

Short abstract
We examined functional and evolutionary patterns in the recently constructed set of 5,873 clusters of predicted orthologs from seven eukaryotic genomes. The analysis reveals a conserved core of largely essential eukaryotic genes as well as major diversification and innovation associated with evolution of eukaryotic genomes.
Abstract
Background
Sequencing the genomes of multiple, taxonomically diverse eukaryotes enables in-depth comparative-genomic analysis which is expected to help in reconstructing ancestral eukaryotic genomes and major events in eukaryotic evolution and in making functional predictions for currently uncharacterized conserved genes.
Results
We examined functional and evolutionary patterns in the recently constructed set of 5,873 clusters of predicted orthologs (eukaryotic orthologous groups or KOGs) from seven eukaryotic genomes: Caenorhabditis elegans, Drosophila melanogaster, Homo sapiens, Arabidopsis thaliana, Saccharomyces cerevisiae, Schizosaccharomyces pombe and Encephalitozoon cuniculi. Conservation of KOGs through the phyletic range of eukaryotes strongly correlates with their functions and with the effect of gene knockout on the organism's viability. The approximately 40% of KOGs that are represented in six or seven species are enriched in proteins responsible for housekeeping functions, particularly translation and RNA processing. These conserved KOGs are often essential for survival and might approximate the minimal set of essential eukaryotic genes. The 131 single-member, pan-eukaryotic KOGs we identified were examined in detail. For around 20 that remained uncharacterized, functions were predicted by in-depth sequence analysis and examination of genomic context. Nearly all these proteins are subunits of known or predicted multiprotein complexes, in agreement with the balance hypothesis of evolution of gene copy number. Other KOGs show a variety of phyletic patterns, which points to major contributions of lineage-specific gene loss and the 'invention' of genes new to eukaryotic evolution. Examination of the sets of KOGs lost in individual lineages reveals co-elimination of functionally connected genes. Parsimonious scenarios of eukaryotic genome evolution and gene sets for ancestral eukaryotic forms were reconstructed. The gene set of the last common ancestor of the crown group consists of 3,413 KOGs and largely includes proteins involved in genome replication and expression, and central metabolism. Only 44% of the KOGs, mostly from the reconstructed gene set of the last common ancestor of the crown group, have detectable homologs in prokaryotes; the remainder apparently evolved via duplication with divergence and invention of new genes.
Conclusions
The KOG analysis reveals a conserved core of largely essential eukaryotic genes as well as major diversification and innovation associated with evolution of eukaryotic genomes. The results provide quantitative support for major trends of eukaryotic evolution noticed previously at the qualitative level and a basis for detailed reconstruction of evolution of eukaryotic genomes and biology of ancestral forms.
Background
Comparative analysis of genomes from distant species provides new insights into gene functions, genome evolution and phylogeny. In particular, the comparative genomics of prokaryotes has revealed previously underappreciated major trends in genome evolution, namely, extensive lineage-specific gene loss and horizontal gene transfer (HGT) [1-7]. To efficiently extract functional and evolutionary information from multiple genomes, rational classification of genes based on homologous relationships is indispensable. The two principal classes of homologs are orthologs and paralogs [8-11]. Orthologs are defined as homologous genes that evolved via vertical descent from a single ancestral gene in the last common ancestor of the compared species. Paralogs are homologous genes, which, at some stage of evolution, have evolved by duplication of an ancestral gene. Orthology and paralogy are intimately linked because, if a duplication (or a series of duplications) occurs after the speciation event that separated the compared species, orthology becomes a relationship between sets of paralogs, rather than individual genes (in which case, such genes are called co-orthologs).
Correct identification of orthologs and paralogs is of central importance for both the functional and evolutionary aspects of comparative genomics [12,13]. Orthologs typically occupy the same functional niche in different organisms; in contrast, paralogs evolve to functional diversification as they diverge after the duplication [14-16]. Therefore, robustness of genome annotation depends on accurate identification of orthologs. A clear demarcation of orthologs and paralogs is also required for constructing evolutionary scenarios, which include, along with vertical inheritance, lineage-specific gene loss and HGT [5,7].
In principle, orthologs, including co-orthologs, should be identified by means of phylogenetic analysis of entire families of homologous proteins, which is expected to define orthologous protein sets as clades [17-19]. However, for genome-wide protein sets, such analysis remains extremely labor-intensive, and error-prone as well. Accordingly, procedures have been developed for identifying sets of likely orthologs without explicit referral to phylogenetic analysis. These procedures are based on the notion of a genome-specific best hit (BeT), that is, the protein from a target genome that is most similar (typically in terms of similarity scores computed using BLAST or another sequence-comparison method) to a given protein from the query genome [20,21]. The assumption central to this approach is that orthologs have a greater similarity to each other than to any other protein from the respective genomes. When multiple genomes are analyzed, pairs of probable orthologs detected on the basis of BeTs are combined into orthologous clusters represented in all or a subset of the analyzed genomes [20,22]. This approach, amended with additional procedures for detecting co-orthologous protein sets and for treating multidomain proteins, was implemented in the database of Clusters of Orthologous Groups (COGs) of proteins [20,23,24]. The current COG set includes approximately 70% of the proteins encoded in 69 genomes of prokaryotes and unicellular eukaryotes [25]. The COGs have been used for functional annotation of new genomes [26-29], target selection in structural genomics [30-32], identification of potential drug targets [33,34] and genome-wide evolutionary studies [4,13,35-38]. Sonnhammer and co-workers independently developed a similar methodology for identification of co-orthologous protein sets from pairwise genome comparisons and applied it to the sequenced eukaryotic genomes [39].
A central notion introduced in the context of the COG analysis is that of a phyletic pattern, that is, the pattern of representation (presence-absence) of analyzed species in each COG [13,20]. Similar concepts have been independently developed and applied by others [40,41]. The COGs show a remarkable scatter of phyletic patterns, with only a small minority represented in all sequenced genomes. A recent quantitative study showed that parsimonious evolutionary scenarios for most COGs involve multiple events of gene loss and HGT [7]. Both similarity and complementarity among the phyletic patterns of COGs, in conjunction with other information, such as conservation of gene order, have been successfully employed to predict gene functions [13,42,43]. The comparison of phyletic pattern has been formalized in set-theoretical algorithms and systematically applied to the computational and experimental analysis of bacterial flagellar systems, which demonstrated the considerable robustness of this approach [44].
We recently extended the system of orthologous protein clusters to complex, multicellular eukaryotes [25]. Here, we examine the phyletic patterns of KOGs in connection with known and predicted protein functions. In-depth analysis of some of these KOGs resulted in prediction of previously uncharacterized, but apparently essential, conserved eukaryotic protein functions. We also reconstruct the parsimonious scenario of evolution of the crown-group eukaryotes by assigning the loss of genes (KOGs) and emergence of new genes to the branches of the phylogenetic tree and explicitly delineate the minimal gene sets for various ancestral forms. To our knowledge, this is the first systematic, genome-wide examination of the sets of orthologous genes in eukaryotes.
Results and discussion
KOGs for seven sequenced eukaryotic genomes: functional and evolutionary implications of phyletic patterns
Eukaryotic KOGs were constructed on the basis of the comparison of proteins encoded in the genomes of three animals (Homo sapiens [45], the fruit fly Drosophila melanogaster [46] and the nematode Caenorhabditis elegans [47]), the green plant Arabidopsis thaliana (thale cress) [48], two fungi (budding yeast Saccharomyces cerevisiae [49] and fission yeast Schizosaccharomyces pombe [50]) and the microsporidian Encephalitozoon cuniculi [51]. The procedure for KOG construction was a modification of the one previously used for COGs [20,24] and is described in greater detail elsewhere ([25]; see also Materials and methods). An important difference stems from the fact that complex eukaryotes encode many more multidomain proteins than prokaryotes and, furthermore, orthologous eukaryotic proteins often differ in domain composition, with additional domains accrued in more complex forms [3,45]. Accordingly, and unlike the original COG construction procedure, probable orthologs with different domain architectures were assigned to one KOG and were not split if they shared a common core of domains. In addition to the KOGs, which consisted of at least three species, clusters of putative orthologs from two species (TWOGs) and lineage-specific expansions (LSEs) of paralogs from each of the analyzed genomes were identified ([25,52]; see also Materials and methods). In most of the analyses discussed below, KOGs and TWOGs are treated together, unless otherwise specified.
Figure 1 shows the assignment of the proteins from each of the analyzed eukaryotes to KOGs with different numbers of species, TWOGs and LSEs. The fraction of proteins assigned to KOGs tends to decrease with the increasing genome size, from 81% for S. pombe to 51% for the largest, the human genome. (For reasons that remain unclear, but might be related to its intracellular parasitic lifestyle, E. cuniculi has a relatively small fraction of conserved proteins that belonged to KOGs: approximately 60%.) The contribution of LSEs shows the opposite trend, being the greatest in the largest genomes, that is, human and Arabidopsis, and minimal in the microsporidian (Figure 1). A notable difference was observed between eukaryotes in terms of their representation in KOGs found in different numbers of species. While the three unicellular organisms are represented mainly in the highly conserved seven- or six-species KOGs, a much larger fraction of the gene set in animals and Arabidopsis is accounted for by LSEs, and by KOGs found in three or four genomes. These include animal-specific genes and genes that are shared by plants and animals but not by fungi and the microsporidian (Figure 1). The large number of KOGs in the latter group (700 KOGs represented in Arabidopsis and at least two animal species) is notable and probably results from massive, lineage-specific loss of genes during eukaryotic evolution (see below).
Figure 1.
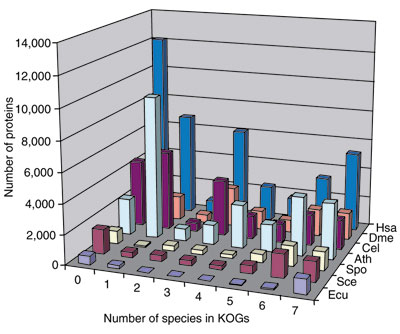
Assignment of proteins from each of the seven analyzed eukaryotic genomes to KOGs with different numbers of species and to LSEs. 0, Proteins without detectable homologs (singletons); 1, LSEs. Species abbreviations: Ath, Arabidopsis thaliana; Cel, Caenorhabditis elegans; Dme, Drosophila melanogaster; Ecu, Encephalitozoon cuniculi; Hsa, Homo sapiens; Sce, Saccharomyces cerevisisae; Spo, Schizosaccharomyces pombe.
The phyletic patterns of KOGs reveal both the existence of a conserved eukaryotic gene core and substantial diversity. The 'pan-eukaryotic' genes, which are represented in each of the seven analyzed genomes, account for around 20% of the KOGs, and approximately the same number of KOGs include all species except for the microsporidian, an intracellular parasite with a highly degraded genome [51]. Among the remaining KOGs, a large group includes representatives of the three analyzed animal species (worm, fly and humans) but a substantial fraction (approximately 30%) are KOGs with unexpected patterns, for example, one animal, one plant and one fungal species (see [53] and examples in Table 1).
Table 1.
KOGs and TWOGs with unexpected phyletic patterns (examples)
| KOG/TWOG number | Phyletic pattern* | (Predicted) structure and function | Prokaryotic homologs | Comments |
| TWOG0892 | ---H--E | Discoidin domain protein, potential regulator of proteasome activity | Detected in a few phylogenetically scattered bacteria, no COG so far [69] | |
| TWOG0263 | A-----E | ATP/ADP translocase | ATP/ADP translocases of chlamydia, rickettsia, Xylella fastidiosa | ATP/ADP translocase is a hallmark of intracellular parasites and symbionts, which allows them to scavenge ATP from the host cell; chloroplast protein in plants. Could be acquired by plants and microsporidia via independent HGT from bacteria. [58] |
| TWOG0689 | ---HY-- | Uncharacterized protein essential for propionate metabolism | PrpD protein of several bacteria and archaea (COG2079) | The yeast and human (and the orthologs from other vertebrates) proteins show the greatest similarity to different subsets of bacterial orthologs, which might suggest independent HGT events. |
| TWOG0871 | ---H-P- | Uncharacterized conserved protein, probably enzyme | COG4336, sporadic representation in several bacterial lineages | The human (and mouse) protein has an additional domain conserved in the archaeon Pyrococcus. Human and S. pombe proteins are most similar to different subsets of bacterial homologs, which suggests the possibility of independent HGT events. |
| TWOG0788 | A----P- | Urease | Ureases of many bacterial species | Highly conserved enzyme present in plants and many fungi but not S. cerevisiae. Plant and fungal ureases have a common domain architecture distinct from that of bacterial orthologs, which suggests monophyletic origin. Might have evolved via early HGT from bacteria (proto-mitochondria?) with subsequent loss in animals and some fungi. |
| 4751 | A--H--E | Recombination repair protein BRCA2, contains varying number of BRCA2 repeats | None | Although sequence conservation is limited to the BRC repeats [101] the number of which varies substantially, statistical significance of the observed sequence similarity and the absence of other homologs suggests that the proteins in this KOG are true orthologs. Apparent orthologs of BRCA2 are detectable also in other species from the taxa represented in the KOGs (mosquito Anopheles gambiae, fungus Ustilago maydis) [102] and in early-branching eukaryotes (Leishmania, Trypanosoma; E.V.K., unpublished work), suggesting that evolution of BRCA2 involved multiple gene losses |
| 4597 | A--H--E | TATA-binding protein 1-interacting protein | None | Probable multiple gene losses |
| 4486 | A--H--E | 3-methyl-adenine DNA glycosylase | Orthologs in many bacteria (COG2094) | The plant protein and those from mammals and microsporidia show the greatest similarity to different subsets of bacterial orthologs. Evolution might have included a combination of gene loss and independent HGT events |
| 1594 | A-D-Y-- | Predicted epimerase related to aldose 1-epimerase | Bacterial orthologs, primarily proteobacteria (COG0676) | Eukaryotic proteins are more closely related to each other than to bacterial orthologs, indicating monophyletic origin. Function remains unknown; might be involved in a distinct and still uncharacterized pathway of polysaccharide biosynthesis. LSE in Arabidopsis (seven paralogs). |
| 4141 | ---HYPE | Rad52/22, protein involved in double-strand break repair | None | Probable gene loss in plants, insects and nematodes |
| 4528 | -CDH--E | Uncharacterized predicted enzyme, possibly a polynucleotide kinase (structure of the ortholog from the bacterium Thermotoga maritima has been determined - pdb code 1j5u) | Conserved in all archaea and several bacteria (COG1371) | Context analysis of archaeal and bacterial genomes suggests functional interaction between proteins of KOG5324 and KOG4246, RNA 3'-terminal phosphate cyclase (KOG4398, COG0430), and tRNA/rRNA cytosine C5-methylase (KOG1299/COG0144) ([103] and E.V.K., unpublished observations). Taken together, the observations appear to implicate KOG5324 and KOG4246 in a still uncharacterized pathway of rRNA and/or tRNA processing and modification. Conservation of these proteins in archaea and early-branching eukaryotes suggests lineage-specific gene loss in plants and fungi. |
| 3833 | -CDH--E | Uncharacterized predicted enzyme, possibly a polynuclotide phosphatase | Conserved in all archaea and several bacteria (COG1690) | See comment for KOG5324 |
*Abbreviations: A, thale cress A. thaliana; C, nematode C. elegans; D, fruit fly D. melanogaster; E, microsporidian Encephalitozoon cuniculi; H, Homo sapiens; S, budding yeast S. cerevisiae; P, fission yeast S. pombe; a letter indicates the presence of the respective species in the given KOG and a dash indicates its absence.
During the manual curation of the KOG set, the KOGs with unexpected patterns were scrutinized in an effort to detect potential highly diverged members from one or more of the analyzed genomes. Some of these unexpected patterns might indicate that a gene is still missing in the analyzed set of protein sequences from one or more of the species included; reports of newly discovered genes have appeared since the release of the initial reports on genome sequences of complex eukaryotes, for example, as a result of massive sequencing of human cDNAs [54], exhaustive annotation of the Drosophila genome [55] and comparative analysis of closely related yeast genomes [56]. The unexpected phyletic patterns seem, however, largely to reflect the extensive, lineage-specific gene loss that is characteristic of eukaryotic evolution [57]; on many occasions, this scenario is supported by the presence of orthologs in other eukaryotic lineages and/or in prokaryotes (Table 1). However, interesting exceptions to the multiple loss explanation might exist as exemplified by the ATP/ADP-translocase, which is present in Arabidopsis and Encephalitozoon and could have evolved via independent HGT from intracellular bacterial parasites ([58] and Table 2).
Table 2.
KOGs represented by exactly one ortholog in seven analyzed eukaryotic genomes (examples)
| KOG number | (Predicted) function | Multiprotein complex | Functional class* | Prokaryotic homologs | Fitness class† | Comments | |
| Yeast‡ | Worm§ | ||||||
| Genes experimentally or computationally characterized previously | |||||||
| 0392 | SNF2 family DNA-dependent ATPase | TBP-DNA complex | Many bacteria and archaea (COG0553) | 0 | 1 | Involved in regulation of transcription from POL II promoters [104] | |
| 0121 | Nuclear cap-binding protein complex, subunit CBP20 (RRM-domain-containing RNA-binding protein) | Cap-binding complex | A | Several bacteria (COG0724) | 1 | X | RRM-domain proteins show scattered presence in bacteria and might have been horizontally transferred from eukaryotes |
| 0213 | U2-snRNP associated splicing factor 3b, subunit 1 | Spliceosome | A | None | 0 | 0 | |
| 0227 | snRNA-associated protein, splicing factor 3a, subunit b (Prp11p) | Spliceosome | A | None | 0 | 0 | |
| 2268 | Predicted nucleic-acid-binding protein kinase of the RIO1 family; 40S ribosomal subunit biogenesis/18S rRNA processing | Pre-40S subunit | A | Orthologs in most archaea but not in bacteria (COG0478) | 0 | X | One of the very small number of protein kinases that show a clear-cut orthologous relationship between all eukaryotes and most archaea, and, apparently, the only one containing a helix-turn-helix nucleic-acid-binding domain. [105] Associated with yeast pre-40S subunit and required for its maturation. [106] |
| 3031 | Protein required for 60S ribosomal subunit biogenesis; [107] contains the IMP4 domain, which is involved in rRNA processing [108]; paralog of KOG3095 and KOG3292, which are also represented in all analyzed genomes. | Processosome | A | Distantly related to COG2136, represented by orthologs in most archaea, but not in bacteria (KSM, unpublished) | 0 | X | The COG2136 proteins appear to be subunits of the predicted archaeal exosome [109]. Apparently, this gene has undergone at least two ancient duplications in eukaryotes |
| 3045 | Predicted RNA methylase involved in rRNA processing | Processosome? | A | Distantly related to numerous Rossmann-fold methylases but prokaryotic orthologs could not be confidently identified | 1 | 1 | This protein (Rrp8p in yeast) has been shown to participate in the processing of rRNA and sequence analysis reveals the presence of a Rossmann-fold methylase domain [110]. Therefore Rrp8p probably methylates either snoRNA or rRNA itself. |
| 3064 | RNA-binding nuclear protein containing a distinct C4 Zn-finger; implicated in the biogenesis of 60S ribosomal subunits [111] | Processosome | A | None | 0 | 0 | Initially identified in yeast as the MAK16 protein required for dsRNA virus reproduction [112] |
| 0291, 0302, 0306, 310, 0319, 0650, 1272 | WD40-repeat proteins, subunits of rRNA processing complexes [69,70] | Processosome | A | WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319) | all 0 | X,X,1,X,1,1,1 | |
| 0284 | Polyadenylation factor I complex, subunit PFS2, WD40-repeat protein | Poly-adenylation complex | A | Same as above (COG2319) | 0 | X | |
| 0337 | RNA helicase involved in 28S rRNA processing | Processosome | A | Most of the archaea and bacteria (COG0513) | 0 | X | |
| 0343 | RNA helicase involved in 28S rRNA processing | Processosome | A | Most of the archaea and bacteria (COG0513) | 0 | X | |
| 1069 | 3'-5' exoribonuclease (RNAse PH), exosome subunit Rrp46 | Exosome | A | Most bacteria and archaea (COG0689) | 0 | 1 | |
| 1070 | Exosome subunit Rrp5 (RNA-binding S1 domain fused to TPR repeats) | Exosome | A | Most bacteria (COG0539, COG0457) | 0 | 1 | |
| 1135 | mRNA cleavage and polyadenylation complex subunit CFT2 (CPSF) | Cleavage and polyadenylation complex | A | Most archaea and some bacteria (COG1236) | 0 | 0 | |
| 1914 | mRNA cleavage and polyadenylation factor I complex, subunit RNA14 | Cleavage and polyadenylation complex | A | None | 0 | X | |
| 1975 | RNA (guanine-7-) methyltransferase (capping enzyme subunit) | Capping enzyme | A | Numerous methyltrans-ferases (COG0500) but no ortholog | 0 | 1 | |
| 2051 | Nonsense-mediated mRNA decay complex, subunit 2 | NMD complex | A | None | 1 | X | |
| 2554 | Pseudouridylate synthase | ? | A | Most archaea and bacteria (COG0101) | 1 | 1 | |
| 2613 | Upf1p-interacting protein, NMD complex subunit Nmd3p | NMD complex | A | All archaea, no bacteria (COG1499) | 0 | X | |
| 2771 | tRNA-specific adenosine-34 deaminase subunit Tad3p | Heterodimeric RNA-specific deaminase | A | Most bacteria and some archaea (COG0590) | 0 | X | |
| 2780 | Protein involved in ribosomal large subunit assembly (RPF1), contains IMP4 domain | Processosome | A | Most archaea, no bacteria (COG2136) | 0 | 1 | |
| 2781 | Subunit of the small (ribosomal) subunit (SSU) processosome (snoRNP), IMP4 | Processosome | A | Most archaea, no bacteria (COG2136) | 0 | 1 | |
| 2874 | Protein involved in rRNA processing and ribosomal assembly | ? | A | All archaea, no bacteria (COG1094) | 0 | 1 | Predicted RNA-binding protein containing KH domain |
| 3013 | Exosome subunit Rrp4 | Exosome | A | Most archaea, on bacteria (COG1097) | 0 | X | |
| 3031 | Protein involved in large ribosome subunit assembly and 28S rRNA processing (Rrf2) | Processosome | A | None | 0 | X | Contains the BRIX domain |
| 3322 | RNAse P/MRP subunit, involved in processing of pre-tRNAs and the 5.8S rRNA | RNAse P/MRP holoenzyme | A | None | 0 | 1 | |
| 3448 | Predicted snRNP core protein | Spliceosome | A | All archaea, no bacteria (COG1958) | 0 | 1 | |
| 3482 | Small nuclear ribonucleoprotein (snRNP) SMF subunit | Spliceosome | A | All archaea, no bacteria (COG1958) | 0 | 0 | |
| 2463 | Predicted RNA-binding protein, consisting of a PIN domain and a Zn-ribbon. Involved in 26S proteasome assembly | 26S proteasome, pre-40S subunit | A,O | Represented by orthologs in all archaea but no bacteria (COG1349) | 0 | X | PIN domain has been detected in exosome subunits and is thought to have RNA-binding properties or even nuclease activity [113,114]. The demonstration of the role of this protein (Nob1p) in proteasome assembly [115], 40S ribosome subunit assembly, and the processing of 18S rRNA 3'-end [116] supports the connection between degradation of RNA and proteins that seems to have been established already in archaea [109]. |
| 3273 | Predicted RNA-binding protein containing KH domain, interacts with Nob1p | 26S proteasome, pre-40S subunit | A,O | Orthologs in all archaea but no bacteria (COG1094) | 0 | 0 | This is the second predicted RNA-binding protein involved in proteasome assembly, [115] which emphasizes the aforementioned link between RNA and protein processing |
| 1831 | Deadenylating 3'-5' exonuclease, negative regulator of PolII transcription | CCR4-NOT core complex | AK | None | 0 | 0 | |
| 1159 | NADP-dependent flavoprotein reductase, probably sulfite reductase subunit | ? | CL | Many bacteria (COG0369) | 0 | X | Genetic evidence of a role in DNA replication [117] |
| 1800 | Ferredoxin/adrenodoxin reductase | ? | C | Most bacteria and some archaea (COG0493) | 0 | X | |
| 1173 | Anaphase-promoting complex (APC), Cdc16 subunit (TPR-repeat protein) | APC | D | Most of archaea and bacteria have TPR-repeat proteins (COG0457) but no orthologs of Cdc16 | 0 | 0 | |
| 3437 | Anaphase-promoting complex (APC), subunit 10 | APC | D | None | 1 | 1 | |
| 1358 | Serine palmitoyltransferase | ? | I | Most bacteria and some archaea (COG0156) | 0 | 0 | |
| 1511 | Mevalonate kinase | ? | I | Most archaea and some bacteria (COG1577) | 0 | X | |
| 3059 | N-acetylglucosaminyltransferase complex, subunit PIG-C/GPI2, involved in phosphatidylinositol biosynthesis | N-acetylglucos-aminyltransferase complex | I | None | 0 | 1 | |
| 0467 | Translation elongation factor 2 paralog (GTPase) | ? | J | All (COG0480) | 0 | X | Involved in 60S ribosomal subunit maturation [118] |
| 1147 | Glutamyl-tRNA synthetase | Multispecificity aminoacyl-tRNA synthetase complex | J | All (COG0008) | 0 | X | |
| 2784 | Phenylalanyl-tRNA synthetase, beta subunit | Heterodimeric phenylalanyl-tRNA synthetase | J | All (COG0016) | 0 | X | |
| 3123 | Diphtamide synthase (methyltransferase) | ? | J | All archaea, no bacteria (COG1798) | 1 | 1 | |
| 0261 | RNA polymerase III, largest subunit | RNAPIII holoenzyme | K | All (COG0086) | 0 | X | |
| 0262 | RNA polymerase I, largest subunit | RNAPI holoenzyme | K | All (COG0086) | 0 | X | |
| 0215 | RNA polymerase III, second largest subunit | RNAPIII holoenzyme | K | All (COG0085) | 0 | X | |
| 0216 | RNA polymerase I, second largest subunit | RNAPI holoenzyme | K | All (COG0085) | 0 | X | |
| 1063 | RNA polymerase II elongator complex, subunit ELP2, WD repeat protein | RNA polymerase II elongator complex | K | WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319) | 1 | X | |
| 1131 | RNA polymerase II transcription initiation/nucleotide excision repair factor TFIIH, 5'-3' helicase subunit RAD3 | RNAPII holoenzyme | K | Most archaea and bacteria (COG1199) | 0 | X | |
| 1920 | RNA polymerase II Elongator subunit | RNAP II elongator complex | K | None | 1 | X | |
| 1932 | TBP-associated factor (Taf2p) | TFIID complex | K | None | 0 | X | |
| 2009 | Transcription initiation factor TFIIIB, Bdp1 subunit (Myb domain) | TFIIIB | K | None | 0 | 0 | |
| 2076 | RNA polymerase III transcription factor TFIIIC, TPR-repeat-containing protein | TFIIIC | K | Most of archaea and bacteria have TPR-repeat proteins (COG0457) but no orthologs of TFIIC | 0 | X | |
| 2487 | RNA polymerase II transcription initiation/nucleotide excision repair factor TFIIH, subunit TFB4 | TFIIH | K | None | 0 | 1 | |
| 2691 | RNA polymerase II subunit 9 | RNAP II holoenzyme | K | Most archaea, no bacteria (COG1594) | 1 | X | |
| 2807 | RNA polymerase II transcription initiation/nucleotide excision repair factor TFIIH, SSL1 subunit | TFIIH | K | No orthologs although von Willebrand A domains are present in a variety of prokaryotic proteins | 0 | 0 | Consists of a von Willebrand A domain most closely related to those in the proteasome subunit RPN10 [119] and a Zn-finger domain |
| 2907 | RNA polymerase I transcription factor TFIIS, subunit A12.2/RPA12 | TFIIS | K | All archaea, no bacteria (COG1594) | 1 | 0 | |
| 3169 | RNA polymerase II transcriptional regulation mediator | Mediator complex [120] | K | None | 0 | X | |
| 3233 | RNA polymerase III subunit C34 | RNAP III holoenzyme | K | None | 0 | 1 | |
| 3297 | RNA polymerase III subunit C25 | RNAP III holoenzyme | K | All archaea, no bacteria (COG1095) | 0 | 0 | |
| 3438 | Subunit common to RNA polymerases I (A) and III (C); Rpc19p | RNAP I and III holoenzymes | K | 0 | 1 | ||
| 3471 | RNA polymerase II transcription initiation/nucleotide excision repair factor TFIIH, subunit TFB2 | TFIIH | K | None | 0 | X | |
| 3490 | Transcription elongation factor SPT4, Zn-ribbon protein | Chromatin-associated transcription complexes | K | None | 1 | 1 | |
| 3497 | RNA polymerase II subunit; Rpb10p | RNAP II holoenzyme | K | All archaea, no bacteria (COG1644) | 0 | X | |
| 3901 | Transcription initiation factor IID subunit (Taf13p) | TFIID | K | None | 0 | X | |
| 3949 | RNA polymerase II elongator complex, subunit ELP4 | RNAP II elongator complex | K | None | 1 | 1 | |
| 4086 | SOH1 protein potentially involved in Pol II transcription regulation and repair | SMCC complex [121] | K | None | 1 | X | |
| 1532 | Predicted GTPase of the XAB1 family [122] | TBP-free TAF(II) complex | L | All archaea and several bacteria (COG1100) | 0 | 0 | XP-A-binding protein in humans, thus implicated in repair ([122] and references therein). |
| 1533 | Predicted GTPase of the XAB1 family (paralog of KOG1757) [122] | TBP-free TAF(II) complex? | L | All archaea and several bacteria (COG1100) | 0 | X | Might have a function in repair given the paralogous relationship with KOG1757. |
| 1625 | DNA polymerase α processivity subunit, inactivated phosphatase | DNA polymerase α holoenzyme | L | Small subunit of archaeal DNA polymerase II (COG1311) | 0 | 0 | The small, regulatory subunit of DNA polymerase α also forms a pan-eukaryotic KOG3044, which is a paralog of KOG0861 (the only recent duplication in KOG3044 is seen in vertebrates). In contrast, another paralog, the small subunit of DNA polymerase ε, is represented in animals, fungi and the early-branching protozoan Plasmodium, but not in plants or Microsporidia. Thus, the history of this polymerase subunit apparently involved inactivation of the phosphatase (or nuclease) inherited from archaea, with subsequent duplications at early stages of eukaryotic evolution [123] |
| 0479 | DNA replication licensing factor MCM3 | Pre-replication complex | L | All archaea, no bacteria (COG1241) | 0 | X | |
| 0481 | DNA replication licensing factor MCM5 | Pre-replication complex | L | All archaea, no bacteria (COG1241) | 0 | X | |
| 0482 | DNA replication licensing factor MCM7 | Pre-replication complex | L | All archaea, no bacteria (COG1241) | 0 | 0 | |
| 0964 | Structural maintenance of chromosome protein 3 (cohesin subunit SMC3) | Sister chromatid cohesion complex | L | Many archaea and bacteria (COG1196) | 0 | X | |
| 0979 | Structural maintenance of chromosome protein 5 (cohesin subunit SMC5) | Sister chromatid cohesion complex | L | Many archaea and bacteria (COG1196) | 0 | X | |
| 1942 | TBP-interacting protein TIP49 (DNA helicase) | chromatin remodeling complex | L | Most of the archaea, no bacteria (COG1224) | 0 | 0 | |
| 1979 | DNA mismatch repair ATPase, MLH1 | Mismatch repair complex | L | Most bacteria and some archaea (COG0323) | 1 | 1 | |
| 2267 | DNA primase, large subunit | DNA polymerase α:primase complex | L | All archaea, no bacteria (COG2219) | 0 | 0 | |
| 2299 | Ribonuclease HI | Replisome | L | All archaea, most bacteria (COG0164) | 1 | X | |
| 2310 | DNA repair exonuclease MRE11 | MRN complex involved in double-strand break repair | L | All archaea, most bacteria (COG0420) | 1 | 1 | |
| 2929 | Origin recognition complex, subunit 2 (ORC2) | ORC | L | None | 1 | 1 | |
| 0179 | 20S proteasome, regulatory subunit beta type PSMB1/PRE7 (paralog of KOG0185) | 20S proteasome | O | All archaea but only actinomycetes among bacteria (COG0638) | 0 | 0 | |
| 0185 | 20S proteasome, regulatory subunit beta type PSMB4/PRE4 (paralog of KOG0179) | 20S proteasome | O | All archaea but only actinomycetes among bacteria (COG0638) | 0 | 0 | |
| 2708 | Predicted metalloprotease with chaperone activity (RNAse H/HSP70 fold) [124] | Putative complex involved in translation regulation [125] | O | Represented by orthologs in all archaea and bacteria (COG0533) | 0 | X | One of the few remaining uncharacterized proteins that are universally conserved in all cellular life forms. The only experimentally demonstrated activity is that of sialoglycoprotease but fusion with a distinct protein kinase in several archaea and analysis of gene neighborhood suggest a fundamental role in signal transduction, possibly translation regulation. [125] |
| 0301 | Protein required for normal rates of ubiquitin-dependent proteolysis, contains WD40 repeats | Proteasome? | O | Same as above (COG2319) | 1 | X | |
| 0358 | Chaperonin complex component, TCP-1 delta subunit (CCT4) | TCP-1 | O | All archaea and nearly all bacteria (COG0459) | 0 | 0 | |
| 0363 | Chaperonin complex component, TCP-1 beta subunit (CCT2) | TCP-1 | O | All archaea and nearly all bacteria (COG0459) | 0 | 0 | |
| 0687 | 26S proteasome regulatory complex, subunit RPN7/PSMD6 | 26S proteasome | O | None | 0 | 0 | |
| 1299 | Vacuolar sorting protein VPS45/Stt10 (Sec1 family) | t-SNARE complex | O | None | 1 | X | Involved in t-SNARE complex assembly [126] |
| 1349 | GPI-anchor transamidase complex, GPI8 subunit | GPI-anchor transamidase complex | O | Distantly related proteases in some bacteria (no COG) | 0 | 1 | |
| 1943 | Beta-tubulin folding cofactor D, involved in chromosome segregation | ? | O | None | 1 | 1 | |
| 2015 | NEDD8-activating complex, UBA3 subunit | NEDD8-activating complex | O | Most bacteria and some archaea (COG0476) | 1 | 1 | |
| 2126 | Phosphoethanolamine N-methyltransferase involved in GPI-anchor biosynthesis | ? | O | Several bacteria and archaea (COG1524) | 0 | X | |
| 2884 | 26S proteasome regulatory complex, subunit RPN10/PSMD4 | 26S proteasome regulatory complex | O | No orthologs although von Willebrand A domains are present in a variety of prokaryotic proteins | 1 | 1 | Contains von Willebrand A domain |
| 2908 | 26S proteasome regulatory complex, subunit RPN9/PSMD13 | 26S proteasome regulatory complex | O | None | 0 | 0 | Contains PINT domain |
| 0209 | Endoplasmic reticulum membrane P-type ATPase | ? | P | Many bacteria and some archaea (COG0474) | 1 | X | |
| 3379 | Uncharacterized member of the histidine triad superfamily of nucleotide hydorlases | ? | R | Most archaea and bacteria (COG0537) | 1 | X | Only biochemical function predicted. |
| 2635 | Coatomer (COPI) complex delta subunit | COPI complex | U | None | 0 | 0 | |
| 2927 | Membrane component of ER protein translocation apparatus (Sec62) | Sec complex | U | None | 0 | 1 | |
| 2978 | Dolichol-phosphate mannosyltransferase | ? | U | All archaea, most bacteria (COG0463) | 0 | X | |
| 3198 | Signal recognition particle, subunit Srp19 | Signal recognition particle | U | All archaea, no bacteria (COG1400) | 0 | X | |
| 3315 | Subunit of the targeting complex (TRAPP) involved in ER to Golgi trafficking | TRAPP | U | None | 0 | X | |
| 3369 | Subunit of the targeting complex (TRAPP) involved in ER to Golgi trafficking | TRAPP | U | None | 0 | X | |
| 1992 | Nuclear export receptor CSE1/CAS (importin beta) | ? | YU | None | 0 | X | |
| New functional predictions | |||||||
| 2316 | PP-loop family ATP pyrophosphatase domain, which in fungi, plants and insects is fused to a duplicated translation inhibitor domain. The fusion, along with the phyletic pattern of the PP-ATPase domain, suggests an essential function in translation regulation | ? | A | Orthologs of the PP-loop domain are present in all archaea (COG2102) but not in bacteria. Orthologs of the translation inhibitor domain are found in most bacteria and several archaea (COG0251) | 1 | X | PP-loop ATPases have been previously implicated in base thiolation in various RNAs [127] and proteins in this K/COG might have a similar function, which is likely to be conserved in eukaryotes and archaea. However, the fusion with translation inhibitor, which has been reported to have endoribonuclease activity [128] is a eukaryote-specific feature |
| 2523 | Predicted RNA-binding protein containing a PUA domain, probable role in RNA modification [129] | Putative novel RNA modification complex | A | Orthologs present in all archaea (COG2016) but not in bacteria | 1 | X | Several of the archaeal orthologs of this protein form fusions with a PP-loop ATPase domain implicated in base thiolation [127]. Thus, the proteins of this KOG might interact with those of KOG2840 (pan-eukaryotic, duplications in Arabidopsis and worm) or KOG2594 (missing in humans and microsporidia) to form a novel enzymatic complex involved in RNA modification |
| 0270, 0271, 1539 | WD40-repeat proteins | Processosome | A | WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319) | all 0 | X,1,X | By analogy with other conserved WD40-repeat proteins, predicted to be subunits of rRNA processing/ribosome assembly complexes |
| 2321 | Nucleolar protein, contains WD40 repeats | rRNA processosome? | A | WD40-repeat proteins are present in several bacterial lineages and are particularly abundant in cyanobacteria but are missing in most archaea; none of them appear to be obvious orthologs of this protein (COG2319) | 0 | 1 | Probable subunit of an rRNA-processing complex |
| 1763 | Uncharacterized conserved protein containing a CCCH Zn-finger; possible role in RNA processing or splicing | ? | A | None | 1 | 1 | CCCH fingers have been shown to bind 3' untranslated regions in various mRNAs [130,131] |
| 2837 | Protein containing a U1-type, RNA-binding C2H2 Zn-finger. Probable role in RNA splicing/processing | Spliceosome? | A | None | 0 | 0 | U1-type fingers are essential for the assembly of U1 RNP [132] |
| 3073 | Predicted RNA-binding protein containing PIN domain and involved in 18S rRNA processing | Pre-40S subunit | A | Most archaea, no in bacteria (COG1412) | 0 | 1 | Interacts with Nop14p and is required for 40S subunit biogenesis and 18S rRNA maturation (11694595). The presence of the PIN domain suggests RNA-binding and, possibly, RNAse activity |
| 3154 | Uncharacterized protein with potential function in translation or ribosomal biogenesis | Pre-40S subunit? | A? | Most archaea, no bacteria (COG2042) | 1 | X | The general functional prediction stems from the observation that the gene for this protein forms a predicted conserved operon with the gene for ribosomal protein L40E in several archaeal genomes |
| 3214 | Small protein containing a Zn-ribbon, possibly RNA-binding; potential role in RNA processing or transcription regulation | ? | A? | Conserved in Crenarchaeota (COG4888) | 1 | 1 | |
| 3800 | Predicted E3 ubiquitin ligase containing RING finger, subunit of transcription/repair factor TFIIH and CDK-activating kinase assembly factor | TFIIH | KO | None | 0 | X | |
| 3176 | Predicted α-helical protein, possibly involved in replication/repair; paralog of KOG3636 | A novel complex with PCNA involved in replication? | L? | Conserved in most (possibly all) archaea but not in bacteria (COG1711) | 0 | X | A function in DNA replication/repair and/or transcription is suggested by the analysis of the genome context of archaeal orthologs which form an evolutionarily conserved association with the genes for replication sliding clamp (PCNA ortholog) (K.S.M. and E.V.K., unpublished work) |
| 3303 | Predicted α-helical protein, possibly involved in replication/repair transcription; paralog of KOG3508 | A novel complex with PCNA involved in replication? | L? | Conserved in most (possibly all) archaea but not in bacteria (COG1711) | 0 | 0 | A function in DNA replication/repair and/or transcription is suggested by the analysis of the genome context of archaeal orthologs which form an evolutionarily conserved association with the genes for replication sliding clamp (PCNA ortholog) (K.S.M. and E.V.K., unpublished.work) |
| 0396 | Predicted E3 ubiquitin ligase | Ub ligase | O | None | 1 | 1 | The proteins in this KOG contain a modified RING domain, which might not be capable of metal-binding similarly to the U-box domain [133] that has been shown to function as E3 [134] |
| 1443 | Multitransmembrane protein, predicted drug/metabolite transporter | ? | R | Most archaea and bacteria (COG0697) | 1 | X | |
| 2647 | Multitransmembrane protein, potential transporter | ? | R | Most bacteria and some archaea (COG0628) | 0 | 1 | |
| 2488 | Predicted N-acetyltransferase | ? | R | Most archaea and bacteria (COG0454) | 1 | X | Putative role in ribosomal maturation? |
| 3347 | Predicted nucleotide kinase; nuclear protein (Fap7p) | ? | R | Conserved in all archaea but not in bacteria (COG1936) | 0 | 1 | Involved in oxidative stress reponse in yeast [135] |
| 3974 | Predicted sugar kinase | Putative novel complex with KOG2585 proteins | R | All archaea and most bacteria (COG0063) | 1 | 1 | Based on fusions seen in prokaryotes, predicted to interact functionally and, possibly, physically with uncharacterized proteins of KOG2585 (represented in all eukaryotes but includes paralogs in some species) |
| No functional prediction | |||||||
| 2318 | Uncharacterized conserved protein | ? | S | None | 0 | 1 | |
| 3237 | Uncharacterized conserved protein containing coiled-coil domain | ? | S | None | 0 | 1 | Coiled-coil domains are often involved in complex assembly; this could be an uncharacterized component of the chromatin or the spliceosome |
Common phyletic patterns of genes that otherwise were not suspected to be functionally linked might suggest the existence of such connections and prompt additional analysis leading to concrete functional predictions [42,59-61]. The pair of KOG5324 and KOG4246 is a case in point that has not been described previously. The initial observation that these KOGs share the same unusual pattern of presence-absence in eukaryotes, and have similar phyletic patterns in prokaryotes, with a ubiquitous presence in archaea, prompted a more detailed examination of the multiple alignments of the respective proteins and the conservation of the (predicted) operon organization in archaea and bacteria (Table 2 and data not shown). The combination of clues from these analyses suggests that the two proteins interact in a still uncharacterized pathway of RNA processing, which also includes RNA 3'-phosphate cyclase (KOG3980)) [62] and cytosine-C5-methylase (NOL1/NOP2 in eukaryotes; KOG1122). The proteins in KOG3833 and KOG4528 are likely to represent novel enzyme families, possibly a kinase-phosphatase pair (E.V.K. and L. Aravind, unpublished data). Notably, these predicted new enzymes are present in animals and E. cuniculi but not in Arabidopsis or yeasts. In contrast, KOG3980 is present in all analyzed eukaryotic genomes except for Arabidopsis, whereas KOG1122 is pan-eukaryotic. These differences in the phyletic patterns of the components of the predicted pathway are concordant with the patterns in eukaryotes in that.
Figure 2 shows the distribution of known and predicted functions of eukaryotic proteins among 20 functional categories for the entire set of KOGs and, separately, for KOGs represented in six or seven species and the animal-specific KOGs. Compared to the functional breakdown of prokaryotic COGs [25], the prevalence of signal transduction is notable among eukaryotes. This feature is particularly prominent in animal-specific KOGs, whereas the highly conserved set is comparatively enriched in proteins that are involved in translation, transcription, chaperone-like functions, cell cycle control and chromatin dynamics (Figure 2). The large number of KOGs for which only general functional prediction was feasible, and those whose functions remain unknown, even among the subset that is represented in six or seven eukaryotic species, emphasizes that our current understanding of eukaryotic biology is seriously lacking with even in respect of the functions of highly conserved genes.
Figure 2.
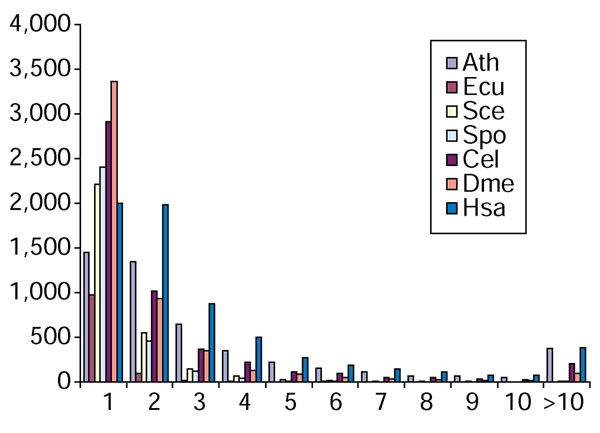
Distribution of the KOGs by the number of paralogs in each of the analyzed eukaryotic genomes. The species abbreviations are as in Figure 1.
The distribution of KOGs by the number of paralogs in each genome is shown in Figure 3. The preponderance of lineage-specific duplication of conserved genes, that is, intra-KOG LSEs, in multicellular eukaryotes is obvious. Cases when a single gene in yeast or, particularly, Encephalitozoon, has two or more co-orthologs in animals and/or plants are most common in KOGs, whereas the reverse situation is rare. These observations support the notion of the major contribution of LSE to the evolution of eukaryotic complexity [52]. However, 131 KOGs are represented by a single ortholog in all genomes compared (Table 2) and a substantial number of KOGs have one member from a majority of the genomes (data not shown). Recent theoretical modeling of the evolution of paralogous families has suggested that, in general, ancient protein families tend to have multiple paralogs [5,63]. Therefore, whenever a KOG has a single member in all or most species, this should be attributed to selection against duplication of this particular gene. A prominent cause of such selection could be the involvement of the respective gene products in essential multisubunit complexes, such that imbalance between subunits leads to deleterious effects [64].
Figure 3.
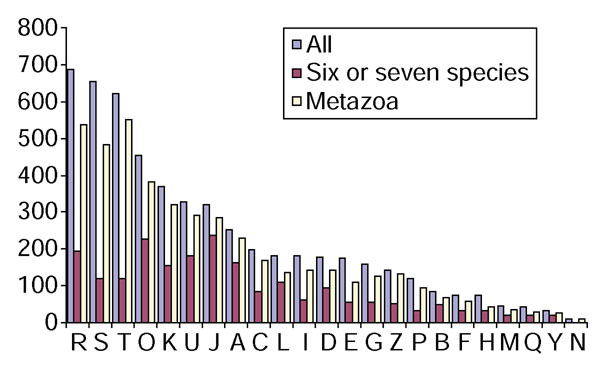
Functional breakdown of the KOGs. Designations of functional categories: A, RNA processing and modification; B, chromatin structure and dynamics; C, energy production and conversion; D, cell-cycle control and mitosis; E, amino acid metabolism and transport; F, nucleotide metabolism and transport; G, carbohydrate metabolism and transport; H, coenzyme metabolism; I, lipid metabolism; J, translation; K, transcription; L, replication and repair; M, membrane and cell wall structure and biogenesis; O, post-translational modification, protein turnover, chaperone functions; P, inorganic ion transport and metabolism; Q, secondary metabolites biosynthesis, transport and catabolism; T, signal transduction; U, intracellular trafficking and secretion; Y, nuclear structure; Z, cytoskeleton; R, general functional prediction only (typically, prediction of biochemical activity), S, function unknown. This breakdown is only for KOGs that included at least three species.
Known and new functions of single-member, pan-eukaryotic KOGs
We examined in greater detail the 131 KOGs that are represented by a single gene in each of the seven genomes (Table 2). As can be envisaged from their presence in diverse eukaryotic taxa, including the 'minimal' genome of Encephalitozoon, and as shown by comparison with the knockout phenotype data (Table 2 and see below), these pan-eukaryotic KOGs are of particular biological importance. For the great majority of these KOGs (113 of the 131), the function has been experimentally determined or confidently predicted to a varying degree of detail using computational methods (Table 2). However, around 20 KOGs from this set remained uncharacterized at the time of this analysis and, for all but two of these, substantial functional inferences could be drawn through a combination of sequence-profile analysis, structure prediction and genomic-context analysis of prokaryotic homologs (Table 2). Some of these predicted new functions are variations on well-known themes, such as two predicted PP-loop ATPases, which are probably involved in novel, essential RNA modifications (KOGs 2522 and 2316) or two predicted E3 components of ubiquitin ligases (KOGs 0396 and 3800). Other predicted functions appear to be completely new, such as proteins in KOG3176 and 3303 which are likely to be essential components of eukaryotic replication and/or repair systems. Each of these uncharacterized but ubiquitous and largely essential eukaryotic genes is an attractive target for experimental studies.
Examination of the experimentally characterized and predicted functions of pan-eukaryotic, single-member KOGs leads to interesting conclusions. Nearly all the functionally characterized KOGs in this set consist of proteins that are subunits of known multiprotein complexes (Table 2). The most prominent of these are the complexes involved in rRNA processing and ribosome assembly, such as the recently discovered rRNA processosome and the pre-40S subunit, as well as the spliceosome, and various complexes involved in transcription (Table 2). Accordingly, this set of KOGs is markedly enriched for proteins involved in various forms of RNA processing, assembly of ribonucleoprotein (RNP) particles and transcription. In addition, KOGs in the single-member pan-eukaryotic set include subunits of molecular complexes that are not directly related to RNA processing, such as the proteasome, the TCP-1 chaperonin complex [65] and the TRAPP complex involved in protein trafficking [66]. Altogether, more than 80% of the yeast proteins in the pan-eukaryotic, single-member KOGs belong to known macromolecular complexes included in the MIPS database [67], as compared to around 64% for all yeast proteins in the KOGs, which is a moderate but statistically highly significant excess (data not shown). This preponderance of multiprotein complex formation among the single-member pan-eukaryotic KOGs is fully compatible with the balance hypothesis [64].
The most unexpected observation regarding the single-member, pan-eukaryotic KOGs, is probably that in 14 of these proteins, the only detectable domain was the WD40 repeat (Table 2). This is particularly notable because WD40-repeat proteins, which are extremely abundant in eukaryotes and are present in several prokaryotic lineages as well [68], are not generally known to form well-defined, one-to-one orthologous relationships. The WD40 proteins in the pan-eukaryotic KOGs listed in Table 2 are exceptions, which is probably due to their unique and essential roles in the assembly of RNA-processing complexes. It has recently been demonstrated that, in S. cerevisiae, seven of these proteins are subunits of the 18S rRNA processosome, or at least are involved in ribosomal assembly [69,70]. Taking these results together with the unusual phyletic pattern, it seems possible to predict with considerable confidence that those WD40 proteins in the 131-KOG set that remain uncharacterized belong to the same or similar RNA-processing complexes (Table 2).
With some notable exceptions, such as the WD40 proteins, the KOGs in the single-member, pan-eukaryotic set show remarkable patterns of evolutionary conservation: they are either (nearly) ubiquitous in the three kingdoms of life, for example, RNA polymerase subunits, or are universally conserved in eukaryotes and archaea but missing in bacteria, such as most of the proteins implicated in RNA processing (Table 2). Thus, it appears that elaborate molecular machines central to the functioning of the eukaryotic cell have evolved, largely from ancestral archaeo-eukaryotic components, at the onset of eukaryotic evolution, and both loss and duplication of the respective genes have been strongly selected against throughout the rest of eukaryotic evolution.
Variation of evolutionary rates among KOGs
Genome-wide analysis of protein evolutionary rates shows a broad range of variation [71]. Here, we investigate the variation of evolutionary rates among the ubiquitous KOGs represented in all seven analyzed genomes and the connection between the evolutionary rate and protein function in the KOG set. The characteristic evolutionary rate of each KOG, which included a member(s) from Arabidopsis, was determined by measuring the mean evolutionary distance from Arabidopsis (the outgroup in the phylogenetic tree; see below) to the other species. Even among the KOGs that include all seven species and, accordingly, appear to represent the conserved core of eukaryotic genes, the evolutionary rates differ by a factor of 20 between the fastest- and the slowest-evolving KOGs. Excluding 5% of the KOGs from each tail of the distribution still leaves almost a fourfold difference in evolutionary rates (Figure 4a).
Figure 4.
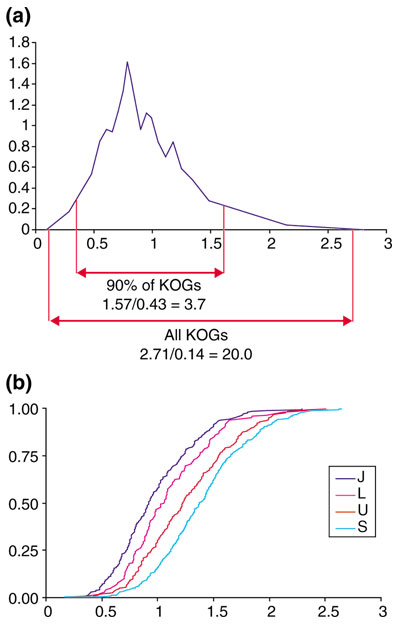
Variation of amino-acid substitution rates among KOGs. (a) Probability-density function for the distribution of evolutionary rates among the set of KOGs including all seven analyzed eukaryotic species. (b) Distribution functions for the evolutionary rates in different functional categories of KOGs. The designations of functional categories are as in Figure 3.
We then compared the distributions of evolutionary rates for different functional categories of KOGs (Tables 3,4 and Figure 4b). Although all the distributions substantially overlapped, there was a statistically highly significant difference between the evolutionary rates for proteins with different functions (Tables 3,4 and Figure 4b). The slowest-evolving proteins are those involved in translation and RNA processing, the fastest-evolving ones are involved in cellular trafficking and transport, whereas components of replication and transcription systems have intermediate evolutionary rates (Tables 3,4 and Figure 4b).
Table 3.
Evolutionary rates in KOGs with different functions: evolutionary rates for different functional categories of KOGs*
| Functional category | Number of KOGs | Mean rate, substitutions per site | Standard deviation |
| J | 227 | 0.98 | 0.37 |
| H | 62 | 0.98 | 0.30 |
| A | 167 | 1.01 | 0.36 |
| C | 140 | 1.01 | 0.43 |
| O | 307 | 1.01 | 0.40 |
| F | 50 | 1.05 | 0.34 |
| E | 130 | 1.07 | 0.38 |
| L | 139 | 1.11 | 0.38 |
| B | 56 | 1.13 | 0.33 |
| Z | 64 | 1.13 | 0.46 |
| K | 209 | 1.15 | 0.42 |
| G | 115 | 1.16 | 0.43 |
| I | 110 | 1.16 | 0.32 |
| T | 200 | 1.18 | 0.39 |
| D | 111 | 1.19 | 0.40 |
| R | 415 | 1.23 | 0.42 |
| M | 33 | 1.26 | 0.47 |
| U | 196 | 1.27 | 0.42 |
| Q | 30 | 1.27 | 0.37 |
| P | 69 | 1.28 | 0.45 |
| N | 2 | 1.30 | 0.78 |
| S | 348 | 1.40 | 0.41 |
| All | 3203 | 1.16 | 0.42 |
*Only the KOGs that included a member(s) from Arabidopsis were analyzed; the evolutionary rates are the average distances between the Arabidopsis representative in the given KOG and the proteins from other species (see Material and methods for details). The functional categories are designated as in Figure 5.
Table 4.
Statistical significance of differences in evolutionary rates between selected functional categories of KOGs (t-test)
| J | L | U | S | |
| J | - | |||
| L | 3 × 10-3 | - | ||
| U | 1 × 10-12 | 3 × 10-4 | - | |
| S | 7 × 10-33 | 5 × 10-13 | 2 × 10-4 | - |
A parsimonious scenario of gene loss and emergence in eukaryotic evolution and reconstruction of ancestral eukaryotic gene sets
Assuming a particular species tree topology, methods of evolutionary parsimony analysis can be used to construct a parsimonious scenario of evolution, that is, mapping of different types of evolutionary events onto the branches of the tree. With prokaryotes, the problem is confounded by the major contributions from both lineage-specific gene loss and HGT to genome evolution, with the relative likelihoods of these events remaining uncertain [5,7]. The possibility of substantial HGT between major lineages of eukaryotes can apparently be safely disregarded, providing for an unambiguous most parsimonious scenario that includes only gene loss and emergence of new genes as elementary events.
Some crucial aspects of the phylogenetic tree of the eukaryotic crown group remain a matter of contention. The consensus of many phylogenetic analyses appears to point to an animal-fungal clade and clustering of microsporidia with the fungi. However, a major uncertainty remains with respect to the topology of the animal tree: the majority of studies on protein phylogenies support a coelomate (chordate-arthropod) clade [72-74], whereas rRNA phylogeny and some protein family trees point to the so-called ecdysozoan (arthropod-nematode) clade [75-78]. We treated the phyletic pattern of each KOG as a string of binary characters (1 for the presence of the given species and 0 for its absence in the given KOG) and constructed the parsimonious scenarios of gene loss and emergence during evolution of the eukaryotic crown group for both the coelomate and the ecdysozoan topologies of the phylogenetic tree. For the purpose of this reconstruction, the Dollo parsimony approach was adopted [79]. Under this approach, gene loss is considered irreversible; thus, a gene (a KOG member) can be lost independently in several evolutionary lineages but cannot be regained. This assumption is justified by the implausibility of HGT between eukaryotes (the Dollo approach is not valid for reconstruction of prokaryotic ancestors).
In the resulting parsimonious scenarios, each branch was associated with both gene loss and emergence of new genes, with the exception of the plant branch and the branch leading to the common ancestor of fungi and animals, to which gene losses could not be assigned with the current set of genomes (Figure 5a,b). There is little doubt that, once genomes of early-branching eukaryotes are included, gene loss associated with these branches will become apparent. The principal features of the reconstructed scenarios include massive gene loss in the fungal clade, with additional elimination of numerous genes in the microsporidian; emergence of a large set of new genes at the onset of the animal clade; and subsequent substantial gene loss in each of the animal lineages, particularly in the nematodes and arthropods (Figure 5a,b). The estimated number of genes lost in S. cerevisiae after its divergence from the common ancestor with the other yeast species, S. pombe, closely agreed with a previous estimate produced by a different approach [57]. The switch from the coelomate topology of the animal sub-tree to the ecdysozoan topology resulted in relatively small changes in the distribution of gains and losses: the most notable difference was the greater number of genes lost in the nematode lineage and the smaller number of genes lost in the insect lineage under the ecdysozoan scenario compared to the coelomate scenario (Figure 5a,b).
Figure 5.
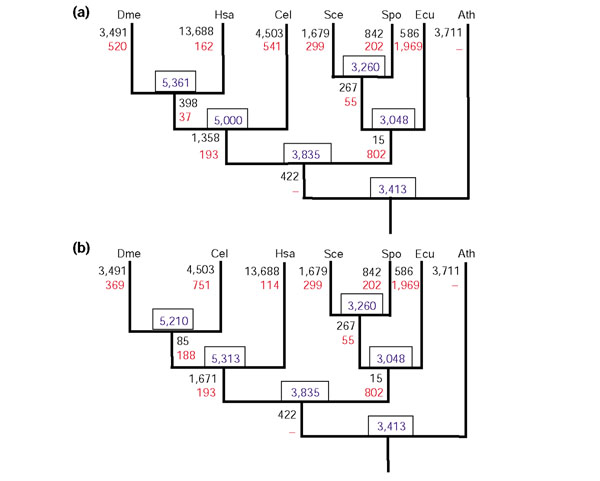
Parsimonious scenarios of loss and emergence of genes (KOGs) in eukaryotic evolution. (a) The coelomate topology of the phylogenetic tree of the eukaryotic crown group. (b) The ecdysozoan topology of the phylogenetic tree of the eukaryotic crown group. The numbers in boxes indicate the inferred number of KOGs in the respective ancestral forms. The numbers next to branches indicate the number of gene gains (emergence of KOGs) (numerator) and gene (KOG) losses (denominator) associated with the respective branches; a dash indicates that the number of losses for a given branch could not be determined. Proteins from each genome that did not belong to KOGs as well as LSEs were counted as gains on the terminal branches. The species abbreviations are as in Figure 1.
The parsimony analysis described above involves explicit reconstruction of the gene sets of ancestral eukaryotic genomes. Under the Dollo parsimony model, which was used for this analysis, an ancestral gene (KOG) set is the union of the KOGs that are shared by the respective outgroup and each of the remaining species. Thus, the gene set for the common ancestor of the crown group includes all the KOGs in which Arabidopsis co-occurs with any of the other analyzed species. Similarly, the reconstructed gene set for the common ancestor of fungi and animals consists of all KOGs in which at least one fungal species co-occurs with at least one animal species. These are conservative reconstructions of ancestral gene sets because, as already indicated, gene losses in the lineages branching off the deepest bifurcation could not be detected. Under this conservative approach, 3,413 genes (KOGs) were assigned to the last common ancestor of the crown group (Figure 5a,b). More realistically, it appears likely that a certain number of ancestral genes have been lost in all, or all but one, of the analyzed lineages during subsequent evolution, such that the gene set of the eukaryotic crown group ancestor might have been close in size to those of modern yeasts. In terms of the functional composition, the reconstructed core gene set of the crown-group ancestor resembled more the highly conserved KOGs than the animal-specific KOGs (Figure 3) in being enriched in housekeeping functions such as translation, transcription and RNA processing (data not shown).
The functional profiles of the gene sets that were lost in different lineages showed substantial differences (Table 5). Thus, for example, in the lineage leading to the common ancestor of the animals, the greatest loss among genes assigned to functional categories was seen in amino acid and coenzyme metabolism; in contrast, in the fly and the nematode, more substantial degradation was observed among transcription factors and proteins with chaperone-like functions. Genes for proteins involved in RNA processing and translation are, in general, not heavily affected by loss except in the highly degraded parasite E. cuniculi. On many occasions, the switch from the coelomate to the ecdysozoan topology replaces two independent, parallel losses in the insect and nematode clades with a single loss at the base of the ecdysozoan branch, although, on the whole, trees based on gene content support the coelomate topology [74]. In particular, the ecdysozoan topology, unlike the coelomate topology, implies early loss of several genes involved in translation, transcription and repair (Table 6). Notably, a large fraction of genes lost in each lineage has only a general functional prediction or no prediction at all (Table 5). This emphasizes the paucity of our current understanding of lineage-specific gene sets.
Table 5.
Functional profiles of genes lost in different eukaryotic lineages
| Functional category | Lost genes (KOGs) | |||||||||
| Hs* | Dm* | Coelomates/ Ecdysozoa | Ce* | Animals | Sc | Sp | Yeasts | Ec | Fungi-Ec | |
| Total | 162/114 | 520/369 | 37/188 | 541/751 | 193 | 299 | 202 | 55 | 1,969 | 802 |
| RNA processing and modification | 2/3 | 9/8 | 1/2 | 10/11 | 4 | 15 | 7 | 1 | 88 | 32 |
| Translation | 3/3 | 16/11 | 0/5 | 13/10 | 9 | 9 | 6 | 3 | 122 | 10 |
| Transcription | 5/2 | 16/12 | 0/4 | 29/33 | 2 | 16 | 9 | 4 | 83 | 40 |
| Replication and repair | 4/5 | 28/14 | 1/15 | 29/14 | 2 | 9 | 7 | 3 | 60 | 16 |
| Chromatin structure and dynamics | 1/1 | 8/6 | 0/2 | 8/6 | 0 | 5 | 3 | 1 | 29 | 11 |
| Energy production and conversion | 7/10 | 9/10 | 5/4 | 12/10 | 7 | 6 | 13 | 1 | 110 | 37 |
| Cell cycle control and mitosis | 3/3 | 11/6 | 0/5 | 15/11 | 3 | 12 | 3 | 1 | 61 | 16 |
| Amino acid metabolism and transport | 5/6 | 16/9 | 1/8 | 15/7 | 38 | 6 | 9 | 0 | 110 | 18 |
| Nucleotide metabolism and transport | 3/3 | 6/3 | 0/3 | 8/5 | 5 | 0 | 3 | 1 | 38 | 9 |
| Carbohydrate metabolism and transport | 3/3 | 13/10 | 1/4 | 18/14 | 8 | 10 | 16 | 3 | 70 | 41 |
| Coenzyme metabolism | 0/2 | 5/5 | 2/2 | 14/12 | 11 | 1 | 1 | 0 | 51 | 12 |
| Lipid metabolism | 1/5 | 27/19 | 4/12 | 18/6 | 4 | 9 | 19 | 2 | 74 | 33 |
| Membrane and cell wall structure and biogenesis | 5/4 | 10/10 | 2/2 | 9/11 | 7 | 5 | 3 | 0 | 37 | 15 |
| Post-translational modification, protein turnover, chaperone functions | 3/5 | 22/15 | 2/9 | 44/40 | 8 | 29 | 21 | 4 | 167 | 69 |
| Inorganic ion transport and metabolism | 2/4 | 8/8 | 2/2 | 8/7 | 9 | 2 | 6 | 4 | 50 | 14 |
| Secondary metabolites biosynthesis, transport and catabolism | 1/2 | 6/5 | 1/2 | 5/3 | 2 | 4 | 1 | 0 | 23 | 5 |
| Signal transduction | 5/3 | 32/22 | 0/10 | 30/37 | 4 | 16 | 7 | 3 | 110 | 52 |
| Intracellular trafficking and secretion | 4/3 | 10/8 | 0/2 | 14/14 | 3 | 5 | 11 | 0 | 116 | 22 |
| Nuclear structure | 0/0 | 3/3 | 0/0 | 5/6 | 0 | 1 | 0 | 0 | 16 | 5 |
| Cytoskeleton | 0/0 | 2/2 | 0/0 | 6/8 | 0 | 9 | 0 | 3 | 44 | 6 |
| General functional prediction only (typically, prediction of biochemical activity) | 14/13 | 79/55 | 5/29 | 88/72 | 30 | 55 | 24 | 11 | 241 | 134 |
| Function unknown | 91/34 | 184/128 | 10/66 | 143/414 | 37 | 75 | 33 | 10 | 269 | 205 |
*For each of the animals, the numerator indicates the number of genes lost under the coelomate topology of the species tree and the denominator indicates the number of genes lost under the ecdysozoan topology of the tree.
Table 6.
Groups of functionally linked genes co-eliminated during evolution of different eukaryotic lineages
| Functional group/ complex | Lost KOGs* | ||||||
| Hs | Dm | Ce | Coelomates/ Ecdysozoa | Animals | Yeasts | Fungi-Ec | |
| Mitochondrial ribosomal proteins | 3331, 3435/ 3331, 3435 | 3505, 4600, 4612/ None | 3505, 4122, 4600, 4612/ 4122 | None/ 3505, 4600, 4612 | 0899, 0938, 1740, 3254, 3278, 4844 | 0408,1686, 1708, 4707 | |
| Spliceosome, including putative associated proteins | 1847, 1960/ 1847 | 1902, 1960, 2991, 3414 | None/ 1960 | 0105, 0107, 0117, 1365, 1588, 1676, 1847, 1996, 2191, 2242, 2548, 2991, 4207, 4211 | |||
| Exosome | 1004, 1613 | ||||||
| Replication origin-recognition complex | 2228, 2538, 4557 | 4557 | |||||
| Mismatch repair system | 0218, 0220, 221, 1977 | 0218, 1977, 4120 | None/ 0218, 1977 | ||||
| Ubiquitin system/ proteasome-signalosome components | 0170, 0428, 1814, 4116, 4185, 4412 | 0168, 0170, 0320, 0421, 0423, 1364, 1571, 1645, 1871, 1873, 1887, 2561, 2932, 3061, 3250, 3268, 4146, 4159, 4275, 4412, 4413, 4414, 4692, 4761 | None/ 0170, 4412 | 0823, 1645, 1734 | 0311, 0423, 0427, 0827, 0895, 1100, 1139, 1464, 1571, 1812, 1887, 2561, 2932, 3011, 3050, 3268, 4185, 4248, 4265, 4275, 4413, 4414, 4427, 4642, 4692, 4761 | ||
| NADH-ubiquinone oxido-reductase/ NADH dehydro-genase | 2865, 2870, 3256, 3300, 3365, 3382, 3389, 3426, 3446, 3456, 3458, 3466, 3468, 4009, 4662, 4668, 4669, 4770, 4845 | ||||||
*For each of the animals, the numerator indicates the KOGs lost under the coelomate topology of the species tree, and the denominator indicates KOGs lost under the ecdysozoan topology.
As noticed previously during the analysis of the genes lost in S. cerevisiae after its divergence from the common ancestor with S. pombe, functionally connected genes tend to be co-eliminated during evolution [57]. The present study generalizes this conclusion as many functionally coherent groups of co-eliminated KOGs become apparent (Table 5). Importantly, different branches of the same complex systems tend to be eliminated in parallel in different lineages, for example, largely non-overlapping sets of genes for proteins of the ubiquitin-proteasome-signalosome systems are lost in the fungal-microsporidial lineage and in the nematodes (Table 6). It seems likely that elimination of these genes reflects independent trends for simplification of regulatory processes in these lineages.
An interesting trend seen in these data is the deterioration of the mitochondrial ribosome, which occurred in several eukaryotic lineages and appears to have been partly parallel (as it occurred independently in fungi-microsporidia and in animals) and partly consecutive: early loss in the ancestral animal line was followed by elimination of additional genes for ribosomal proteins in individual lineages (Table 6). C. elegans has one of the shortest mitochondrial rRNAs and might have a 'minimal' mitochondrial ribosome [80]; the present analysis details the stages leading to this ultimate degradation of the mitochondrial ribosome.
An exhaustive analysis of the patterns of gene loss is beyond the scope of this work. It seems clear that it has potential of improving our understanding of eukaryotic evolution and functional predictions through examination of co-eliminated gene groups.
Evolutionary relationships between eukaryotic and prokaryotic orthologous gene sets
The prokaryotic COGs and eukaryotic KOGs were identified in separate genome comparisons, although an overlap existed because both sets included the unicellular eukaryotes, namely two yeasts and the microsporidian. To identify the prokaryotic counterparts of the KOGs, the sequences of the eukaryotic proteins included in the KOGs were compared using the RPS-BLAST program to the position-specific scoring matrices (PSSMs) constructed for all prokaryotic COGs ([81] see Materials and methods for details). The results were checked manually and also by comparing the assignment of proteins from unicellular eukaryotes to each of the orthologous gene sets. Altogether, probable orthologous relationships were established between 2,456 eukaryotic KOGs and TWOGs (44% of the total) and 1,516 prokaryotic COGs. A more detailed breakdown of the relationships between eukaryotic and prokaryotic orthologous gene clusters could reveal important evolutionary trends. Figure 6a compares the occurrence of prokaryotic counterparts for the entire set of eukaryotic KOGs and its subsets conserved at different levels. Clearly, the reconstructed gene set of the common ancestor of the crown group and, particularly, the pan-eukaryotic KOGs are enriched in ancient KOGs (those with prokaryotic counterparts) as compared to the full KOG collection. In contrast, among KOGs that are inferred to have evolved in individual lineages within the crown group, a significantly lower fraction has detectable prokaryotic counterparts (Figure 6a).
Figure 6.
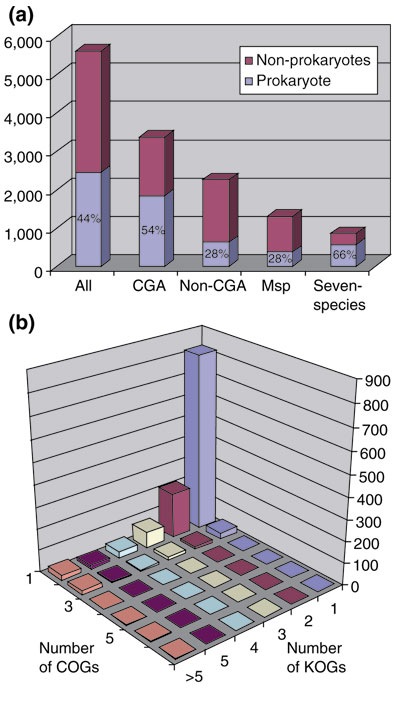
Correspondence between eukaryotic and prokaryotic orthologous gene sets. (a) Representation of prokaryotic counterparts in different subsets of KOGs. CGA, crown group ancestor; non-CGA, KOGs not represented in the crown group ancestor; MSP, metazoa-specific KOGs. (b) Evidence of ancient duplications of eukaryotic genes revealed by the KOGs against COGs comparison. The connections between KOGs and COGs detected by using RPS-BLAST (see text) were analyzed by single linkage clustering.
Early evolution of eukaryotes is known to have involved duplication of ancient genes inherited from prokaryotes [82], and this was apparent in the KOGs against COGs comparison. Although one-to-one relationships were predominant, in around 30% of cases, two or more eukaryotic KOGs corresponded to the same prokaryotic COG (Figure 6b). This indicates extensive duplication of ancestral genes at early stages of eukaryotic evolution; moreover, a substantial fraction of these genes have undergone repeated duplications, resulting in a one-to-many relationship between prokaryotic and eukaryotic orthologs (Figure 6b).
An in-depth analysis of the relationships between eukaryotic and prokaryotic orthologous gene clusters should include an attempt to decipher their evolutionary history, that is, classification of the C/KOGs represented both in eukaryotes and prokaryotes into: those that have been inherited from the last universal common ancestor; the archaeo-eukaryotic subset; and those that are shared because of HGT between bacteria and eukaryotes at various stages of eukaryotic evolution. This analysis is beyond the scope of the present work. Perhaps the principal message to stress here is that, using a fairly sensitive sequence comparison method, prokaryotic homologs could be detected for only some 44% of the eukaryotic KOGs, and this fraction increased to around 54% for those genes that could be traced to the last common ancestor of the crown group (Figure 6a). This observation emphasizes the major amount of innovation that accompanied the emergence and early evolution of eukaryotes; even those KOGs for which prokaryotic counterparts will be eventually identified through more sensitive sequence and structure comparison apparently experienced rapid evolution during the prokaryote-eukaryote transition.
Phyletic patterns of KOGs and dispensability of yeast and worm genes
There are 860 KOGs with at least one representative from each of the seven analyzed genomes. In accord with the 'knockout rate' hypothesis [83], which has been largely supported by recent, genome-wide analysis of gene conservation [38,84], it could be expected that these highly conserved genes were essential for the survival of eukaryotic organisms. This appears particularly plausible given the near-minimal eukaryotic gene complement of the microsporidian. The prediction was put to the test using the recently published functional profile of the yeast S. cerevisiae genome, which includes the data on the growth rates of homozygous deletion strains for 96% of the open reading frames (ORFs) in the yeast genome [85]. Growth rates have been previously interpreted as a measure of fitness [84].
When the phyletic patterns of the KOGs were superimposed on the data on gene dispensability (with essential genes operationally defined as those whose deletion had a lethal effect in a rich medium) [85], it was found that 45% of the essential genes were conserved in all seven species and 25% were represented in six species (typically with the exception of E. cuniculi); 15% of the essential yeast genes had no orthologs in the other analyzed genomes (Figure 7a). In a striking contrast, among non-essential genes, only 16.5% were represented in all compared genomes and 28.5% had no detectable orthologs (Figure 7a). The reciprocal comparison is equally illustrative: essential genes composed 18.5% of the entire set of yeast genes but 35% of the genes (KOGs) represented in all seven species. This translates into a statistically highly significant dependence between a gene's (in)dispensability and conservation over long evolutionary distances. The probability of the set of highly conserved genes being so enriched for essential genes as a result of chance was estimated at <<10-10. Notably, an even greater enrichment for essential genes was seen among the KOGs that were represented by one, and only one, ortholog in each of the seven analyzed genomes: of the 131 such KOGs, 98 (75%) included an essential yeast gene (Table 2). Such preponderance of essential genes could be expected because, in this set of KOGs, the indispensability of the respective function could not have been masked by the presence of paralogs.
Figure 7.
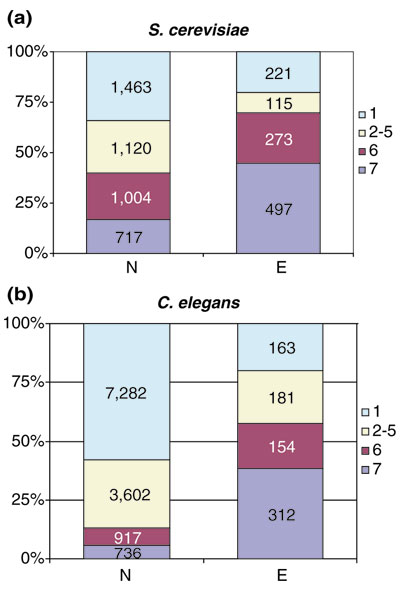
Gene dispensability in yeast and worm and phyletic patterns of the respective KOGs. (a) Distribution of essential and non-essential genes among different size classes of KOGs and LSEs in yeast Saccharomyces cerevisiae. (b) Distribution of essential and non-essential genes among different size classes of KOGs and LSEs in the nematode C. elegans. The number of species in the KOGs and LSEs is color-coded as indicated to the right of each plot.
For an additional set of around 15% non-essential yeast genes, knockout results in a measurable retardation of growth [85]. Unexpectedly and in contrast to the result obtained with the essential genes, we failed to observe a correlation between the magnitude of a gene's knockout effect on yeast growth and the phyletic pattern (data not shown). This seems to indicate that the measured effect on yeast growth might not translate into an effect on fitness that the loss of the ortholog of the given gene has in distant species.
In C. elegans, much as in yeast, essentiality of genes appears to correlate with strong evolutionary conservation, as already noticed in the recent genome-wide study on inhibition of worm gene expression by RNA interference (RNAi) [86]. We compared this dataset, which covers around 86% of C. elegans genes, to the phyletic patterns of the respective KOGs. Of the essential worm genes, 38% were conserved in all seven compared species and 19% were conserved in six species (Figure 7b). In contrast, only 6% of the non-essential C. elegans genes were represented in seven species and 7% were conserved in six species (Figure 7b). Thus, there seems to be a strong and robust connection between a gene's essentiality and its tendency to be conserved in evolution over a wide span of taxa; this connection was established using two independent datasets from biologically extremely different model organisms.
Domain accretion in orthologous sets of eukaryotic proteins
As noticed previously, the complexity of domain architecture of proteins in some orthologous sets increases with increasing organismic complexity; this phenomenon has been dubbed domain accretion [3]. With the KOG set in hand, we sought to assess the extent of accretion quantitatively by using the data on the presence of domains from the CDD (conserved domain alignments database) collection in each of the KOG members. The results summarized in Table 7 show a relatively small but statistically significant excess of domains in proteins from multicellular organisms compared to the orthologs from unicellular organisms. Furthermore, among the multicellular eukaryotes, human proteins have the greatest complexity of domain architectures, followed by Drosophila and Arabidopsis (Table 6), in agreement with preliminary results reported previously. Among the unicellular eukaryotes, Encephalitozoon had by far the least complex domain architectures (Table 6), which reflects the general genome reduction in this intracellular parasite.
Table 7.
Domain accretion in complex eukaryotes
| Hsa | Dme | Ath | Cel | Sce | Spo | Ecu | |
| Hsa | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | |
| 470 | |||||||
| Dme | 3214 | 2 × 10-1 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | |
| 805 | |||||||
| 327 | 354 | ||||||
| Ath | 2224 | 2085 | 3 × 10-1 | <1 × 10-10 | <1 × 10-10 | <1 × 10-10 | |
| 530 | 403 | ||||||
| 347 | 428 | 334 | |||||
| Cel | 2986 | 2962 | 2052 | 1 × 10-8 | <1 × 10-10 | <1 × 10-10 | |
| 880 | 650 | 376 | |||||
| 149 | 161 | 183 | 197 | ||||
| Sce | 1789 | 1704 | 1769 | 1715 | 1 × 10-2 | <1 × 10-10 | |
| 504 | 411 | 374 | 336 | ||||
| 100 | 123 | 135 | 150 | 158 | |||
| Spo | 1880 | 1807 | 1886 | 1808 | 2360 | <1 × 10-10 | |
| 549 | 426 | 388 | 359 | 216 | |||
| 10 | 17 | 12 | 14 | 13 | 19 | ||
| Ecu | 700 | 738 | 739 | 748 | 816 | 835 | |
| 332 | 254 | 235 | 244 | 158 | 140 |
For a given pair of species the numbers in each cell below the diagonal represent, from top to bottom: the number of KOGs in which the average number of detected domains from the CDD collection (cut-off E = 10-3) in the proteins from the species to the left is greater than that for the species to the right; the number of KOGs with equal average number of domains; the number of KOGs in which the average number of domains is greater for the species to the right (for example, D. melanogaster has a greater number of detected domains than H. sapiens in 470 KOGs, the same number in 3,214 KOGs, and a smaller number in 805 KOGs). The numbers above the diagonal are the statistical significance of the difference, P(χ2).
Conclusions
The present analysis of KOGs provides quantitative backing for many trends in the evolution of eukaryotic genomes that previously have been noticed on the general, qualitative level. The important quantities reported here include the size of the conserved core of eukaryotic genes, the conservative reconstructions of ancestral gene sets, the numbers of genes that appear to have been lost and gained in individual eukaryotic lineages, and the extent of correlation between gene dispensability and evolutionary conservation, which is reflected in phyletic patterns. In addition, we evaluated the range of variation of evolutionary rates of genes in different functional categories and obtained statistical support for the important evolutionary phenomenon of domain accretion. Furthermore, we observed that only a minority of eukaryotic KOGs have readily detectable prokaryotic counterparts, which emphasizes the extent of innovation linked to the origin of eukaryotes and subsequent major transitions in eukaryotic evolution, such as the origin of multicellularity and the origin of animals.
The case study of the KOGs that are represented by just one member in all eukaryotic genomes compared shows the potential of KOGs for functional prediction by inferring the probable functions for almost all KOGs in this set that had remained uncharacterized. This analysis also revealed unexpected facets of evolution of widespread and essential eukaryotic proteins, such as the counterintutitive preponderance of WD40-repeat proteins among the single-member pan-eukaryotic KOGs.
The current KOG set includes proteins from seven genomes whose sequences were available as of 1 July, 2002. The genomes of the mouse [87], the fugu fish [88], the Anopheles mosquito [89], the urochordate Ciona instestinalis [90] and the malarial parasite Plasmodium falciparum [91] have become available since then but were not included, partly because of problems with protein annotation for some of these genomes, and partly due to the time-consuming and labor-intensive nature of KOG analysis. Inclusion of these and other newly sequenced genomes should proceed at a faster rate once the system itself is established, and will enable further, deeper studies into the functional and evolutionary patterns of eukaryotic life.
Materials and methods
Construction and annotation of KOGs
A more detailed description of the procedures employed for this purpose is presented elsewhere [25]. The protein sets for all eukaryotic species, with the exception of C. elegans and H. sapiens, were from the genome division of the National Center for Biotechnology Information (NCBI). The protein sequences for C. elegans were from the WormPep67 database and the human sequences were from NCBI build 30. Briefly, the KOG construction protocol included: First, the detection and masking of common, repetitive domains using the RPS-BLAST program and the PSSMs for the respective domains from the CDD collection [81]; second, all-against-all comparison of protein sequences from the analyzed genomes using the BLASTP program [92], with masking of low sequence complexity regions using the SEG program [93]; third, identification of triangles of mutually consistent BeTs; merging triangles of BeTs with a common side to form preliminary KOGs; forth, adding members of co-orthologous sets missed at previous step using the COGNITOR procedure [24]; fifth, manual examination of each candidate KOG, aimed at eliminating the false positives incorporated into the KOGs by the automatic procedure and inclusion of false negatives that were missed originally; sixth, assignment of proteins containing promiscuous domains masked at the first step to Fuzzy Orthologous Groups (FOGs), named after the respective domains (when a sequence assigned to a KOG contained one or more masked domains, the sequences of these domains were restored); and finally, examination of the largest preliminary KOGs, which included numerous proteins from all or several genomes by using phylogenetic trees, cluster analysis with the BLASTCLUST program [94], comparison of domain architectures, and visual inspection of alignments. As a result, some of these preliminary KOGs were split into two or more smaller final KOGs.
Annotation of KOGs included critical assessment of the annotations available through GenBank, other public databases and the primary literature and additional, in-depth sequence analysis aimed at detection of previously unnoticed homologous relationships. The annotated functions of KOGs were classified into 23 categories (see legend to Figure 3), which were adapted from the functional classification previously used for COGs [24] by including several specific eukaryotic categories.
Other sequence analysis procedures
During KOG annotation, proteins that are currently annotated as 'hypothetical' or 'unknown', or otherwise had a vague or suspect annotation, were subject to additional sequence analysis, which included iterative sequence similarity searches with the PSI-BLAST program [92], RPS-BLAST searches for conserved domains [80], and additional domain architecture analysis using the SMART system [95]. To estimate sequence evolution rates, multiple alignments of KOGs were constructed using the MAP program [96] and the pairwise evolutionary distances were calculated with the maximum likelihood method under the PAM model by using the PROTDIST program of the PHYLIP package [97]. When a KOG included more than one member from the given species, the paralog with the greatest average similarity to proteins from other organisms was selected to represent the species in the given KOG. Since A. thaliana is the most likely outgroup species for the analyzed set of eukaryotes, distances from the Arabidopsis representative to proteins from all other species were averaged to estimate the characteristic evolutionary distance for the given KOG. Data from KOGs with excessive variability of the distances between A. thaliana and other species (standard deviation to mean ratio > 0.5) were discarded. As the divergence times for all KOGs are presumed to be the same (and equal to the time elapsed since the last common ancestor for the eukaryotic crown group), the mean evolutionary distance in a KOG is a measure of the KOG's evolutionary rate.
The parsimonious evolutionary scenario, which included gene losses and emergence of KOGs mapped to the branches of the eukaryotic phylogenetic tree, was constructed by using the DOLLOP program of the PHYLIP package [97]; this program is based on the Dollo parsimony method, which assumes irreversibility of character loss [79].
For the analysis of domain accretion, conserved domains from the NCBI CDD database were detected in the eukaryotic proteins that belonged to the KOGs by using the RPS-BLAST program [81] with an E-value cut-off of 0.001. Domains with biased amino acid sequence composition, which tend to produce a high false-positive rate in RPS-BLAST searches, were excluded from the analysis.
The eukaryotic KOG set is accessible at [98] and via ftp at [99]. The reconstructed ancestral gene sets are available at [100].
Acknowledgments
Acknowledgements
We thank Roman Tatusov for his major contribution to the construction of the KOGs, Igor Garkavtsev for his participation in the initial stages of the KOG project, and L. Aravind and Wei Yang for useful discussions and sharing their unpublished observations.
References
- Doolittle WF. Lateral genomics. Trends Cell Biol. 1999;9:M5–M8. doi: 10.1016/S0962-8924(99)01664-5. [DOI] [PubMed] [Google Scholar]
- Doolittle WF. Phylogenetic classification and the universal tree. Science. 1999;284:2124–2129. doi: 10.1126/science.284.5423.2124. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Aravind L, Kondrashov AS. The impact of comparative genomics on our understanding of evolution. Cell. 2000;101:573–576. doi: 10.1016/s0092-8674(00)80867-3. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel B, Bork P, Huynen MA. Genomes in flux: the evolution of archaeal and proteobacterial gene content. Genome Res. 2002;12:17–25. doi: 10.1101/gr.176501. [DOI] [PubMed] [Google Scholar]
- Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19:2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- Mirkin BG, Fenner TI, Galperin MY, Koonin EV. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol Biol. 2003;3:2. doi: 10.1186/1471-2148-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch WM. Distinguishing homologous from analogous proteins. Syst Zool. 1970;19:99–106. [PubMed] [Google Scholar]
- Fitch WM. Homology a personal view on some of the problems. Trends Genet. 2000;16:227–231. doi: 10.1016/S0168-9525(00)02005-9. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Greene EA, Pietrokovski S, Bork P, Attwood TK, Hood L. Gene families: the taxonomy of protein paralogs and chimeras. Science. 1997;278:609–614. doi: 10.1126/science.278.5338.609. [DOI] [PubMed] [Google Scholar]
- Sonnhammer EL, Koonin EV. Orthology, paralogy and proposed classification for paralog subtypes. Trends Genet. 2002;18:619–620. doi: 10.1016/S0168-9525(02)02793-2. [DOI] [PubMed] [Google Scholar]
- Wilson CA, Kreychman J, Gerstein M. Assessing annotation transfer for genomics: quantifying the relations between protein sequence, structure and function through traditional and probabilistic scores. J Mol Biol. 2000;297:233–249. doi: 10.1006/jmbi.2000.3550. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Galperin MY. Sequence-Evolution-Function Computational Approaches in Comparative Genomics. New York: Kluwer Academic Publishers; 2002. [PubMed] [Google Scholar]
- Pauling L, Zuckerkandl E. Chemical paleogenetics. Molecular "restoration studies" of extinct forms of life. Acta Chem Scand. 1963;17:S9–S16. [Google Scholar]
- Ohno S. Evolution by Gene Duplication. Berlin-Heidelberg-New York: Springer-Verlag; 1970. [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicheritz-Ponten T, Andersson SG. A phylogenomic approach to microbial evolution. Nucleic Acids Res. 2001;29:545–552. doi: 10.1093/nar/29.2.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmasek CM, Eddy SR. RIO: Analyzing proteomes by automated phylogenomics using resampled inference of orthologs. BMC Bioinformatics. 2002;3:14. doi: 10.1186/1471-2105-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm CE, Sonnhammer EL. Automated ortholog inference from phylogenetic trees and calculation of orthology reliability. Bioinformatics. 2002;18:92–99. doi: 10.1093/bioinformatics/18.1.92. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Huynen MA, Bork P. Measuring genome evolution. Proc Natl Acad Sci USA. 1998;95:5849–5856. doi: 10.1073/pnas.95.11.5849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague MG, Hutchison CA., 3rd Gene content phylogeny of herpesviruses. Proc Natl Acad Sci USA. 2000;97:5334–5339. doi: 10.1073/pnas.97.10.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, Kiryutin B, Galperin MY, Fedorova ND, Koonin EV. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–28. doi: 10.1093/nar/29.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R, Mekhedov SL, Nikolskaya AN, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics. 2003;4:41. doi: 10.1186/1471-2105-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale DA, Shankavaram UT, Galperin MY, Wolf YI, Aravind L, Koonin EV. Towards understanding the first genome sequence of a crenarchaeon by genome annotation using clusters of orthologous groups of proteins (COGs). Genome Biol. 2000;1:research0009.1–0009.19. doi: 10.1186/gb-2000-1-5-research0009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolling J, Breton G, Omelchenko MV, Makarova KS, Zeng Q, Gibson R, Lee HM, Dubois J, Qiu D, Hitti J, et al. Genome sequence and comparative analysis of the solvent-producing bacterium Clostridium acetobutylicum. J Bacteriol. 2001;183:4823–4838. doi: 10.1128/JB.183.16.4823-4838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland M, Sanderson KE, Spieth J, Clifton SW, Latreille P, Courtney L, Porwollik S, Ali J, Dante M, Du F, et al. Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature. 2001;413:852–856. doi: 10.1038/35101614. [DOI] [PubMed] [Google Scholar]
- Slesarev AI, Mezhevaya KV, Makarova KS, Polushin NN, Shcherbinina OV, Shakhova VV, Belova GI, Aravind L, Natale DA, Rogozin IB, et al. The complete genome of hyperthermophile Methanopyrus kandleri AV19 and monophyly of archaeal methanogens. Proc Natl Acad Sci USA. 2002;99:4644–4649. doi: 10.1073/pnas.032671499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cort JR, Koonin EV, Bash PA, Kennedy MA. A phylogenetic approach to target selection for structural genomics: solution structure of YciH. Nucleic Acids Res. 1999;27:4018–4027. doi: 10.1093/nar/27.20.4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner SE. Target selection for structural genomics. Nat Struct Biol. 2000;7(Suppl):967–969. doi: 10.1038/80747. [DOI] [PubMed] [Google Scholar]
- Gerstein M. Integrative database analysis in structural genomics. Nat Struct Biol. 2000;7(Suppl):960–963. doi: 10.1038/80739. [DOI] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Searching for drug targets in microbial genomes. Curr Opin Biotechnol. 1999;10:571–578. doi: 10.1016/S0958-1669(99)00035-X. [DOI] [PubMed] [Google Scholar]
- Buysse JM. The role of genomics in antibacterial target discovery. Curr Med Chem. 2001;8:1713–1726. doi: 10.2174/0929867013371699. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Kondrashov FA, Rogozin IB, Tatusov RL, Wolf YI, Koonin EV. Constant relative rate of protein evolution and detection of functional diversification among bacterial, archaeal and eukaryotic proteins. Genome Biol. 2001;2:research0053.1–0053.9. doi: 10.1186/gb-2001-2-12-research0053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanai I, Derti A, DeLisi C. Genes linked by fusion events are generally of the same functional category: a systematic analysis of 30 microbial genomes. Proc Natl Acad Sci USA. 2001;98:7940–7945. doi: 10.1073/pnas.141236298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecompte O, Ripp R, Puzos-Barbe V, Duprat S, Heilig R, Dietrich J, Thierry JC, Poch O. Genome evolution at the genus level: comparison of three complete genomes of hyperthermophilic archaea. Genome Res. 2001;11:981–993. doi: 10.1101/gr.GR1653R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan IK, Rogozin IB, Wolf YI, Koonin EV. Essential genes are more evolutionarily conserved than are nonessential genes in bacteria. Genome Res. 2002;12:962–968. doi: 10.1101/gr.87702. 10.1101/gr.87702. Article published online before print in May 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CE, Sonnhammer EL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J Mol Biol. 2001;314:1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Gaasterland T, Ragan MA. Microbial genescapes: phyletic and functional patterns of ORF distribution among prokaryotes. Microb Comp Genomics. 1998;3:199–217. doi: 10.1089/omi.1.1998.3.199. [DOI] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci USA. 1999;96:4285–4288. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galperin MY, Koonin EV. Who's your neighbor? New computational approaches for functional genomics. Nat Biotechnol. 2000;18:609–613. doi: 10.1038/76443. [DOI] [PubMed] [Google Scholar]
- Myllykallio H, Lipowski G, Leduc D, Filee J, Forterre P, Liebl U. An alternative flavin-dependent mechanism for thymidylate synthesis. Science. 2002;297:105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- Levesque M, Shasha D, Kim W, Surette MG, Benfey PN. Trait-to-Gene. A computational method for predicting the function of uncharacterized genes. Curr Biol. 2003;13:129–133. doi: 10.1016/S0960-9822(03)00009-5. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- The C. elegans Sequencing Consortium Genome sequence of the nematode C. elegans : a platform for investigating biology. Science. 1998;282:2012–2018. doi: 10.1126/science.282.5396.2012. [DOI] [PubMed] [Google Scholar]
- Arabidopsis Genome Initiative Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature. 2000;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Goffeau A, Barrell BG, Bussey H, Davis RW, Dujon B, Feldmann H, Galibert F, Hoheisel JD, Jacq C, Johnston M, et al. Life with 6000 genes. Science. 1996;274:563–547. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- Wood V, Gwilliam R, Rajandream MA, Lyne M, Lyne R, Stewart A, Sgouros J, Peat N, Hayles J, Baker S, et al. The genome sequence of Schizosaccharomyces pombe. Nature. 2002;415:871–880. doi: 10.1038/nature724. [DOI] [PubMed] [Google Scholar]
- Katinka MD, Duprat S, Cornillot E, Metenier G, Thomarat F, Prensier G, Barbe V, Peyretaillade E, Brottier P, Wincker P, et al. Genome sequence and gene compaction of the eukaryote parasite Encephalitozoon cuniculi. Nature. 2001;414:450–453. doi: 10.1038/35106579. [DOI] [PubMed] [Google Scholar]
- Lespinet O, Wolf YI, Koonin EV, Aravind L. The role of lineage-specific gene family expansion in the evolution of eukaryotes. Genome Res. 2002;12:1048–1059. doi: 10.1101/gr.174302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clusters of orthologous groups for eukaryotic complete genomes http://www.ncbi.nlm.nih.gov/COG/new/shokog.cgi
- Yudate HT, Suwa M, Irie R, Matsui H, Nishikawa T, Nakamura Y, Yamaguchi D, Peng ZZ, Yamamoto T, Nagai K, et al. HUNT: launch of a full-length cDNA database from the Helix Research Institute. Nucleic Acids Res. 2001;29:185–188. doi: 10.1093/nar/29.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra S, Crosby MA, Mungall CJ, Matthews BB, Campbell KS, Hradecky P, Huang Y, Kaminker JS, Millburn GH, Prochnik SE, et al. Annotation of the Drosophila melanogaster euchromatic genome: a systematic review. Genome Biol. 2002;3:research0083.1–0083.22. doi: 10.1186/gb-2002-3-12-research0083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Aravind L, Watanabe H, Lipman DJ, Koonin EV. Lineage-specific loss and divergence of functionally linked genes in eukaryotes. Proc Natl Acad Sci USA. 2000;97:11319–11324. doi: 10.1073/pnas.200346997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Aravind L, Koonin EV. Rickettsiae and Chlamydiae: evidence of horizontal gene transfer and gene exchange. Trends Genet. 1999;15:173–175. doi: 10.1016/S0168-9525(99)01704-7. [DOI] [PubMed] [Google Scholar]
- Marcotte EM, Pellegrini M, Thompson MJ, Yeates TO, Eisenberg D. A combined algorithm for genome-wide prediction of protein function. Nature. 1999;402:83–86. doi: 10.1038/47048. [DOI] [PubMed] [Google Scholar]
- Huynen MJ, Snel B. Gene and context: integrative approaches to genome analysis. Adv Protein Chem. 2000;54:345–379. doi: 10.1016/s0065-3233(00)54010-8. [DOI] [PubMed] [Google Scholar]
- Aravind L. Guilt by association: contextual information in genome analysis. Genome Res. 2000;10:1074–1077. doi: 10.1101/gr.10.8.1074. [DOI] [PubMed] [Google Scholar]
- Billy E, Wegierski T, Nasr F, Filipowicz W. Rcl1p, the yeast protein similar to the RNA 3'-phosphate cyclase, associates with U3 snoRNP and is required for 18S rRNA biogenesis. EMBO J. 2000;19:2115–2126. doi: 10.1093/emboj/19.9.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karev GP, Wolf YI, Rzhetsky AY, Berezovskaya FS, Koonin EV. Birth and death of protein domains: A simple model of evolution explains power law behavior. BMC Evol Biol. 2002;2:18. doi: 10.1186/1471-2148-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp B, Pal C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003;424:194–197. doi: 10.1038/nature01771. [DOI] [PubMed] [Google Scholar]
- Kubota H, Hynes G, Willison K. The chaperonin containing t-complex polypeptide 1 (TCP-1). Multisubunit machinery assisting in protein folding and assembly in the eukaryotic cytosol. Eur J Biochem. 1995;230:3–16. doi: 10.1111/j.1432-1033.1995.tb20527.x. [DOI] [PubMed] [Google Scholar]
- Jones S, Newman C, Liu F, Segev N. The TRAPP complex is a nucleotide exchanger for Ypt1 and Ypt31/32. Mol Biol Cell. 2000;11:4403–4411. doi: 10.1091/mbc.11.12.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewes HW, Frishman D, Guldener U, Mannhaupt G, Mayer K, Mokrejs M, Morgenstern B, Munsterkotter M, Rudd S, Weil B. MIPS: a database for genomes and protein sequences. Nucleic Acids Res. 2002;30:31–34. doi: 10.1093/nar/30.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP, Aravind L, Schultz J, Bork P, Koonin EV. Eukaryotic signalling domain homologues in archaea and bacteria. Ancient ancestry and horizontal gene transfer. J Mol Biol. 1999;289:729–745. doi: 10.1006/jmbi.1999.2827. [DOI] [PubMed] [Google Scholar]
- Pestov DG, Stockelman MG, Strezoska Z, Lau LF. ERB1, the yeast homolog of mammalian Bop1, is an essential gene required for maturation of the 25S and 5.8S ribosomal RNAs. Nucleic Acids Res. 2001;29:3621–3630. doi: 10.1093/nar/29.17.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragon F, Gallagher JE, Compagnone-Post PA, Mitchell BM, Porwancher KA, Wehner KA, Wormsley S, Settlage RE, Shabanowitz J, Osheim Y, et al. A large nucleolar U3 ribonucleoprotein required for 18S ribosomal RNA biogenesis. Nature. 2002;417:967–970. doi: 10.1038/nature00769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishin NV, Wolf YI, Koonin EV. From complete genomes to measures of substitution rate variability within and between proteins. Genome Res. 2000;10:991–1000. doi: 10.1101/gr.10.7.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges SB. The origin and evolution of model organisms. Nat Rev Genet. 2002;3:838–849. doi: 10.1038/nrg929. [DOI] [PubMed] [Google Scholar]
- Blair JE, Ikeo K, Gojobori T, Hedges SB. The evolutionary position of nematodes. BMC Evol Biol. 2002;2:7. doi: 10.1186/1471-2148-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Koonin EV. Coelomata and not Ecdysozoa: evidence from genome-wide phylogenetic analysis. Genome Res. 2004;14:29–36. doi: 10.1101/gr.1347404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, Lake JA. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387:489–493. doi: 10.1038/387489a0. [DOI] [PubMed] [Google Scholar]
- de Rosa R, Grenier JK, Andreeva T, Cook CE, Adoutte A, Akam M, Carroll SB, Balavoine G. Hox genes in brachiopods and priapulids and protostome evolution. Nature. 1999;399:772–776. doi: 10.1038/21631. [DOI] [PubMed] [Google Scholar]
- Mallatt J, Winchell CJ. Testing the new animal phylogeny: first use of combined large-subunit and small-subunit rRNA gene sequences to classify the protostomes. Mol Biol Evol. 2002;19:289–301. doi: 10.1093/oxfordjournals.molbev.a004082. [DOI] [PubMed] [Google Scholar]
- Peterson KJ, Eernisse DJ. Animal phylogeny and the ancestry of bilaterians: inferences from morphology and 18S rDNA gene sequences. Evol Dev. 2001;3:170–205. doi: 10.1046/j.1525-142x.2001.003003170.x. [DOI] [PubMed] [Google Scholar]
- Farris JS. Phylogenetic analysis under Dollo's Law. Syst Zool. 1977;26:77–88. [Google Scholar]
- Mears JA, Cannone JJ, Stagg SM, Gutell RR, Agrawal RK, Harvey SC. Modeling a minimal ribosome based on comparative sequence analysis. J Mol Biol. 2002;321:215–234. doi: 10.1016/S0022-2836(02)00568-5. [DOI] [PubMed] [Google Scholar]
- Marchler-Bauer A, Anderson JB, DeWeese-Scott C, Fedorova ND, Geer LY, He S, Hurwitz DI, Jackson JD, Jacobs AR, Lanczycki CJ, et al. CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res. 2003;31:383–387. doi: 10.1093/nar/gkg087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JR, Doolittle WF. Archaea and the prokaryote-to-eukaryote transition. Microbiol Mol Biol Rev. 1997;61:456–502. doi: 10.1128/mmbr.61.4.456-502.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Carlson SS, White TJ. Biochemical evolution. Annu Rev Biochem. 1977;46:573–639. doi: 10.1146/annurev.bi.46.070177.003041. [DOI] [PubMed] [Google Scholar]
- Hirsh AE, Fraser HB. Protein dispensability and rate of evolution. Nature. 2001;411:1046–1049. doi: 10.1038/35082561. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, Dow S, Lucau-Danila A, Anderson K, Andre B, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421:231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A, et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. doi: 10.1126/science.1072104. [DOI] [PubMed] [Google Scholar]
- Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- Dehal P, Satou Y, Campbell RK, Chapman J, Degnan B, De Tomaso A, Davidson B, Di Gregorio A, Gelpke M, Goodstein DM, et al. The draft genome of Ciona intestinalis: insights into chordate and vertebrate origins. Science. 2002;298:2157–2167. doi: 10.1126/science.1080049. [DOI] [PubMed] [Google Scholar]
- Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootton JC, Federhen S. Analysis of compositionally biased regions in sequence databases. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- NCBI BLAST server ftp://ftp.ncbi.nih.gov/blast
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. On global sequence alignment. Comput Appl Biosci. 1994;10:227–235. doi: 10.1093/bioinformatics/10.3.227. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods. Methods Enzymol. 1996;266:418–427. doi: 10.1016/s0076-6879(96)66026-1. [DOI] [PubMed] [Google Scholar]
- Clusters of orthologous groups for eukaryotic complete genomes http://www.ncbi.nlm.nih.gov/COG/new/shokog.cgi
- The Eukaryotic Clusters of Orthologous Groups of proteins (KOGs): download ftp://ftp.ncbi.nih.gov/pub/COG/KOG
- Reconstructed KOG sets for eukaryotic ancestral forms ftp://ftp.ncbi.nih.gov/pub/koonin/Ancestors/
- Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH. The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment. Proc Natl Acad Sci USA. 1998;95:5287–5292. doi: 10.1073/pnas.95.9.5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojic M, Kostrub CF, Buchman AR, Holloman WK. BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis. Mol Cell. 2002;10:683–691. doi: 10.1016/s1097-2765(02)00632-9. [DOI] [PubMed] [Google Scholar]
- Genschik P, Drabikowski K, Filipowicz W. Characterization of the Escherichia coli RNA 3'-terminal phosphate cyclase and its sigma54-regulated operon. J Biol Chem. 1998;273:25516–25526. doi: 10.1074/jbc.273.39.25516. [DOI] [PubMed] [Google Scholar]
- Dasgupta A, Darst RP, Martin KJ, Afshari CA, Auble DT. Mot1 activates and represses transcription by direct, ATPase-dependent mechanisms. Proc Natl Acad Sci USA. 2002;99:2666–2671. doi: 10.1073/pnas.052397899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard CJ, Aravind L, Koonin EV. Novel families of putative protein kinases in bacteria and archaea: evolution of the "eukaryotic" protein kinase superfamily. Genome Res. 1998;8:1038–1047. doi: 10.1101/gr.8.10.1038. [DOI] [PubMed] [Google Scholar]
- Vanrobays E, Gelugne JP, Gleizes PE, Caizergues-Ferrer M. Late cytoplasmic maturation of the small ribosomal subunit requires RIO proteins in Saccharomyces cerevisiae. Mol Cell Biol. 2003;23:2083–2095. doi: 10.1128/MCB.23.6.2083-2095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P, Echeverri C, Oegema K, Coulson A, Jones SJ, Copley RR, Duperon J, Oegema J, Brehm M, Cassin E, et al. Functional genomic analysis of cell division in C. elegans using RNAi of genes on chromosome III. Nature. 2000;408:331–336. doi: 10.1038/35042526. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Baserga SJ. Imp3p and Imp4p, two specific components of the U3 small nucleolar ribonucleoprotein that are essential for pre-18S rRNA processing. Mol Cell Biol. 1999;19:5441–5452. doi: 10.1128/mcb.19.8.5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Wolf YI, Aravind L. Prediction of the archaeal exosome and its connections with the proteasome and the translation and transcription machineries by a comparative-genomic approach. Genome Res. 2001;11:240–252. doi: 10.1101/gr.162001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Vanrobays E, Gelugne JP, Caizergues-Ferrer M, Henry Y. Rrp8p is a yeast nucleolar protein functionally linked to Gar1p and involved in pre-rRNA cleavage at site A2. RNA. 2000;6:826–843. doi: 10.1017/S1355838200992288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake Y, Wickner RB. Yeast virus propagation depends critically on free 60S ribosomal subunit concentration. Mol Cell Biol. 1995;15:2772–2781. doi: 10.1128/mcb.15.5.2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickner RB, Leibowitz MJ. Mak mutants of yeast: mapping and characterization. J Bacteriol. 1979;140:154–160. doi: 10.1128/jb.140.1.154-160.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Aravind L, Galperin MY, Grishin NV, Tatusov RL, Wolf YI, Koonin EV. Comparative genomics of the Archaea (Euryarchaeota): evolution of conserved protein families, the stable core, and the variable shell. Genome Res. 1999;9:608–628. [PubMed] [Google Scholar]
- Clissold PM, Ponting CP. PIN domains in nonsense-mediated mRNA decay and RNAi. Curr Biol. 2000;10:R888–R890. doi: 10.1016/S0960-9822(00)00858-7. [DOI] [PubMed] [Google Scholar]
- Tone Y, Toh EA. Nob1p is required for biogenesis of the 26S proteasome and degraded upon its maturation in Saccharomyces cerevisiae. Genes Dev. 2002;16:3142–3157. doi: 10.1101/gad.1025602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Oeffinger M, Dlakic M, Tollervey D. Nob1p is required for cleavage of the 3' end of 18S rRNA. Mol Cell Biol. 2003;23:1798–1807. doi: 10.1128/MCB.23.5.1798-1807.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanet R, Heude M. Characterization of mutations that are synthetic lethal with pol3-13, a mutated allele of DNA polymerase delta in Saccharomyces cerevisiae. Curr Genet. 2003;43:337–350. doi: 10.1007/s00294-003-0407-2. [DOI] [PubMed] [Google Scholar]
- Becam AM, Nasr F, Racki WJ, Zagulski M, Herbert CJ. Ria1p (Ynl163c), a protein similar to elongation factors 2, is involved in the biogenesis of the 60S subunit of the ribosome in Saccharomyces cerevisiae. Mol Genet Genomics. 2001;266:454–462. doi: 10.1007/s004380100548. [DOI] [PubMed] [Google Scholar]
- Whittaker CA, Hynes RO. Distribution and evolution of von Willebrand/integrin A domains: widely dispersed domains with roles in cell adhesion and elsewhere. Mol Biol Cell. 2002;13:3369–3387. doi: 10.1091/mbc.E02-05-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers LC, Kornberg RD. Mediator of transcriptional regulation. Annu Rev Biochem. 2000;69:729–749. doi: 10.1146/annurev.biochem.69.1.729. [DOI] [PubMed] [Google Scholar]
- Gu W, Malik S, Ito M, Yuan CX, Fondell JD, Zhang X, Martinez E, Qin J, Roeder RG. A novel human SRB/MED-containing cofactor complex, SMCC, involved in transcription regulation. Mol Cell. 1999;3:97–108. doi: 10.1016/s1097-2765(00)80178-1. [DOI] [PubMed] [Google Scholar]
- Leipe DD, Wolf YI, Koonin EV, Aravind L. Classification and evolution of P-loop GTPases and related ATPases. J Mol Biol. 2002;317:41–72. doi: 10.1006/jmbi.2001.5378. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Phosphoesterase domains associated with DNA polymerases of diverse origins. Nucleic Acids Res. 1998;26:3746–3752. doi: 10.1093/nar/26.16.3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Gleaning non-trivial structural, functional and evolutionary information about proteins by iterative database. J Mol Biol. 1999;287:1023–1040. doi: 10.1006/jmbi.1999.2653. [DOI] [PubMed] [Google Scholar]
- Wolf YI, Rogozin IB, Kondrashov AS, Koonin EV. Genome alignment, evolution of prokaryotic genome organization and prediction of gene function using genomic context. Genome Res. 2001;11:356–372. doi: 10.1101/gr.GR-1619R. [DOI] [PubMed] [Google Scholar]
- Bryant NJ, James DE. Vps45p stabilizes the syntaxin homologue Tlg2p and positively regulates SNARE complex formation. EMBO J. 2001;20:3380–3388. doi: 10.1093/emboj/20.13.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Koonin EV, Aravind L. Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita R, Kawagoshi A, Sawasaki T, Madin K, Ogasawara T, Oka T, Endo Y. Ribonuclease activity of rat liver perchloric acid-soluble protein, a potent inhibitor of protein synthesis. J Biol Chem. 1999;274:20688–20692. doi: 10.1074/jbc.274.29.20688. [DOI] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. Novel predicted RNA-binding domains associated with the translation machinery. J Mol Evol. 1999;48:291–302. doi: 10.1007/pl00006472. [DOI] [PubMed] [Google Scholar]
- Bai C, Tolias PP. Cleavage of RNA hairpins mediated by a developmentally regulated CCCH zinc finger protein. Mol Cell Biol. 1996;16:6661–6667. doi: 10.1128/mcb.16.12.6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Kato N, Wang W, Li J, Chen X. Two RNA binding proteins, HEN4 and HUA1, act in the processing of AGAMOUS pre-mRNA in Arabidopsis thaliana. Dev Cell. 2003;4:53–66. doi: 10.1016/s1534-5807(02)00399-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelissen RL, Heinrichs V, Habets WJ, Simons F, Luhrmann R, van Venrooij WJ. Zinc finger-like structure in U1-specific protein C is essential for specific binding to U1 snRNP. Nucleic Acids Res. 1991;19:449–454. doi: 10.1093/nar/19.3.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L, Koonin EV. The U box is a modified RING finger - a common domain in ubiquitination. Curr Biol. 2000;10:R132–R134. doi: 10.1016/S0960-9822(00)00398-5. [DOI] [PubMed] [Google Scholar]
- Cyr DM, Hohfeld J, Patterson C. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci. 2002;27:368–375. doi: 10.1016/S0968-0004(02)02125-4. [DOI] [PubMed] [Google Scholar]
- Juhnke H, Charizanis C, Latifi F, Krems B, Entian KD. The essential protein fap7 is involved in the oxidative stress response of Saccharomyces cerevisiae. Mol Microbiol. 2000;35:936–948. doi: 10.1046/j.1365-2958.2000.01768.x. [DOI] [PubMed] [Google Scholar]