2006). Autophagy occurs upstream or parallel to the apoptosome during histolytic cell death. Development 133, 1457–1465 [DOI] [PubMed] [Google Scholar]
- Ashburner M., Thompson P., Roote J., Lasko P. F., Grau Y., el Messal M., Roth S., Simpson P. (1990). The genetics of a small autosomal region of Drosophila melanogaster containing the structural gene for alcohol dehydrogenase. VII. Characterization of the region around the snail and cactus loci. Genetics 126, 679–694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakhrat A., Pritchett T., Peretz G., McCall K., Abdu U. (2010). Drosophila Chk2 and p53 proteins induce stage-specific cell death independently during oogenesis. Apoptosis 15, 1425–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee T., Nath S., Roychoudhury S. (2009). DNA damage induced p53 downregulates Cdc20 by direct binding to its promoter causing chromatin remodeling. Nucleic Acids Res. 37, 2688–2698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basto R., Gomes R., Karess R. E. (2000). Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nat. Cell Biol. 2, 939–943 [DOI] [PubMed] [Google Scholar]
- Bello B., Holbro N., Reichert H. (2007). Polycomb group genes are required for neural stem cell survival in postembryonic neurogenesis of Drosophila. Development 134, 1091–1099 [DOI] [PubMed] [Google Scholar]
- Belote J. M., Fortier E. (2002). Targeted expression of dominant negative proteasome mutants in Drosophila melanogaster. Genesis 34, 80–82 [DOI] [PubMed] [Google Scholar]
- Berger C., Harzer H., Burkard T. R., Steinmann J., van der Horst S., Laurenson A. S., Novatchkova M., Reichert H., Knoblich J. A. (2012). FACS purification and transcriptome analysis of drosophila neural stem cells reveals a role for Klumpfuss in self-renewal. Cell Rep 2, 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J., Maeda R. K., Hediger M., Karch F., Basler K. (2007). An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proc. Natl. Acad. Sci. USA 104, 3312–3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutros M., Paricio N., Strutt D. I., Mlodzik M. (1998). Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell 94, 109–118 [DOI] [PubMed] [Google Scholar]
- Brodsky M. H., Nordstrom W., Tsang G., Kwan E., Rubin G. M., Abrams J. M. (2000). Drosophila p53 binds a damage response element at the reaper locus. Cell 101, 103–113 [DOI] [PubMed] [Google Scholar]
- Cabal-Hierro L., Lazo P. S. (2012). Signal transduction by tumor necrosis factor receptors. Cell. Signal. 24, 1297–1305 [DOI] [PubMed] [Google Scholar]
- Cenci G., Siriaco G., Raffa G. D., Kellum R., Gatti M. (2003). The Drosophila HOAP protein is required for telomere capping. Nat. Cell Biol. 5, 82–84 [DOI] [PubMed] [Google Scholar]
- Chao W. C., Kulkarni K., Zhang Z., Kong E. H., Barford D. (2012). Structure of the mitotic checkpoint complex. Nature 484, 208–213 [DOI] [PubMed] [Google Scholar]
- Chew S. K., Akdemir F., Chen P., Lu W. J., Mills K., Daish T., Kumar S., Rodriguez A., Abrams J. M. (2004). The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev. Cell 7, 897–907 [DOI] [PubMed] [Google Scholar]
- Cho Y. S., Challa S., Moquin D., Genga R., Ray T. D., Guildford M., Chan F. K. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S. (1995). Control of proliferation activation in quiescent neuroblasts of the Drosophila central nervous system. Development 121, 1173–1182 [DOI] [PubMed] [Google Scholar]
- Dawson I. A., Roth S., Akam M., Artavanis-Tsakonas S. (1993). Mutations of the fizzy locus cause metaphase arrest in Drosophila melanogaster embryos. Development 117, 359–376 [DOI] [PubMed] [Google Scholar]
- Dawson I. A., Roth S., Artavanis-Tsakonas S. (1995). The Drosophila cell cycle gene fizzy is required for normal degradation of cyclins A and B during mitosis and has homology to the CDC20 gene of Saccharomyces cerevisiae. J. Cell Biol. 129, 725–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckbar D., Jeggo P. A., Löbrich M. (2011). Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 46, 271–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delavallée L., Cabon L., Galán-Malo P., Lorenzo H. K., Susin S. A. (2011). AIF-mediated caspase-independent necroptosis: a new chance for targeted therapeutics. IUBMB Life 63, 221–232 [DOI] [PubMed] [Google Scholar]
- Eguren M., Manchado E., Malumbres M. (2011). Non-mitotic functions of the Anaphase-Promoting Complex. Semin. Cell Dev. Biol. 22, 572–578 [DOI] [PubMed] [Google Scholar]
- Eissenberg J. C., Morris G. D., Reuter G., Hartnett T. (1992). The heterochromatin-associated protein HP-1 is an essential protein in Drosophila with dosage-dependent effects on position-effect variegation. Genetics 131, 345–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs Y., Steller H. (2011). Programmed cell death in animal development and disease. Cell 147, 742–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Vitale I., Vacchelli E., Kroemer G. (2011) Cell death signaling and anticancer therapy. Front. Oncol. 1, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L., Kepp O., Kroemer G. (2012a). Mitochondria: master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 13, 780–788 [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Vitale I., Abrams J. M., Alnemri E. S., Baehrecke E. H., Blagosklonny M. V., Dawson T. M., Dawson V. L., El-Deiry W. S., Fulda S., et al. (2012b). Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J., Zhong C. Q., Zhang D. W. (2011). Programmed necrosis: backup to and competitor with apoptosis in the immune system. Nat. Immunol. 12, 1143–1149 [DOI] [PubMed] [Google Scholar]
- Hay B. A., Wolff T., Rubin G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120, 2121–2129 [DOI] [PubMed] [Google Scholar]
- He S., Wang L., Miao L., Wang T., Du F., Zhao L., Wang X. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 [DOI] [PubMed] [Google Scholar]
- Hitomi J., Christofferson D. E., Ng A., Yao J., Degterev A., Xavier R. J., Yuan J. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135, 1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J. R., Meireles A. M., Fisher K. H., Garcia A., Antrobus P. R., Wainman A., Zitzmann N., Deane C., Ohkura H., Wakefield J. G. (2008). A microtubule interactome: complexes with roles in cell cycle and mitosis. PLoS Biol. 6, e98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T., Kanda H., Okano H., Xu T., Miura M. (2011). Eiger and wengen: the Drosophila orthologs of TNF/TNFR. Adv. Exp. Med. Biol. 691, 45–50 [DOI] [PubMed] [Google Scholar]
- Irwin D. J., Lee V. M., Trojanowski J. Q. (2013). Parkinson’s disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 14, 626–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia L., Kim S., Yu H. (2013). Tracking spindle checkpoint signals from kinetochores to APC/C. Trends Biochem. Sci. 38, 302–311 [DOI] [PubMed] [Google Scholar]
- Joza N., Galindo K., Pospisilik J. A., Benit P., Rangachari M., Kanitz E. E., Nakashima Y., Neely G. G., Rustin P., Abrams J. M., et al. (2008). The molecular archaeology of a mitochondrial death effector: AIF in Drosophila. Cell Death Differ. 15, 1009–1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H., Igaki T., Okano H., Miura M. (2011). Conserved metabolic energy production pathways govern Eiger/TNF-induced nonapoptotic cell death. Proc. Natl. Acad. Sci. USA 108, 18977–18982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karres J. S., Hilgers V., Carrera I., Treisman J., Cohen S. M. (2007). The conserved microRNA miR-8 tunes atrophin levels to prevent neurodegeneration in Drosophila. Cell 131, 136–145 [DOI] [PubMed] [Google Scholar]
- Kidokoro T., Tanikawa C., Furukawa Y., Katagiri T., Nakamura Y., Matsuda K. (2008). CDC20, a potential cancer therapeutic target, is negatively regulated by p53. Oncogene 27, 1562–1571 [DOI] [PubMed] [Google Scholar]
- Kuranaga E., Kanuka H., Igaki T., Sawamoto K., Ichijo H., Okano H., Miura M. (2002). Reaper-mediated inhibition of DIAP1-induced DTRAF1 degradation results in activation of JNK in Drosophila. Nat. Cell Biol. 4, 705–710 [DOI] [PubMed] [Google Scholar]
- Laster S. M., Wood J. G., Gooding L. R. (1988). Tumor necrosis factor can induce both apoptic and necrotic forms of cell lysis. J. Immunol. 141, 2629–2634 [PubMed] [Google Scholar]
- Lee C. Y., Robinson K. J., Doe C. Q. (2006). Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature 439, 594–598 [DOI] [PubMed] [Google Scholar]
- Lee T. V., Ding T., Chen Z., Rajendran V., Scherr H., Lackey M., Bolduc C., Bergmann A. (2008). The E1 ubiquitin-activating enzyme Uba1 in Drosophila controls apoptosis autonomously and tissue growth non-autonomously. Development 135, 43–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leismann O., Lehner C. F. (2003). Drosophila securin destruction involves a D-box and a KEN-box and promotes anaphase in parallel with Cyclin A degradation. J. Cell Sci. 116, 2453–2460 [DOI] [PubMed] [Google Scholar]
- Leismann O., Herzig A., Heidmann S., Lehner C. F. (2000). Degradation of Drosophila PIM regulates sister chromatid separation during mitosis. Genes Dev. 14, 2192–2205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulier F., Rodriguez A., Khush R. S., Abrams J. M., Lemaitre B. (2000). The Drosophila caspase Dredd is required to resist gram-negative bacterial infection. EMBO Rep. 1, 353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Rodriguez J., Yoo Y., Shareef M. M., Badugu R., Horabin J. I., Kellum R. (2011). Cooperative and antagonistic contributions of two heterochromatin proteins to transcriptional regulation of the Drosophila sex determination decision. PLoS Genet. 7, e1002122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean J. R., Chaix D., Ohi M. D., Gould K. L. (2011). State of the APC/C: organization, function, and structure. Crit. Rev. Biochem. Mol. Biol. 46, 118–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy O., Pál M., Udvardy A., Shirras C. A., Boros I., Shirras A. D., Deák P. (2012). lemmingA encodes the Apc11 subunit of the APC/C in Drosophila melanogaster that forms a ternary complex with the E2-C type ubiquitin conjugating enzyme, Vihar and Morula/Apc2. Cell Div. 7, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nüsslein-Volhard C., Wieschaus E. (1980). Mutations affecting segment number and polarity in Drosophila. Nature 287, 795–801 [DOI] [PubMed] [Google Scholar]
- Ouyang Y., Song Y., Lu B. (2011). dp53 Restrains ectopic neural stem cell formation in the Drosophila brain in a non-apoptotic mechanism involving Archipelago and cyclin E. PLoS ONE 6, e28098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owusu-Ansah E., Yavari A., Mandal S., Banerjee U. (2008). Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat. Genet. 40, 356–361 [DOI] [PubMed] [Google Scholar]
- Park J., Lee S. B., Lee S., Kim Y., Song S., Kim S., Bae E., Kim J., Shong M., Kim J. M., et al. (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161 [DOI] [PubMed] [Google Scholar]
- Pereanu W., Shy D., Hartenstein V. (2005). Morphogenesis and proliferation of the larval brain glia in Drosophila. Dev. Biol. 283, 191–203 [DOI] [PubMed] [Google Scholar]
- Pesin J. A., Orr-Weaver T. L. (2008). Regulation of APC/C activators in mitosis and meiosis. Annu. Rev. Cell Dev. Biol. 24, 475–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J. (2011). Cubism and the cell cycle: the many faces of the APC/C. Nat. Rev. Mol. Cell Biol. 12, 427–438 [DOI] [PubMed] [Google Scholar]
- Price D. M., Jin Z., Rabinovitch S., Campbell S. D. (2002). Ectopic expression of the Drosophila Cdk1 inhibitory kinases, Wee1 and Myt1, interferes with the second mitotic wave and disrupts pattern formation during eye development. Genetics 161, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffa G. D., Ciapponi L., Cenci G., Gatti M. (2011). Terminin: a protein complex that mediates epigenetic maintenance of Drosophila telomeres. Nucleus 2, 383–391 [DOI] [PubMed] [Google Scholar]
- Reed B. H., Orr-Weaver T. L. (1997). The Drosophila gene morula inhibits mitotic functions in the endo cell cycle and the mitotic cell cycle. Development 124, 3543–3553 [DOI] [PubMed] [Google Scholar]
- Ring J. M., Martinez Arias A. (1993). puckered, a gene involved in position-specific cell differentiation in the dorsal epidermis of the Drosophila larva. Dev. Suppl. 1993, 251–259 [PubMed] [Google Scholar]
- Ritson G. P., Custer S. K., Freibaum B. D., Guinto J. B., Geffel D., Moore J., Tang W., Winton M. J., Neumann M., Trojanowski J. Q., et al. (2010). TDP-43 mediates degeneration in a novel Drosophila model of disease caused by mutations in VCP/p97. J. Neurosci. 30, 7729–7739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong Y. S., Titen S. W., Xie H. B., Golic M. M., Bastiani M., Bandyopadhyay P., Olivera B. M., Brodsky M., Rubin G. M., Golic K. G. (2002). Targeted mutagenesis by homologous recombination in D. melanogaster. Genes Dev. 16, 1568–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist S. E., Haque N. S., Chen C. H., Hay B. A., Hariharan I. K. (2010). Inactivation of both Foxo and reaper promotes long-term adult neurogenesis in Drosophila. Curr. Biol. 20, 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr D. A., Williams B. C., Hays T. S., Goldberg M. L. (1998). ZW10 helps recruit dynactin and dynein to the kinetochore. J. Cell Biol. 142, 763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su T. T., O’Farrell P. H. (1997). Chromosome association of minichromosome maintenance proteins in Drosophila mitotic cycles. J. Cell Biol. 139, 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsu Y., Nakamura M., Stapleton M., Danos M. C., Matsumoto K., O’Connor M. B., Shibuya H., Ueno N. (2000). TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol. Cell. Biol. 20, 3015–3026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L., Hires S. A., Mao T., Huber D., Chiappe M. E., Chalasani S. H., Petreanu L., Akerboom J., McKinney S. A., Schreiter E. R., et al. (2009). Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda H., Mochizuki H., Flores R., III, Josowitz R., Krasieva T. B., Lamorte V. J., Suzuki E., Gindhart J. G., Furukubo-Tokunaga K., Tomoda T. (2008). UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev. 22, 3292–3307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenabeele P., Declercq W., Van Herreweghe F., Vanden Berghe T. (2010). The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal. 3, re4 [DOI] [PubMed] [Google Scholar]
- Vaskova M., Bentley A. M., Marshall S., Reid P., Thummel C. S., Andres A. J. (2000) Genetic analysis of the Drosophila 63F early puff. Characterization of mutations in E63-1 and maggie, a putative Tom22. Genetics 156, 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercammen D., Beyaert R., Denecker G., Goossens V., Van Loo G., Declercq W., Grooten J., Fiers W., Vandenabeele P. (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187, 1477–1485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Cai Y., Chia W., Yang X. (2006). Drosophila homologs of mammalian TNF/TNFR-related molecules regulate segregation of Miranda/Prospero in neuroblasts. EMBO J. 25, 5783–5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Golden K. L., Lee C. Y. (2010). dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila. Dev. Cell 18, 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng M., Komori H., Lee C. Y. (2012). Identification of neural stem cells in the Drosophila larval brain. Methods Mol. Biol. 879, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Hou L., Li Y., Ni J., Liu L. (2013). Neuronal necrosis and spreading death in a Drosophila genetic model. Cell Death Dis. 4, e723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. (2007). Cdc20: a WD40 activator for a cell cycle degradation machine. Mol. Cell 27, 3–16 [DOI] [PubMed] [Google Scholar]
- Zhang D. W., Shao J., Lin J., Zhang N., Lu B. J., Lin S. C., Dong M. Q., Han J. (2009). RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 [DOI] [PubMed] [Google Scholar]
- Zheng L., Jia J., Finger L. D., Guo Z., Zer C., Shen B. (2011). Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 39, 781–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivotovsky B., Orrenius S. (2011). Calcium and cell death mechanisms: a perspective from the cell death community. Cell Calcium 50, 211–221 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.