

Abstract
Disruptions in polarity and mitotic spindle orientation contribute to the progression and evolution of tumorigenesis. However, little is known about the molecular mechanisms regulating these processes in vivo. Here, we demonstrate that Polo-like kinase 2 (Plk2) regulates mitotic spindle orientation in the mammary gland and that this might account for its suggested role as a tumor suppressor. Plk2 is highly expressed in the mammary gland and is required for proper mammary gland development. Loss of Plk2 leads to increased mammary epithelial cell proliferation and ductal hyperbranching. Additionally, a novel role for Plk2 in regulating the orientation of the mitotic spindle and maintaining proper cell polarity in the ductal epithelium was discovered. In support of a tumor suppressor function for Plk2, loss of Plk2 increased the formation of lesions in multiparous glands. Collectively, these results demonstrate a novel role for Plk2 in regulating mammary gland development.
Keywords: Plk2, Mammary gland development, Spindle orientation
INTRODUCTION
Mammary gland development is a precisely coordinated process that occurs primarily after birth. Pubertal mouse mammary gland development commences at 3-4 weeks of age and is stimulated by estrogen, progesterone and growth hormone. These circulating hormones prompt the formation of highly proliferative terminal end bud (TEB) structures that invade the stromal fat pad in a process termed ductal elongation. The mammary epithelial cells (MECs) of the TEBs maintain a carefully regulated balance between proliferation and apoptosis to generate hollow ducts composed of a single layer of luminal epithelium surrounded by myoepithelial cells (Humphreys et al., 1996). The luminal epithelial cells establish apical-basal polarity that is crucial for maintaining the integrity and function of the glandular epithelium (St Johnston and Ahringer, 2010). Upon reaching the end of the fat pad, the TEBs regress, leaving behind a branched, ductal tree that remains mostly quiescent until pregnancy (Harris, 2010). Disruption of any of these highly regulated developmental processes, including proliferation, apoptosis and polarity, can influence mammary tumorigenesis.
Breast cancer is a heterogeneous disease that can be classified into distinct subtypes based primarily on the expression pattern of estrogen (ER) and progesterone (PR) hormone receptors, as well as the human epidermal growth factor receptor 2 (HER2) (Perou et al., 2000). Treating individuals with breast cancer presents a significant challenge due to the lack of targeted therapies, especially of triple-negative breast cancer, and the relatively minimal understanding of the signaling networks that regulate each specific subtype (Di Cosimo and Baselga, 2010). Therefore, it is imperative to identify signaling pathways that contribute to the etiology of breast cancer in an attempt to discover novel therapeutic targets. RNA interference (RNAi) has been used successfully in high-throughput screens to identify potential drug targets (Gargiulo et al., 2013; Liu et al., 2013). In particular, RNAi has been used to identify putative tumor suppressors in forward genetic screens (Westbrook et al., 2005; Krastev et al., 2011; Iorns et al., 2012). Recently, our laboratory performed RNAi screens designed to specifically target kinases and phosphatases, characterizing PTPN12 as a tumor suppressor (Sun et al., 2011). In these same screens, Polo-like kinase 2 (Plk2) was identified as a potential tumor suppressor in epithelial-derived cancers. Frequent focal deletion of Plk2 has been reported to occur in numerous solid cancers, including breast cancer (Beroukhim et al., 2010).
Polo-like kinases are crucial regulators of various aspects of cell division, including mitosis, cytokinesis and the centrosome cycle (Archambault and Glover, 2009). The polo-like kinase family consists of five members, Plk1-Plk5, that are each characterized by their conserved C-terminal polo box and N-terminal kinase domains (Glover et al., 1998; Barr et al., 2004, Andrysik et al., 2010). Plk2 (also referred to as serum-inducible kinase, or Snk) was originally identified as an early growth response gene whose mRNA expression increases in response to serum (Simmons et al., 1992). During cell cycle progression, Plk2 is activated in the early G1 phase and is required for centriole duplication (Warnke et al., 2004). Targeted germline deletion of Plk2 in a mouse model results in embryonic growth abnormalities, but does not cause gross morphological phenotypes postnatally, perhaps owing to compensation from other Plk family members (Ma et al., 2003). Plk2 is expressed in a tissue-specific manner, with relatively high expression levels in the mammary gland, perhaps providing an additional explanation for the paucity of overt abnormalities in the Plk2-null mice (Liby et al., 2001; Winkles and Alberts, 2005). In the pubertal mammary gland, Plk2 is enriched in TEBs compared with the ductal epithelium, but the function of Plk2 in the developing gland is unknown (Kouros-Mehr and Werb, 2006).
In this study, a germline knockout mouse was employed to determine the effect of loss of Plk2 on mammary gland development and tumorigenesis. During puberty, Plk2 deletion resulted in increased proliferation of MECs, increased ductal side branching and delayed ductal elongation. In the adult, Plk2-null epithelial ducts have reached the end of the mammary fat pad; however, the increased proliferation and hyperbranching phenotypes persist. Expression profiling of Plk2-null MECs revealed disrupted expression of genes that regulate mitotic spindle assembly. Accordingly, loss of Plk2 led to misorientation of the mitotic spindle and loss of apical-basal polarity in the luminal epithelium, as well as luminal filling, suggesting that Plk2 is required to maintain proper cell division and polarity in the mammary luminal epithelial compartment. Additionally, loss of Plk2 resulted in an increase in the formation of lesions in the mammary glands of multiparous mice. Collectively, these results show that Plk2 is required for proper mammary gland development.
RESULTS
Deletion of Plk2 results in a delay in ductal elongation and increased branching
To elucidate the role of Plk2 in mammary development and breast cancer, we characterized a Plk2 germline knockout mouse model (Inglis et al., 2009) for defects in mammary gland development and homeostasis. PCR was performed to confirm deletion of the Plk2 gene in Plk2-/- compared with Plk2+/+ mice (supplementary material Fig. S1A). To detect morphological defects, mammary glands were harvested at 6, 8, 10 and 12 weeks of age and analyzed initially as whole mounts. A reduction in ductal elongation through the mammary fat pad in the Plk2-null glands was observed beginning at 6 weeks of age that persisted through 10 weeks of age (Fig. 1A). To quantify the defects in ductal elongation, the percentage of the fat pad occupied by epithelium (% fat pad filled) was quantified. Throughout development, Plk2-/- mammary glands showed a reduced percentage of fat pad filled by epithelium compared with the Plk2+/+ mammary glands (Fig. 1B). However, by 12 weeks of age, Plk2-/- ducts reached the ends of the mammary fat pad, indicating that the impairment in ductal elongation was transient (Fig. 1B). Whole-mount analysis of Plk2-/- mammary glands also revealed a hyperbranched phenotype observed at all stages of development (Fig. 1C). Quantitative analysis showed a significant increase in side branching in the Plk2-null mammary glands beginning at 6 weeks of age (Fig. 1D). At 8 weeks of age, the Plk2-null glands showed a 2.1-fold increase in branchpoints per millimeter of epithelial duct compared with control glands. This hyperbranched phenotype persisted in the glands of 12-week-old Plk2-/- mice. Taken together, these data suggest that Plk2 is an important regulator of ductal elongation and branching morphogenesis in the mammary gland.
Fig. 1.

Plk2 loss leads to delayed ductal elongation and increased branching. (A) Carmine stained whole-mount analyses during virgin mammary gland development at 8, 10 and 12 weeks of age denote a delay in ductal elongation in Plk2-/- compared with Plk2+/+. The red line indicates the distance from the lymph node to the leading end bud. (B) Plk2-/- mammary glands exhibit a transient delay in development, appearing from 6 to 10 weeks of age. At 10 weeks of age, the Plk2+/+ mammary glands have reached the end of the fat pad. Plk2-/- reach the end of the fat pad at 12 weeks of age. (C,D) Plk2-/- mammary glands display increased branching that appears as early as 6 weeks of age and persists throughout development. Scale bars: 10 mm. Statistical significance was determined by Student’s t-test. *P<0.01, **P<0.001, ***P<0.0001. Error bars represent s.e.m. with a sample size n>3.
Loss of Plk2 leads to increased proliferation of mammary epithelial cells without a concomitant increase in apoptosis
Next, we sought to determine whether the increase in branching observed in Plk2-/- mammary glands was due to alterations in proliferation. To quantify proliferation, mice were injected with BrdU at several time points throughout mammary gland development as a measure of cells in S phase. Notably, there was a substantial increase in proliferation at 8 weeks of age in Plk2-/- TEBs (Fig. 2A,B,G). At 10 and 12 weeks of age, when the epithelial ducts of the control mammary glands had reached the ends of the mammary fat pad (Fig. 1B), the Plk2+/+ ductal epithelial cells exhibited very low levels of proliferation, as expected (Fig. 2E,G). Conversely, the Plk2-null glands showed significantly increased levels of proliferation at 10 weeks of age when compared with controls. By 12 weeks of age, the Plk2-null epithelium had filled the mammary fat pad (Fig. 1B); however, the Plk2-/- ductal epithelial cells continued to proliferate, as evidenced by the presence of ∼10% BrdU-positive cells. The Plk2+/+ control glands had no detectable BrdU-positive cells at 12 weeks of age (Fig. 2E,F). Interestingly, the increase in proliferation in the absence of Plk2 was maintained at 16 weeks of age (Fig. 2G). To further confirm the proliferative phenotype, we stained for pH3 (phosphorylated histone 3) a marker for chromatin condensation, which is a key process during mitosis. Not surprisingly, a similar increase of proliferation at 8, 10, 12 and 16 weeks of age was observed indicating that the increase in proliferation is not due to cells prolonged in S phase, as previously reported (Matthew et al., 2007) (supplementary material Fig. S2A). These data suggest that Plk2 negatively regulates proliferation in mammary epithelial cells, and that loss of Plk2 leads to a hyperproliferative phenotype.
Fig. 2.

Increase in proliferation in Plk2-/- mammary glands. (A,B) Immunofluorescence for BrdU incorporation on paraffin-embedded sections of mammary glands at 8 weeks of age (E,F) and 12 weeks of age demonstrates that loss of Plk2 causes an increase in proliferation during puberty as well as in a mature virgin mammary gland (G). Immunofluorescence on 8-week-old mammary gland sections using cleaved caspase 3 (C,D) showing Plk2 had no significant effect on apoptosis (H). Scale bars: 40 μm. Statistical significance was determined by Student’s t-test. *P<0.01, **P<0.001. Error bars represent s.e.m. with a sample size n>3.
Many oncogenic insults that increase proliferation also result in a compensatory increase in apoptosis (Debnath et al., 2002). To test whether the elevated proliferation in Plk2-/- glands lead to a concomitant increase in apoptosis, immunofluorescent detection of cleaved caspase 3(CC3), a marker for apoptosis was performed. Consistent with normal developmental timing, we observed higher levels of apoptosis during ductal elongation at 6 and 8 weeks of age, as epithelial cells in the TEBs undergo apoptosis to form the lumen. However, there were no significant differences in apoptosis between Plk2-null mammary glands and controls at these time points or later in development (Fig. 2C,D,H). An additional method for detection of apoptosis was employed. TUNEL assays on 8-week mammary glands from Plk2+/+ and Plk2-/- revealed similar results with no significant difference in apoptosis (supplementary material Fig. S2B-D). These data demonstrate that Plk2 loss leads to hyper-proliferation without a concomitant increase in apoptosis, and suggest that Plk2 regulates mammary gland homeostasis. Accordingly, Plk2 loss may potentiate mammary hyperplasia.
Loss of Plk2, specifically in the mammary epithelial cells, recapitulates the delay in ductal elongation, the hyperbranching and increased proliferation
Plk2 expression was detected by immunohistochemistry in both the basal and luminal compartments of the TEBs and ducts (supplementary material Fig. S1B). To determine whether the mammary epithelial cell defects were cell-autonomous or due to Plk2 loss in the stromal compartment and/or secondary systemic effects, mammary epithelial transplants were performed. Plk2+/+ and Plk2-/- MECs were injected into the contralateral fat pads of wild-type mice that were cleared of endogenous epithelium and the resultant outgrowths were analyzed 6 and 8 weeks post-transplantation. A delay in ductal elongation was observed in Plk2-/- outgrowths 6 weeks post-transplantation with only ∼20% of the fat pad occupied, in contrast to 80% of the fat pad filled in Plk2+/+ outgrowths (supplementary material Fig. S3A,B,G). Whole-mount analysis showed that both wild-type and Plk2-null MECs completed mammary gland development 8 weeks post-transplantation (supplementary material Fig. S3G). Whole mounts and Hematoxylin and Eosin staining also showed that Plk2-null outgrowths appeared hyperbranched compared with contralateral wild-type outgrowths (supplementary material Fig. S3C-F,H). Branching quantification demonstrated that Plk2-null outgrowths had an almost twofold increase in branch points per millimeter when compared with wild-type outgrowths at 6 and 8 weeks post-transplantation (supplementary material Fig. S3F). The proliferative phenotype observed in the Plk2-/- endogenous mammary gland was further investigated to determine whether it was due to the absence of Plk2 in MECs. Not surprisingly an increase in proliferation in the Plk2-/- outgrowths isolated 6 weeks post-transplantation was detected (supplementary material Fig. S4A,B,E). Although an increase in proliferation was also observed at 8 weeks post-transplantation in Plk2-/- outgrowths, this difference was not statistically significant. Owing to the low levels of proliferation observed in the resultant outgrowths insufficient numbers of mitotic events were detected to accurately measure spindle orientation. Taken together, these results provide evidence that the delay in ductal elongation, hyperbranching and increase in proliferation phenotypes are a direct consequence of Plk2 loss in the mammary epithelium.
Plk2 inactivation leads to misregulation of genes that regulate mitotic spindle assembly
To determine how Plk2 may restrain proliferation in the mammary gland, gene expression analyses were performed on RNA isolated from Plk2+/+ and Plk2-/- MECs. The Database for Annotation, Visualization and Integrated Discovery (DAVID) was employed to identify cellular processes that are misregulated upon Plk2 loss. Additionally microarray results were validated using genes known to be abundantly expressed in the mammary gland in addition to cell cycle-specific genes (supplementary material Fig. 5B,C). Interestingly, many genes involved in cell cycle control were dysregulated in the Plk2-compromised state (supplementary material Fig. S5A). Notably, Plk2 inactivation led to dysregulation of many genes involved in proper assembly and integrity of the kinetochore and mitotic spindle, such as Cenpo (1.63), Dsn1 (1.61) and Spc25 (4.98) (supplementary material Fig. S5A). Spc25 is a component of the Ndc80 complex, which is a regulator of kinetochore/microtubule attachment a process that must be tightly regulated to establish proper spindle orientation (Tanaka and Desai, 2008; Sun et al., 2010). Interestingly, a threefold increase in Spc25 protein levels was observed in the absence of Plk2 (supplementary material Fig. S5D). In addition, Haus1 (2.50) was significantly upregulated in Plk2-null MECs. Haus1 is involved in microtubule nucleation, which is crucial for the assembly of the mitotic spindle and is also a regulator of NuMA, a key protein involved in spindle orientation (Bowman et al., 2006; Lawo et al., 2009) (supplementary material Fig. S5A). These results suggest that Plk2 inactivation may be important for regulating the assembly, and hence orientation of the mitotic spindle in the mammary epithelium.
Plk2 is required for proper orientation of the mitotic spindle and polarization of the epithelium
Our results suggest Plk2 regulates proper assembly and function of the mitotic spindle in the mammary epithelium. Thus, we tested whether Plk2 inactivation led to defects in spindle orientation in ductal luminal epithelial cells in vivo. In most epithelia, planar cell division and proper mitotic spindle orientation are necessary to maintain the organization of the epithelial monolayer and tissue integrity. In the mammary gland, mitotic spindle orientation has not been well-studied, but in vitro studies in three-dimensional cultures have suggested that luminal MECs normally divide within the plane of the luminal compartment. Upon further examination of ductal mitotic cells, we frequently observed luminal cells dividing in a non-parallel orientation to the plane of the luminal compartment and into the ductal luminal space in the absence of Plk2. To examine this phenotype further, we treated 10- to 12-week-old wild-type and Plk2-null mice with estrogen and progesterone for 2 days to induce proliferation of the MECs, thus increasing the number of mitotic figures for quantification. Plk2+/+ were used as controls to rule out any effect that might occur as a consequence of estrogen and progesterone treatment alone. We performed immunofluorescence to detect nuclear mitotic apparatus protein 1 (NuMA), a protein that is concentrated at the spindle poles during mitosis, to determine the orientation of the mitotic spindle in mammary ducts. As expected, in Plk2+/+ mammary glands the orientation of the mitotic spindle was parallel to the basement membrane (Fig. 3A), whereas in Plk2-/- mammary glands the mitotic spindle was altered and most frequently not parallel to the basement membrane (Fig. 3B), which confirms previous observations. Abnormal spindle orientation in Plk2-/- mammary glands was also observed using phosphorylated histone 3 (pH3), a marker of condensed chromatin in mitotic cells (supplementary material Fig. S6A,B,E). To quantify the orientation of the mitotic spindle in the wild-type and Plk2-null mammary ducts, the angle between the basement membrane and the plane of the mitotic spindle was measured and the measurements were classified into three categories: 0-10°, 10-45° and 45-90° (Fig. 3I). In control mammary glands, almost 90% of the mitotic cells had their spindles oriented parallel to the basement membrane (0-10°), whereas only 15% of Plk2-null mitotic cells fell into this category (Fig. 3I). The majority of Plk2-null mitotic cells (almost 80%) exhibited abnormal spindle orientation with an angle greater than 10° compared with controls (Fig. 3I). Interestingly, Plk2-null mammary ducts displayed an almost equal distribution of cells with angles of 10-45° and 45-90°, suggesting a random orientation of the mitotic spindle relative to the control mammary ducts (Fig. 3I). These data suggest that loss of Plk2 results in misorientation of the mitotic spindle in luminal epithelial cells.
Fig. 3.
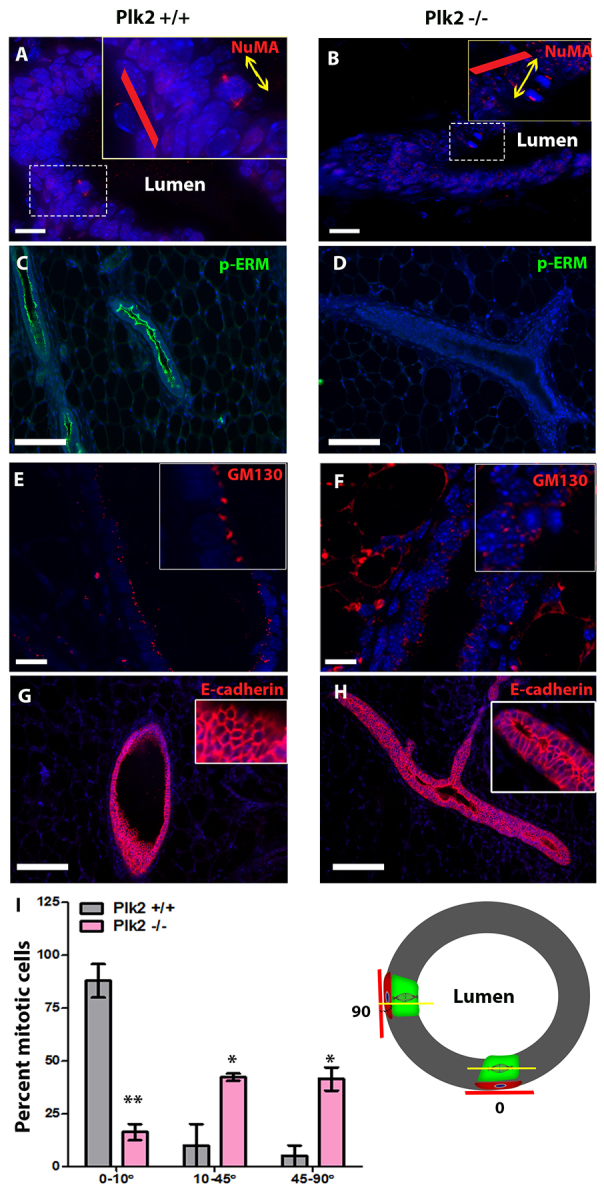
Plk2 is required for proper spindle orientation and proper formation of the polarized epithelium. (A,B) Nuclear mitotic apparatus protein 1 (NuMA) staining showing the disruption of spindle orientation upon Plk2 loss. The red line indicates the proper plane of cell division and the yellow arrows denote the actual plane of division occurring. The angle between the basement membrane and plane of cell division was measured for Plk2+/+ and Plk2-/- mitotic epithelial cells (I). The majority of Plk2-/- epithelial cells undergoing cell division portrayed an angle greater than 10°, indicating that the cells were dividing perpendicular to the basement membrane (I). (C-F) Immunofluorescence of paraffin embedded mammary gland sections using the apical marker phosphorylated ERM and GM130 (a Golgi marker), both of which display the disruption of apical polarization in Plk2-/- mammary glands. (G,H) No alterations in E-cadherin staining on 8-week-old mammary gland sections. Scale bars: 40 μm. Statistical significance was determined by Student’s t-test. *P<0.01, **P<0.001. Error bars represent s.e.m. with a sample size n>3.
Proper orientation of the mitotic spindle and the maintenance of polarization are crucial components required to preserve the integrity of epithelial tissue. In order to determine whether polarization of the epithelium was compromised, the distribution of apical-basal polarity markers in wild-type and Plk2-null mammary ducts was analyzed. First immunofluorescent detection of phospho-ERM (pERM), which localizes to the apical surface of luminal epithelial cells, as shown in the wild-type mammary ducts was performed (Fig. 3C). Surprisingly, Plk2-/- mammary ducts showed an almost complete loss of pERM staining (Fig. 3D). To further investigate a defect in apical polarity, the Golgi apparatus marker GM130, which is restricted to the apical surface of luminal epithelial cells in the mammary gland was also employed. As expected, Plk2+/+ mammary ducts showed a well-aligned, continuous layer of GM130 staining localized to the apical surface of luminal cells (Fig. 3E). Conversely, GM130 was randomly distributed in the Plk2-/- mammary ducts (Fig. 3F). Lastly, immunofluorescence was performed for ZO-1, a tight junction protein localized apically on luminal cells. A lack of ZO-1 staining was observed in our Plk2-/- mammary glands (supplementary material Fig. S6C,D). To rule out a non-cell autonomous effect, the outgrowths from the previously mentioned transplantation studies also were analyzed for pERM levels and not surprisingly a significant decrease in pERM staining in Plk2-/- outgrowths was observed (supplemental material Fig. S4C,D). Disruption of the distribution of these apical polarity markers suggests that Plk2 is required to maintain proper luminal apical-basal polarity in the mammary gland.
To determine whether the loss of apical polarity was due to, or accompanied by, a loss of cell-cell adhesion, we performed immunofluorescence to detect alterations in E-cadherin localization. E-cadherin is crucial for maintaining cell-cell contacts in epithelial tissues and localizes to adherens junctions as shown in wild-type mammary ducts (Fig. 3G). Loss of Plk2 did not affect the localization of E-cadherin (Fig. 3H), suggesting that the disrupted apical polarity in Plk2-null luminal epithelium is not due to the inability of the luminal cells to maintain proper cell-cell contact. Collectively, these data suggest that loss of Plk2 results in alterations of spindle orientation and compromises the integrity of the epithelium by disrupting polarization.
Plk2-null mammary glands show disrupted lumen formation
Do these alterations in spindle orientation and polarization have further consequences on tissue architecture? Hematoxylin and Eosin (H&E) staining performed on mammary sections from Plk2-/- and control glands confirmed the hyperbranched phenotype observed by whole-mount analysis and further revealed a luminal filling phenotype in the Plk2-/- mammary glands (Fig. 4A-D). To determine whether the failure to form lumens was accompanied by a disruption in the luminal and/or myoepithelial layer organization, immunofluorescent staining using antibodies against keratin 8 (K8; Krt8 - Mouse Genome Informatics) and keratin 5 (K5; Krt5 - Mouse Genome Informatics), which are well-established markers for luminal and myoepithelial compartments, respectively, was performed. In Plk2+/+ mammary glands a single layer of K8+ luminal cells surrounded by a continuous, single layer of K5+ myopeithelial cells was observed, as expected (Fig. 4E). In Plk2-/- mammary ducts, the K8+ luminal compartment was multi-layered and revealed K8+ cells accumulating in the lumen. No alterations were detected in the myoepithelial cell layer in Plk2-/- mammary glands (Fig. 4F). These data suggest that Plk2 is required for proper lumen formation during ductal morphogenesis.
Fig. 4.
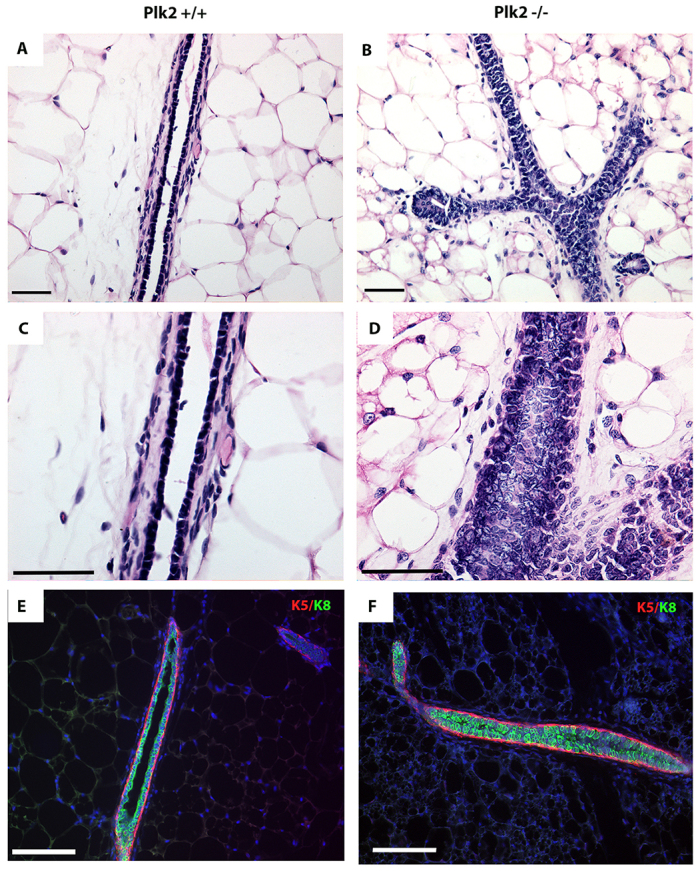
Plk2 deletion disrupts lumen formation and stromal architecture. (A,B) H&E staining denoting the increase in branching as well as lack of lumens (C,D) in 12-week-old Plk2-/- mammary glands. (E,F) Immunofluorescence using K5 (myoepithelial) and K8 (luminal) markers shows an increase in luminal cells that leads to a lack of ductal lumen. Scale bars: 40 μm.
Increase in lesions in the absence of Plk2 in parous gland
The phenotypes observed in the Plk2-null mammary gland, including hyperproliferation, disrupted luminal polarity and luminal filling, are also common features of early mammary tumorigenesis. To determine whether Plk2 functions as a tumor suppressor, nulliparous Plk2+/+ and Plk2-/- females were aged for 12-15 months and monitored for tumor formation. In the absence of any other genetic changes, such as alterations in p53 to provide a sensitized background, Plk2-/- mice failed to develop palpable mammary tumors and whole-mount analysis also revealed an absence of macroscopic lesions. This result was not unexpected as C57Bl/6 mice are known to be a low incidence mammary tumor strain. Pregnancy and involution results in significant increases in proliferation and apoptosis of MECs, respectively, thus potentially increasing their susceptibility to mutagenic events. To determine whether there was an effect of parity on tumorigenesis, Plk2+/+ and Plk2-/- female mice were mated and allowed to progress through several rounds of pregnancy and lactation followed by complete involution. Again, Plk2-null mice did not develop palpable mammary tumors. However, macroscopic lesions were detected by whole-mount analysis of mammary glands from parous females following involution. Although there were lesions present in both Plk2+/+ and Plk2-/- mammary glands, the Plk2-null mice showed a twofold increase in the number of lesions per mammary gland when compared with parous age-matched wild-type mice (Fig. 5A,B,G). In addition, Plk2-null lesions appeared to have decreased epithelial organization in H&E stained sections of parous mammary glands (Fig. 5C,D). Interestingly, Plk2-/- parous mammary glands contained a twofold increase in keratin pearls, indicating that the mammary ductal epithelium had undergone squamous differentiation (Fig. 5E,F; supplementary material Fig. S7A). To better visualize alterations in the stroma, we stained parous sections with Masson’s Trichrome and found that there was abundant collagen deposition in both Plk2+/+ and Plk2-/- lesions (Fig. 5E,F). These data suggest that absence of Plk2 cooperates with parity, resulting in an increased number of lesions.
Fig. 5.
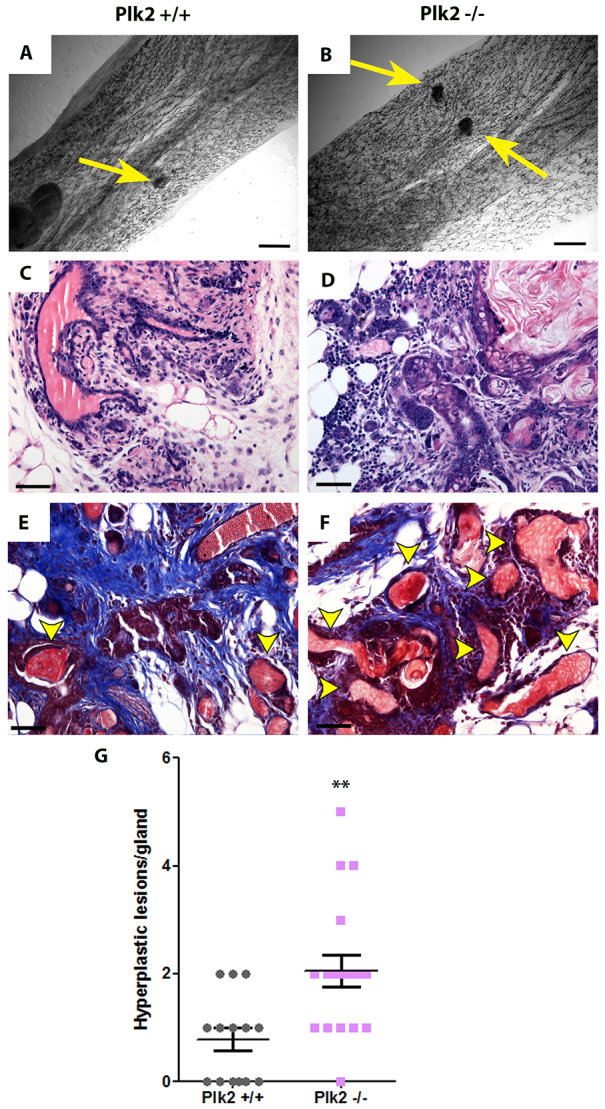
Increase in lesions upon Plk2 loss in parous glands. (A,B) Carmine stained whole-mount analysis of parous Plk2+/+ and Plk2-/- mammary glands revealed the existence of lesions (yellow arrows). H&E (C,D) and Masson’s Trichrome staining (E,F) on paraffin-embedded mammary gland sections of parous lesions illustrates the process of keratinization occurring indicated by yellow arrowheads. Quantitative analysis of lesions indicates an increase in lesions in Plk2-/- parous glands (G). Scale bars: 10 mm in A,B; 40 μm in C-F. Statistical significance was determined by Student’s t-test. **P<0.001. Error bars represent s.e.m. with a sample size n>3.
Plk2-null lesions are less differentiated and exhibit decreased expression of estrogen and progesterone receptors
To further characterize the lesions in Plk2+/+ and Plk2-/- parous mammary glands, K8 and K5 immunofluorescent staining was used to examine organization of the luminal and basal epithelial layers. Plk2+/+ lesions displayed intact K5+ myoepithelial cell layers surrounding the K8+ luminal compartments, indicating the proper organization of the two epithelial compartments (Fig. 6A). In stark contrast, Plk2-/- lesions lacked proper epithelial organization. Both K5+ cells, as well as cells that were double-positive for K8+/K5+ were observed, within the lumens of the ducts (Fig. 6B). Quantitation of Plk2+/+ revealed there were no double-positive cells present in lesions and Plk2-/- had 4% of total cells in lesions that were positive for K8 and K5 (supplementary material Fig. S7B). Collectively, these results suggest that lesions that arise as a consequence of Plk2 loss appear less differentiated, perhaps as an expansion of a multipotent progenitor population.
Fig. 6.
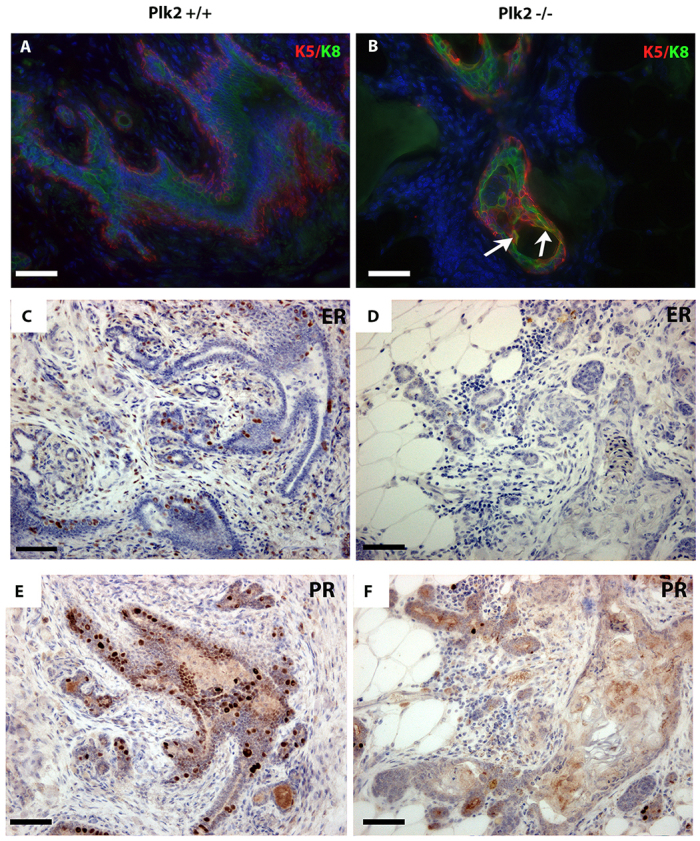
Plk2-/- lesions are less differentiated, have lost detectable estrogen receptor expression and have decreased progesterone receptor levels. (A,B) Immunofluorescence of paraffin-embedded mammary gland sections using K5 and K8 indicates a less differentiated lesion in Plk2-/- parous mammary gland (arrows). Epithelial cells positive for K5 and K8 were identified in Plk2-/- lesions indicated by white arrows. Plk2-/- parous lesions have lost detectable ER expression (C,D) and have a marked decrease in PR (E,F). Scale bars: 40 μm.
Because loss of estrogen and progesterone often is associated with a less differentiated status of mammary epithelium (Lapidus et al., 1998) the estrogen receptor (ER) and progesterone receptor (PR) status of the parous lesions was investigated by immunohistochemical staining. The Plk2+/+ lesions maintained expression of ER and PR in the luminal epithelial cells (Fig. 6C,E), whereas Plk2-/- lesions showed a prominent decrease of ER expression and a decrease in the number of cells expressing PR (Fig. 6D,F; supplementary material Fig S7C,D). These data further suggest that lesions that arise as a consequence of Plk2 loss exhibit a less differentiated phenotype.
DISCUSSION
In this study, we identified a novel role for Plk2 in regulating mammary gland development. In the absence of Plk2, we observed increased proliferation of MECs, hyperbranching of the ductal tree and a transient delay in ductal elongation. Levels of apoptosis were not significantly altered and hyperplastic lesions or tumors were not observed in virgin mice, despite the increase in proliferation in Plk2-/- mammary glands. This indicates that any increase in cell number that might occur due to the increase in proliferation is possibly compensated for by removal of cells from ductal lumens sometime after 16 weeks and prior to lactation, as has been observed in several other knockout mice models by an unknown mechanism (Mailleux et al., 2007; Moraes et al., 2009). We were able to recapitulate the ductal elongation, hyperbranching and proliferative phenotypes in transplantation studies into wild-type fat pads, indicating that the alterations in ductal elongation, increase in branching and increase in proliferation are a consequence of Plk2 loss specifically in the epithelium. Additionally, we find that the proliferative phenotype is observed in transplants during pubertal development and although there are more proliferating cells in Plk2-/- upon completion of development, these differences were not statistically significant. The maintenance of proliferation in post-pubertal development in the endogenous gland may be due to alterations in systemic hormonal regulation present only in the germline knockout. Interestingly, expression profiling of Plk2-null MECs showed alterations in key genes involved in regulating the mitotic spindle. Moreover, we found that Plk2 is required to maintain the integrity of the ductal epithelium, as loss of Plk2 led to mitotic spindle misorientation and disruption of apical-basal polarity in the luminal epithelium. Finally, we observed an increased number of lesions in multiparous Plk2-null mammary glands, providing evidence for the role of Plk2 in suppression of mammary tumorigenesis.
We identified a novel role for Plk2 in regulating the orientation of the mitotic spindle. Previous data suggested that Plk2 is important for the G1/S transition of the cell cycle and in the DNA damage response (Warnke et al., 2004; Matthew et al., 2007). Plk2 has also been reported to be crucial for centriole duplication during the G1/S transition. Interestingly, no defect in pericentrin and γ-tubulin centrosomal staining was observed in vivo in the absence of Plk2 (supplementary material Fig. S8A-D), but we cannot completely rule out the possibility that defective centriole duplication and/or function is involved in the mechanism by which Plk2 regulates spindle orientation due to the inability to stain centrioles in mammary gland tissue. Our studies use a genetically engineered mouse model with germline ablation of Plk2, and therefore, nontransformed mammary epithelial cells are being examined in the context of the microenvironment. Thus, it is not surprising that some functions of Plk2 may differ from those observed in previous in vitro studies performed using transformed U2OS osteosarcoma cells with genetic alterations such as the loss of p16 and cultured in the presence of serum (Warnke et al., 2004; Cizmecioglu et al., 2012). Here, we report for the first time a potential role for Plk2 during early mitosis in organizing the proper orientation of the mitotic spindle in non-transformed mammary epithelial cells. We observed that in the absence of Plk2, the plane of the mitotic spindle in luminal epithelial cells was most frequently perpendicular to the basement membrane, leading to cells dividing into the ductal lumen and a luminal filling phenotype. This result provides a new area of investigation to elucidate Plk2 function during mitosis. One crucial question that remains is how does Plk2 regulate the orientation of the mitotic spindle? One possibility is that Plk2 regulates the mitotic spindle by influencing the expression of key mitotic spindle genes that are important for key processes during spindle assembly, such as Spc25 and Haus1, most likely by an indirect mechanism. Spc25 and Haus1 mRNAs were both significantly upregulated in Plk2-null MECs and although this could be explained by the increase in proliferation, this possibility is unlikely because we did not observe a proliferation signature upon DAVID analyses. Spc25 is a key component important in the assembly of the kinetochore, and it is likely that upregulation of Spc25 could result in mislocalization of Spc25 protein and resulting alterations in kinetochore assembly (Cheeseman et al., 2008). Establishing a correctly oriented mitotic spindle depends on initial key processes such as orientation of the centrosomes to opposite poles and proper microtubule nucleation, Haus1 is a component of the Augmin complex, a key regulator of microtubule nucleation (Lawo et al., 2009). Disruption of the previously mentioned processes can ultimately alter spindle assembly dynamics. Previous studies have shown important regulators of spindle orientation in other epithelial tissues, such as ABL1 in the skin, IFT20 in the kidney and APC in colon cancer (Jonassen et al., 2008; Fleming et al., 2009; Matsumura et al., 2012). We describe for the first time that Plk2 possibly acts as a tumor suppressor to regulate the orientation of the mitotic spindle in the developing mammary gland. The analysis of somatic copy number alterations from over 3000 cancer specimens belonging to 26 histological types has identified focal loss of Plk2 as a frequent event, and this has been confirmed specifically in basal-like breast cancers using the TCGA web portal (Beroukhim et al., 2010) (C. Shaw, C. Perou and J. M. Rosen, unpublished observations). Interestingly, several tumor suppressors have been reported to alter the orientation of the mitotic spindle, such as APC, E-cadherin and VHL. These proteins also regulate epithelial polarity and microtubule dynamics, which suggests that these processes are not mutually exclusive (Pease and Tirnauer, 2011). Although misoriented spindles do not necessarily directly result in tumorigenesis, they do contribute to several aspects of tumor biology such as tissue hypertrophy and metastasis (Pease and Tirnauer, 2011). Collectively, our studies suggest an important role for Plk2 in regulating both spindle orientation and consequently epithelial tissue integrity.
A role for Plk2 in regulating embryonic polarity in C. elegans by directly binding key polarity proteins via the polo box domains was identified previously (Nishi et al., 2008). Here, we report a role for Plk2 in the maintenance of proper apical polarization in mammary gland ductal epithelium. Plk2 could be participating in the maintenance of polarity by directly interacting with polarity proteins as reported in other model systems. Alternatively, the loss of apical polarity could also be explained by the increased proliferation of luminal MECs. These cells are initially restricted to the single-layered luminal compartment, but as they proliferate the layer is disrupted and the MECs enter the luminal space, thus disrupting apical polarization. Although it would be interesting to determine whether the loss of polarization is a cause or consequence of altered mitotic spindle orientation, unfortunately these two phenomenon are observed simultaneously, which makes it difficult to decipher which arises initially.
Previous studies have correlated the chromosomal loss of Plk2 with both ER-negative and the basal subtype of breast cancer (Weigman et al., 2012). Interestingly, in our studies we observed that in the absence of Plk2, multiparous mice develop an increased number of less differentiated lesions, as indicated by the disorganized epithelial compartments, the increase in K8+/K5+ double-positive cells and the decreased expression of ER and PR. We cannot rule out the possibility that the increase in lesions could be due to an increase in cellularity as a result of increased proliferation. Collectively, these studies suggest that Plk2 may function as a putative tumor suppressor in mammary tumorigenesis. However, other genetic and epigenetic changes are most likely required for the development of palpable tumors.
Based upon previous studies performed in vitro in U2OS cells, our expectations when these studies were initiated were that Plk2 loss in the mammary gland would cause defective centriole duplication that would result in monopolar spindles, decreased proliferation and increased apoptosis compromising outgrowth potential, as has been reported to occur in cells with compromised spindles (Cho et al., 2006; Pan et al., 2008; Xu et al., 2011). Contrary to these expectations, we observed hyper-proliferation, increased branching, altered spindle orientation, ductal luminal filling and hyperplastic lesions. These unexpected results provide novel insight into the functional role of Plk2 in vivo during postnatal mammary gland development. Although these studies suggest that Plk2 may be an important tumor suppressor in breast cancer, direct proof of this hypothesis may require studying Plk2 function in a sensitized genetic background, such as Balb/c mice expressing a mutant p53, which display an increased susceptibility to breast cancer. The original genetic screen in which Plk2 was identified employed HMECs that overexpress Myc, so the role of Plk2 may cooperate with Myc in tumorigenesis. Finally, additional studies are required to identify additional substrates that may be targeted to restore Plk2 function. The reagents necessary to perform these experiments are currently under development.
MATERIALS AND METHODS
Mouse strain
For mammary gland developmental studies, we used a Plk2+/+ and Plk2-/- C57BL6/129s mixed background germline knockout obtained from Elan Pharmaceuticals (Inglis et al., 2009). These mice were maintained in a mixed genetic background (C57BL6/129s). For transplantation experiments, we used SCID/beige purchased from Harlan Laboratories (Houston, TX, USA). All animals were housed and maintained in accordance with guidelines of the Institutional Animal Care and Use Committee of Baylor College of Medicine. All experiments were performed with age-matched littermates.
Tissue harvest
The fourth pair of mammary glands was harvested at 6, 8, 10 and 12 weeks of age for the developmental studies. Additionally, we harvested the fourth pair of glands from multiparous females post natural involution. BrdU was injected two hours prior to tissue harvest at a concentration of 10 μl/g of total body weight from a stock of (3 mg/ml); this allowed for proliferation analysis.
Whole-mount and branching analysis
For mammary gland whole-mount analysis, tissue was mounted on glass slides, fixed in Carnoy’s fixative, stained with carmine red then dehydrated and cleared in xylene. Plk2+/+ control mammary glands were isolated at 6 weeks (n=4), 8 weeks (n=5), 10 weeks (n=3), 12 weeks (n=4) and 16 weeks (n=3), and Plk2-/- mammary glands were also isolated at 6 weeks (n=3), 8 weeks (n=3), 10 weeks (n=4), 12 weeks (n=7) and 16 weeks (n=3). These glands were analyzed for percent fat pad filled and branching analysis. To evaluate the percent fat pad filled 0.71× images, and for branching quantitation 1.6× images were analyzed using a Leica MZ16F stereoscope. Following these analyses all mammary glands were paraffin embedded. Proliferation, apoptosis and polarity experiments were performed using a minimum of three animals per group. Multiparous studies were performed using Plk2+/+ (n=6) and Plk2-/- (n=8) parous animals.
Primary mammary epithelial cell isolation and transplantation
Primary mammary epithelial cells (MECS) were isolated from 8-week-old mice for transplantation experiments. MECS were isolated from #3, #4 and #5 mammary glands by mincing freshly isolated glands into 1 mm3 fragments using a Vibratome Series 800-Mcllwain Tissue Chopper. The tissue fragments were digested in DMEM/F12, which contained 2 mg/ml collagenase A (Roche Applied Science) for 1 hour at 37°C shaking at 120 rpm. The adipocytes were removed from the organoids by centrifuging at 357 g for 5 minutes following this centrifugation step, the remaining stromal cells were removed by sequential centrifugation at 357 g for 5 seconds. The organoids were then subjected to trypsinization by resuspending them in 0.25% Trypsin-EDTA for 5 minutes at 37°C then washing and filtering using a 0.40 μm cell strainer to obtain a single cell suspension. Single cells were resuspended in DMEM/F12 containing 20% Matrigel at a concentration of 20,000 cells/μl and kept on ice until transplantation. For transplants, 200,000 cells were injected into cleared contralateral fat pads of 3-week-old SCID/Beige host mice. Mammary glands were harvested 8 weeks post-transplantation and whole mount as well as histological analyses were performed.
Immunostaining
Sections (5 μm) were cut from paraffin embedded tissue, deparafinized and rehydrated through a graded series of ethanols. Antigen retrieval was performed by boiling 10 nM sodium citrate buffer for 20 minutes. Washes were performed with 1×PBS and primary antibodies were incubated at 4°C overnight in a humidified chamber. All primary antibodies were diluted in 5% BSA, 0.5% Tween-20 blocking buffer. For immunofluorescence BrdU (1:10, BD Biosciences), p-H3 (1:300, Upstate Biotech), cleaved caspase 3 (1:200, Cell Signaling), K5 (1:5000, Covance), K8 (1:250, Developmental Studies Hybridoma Bank), pERM (1:500, Cell Signaling), NuMA (1:250, Abcam), Pericentrin (1:500, BD Biosciences), γ-tubulin (1:500, Sigma) and ZO-1 (1:400, Millipore) were employed. MOM block (Vector Laboratories) was used as a diluent for GM130 (1:150, BD Biosciences) and E-cadherin (1:1000, Zymed). TUNEL assay was performed as described (Kingsley-Kallesen et al., 2002). Plk2 (1:300, Abcam) was used for immunohistochemistry. Estrogen receptor (1:400, Santa Cruz) and progesterone receptor (1:100, DAKO) staining was performed by the Lester and Sue Smith Breast Center, Baylor College of Medicine (Houston, TX, USA).
For immunohistochemistry, Vectastain Elite ABC and diaminobensidine (DAB) substrate kits (Vector Laboratories) were used. Upon detection, sections were counterstained with Hematoxylin and slides were coverslipped using Permount (Fisher).
RNA isolation
The fourth pair of mammary glands was isolated from Plk2+/+ and Plk2-/- mice, and three mice were used for each genotype. MECs were purified from these glands and total RNA was isolated using Trizol Reagent (Invitrogen) according to manufacturer’s protocols. Additionally, total RNA was isolated from whole-gland samples. Total RNA was DNase-treated (Invitrogen) and purified using RNeasy MinElute Cleanup Kit (Qiagen).
Gene expression profiling
Microarray analysis were performed on biological triplicates from Plk2+/+ and Plk2-/- on MECs. RNA was isolated and treated as previously described in RNA isolation. RNA samples were used to conduct sample quality checks by BCM Genomic and RNA Profiling Core using the Nandrop ND-1000 and Agilent Bioanalyzer Nano Chip. RNA was amplified and labeled with Cy-3 using Agilent Quick Amp Labeling Kit Protocol Version 6.5. Samples were hybridized to Agilent Sure Print 3 Mouse GE 8×60K Microarrays by the BCM Genomic and RNA Profiling Core. Data were quantile normalized, significantly regulated genes were identified by comparing Plk2+/+ with Plk2-/- using t-test and fold change. False Discovery Rate (FDR) was estimated using the Storey method for the set of genes with nominal P<0.01, FDR was estimated at 27% (suggesting ∼73% true positives) (Storey and Tibshirani, 2003). Data were further analyzed using the Database for Annotation, Visualization and Integrated Discovery (DAVID) to identify alterations in biological processes. All gene expression data has been deposited in Gene Expression Omnibus (GEO) with accession number GSE50003.
Quantitative PCR
To validate microarray data RNA was isolated from Plk2+/+ and Plk2-/- mice. RNA was isolated using TRIzol (Life Technologies), treated with DNase using DNA-Free kit (Ambion) and reverse transcribed using the High Capacity RNA to cDNA kit (Applied Biosystems). qPCR was performed on StepOnePlus Real Time PCR System (Applied Biosystems) using SYBR Green as a marker for DNA amplification. Primers to PR (mPRf-tgcacctgatctaatcctaaatga, mPRr-ggtaaggcacagcgagtagaa), Lef1 (mLef1f-tcctgaaatccccaccttct, mLef1r-tgggataaacaggctgacct), Elf5 (mElf5f-ggacctagccaccacttgtc, mElf5r-atcagggggtcacagaagg), Plk2 (mPlk2f-catcaccaccattcccact, mPlk2r-tcgtaacactttgcaaatcca), Spc25 (mSPC25f-tttccatgagcataaatgaagc, mSPC25r-gctgaaaaattgttggtcttcc) and Haus1 (mHAUS1f-aaagctgcagaggagcaact, mHAUS1r-atagtctgctcttttaactccgaga) were designed using Universal Probe Library program (Roche). Primers to Gapdh (mGAPDHf - ggagaaacctgccaagtatga, mGAPDHr-tcctcagtgtagcccaaga) were obtained from Integrated DNA Technologies. All primer sets were tested for primer efficiency using a fivefold dilution series containing five dilutions. To determine relative levels of gene expression, we used the ΔΔCT method. To further validate cell cycle-specific genes we used Spc25 and Haus1 primer sets. RNA was isolated from MECs of ten individual mice (Plk2+/+ n=6 and Plk2-/- n=4) and utilized in microarray validation analysis. 18SrRNA (18SrRNAf-gagggagcctgagaaacgg, 18SrRNAr-gtcgggagtgggtaatttgc) was used as internal reference gene. The samples used for validation were from a separate cohort of mice distinct from the samples used for microarray analysis accounting for the variability observed.
Estrogen and progesterone treatment
Estrogen and progesterone treatment was performed on 12-week-old mice as follows. Mice were injected with 100 μl of estrogen and progesterone sesame oil solution with a final concentration of 1 μg of E2 and 1 mg of progesterone under the skin between the shoulder blades. Mice were injected intraperitoneally with BrdU 2 days post-estrogen and -progesterone treatment 2 hours prior to sacrificing (Grimm et al., 2002).
Quantitation of spindle orientation
Plk2+/+ (n=5) and Plk2-/- (n=5) mice were treated with estrogen and progesterone and used for spindle orientation analysis. Immunofluorescence was performed for NuMA and p-H3 on paraffin-embedded sections. We measured the angle between the basement membrane and the plane of cell division of Plk2+/+ (110 events) and Plk2-/- (115 events) mitotic cells during the stages of metaphase, anaphase and telophase cells.
Protein extraction and immunoblot analysis
Protein was isolated using an online protocol (https://www.bcm.edu/rosenlab/index.cfm?pmid=12991). Protein was quantified using a colorimetric assay kit (Bio-Rad). SDS-PAGE was employed to separate proteins that were transferred onto a nitrocellulose membrane. Spc25 1:1000 (Abcam) and Gapdh 1:1000 (Cell Signaling) antibodies were employed. Secondary antibodies used were IRDye 700 anti-rabbit, IRDye 700 anti-mouse, IRDye 800 anti-rabbit and IRDye 800 anti-mouse and were incubated in Odyssey Blocking Buffer (Li-Cor). Protein levels were quantitated using the Odyssey software.
Supplementary Material
Acknowledgments
The authors would like to thank the following core laboratories and directors ‘Genome-wide shRNA Screening C-BASS’, ‘Dan Liu’, ‘Genomic and RNA Profiling’, ‘Lisa White’, ‘Integrated Microscopy’ and ‘Michael Mancini’. The authors also thank Li-Yuan Yu-Lee for providing antibodies. Yiqun Zhang provided technical assistance with gene array analyses. We also thank Li-Yuan, Yu-Lee, Dan Medina, Kevin Roarty and Sarah Kurley for critical reading and editing of the manuscript. Finally, we thank Maria Gonzalez-Rimbau and Alvenia Daniels for lab management, and Shirley Small for mouse colony maintenance.
Footnotes
Competing interests
The authors declare no competing financial interests.
Author contributions
E.V. conceived and performed experiments, and wrote the manuscript; E.B.K. performed experiments. A.N.S. performed experiments and edited the manuscript; C.J.C. performed statistical analysis; T.F.W. and J.M.R. contributed important intellectual insight to the development of the study, provided funding for research and wrote the manuscript.
Funding
C.J.C. was supported in part by the National Institutes of Health [NIH P30 CA125123]. This work was supported by the Komen Grant (J.M.R.) (SAC 110031) and E.V. was supported by a Department of Defense Pre-doctoral Fellowship Award [W81XWH-11-1-0079]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.108258/-/DC1
References
- Andrysik Z., Bernstein W. Z., Deng L., Myer D. L., Li Y. Q., Tischfield J. A., Stambrook P. J., Bahassi M. (2010). The novel mouse Polo-like kinase 5 responds to DNA damage and localizes in the nucleolus. Nucleic Acids Res. 38, 2931–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archambault V., Glover D. M. (2009). Polo-like kinases: conservation and divergence in their functions and regulation. Nat. Rev. Mol. Cell Biol. 10, 265–275 [DOI] [PubMed] [Google Scholar]
- Barr F. A., Silljé H. H., Nigg E. A. (2004). Polo-like kinases and the orchestration of cell division. Nat. Rev. Mol. Cell Biol. 5, 429–441 [DOI] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C. H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J. S., Dobson J., Urashima M., et al. (2010). The landscape of somatic copy-number alteration across human cancers. Nature 463, 899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman S. K., Neumüller R. A., Novatchkova M., Du Q., Knoblich J. A. (2006). The Drosophila NuMA Homolog Mud regulates spindle orientation in asymmetric cell division. Dev. Cell 10, 731–742 [DOI] [PubMed] [Google Scholar]
- Cheeseman I. M., Hori T., Fukagawa T., Desai A. (2008). KNL1 and the CENP-H/I/K complex coordinately direct kinetochore assembly in vertebrates. Mol. Biol. Cell 19, 587–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J. H., Chang C. J., Chen C. Y., Tang T. K. (2006). Depletion of CPAP by RNAi disrupts centrosome integrity and induces multipolar spindles. Biochem. Biophys. Res. Commun. 339, 742–747 [DOI] [PubMed] [Google Scholar]
- Cizmecioglu O., Krause A., Bahtz R., Ehret L., Malek N., Hoffmann I. (2012). Plk2 regulates centriole duplication through phosphorylation-mediated degradation of Fbxw7 (human Cdc4). J. Cell Sci. 125, 981–992 [DOI] [PubMed] [Google Scholar]
- Debnath J., Mills K. R., Collins N. L., Reginato M. J., Muthuswamy S. K., Brugge J. S. (2002). The role of apoptosis in creating and maintaining luminal space within normal and oncogene-expressing mammary acini. Cell 111, 29–40 [DOI] [PubMed] [Google Scholar]
- Di Cosimo S., Baselga J. (2010). Management of breast cancer with targeted agents: importance of heterogeneity. [corrected]. Nat. Rev. Clin. Oncol. 7, 139–147 [DOI] [PubMed] [Google Scholar]
- Fleming E. S., Temchin M., Wu Q., Maggio-Price L., Tirnauer J. S. (2009). Spindle misorientation in tumors from APC(min/+) mice. Mol. Carcinog. 48, 592–598 [DOI] [PubMed] [Google Scholar]
- Gargiulo G., Cesaroni M., Serresi M., de Vries N., Hulsman D., Bruggeman S. W., Lancini C., van Lohuizen M. (2013). In vivo RNAi screen for BMI1 targets identifies TGF-β/BMP-ER stress pathways as key regulators of neural- and malignant glioma-stem cell homeostasis. Cancer Cell 23, 660–676 [DOI] [PubMed] [Google Scholar]
- Glover D. M., Hagan I. M., Tavares A. A. (1998). Polo-like kinases: a team that plays throughout mitosis. Genes Dev. 12, 3777–3787 [DOI] [PubMed] [Google Scholar]
- Grimm S. L., Seagroves T. N., Kabotyanski E. B., Hovey R. C., Vonderhaar B. K., Lydon J. P., Miyoshi K., Hennighausen L., Ormandy C. J., Lee A. V., et al. (2002). Disruption of steroid and prolactin receptor patterning in the mammary gland correlates with a block in lobuloalveolar development. Mol. Endocrinol. 16, 2675–2691 [DOI] [PubMed] [Google Scholar]
- Harris J. R. (2010). Diseases of The Breast. Philadelphia, PA: Lippincott Williams & Wilkins; [Google Scholar]
- Humphreys R. C., Krajewska M., Krnacik S., Jaeger R., Weiher H., Krajewski S., Reed J. C., Rosen J. M. (1996). Apoptosis in the terminal endbud of the murine mammary gland: a mechanism of ductal morphogenesis. Development 122, 4013–4022 [DOI] [PubMed] [Google Scholar]
- Inglis K. J., Chereau D., Brigham E. F., Chiou S. S., Schöbel S., Frigon N. L., Yu M., Caccavello R. J., Nelson S., Motter R., et al. (2009). Polo-like kinase 2 (PLK2) phosphorylates alpha-synuclein at serine 129 in central nervous system. J. Biol. Chem. 284, 2598–2602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorns E., Ward T. M., Dean S., Jegg A., Thomas D., Murugaesu N., Sims D., Mitsopoulos C., Fenwick K., Kozarewa I., et al. (2012). Whole genome in vivo RNAi screening identifies the leukemia inhibitory factor receptor as a novel breast tumor suppressor. Breast Cancer Res. Treat. 135, 79–91 [DOI] [PubMed] [Google Scholar]
- Jonassen J. A., San Agustin J., Follit J. A., Pazour G. J. (2008). Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J. Cell Biol. 183, 377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsley-Kallesen M., Mukhopadhyay S. S., Wyszomierski S. L., Schanler S., Schütz G., Rosen J. M. (2002). The mineralocorticoid receptor may compensate for the loss of the glucocorticoid receptor at specific stages of mammary gland development. Mol. Endocrinol. 16, 2008–2018 [DOI] [PubMed] [Google Scholar]
- Kouros-Mehr H., Werb Z. (2006). Candidate regulators of mammary branching morphogenesis identified by genome-wide transcript analysis. Dev. Dyn. 235, 3404–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krastev D. B., Slabicki M., Paszkowski-Rogacz M., Hubner N. C., Junqueira M., Shevchenko A., Mann M., Neugebauer K. M., Buchholz F. (2011). A systematic RNAi synthetic interaction screen reveals a link between p53 and snoRNP assembly. Nat. Cell Biol. 13, 809–818 [DOI] [PubMed] [Google Scholar]
- Lapidus R. G., Nass S. J., Davidson N. E. (1998). The loss of estrogen and progesterone receptor gene expression in human breast cancer. J. Mammary Gland Biol. Neoplasia 3, 85–94 [DOI] [PubMed] [Google Scholar]
- Lawo S., Bashkurov M., Mullin M., Ferreria M. G., Kittler R., Habermann B., Tagliaferro A., Poser I., Hutchins J. R., Hegemann B., et al. (2009). HAUS, the 8-subunit human Augmin complex, regulates centrosome and spindle integrity. Curr. Biol. 19, 816–826 [DOI] [PubMed] [Google Scholar]
- Liby K., Wu H., Ouyang B., Wu S., Chen J., Dai W. (2001). Identification of the human homologue of the early-growth response gene Snk, encoding a serum-inducible kinase. DNA Seq. 11, 527–533 [DOI] [PubMed] [Google Scholar]
- Liu Y., Marks K., Cowley G. S., Carretero J., Liu Q., Nieland T. J., Xu C., Cohoon T. J., Gao P., Zhang Y., et al. (2013). Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1 mutant lung cancer. Cancer Discov. 3, 870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S., Charron J., Erikson R. L. (2003). Role of Plk2 (Snk) in mouse development and cell proliferation. Mol. Cell. Biol. 23, 6936–6943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailleux A. A., Overholtzer M., Schmelzle T., Bouillet P., Strasser A., Brugge J. S. (2007). BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev. Cell 12, 221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S., Hamasaki M., Yamamoto T., Ebisuya M., Sato M., Nishida E., Toyoshima F. (2012). ABL1 regulates spindle orientation in adherent cells and mammalian skin. Nat. Commun. 3, 626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthew E. M., Yen T. J., Dicker D. T., Dorsey J. F., Yang W., Navaraj A., El-Deiry W. S. (2007). Replication stress, defective S-phase checkpoint and increased death in Plk2-deficient human cancer cells. Cell Cycle 6, 2571–2578 [DOI] [PubMed] [Google Scholar]
- Moraes R. C., Chang H., Harrington N., Landua J. D., Prigge J. T., Lane T. F., Wainwright B. J., Hamel P. A., Lewis M. T. (2009). Ptch1 is required locally for mammary gland morphogenesis and systemically for ductal elongation. Development 136, 1423–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi Y., Rogers E., Robertson S. M., Lin R. (2008). Polo kinases regulate C. elegans embryonic polarity via binding to DYRK2-primed MEX-5 and MEX-6. Development 135, 687–697 [DOI] [PubMed] [Google Scholar]
- Pan C., Yan M., Yao J., Xu J., Long Z., Huang H., Liu Q. (2008). Aurora kinase small molecule inhibitor destroys mitotic spindle, suppresses cell growth, and induces apoptosis in oral squamous cancer cells. Oral Oncol. 44, 639–645 [DOI] [PubMed] [Google Scholar]
- Pease J. C., Tirnauer J. S. (2011). Mitotic spindle misorientation in cancer—out of alignment and into the fire. J. Cell Sci. 124, 1007–1016 [DOI] [PubMed] [Google Scholar]
- Perou C. M., Sørlie T., Eisen M. B., van de Rijn M., Jeffrey S. S., Rees C. A., Pollack J. R., Ross D. T., Johnsen H., Akslen L. A., et al. (2000). Molecular portraits of human breast tumours. Nature 406, 747–752 [DOI] [PubMed] [Google Scholar]
- Simmons D. L., Neel B. G., Stevens R., Evett G., Erikson R. L. (1992). Identification of an early-growth-response gene encoding a novel putative protein kinase. Mol. Cell. Biol. 12, 4164–4169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- St Johnston D., Ahringer J. (2010). Cell polarity in eggs and epithelia: parallels and diversity. Cell 141, 757–774 [DOI] [PubMed] [Google Scholar]
- Storey J. D., Tibshirani R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 100, 9440–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. C., Lee S. E., Xu Y. N., Kim N. H. (2010). Perturbation of Spc25 expression affects meiotic spindle organization, chromosome alignment and spindle assembly checkpoint in mouse oocytes. Cell Cycle 9, 4552–4559 [DOI] [PubMed] [Google Scholar]
- Sun T., Aceto N., Meerbrey K. L., Kessler J. D., Zhou C., Migliaccio I., Nguyen D. X., Pavlova N. N., Botero M., Huang J., et al. (2011). Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 144, 703–718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. U., Desai A. (2008). Kinetochore-microtubule interactions: the means to the end. Curr. Opin. Cell Biol. 20, 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnke S., Kemmler S., Hames R. S., Tsai H. L., Hoffmann-Rohrer U., Fry A. M., Hoffmann I. (2004). Polo-like kinase-2 is required for centriole duplication in mammalian cells. Curr. Biol. 14, 1200–1207 [DOI] [PubMed] [Google Scholar]
- Weigman V. J., Chao H. H., Shabalin A. A., He X., Parker J. S., Nordgard S. H., Grushko T., Huo D., Nwachukwu C., Nobel A., et al. (2012). Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res. Treat. 133, 865–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook T. F., Martin E. S., Schlabach M. R., Leng Y., Liang A. C., Feng B., Zhao J. J., Roberts T. M., Mandel G., Hannon G. J., et al. (2005). A genetic screen for candidate tumor suppressors identifies REST. Cell 121, 837–848 [DOI] [PubMed] [Google Scholar]
- Winkles J. A., Alberts G. F. (2005). Differential regulation of polo-like kinase 1, 2, 3, and 4 gene expression in mammalian cells and tissues. Oncogene 24, 260–266 [DOI] [PubMed] [Google Scholar]
- Xu D. R., Huang S., Long Z. J., Chen J. J., Zou Z. Z., Li J., Lin D. J., Liu Q. (2011). Inhibition of mitotic kinase Aurora suppresses Akt-1 activation and induces apoptotic cell death in all-trans retinoid acid-resistant acute promyelocytic leukemia cells. J. Transl. Med. 9, 74 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.