

Abstract
We report two prenatal and two postnatal diagnosed cases (the latter monozygotic twins) with ring chromosomes after GTG banding. All four, de novo r(18), cases turned out to be more complex after application of high-resolution molecular cytogenetics techniques such as use of fluorescence in situ hybridization, centromeric probes, multicolor banding, and locus-specific probes for chromosome 18. All four cases are mosaics involving chromosome 18 in up to five different cell lines, including 46,r(18); 46,dr(18); 47,r(18)x2; 46,mar(18); and 45,-18. Mosaicism sharing both numerical and structural anomalies is rare, but rings often appear as mosaics due to their mitotic instability. Overall, patients with ring chromosome 18 usually share clinical features of 18q- syndrome and, less frequently, those of 18p- syndrome. High-resolution molecular cytogenetics techniques were useful in the characterization of cases with dynamic mosaicism and in establishing the relationship between loss or gain of chromosomal material and the phenotype.
Keywords: mosaicism, ring chromosome, mitotic instability, fluorescence in situ hybridization, multicolor banding
The occurrence of structural abnormalities that involve chromosome 18 is relatively high (Baumer et al. 2002; Miller et al. 2003). The most frequent of these abnormalities are deletions and ring chromosome formation. About 70 cases have been reported with ring chromosome 18, and most of these were female (Schinzel 2001). The phenotypic variability observed in patients with r(18) depends primarily on the extension of the deleted chromosome segment (Stankiewicz et al. 2001). In most cases, patients that present r(18) show a phenotype such as the 18q-, which is highly variable, probably depending on the extent of the terminal or interstitial 18q deletions (Brkanac et al. 1998; Cody et al. 1999; Engelen et al. 2003). Few cases share the phenotype like 18p- syndrome or a combination of these two syndromes (Bird et al. 1997; Schinzel 2001; Stankiewicz et al. 2001). In general, it is thought that malformations in these patients result from terminal deletions of both arms of the chromosome (Vermeesch et al. 2002). Rings often appear as mosaics, and, in some cases, loss of the ring, double-sized rings, or multiple copies of the ring have been observed, as a consequence of the structural instability of the ring during cell division (Baumer et al. 2002; Miller et al. 2003). Mosaicism involving both structural and numerical chromosome anomalies in a single individual is rare (Eiben et al. 1992; Sutcliffe et al. 2001). We present two prenatal and two postnatal cases (twins) showing mosaicism for rings, marker, and monosomy 18. In these cases, the application of several high-resolution molecular cytogenetics techniques, such as fluorescence in situ hybridization (FISH) using centromeric probes, multicolor banding (MCB), and locus-specific probes for chromosome 18, were important for increasing the precision of the definition of the rearrangement itself and for defining more accurately the level of mosaicism, attained by the evaluation of a larger number of cells.
Materials and Methods
Case Reports
Case A. A prenatal diagnosis was performed in a 34-year-old pregnant woman in the 14th week of gestation because of malformations shown by ultrasound, including holoprosencephaly (HPE) and hypotelorism (Figure 1A). It was the first gestation of a non-consanguineous healthy couple. After cytogenetic evaluation of the amniocytes (for results, see below), the parents chose, after genetic counselling, to terminate the pregnancy in the 15th week of gestation. An autopsy was performed, and the anatomopathologic study revealed a fetus with female-like gonads, HPE (Figure 1B), low-set ears, and without other internal or external malformations.
Figure 1.
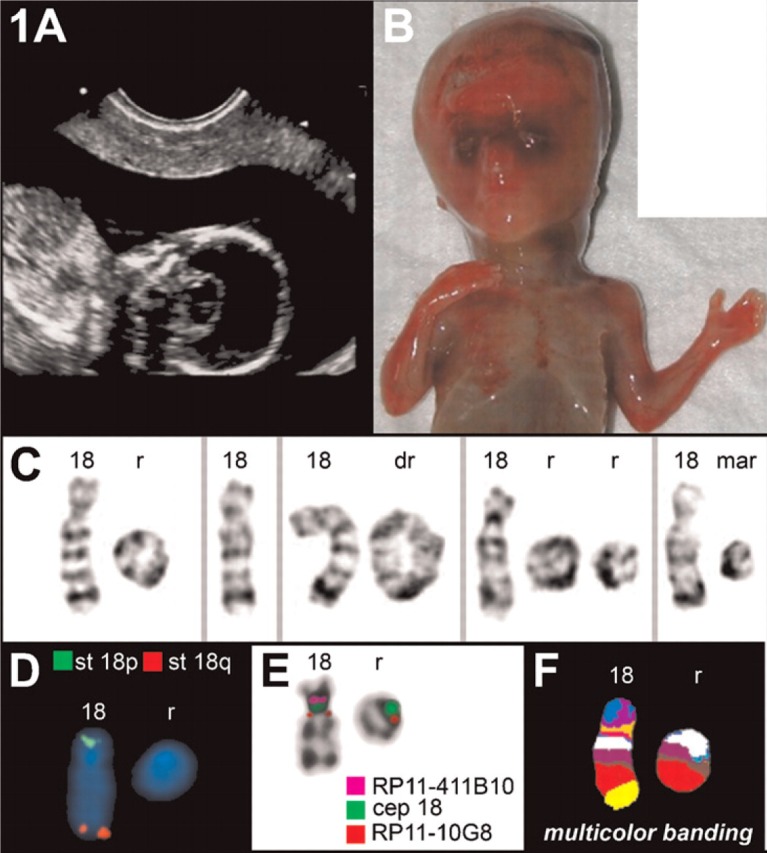
Case A. (A) Sonography revealed a holoprosencephaly (HPE). (B) Frontal view of the autopsy of the fetus at 17 weeks, showing the HPE. (C) Partial G-banded karyotype showing the normal chromosome 18, the single ring (r), double ring (dr), two rings, and the marker (mar) (D) Commercially available FISH subtelomeric probes (Abbott/Vysis) for chromosomes 18p (st 18p) and 18q (st 18q) demonstrated that both corresponding regions are deleted in the ring chromosome. (E) Fluorescence in situ hybridization applying subcentromeric probes revealed that the centromere-near region in 18p11.21 was absent and the centromere-near region in 18q11.21 was present on the ring. (F) Multicolor banding (MCB) confirmed the aforementioned findings.
Case B. A prenatal diagnosis was performed in a 27-year-old pregnant woman in the 28th week of gestation because of abnormal ultrasound results of the heart, suggesting a vitium cordis, i.e., pulmonar stenosis with poststenotic dilatation and hypoplastic right ventricle. The pregnancy was terminated in the 34th gestational week. Autopsy revealed deepset ears, large back of the head, and flat arch of the feet. Moreover, the presence of a valvulary pulmonar stenosis with poststenotic dilatation was confirmed; also detected was a premature closure of ductus botalii, and an obstructive fetal uropathy with left-sided ureterstenosis including dilatation of renal pelvis and urethra dilatation plus fibrosis.
Cases C1 and C2. Monozygotic twins were born to healthy Caucasian, non-consanguineous parents. The mother, a 1st gravida, was 28 years old at delivery. After an uneventful pregnancy, the girls were delivered by caesarean section at 36 weeks of gestation. At birth, auxological data for the first twin were: weight, 2.180 g (<3rd percentile); length, 43 cm (4th percentile); occipital-frontal circumference (OFC), 31.5 cm (20th percentile); and for the second twin: weight, 1.460 g; length, 40 cm; OFC, 29.5 cm (all <3rd percentile); Apgar scores were 9/10/10, respectively.
Because of similar abnormal clinical features observed in both girls, such as minimal dysmorphisms including a smooth philtrum, a thin upper lip, low-set ears, widely spaced nipples, and clinodactyly of the fifth finger, cytogenetic analysis from lymphocytes was done.
Developmental delay was first observed at 6 months. Independent walking was achieved at the age of 20 months. At the age of 4 years, they spoke short sentences of three to four words. At the age of 8 years, they were able to read single words, and it was expected that they would acquire adequate reading abilities. Both girls displayed growth retardation, with all growth parameters developing below the 3rd percentile. At the age of 8 4/12 years, height was 107.8 cm [−4, 7 standard deviation score (SDS)] for the first twin and 103.7 cm (−5, 5 SDS) for the second twin. Ossification was delayed to the level of 5 9/12 years and 5 years, respectively. Growth hormone and related factors [insulin-like growth factor (IGF) 1 and IGF binding protein 3] measured in the low-normal range on several occasions. Ultrasound examination excluded major malformations of internal organs. Magnetic resonance imaging of the brain performed in the first twin at the age of 4 years showed an Arnold-Chiari-I malformation without hydrocephalus and delayed myelination. Seizures have not been observed. Major clinical problems were recurrent infections, bronchial hyperreactivity, and dry and eczematic skin. All studies followed the principles outlined in the Declaration of Helsinki.
Cytogenetics
Metaphase spreads were prepared, using standard procedures (Rooney and Czepulkowski, 1992), from cultured amniotic fluid cells (Cases A and B) and peripheral lymphocytes (Cases C1 and C2). Chromosomes were GTG banded (550 bands per haploid genome).
Molecular cytogenetics was performed using centromeric (D18Z1; Vysis/Abbott, Inc., Des Plaines, IL) and subtelomeric probes (D18S552 and D18S1390; Vysis/Abbott), subcentromere-specific mFISH [subcenM-FISH, with BAC probes RP11-411B10 and RP11-10G8 (Starke et al. 2003)] and MCB (Liehr et al. 2002).
SubcenM-FISH and MCB were analysed using a Zeiss Axioplan fluorescence microscope (Zeiss; Jena, Germany) with MetaSystems (Isis) software (Altlussheim, Germany). The other FISH studies were done using a Nikon Eclipse fluorescence microscope (Nikon Instruments Europe B.V.; Badhoevedorp, The Netherlands) coupled with the Cytovision system (Applied Imaging International Lda; Newcastle upon Tyne, UK).
Results
Case A
Cytogenetic analysis of G-banded chromosomes after the first passage from different culture lines of amniotic fluid cells, using flask technique, revealed a mosaicism involving two cell lines: mos 46,XY,r(18)[36]/45,XY,-18[7]. FISH was performed in order to clarify the structural rearrangement and the distribution of the different cell lines. Chromosome 18 centromeric probe D18Z1 showed signals on both the normal chromosome 18 and the ring chromosomes. With this probe, it was also possible to identify a metaphase with a duplicated ring and another with a double ring. Hybridization with subtelomeric probes D18S552 (18p) and D18S1390 (18q) showed normal signals on the normal chromosome and deletion of both terminal regions in the ring chromosomes (data not shown). With these FISH studies, it was possible to identify two extra cell lines, so that the karyotype was: mos 46,XY,r(18)[36]/45,XY,-18[7]/47,XY, + r(18) x2 [1]/46,XY,dupr(18)[1] (Figure 1C; Table 1).
Table 1.
Karyotype after application of MCB, subtelomeric probes 18pter and 18qter in combination with the centromeric probe for chromosome 18, and subcenM-FISH in the four cases
| Case | Karyotype |
| Case A | |
| Amniocytes | |
| Routine cytogenetics | mos 46,XY,r(18)[36]/45,XY-18[7] |
| Results after MCB | mos 46,XY,r(18)(::p11.1→q22)/45,XY,-18/46,XY,dupr(18) (::p11.1→q22::)/46,XY,del(18)(:p11.1→q12:) |
| Results after subtelomeric and centromeric probes | mos 46,XY,r(18)(::p11.1→::q22)[36]/45,XY,-18[7]/47,XY,-18,+r(18)(::p11.1→::q22)x2[1]/46,XY,dupr(18)(::p11.1→q22::)[1] |
| Fibroblasts | |
| Routine cytogenetics | mos 46,XY,r(18)[89]/45,XY-18[22]/46,XY,dupr(18)[2]/46,XY,mar(18)[4] |
| Results after subtelomeric and centromeric probes | mos 46,XY,r(18)(::p11.1→::q22)[89]/45,XY,-18[22]/46,XY,dupr(18)(::p11.1→q22::)[2]/46,XY,mar(18) |
| Case B | |
| Routine cytogenetics | 46,XY,r(18)[90%]/45,XY,-18[10%] |
| Results after MCB | r(18)(::p11.2?2→q21.?2::)[12]/r(18;18)(::p11.2?2→q21.?2::p11.2?2→q21.?2::)[4]/r(18;18)(::p11.2?2→q21.?2:: p11.2?2→q21.?2:)x2[1]/del(18)(:p11.21→q11.2)[1]/ace(18)(:q11.2→q12.?2)[1] |
| Case C1 | |
| Routine cytogenetics | 46,XX,r(18)(p11.32q22.3) |
| Results after MCB | 46,XX,r(18)(::p11.32→q23::)[8]/46,XX,r(18)(::p11.32→q23::p11.32→q23::)[4]/45,XX,-18[2] |
| Results after subtelomeric and centromeric probes | 46,XX,r(18)(::p11.32→q23::)[11]/46,XX,r(18)(::p11.32→q23::p11.32→q23::)[3]/45,XX,-18[1]/47,XX,r(18)(::p11.32→q23::p11.32→q23::),+del(18)(q?11.2)[1]/47,XX,-18,+del(18)(:p11.?1→q11.?1:)x2[1]/46,XX,min(18)(:p11.?1→q11.?1:)[1] |
| Result after subcenM-FISH | 46,XX,r(18)(::p11.32→q23::)[6]/46,XX,r(18)(::p11.32→q23::p11.32→q23::)[4] |
| Case C2 | |
| Routine cytogenetics | 46,XX,r(18)(p11.32q22.3) |
| Result after MCB | 46,XX,r(18)(::p11.32→q23::p11.32→q23::)[7]/46,XX,r(18)(::p11.32→q23::)[4]/45,XX,-18[3]/46,XX,r(18)(::p11.32→q22.1::)[1]/45,XX,-18,r(18)(::p11.32rarr;q23::)[1] |
| Result after subtelomeric and centromeric probes | 46,XX,r(18)(::p11.32→q23::)[7]/46,XX,r(18)(::p11.32→q23::p11.32→q23::)[3]/45,XX,-18[1]/45,XX,-18,r(18)(::p11.32→q23::)[1]46,XX,-18,r(18)(::p11.32→q23::)x2[1]/46,XX,del(18)(:p11.?1→q11.?1:)[1] |
| Result after subcenM-FISH | 46,XX,r(18)(::p11.32→q23::)[3]/46,XX,r(18)(::p11.32→q23::p11.32→q23::)[3]/46,XX,-18×2,+r(18)(::p11.32->q23::p11.32→q23::)x2[3]/47,XX,del(18)(:p11.2→q11.2:)x2[1] |
FISH, fluorescence in situ hybridization; MCB, multicolor banding. Because highly complex karyotypes were present in all cases, slightly different results were obtained for the probe sets used, as listed here.
After termination of the pregnancy, a cytogenetic analysis and FISH studies were performed in the fibroblasts of the expelled fetus after the third passage. A new cell line with a small marker chromosome was detected, as well as three of the cell lines observed in the amniocytes. The line with two rings, observed in the amniocytes, was not found in the fibroblasts. Thus, the result of the cytogenetic analysis in this tissue was: mos 46,XY,r(18)[89]/45,XY,-18[22]/46,XY,dupr(18)[2]/46,XY,mar(18)[4]. FISH with a centromere 18—specific probe was also performed to determine the origin of the marker chromosome, which was found to contain material from the pericentromeric region of chromosome 18 (Figure 1E).
Applying subtelomeric probes 18p and 18q (Figure 1D), subcenM-FISH (Figure 1E), and MCB (Figure 1F) analysis, only the most frequent variant of the derivative chromosome 18 could be detected. Thus, a final karyotype such as the following can be suggested: mos 46,XY,r(18)(::p11.1→::q22)/45,XY,-18/46,XY, dupr(18)(::p11.1→q22::)/46,XY,del(18)(:p11.1→q12:) (Table 1). Karyotypes of both parents were normal.
Case B
After G-banding the amniocytes, two cell lines were accounted for with the following karyotype: 46,XY,r(18)[90%]/45,XY,-18[10%].
MCB analyses revealed a complex karyotype with five cell lines: r(18)(::p11.2?2→q21.?2::)[12]/r(18;18)(::p11.2?2→q21.?2::p11.2?2→q21.?2::)[4]/r(18;18)(::p11.2?2→q21.?2::p11.2?2→q21.?2::)x2[1]/del(18)(:p11.21→q11.2:)[1]/ace(18)(:q11.2→q12.?2:)[1] (Figure 2; Table 1). Karyotypes of both parents were normal.
Figure 2.
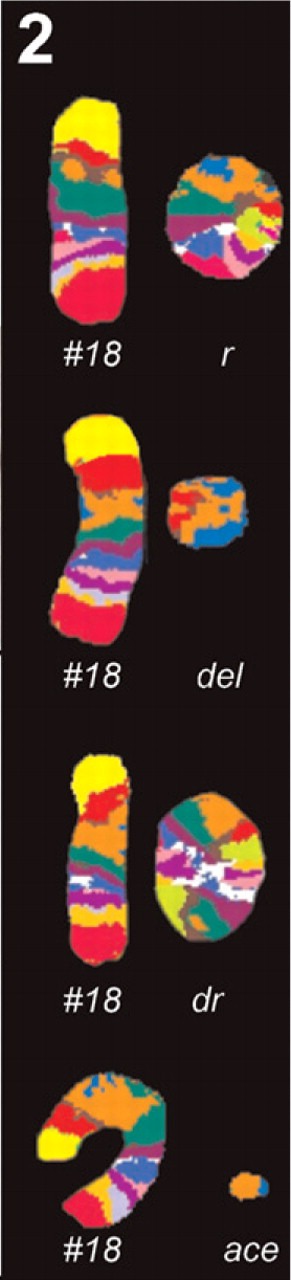
Case B, MCB results. In addition to a normal chromosome 18, either a ring chromosome (r), a double ring (dr), a minute shaped chromosome (del), or an acentric fragment (ace) was present.
Cases C1 and C2
Structurally abnormal female karyotypes with ring chromosomes 18 [karyotype 46,XX,r(18)(p11.32q22.3) de novo] were observed in both girls without evidence for mosaicism. MCB, subcenM-FISH, and application of subtelomeric probes in combination with centromeric probes were very useful, because they allowed the identification of additional cell lines and a variety of rearrangemnts revealing differing complex karyotypes in each of the twins (see Table 1). Parental karyotypes were normal. Two younger sisters of the twins are healthy.
Discussion
We report here four cases, two prenatal and two postnatal, with complex karyotypic changes involving the formation of rings of chromosome 18.
There are reports of rings for all chromosomes, although the most frequent are those involving autosomes 13 and 18 (Mohamed et al. 2001). It is suggested that chromosome 18 might carry an elevated number of certain repeated sequences susceptible to interchromosomal and intrachromosomal rearrangements (Pater et al. 2003). The most frequent structural rearrangements that involve chromosome 18 are in deletions and ring chromosomes.
One of the mechanisms of formation of a ring chromosome involves breakage of the chromosome at both ends and the joining back of both extremities, with loss of terminal regions (Sigurdardottir et al. 1999). Depending on the size of the deletion at each end of the chromosome, the phenotypic appearance ranges from normal fertile carriers in a minority of cases to those with severe malformations. Incidence and types of congenital malformations are similar to those carrying the del(18)(q21-qter) (Schinzel 2001).
There is a difficulty in establishing a genotype—phenotype correlation in a ring carrier. It is necessary to determine primary deletions associated with ring chromosome formation and also secondary loss or gain of material that may occur due to the instability of ring chromosomes. This can cause a dilemma in prenatal diagnosis (Tümer et al. 2004). Rings often appear as mosaics because of sister chromatid exchange within a ring in mitotic crossing-over events, which generates aneuploid cells with increased mortality (Sigurdardottir et al. 1999; Vermeesch et al. 2002).
Patients with ring chromosome 18 usually share clinical features of the 18q- syndrome, such as hypotonia, poor coordination, microcephaly, hearing abnormalities, and malformation of genitalia (Brkanac et al. 1998; Cody et al. 1999; Schinzel 2001). Less frequently, these patients show characteristics in common with the 18p-syndrome, such as mild to moderate growth deficiency, microcephaly, ptosis and, more occasionally, HPE (Schinzel 2001). Case A shares features of both syndromes. Cytogenetic analysis revealed a male fetus, and the anatomopathologic study reported a fetus with female-like gonads. This discrepancy may be related to malformation of genitalia, a clinical feature of 18q-syndrome. Low-set ears is also an abnormality associated with deletion of the long arm of chromosome 18. HPE (also found in the autopsy) is probably due to deletion of 18p (Muenke 1989; Bird et al. 1997). The genetic causes of HPE can be cytogenetic anomalies or monogenic syndromes (Cohen 1989; Muenke 1989; Croen et al. 1996; Olsen et al. 1997). The association between HPE and the deletion of chromosome 18p was first described in a child with HPE and monosomy 18p due to an unbalanced translocation (Johnson and Bachman 1976). The region 18p11.3 is defined as one of the critical regions for HPE (Overhauser et al. 1995). Despite the fact that all of the cases reported here showed deletions on 18p11.3, HPE was only found in Case A. This could mean that the HPE-critical region was most probably not disrupted in the other three cases; i.e., the chromosome breaks were more distal, toward the telomere. According to Strenge and Froster (2004), HPE occurs in only 10–20% of the cases showing the terminal deletion.
The occurrence of an r(18) together with a duplication of a segment of chromosome 18 or with a small marker chromosome is rare (Madan et al. 1981; Stankiewicz et al. 2001). In each of the four cases reported here, a marker was detected in a small percentage of the cells observed from different cultures, suggesting that it is not a culture artefact (Table 1). The most likely explanation for the mosaic occurrence of the small marker in such small proportions could be that its loss would not be associated with a decreased cell survival as it would be if a complete ring were lost (Sigurdardottir et al. 1999).
Cytogenetic analysis of Case A revealed a mosaicism involving a total of five cell lines (four in the amniotic fluid and four in post mortem fibroblasts, with two lines that only appear each in one or the other tissue) showing the mitotic instability of the ring (Figure 1; Table 1). Mosaicism of monosomy 18 was considered as a culture artefact, due to mitotic loss of the ring (Fischer et al. 2001; Schinzel 2001). However, there are reasons that make us think otherwise and that lead us to agree with Yardin et al. (2001): two different cultures of the amniotic fluid in Case A gave approximately the same level of mosaicim, and the monosomic cell line was also present in fibroblasts. In both tissues, the percentage of this cell line was similar (16% and 18%, respectively). Also in all four cases, a cell line with −18 was observed (Table 1). One could suggest that this cell line may have started as an r(18), which, because of its mitotic instability and crossing-over events between the ring sister chromatids, led to its loss during cell division, generating the monosomy. Therefore, the monosomic cell line may be part of the biological mechanism associated with the propagation of the ring during cell division. These events are also responsible for loss or duplications of ring chromosomes.
The growth retardation observed in both twins (Cases C1 and C2) could be associated with the occurrence of sister chromatid exchange within the rings, which generates aneuploid cells with double rings and, consequently, serious genetic imbalance. This process may lead to increased cell death, which can lead to a decreased number of viable cells at any given interval of development and subsequently to growth deficiencies in the carriers. Generally, severe growth failure is seen more often in patients with larger rings than among patients with smaller ones, because of the probability of the higher frequency of sister chromatid exchanges in the former (Kosztolányi 1987).
In all the cases presented here, a de novo r(18) was established, showing mitotic instability that seems to be an important tool for the investigation of the dynamics of ring chromosome mosaicism, through multiple cell divisions. Mosaicism in all four reported cases can be explained by post-zygotic errors during mitosis. The variability of the cell lines observed and in the structure of the ring chromosomes found highlights the importance of analyzing a large number of cells and of using various cytogenetic techniques.
The use of molecular cytogenetics is also of great value in the establishment of the presence of either loss or gain of chromosomal material in the ring structure, because it will help to elucidate the role played by that material in determining the phenotypes, facilitating the search for candidate genes in those regions.
Acknowledgments
This work was supported in part by the Deutsche For-schungsgemeinschaft (436 RUS 17/22/06, LI820/11-1) and the Ernst-Abbe-Stiftung.
Literature Cited
- Baumer A, Uzielli MLG, Guarducci S, Lapi E, Rothlisberger B, Shinzel A. (2002) Meiotic origin of two ring chromosomes 18 in a girl with developmental delay. Am J Med Genet 113: 101–104 [DOI] [PubMed] [Google Scholar]
- Bird LM, Pretorius DH, Mendonza AE, Jones MC. (1997) Anencephaly with holoprosencephalic facies due to ring chromosome 18. Clin Dysmorphol 6: 351–358 [DOI] [PubMed] [Google Scholar]
- Brkanac Z, Cody JD, Leach RJ, DuPont BR. (1998) Identification of cryptic rearrangements in patients with 18q- deletion syndrome. Am J Hum Genet 62: 1500–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cody JD, Ghidoni PD, DuPont BR, Hale DE, Hilsenbeck SG, Stratton RF, Hoffman DS, et al. (1999) Congenital anomalies and anthromometry of 42 individuals with deletions of chromosome 18q. Am J Med Genet 85: 455–462 [DOI] [PubMed] [Google Scholar]
- Cohen MM. (1989) Perspectives on holoprosencephaly: Part I. Epidemiology, genetics, and syndromology. Teratology 40: 211–235 [DOI] [PubMed] [Google Scholar]
- Croen LA, Shaw GM, Lammer EJ. (1996) Holoprosencephaly: epidemiology, and clinical characteristics of a California population. Am J Med Genet 64: 465–472 [DOI] [PubMed] [Google Scholar]
- Eiben B, Unger M, Stoltenberg G, Rutt G, Goebel R, Meyer A, Gamerdinger U, et al. (1992) Prenatal diagnosis of monosomy 18 and ring chromosome 18 mosaicism. Prenat Diagn 12: 945–950 [DOI] [PubMed] [Google Scholar]
- Engelen JJM, Moog U, Weber J, Haagen AAM, Van Uum CMJ, Hamers AJH. (2003) Deletion of chromosome region 18q21,1→18q21.3 in a patient without clinical features of the 18q- phenotype. Am J Med Genet 119: 356–359 [DOI] [PubMed] [Google Scholar]
- Fischer W, Dermitzel A, Osmers R, Pruggmayer M. (2001) Complete karyotype discrepancy between placental and fetal cells in a case of ring chromosome 18. Prenat Diagn 21: 481–483 [DOI] [PubMed] [Google Scholar]
- Johnson G, Bachman R. (1976) A 46, XY, del(18)(pter leads to p1 100:) cebocephalic child from a 46, XX, t(12;18)(18pter leads to 18 p 1100:: 12qter leads to 12pter) normal parent. Hum Genet 34: 103–106 [DOI] [PubMed] [Google Scholar]
- Kosztolányi G. (1987) Does “ring syndrome” exist? An analysis of 207 case reports on patients with a ring autosome. Hum Genet 75: 174–179 [DOI] [PubMed] [Google Scholar]
- Liehr T, Heller A, Starke H, Rubtsov N, Trifonov V, Mrasek K, Weise A, et al. (2002) Microdissection based high resolution multicolor banding for all 24 human chromosomes. Int J Mol Med 9: 335–339 [PubMed] [Google Scholar]
- Madan K, Vlasveld L, Barth PG. (1981) Ring-18 and isopseudodicentric-18 in the same child: a hypothesis to account for common origin. Ann Genet 24: 12–16 [PubMed] [Google Scholar]
- Miller K, Pabst B, Rilter H, Nürnberg P, Siebert R, Schmidtke J, Arslan-Kirchner M. (2003) Chromosome 18 replaced by two ring chromosomes of chromosome 18 origin. Hum Genet 112: 343–347 [DOI] [PubMed] [Google Scholar]
- Mohamed AN, Ebrahim SA, Aatre R, Qureshi F, Jacques SM, Evans MI. (2001) Prenatal diagnosis of a de novo ring chromosome 11. Am J Med Genet 102: 368–371 [DOI] [PubMed] [Google Scholar]
- Muenke M. (1989) Clinical, cytogenetic and molecular approaches to the genetic heterogeneity of holoprosencephaly. Am J Med Genet 34: 237–245 [DOI] [PubMed] [Google Scholar]
- Olsen CL, Hughes JP, Youngblood LG, Sharp-Stimac M. (1997) Epidemiology of holoprosencephaly and phenotypic characteristics of affected children: New York State, 1984-1989. Am J Med Genet 73: 217–226 [DOI] [PubMed] [Google Scholar]
- Overhauser J, Mitchell HF, Zackai E, Tick D, Rojas K, Muenke M. (1995) Physical mapping of the holoprosencephaly critical region in 18p11.3. Am J Hum Genet 57: 1080–1085 [PMC free article] [PubMed] [Google Scholar]
- Pater JM, Smeets DFCM, Scheres JMJC. (2003) Unique mosaicism of structural chromosomal rearrangement: is chromosome 18 preferentially involved? Am J Med Genet A 119: 26–31 [DOI] [PubMed] [Google Scholar]
- Rooney DE, Czepulkowski BH, eds (1992) Human Cytogenetics: A Practical Approach. 2nd ed. New York, Oxford University Press [Google Scholar]
- Schinzel A. (2001) Catalogue of Unbalanced Chromosome Aberrations in Man. 2nd ed. Berlin and New York, Walter de Gruyter [Google Scholar]
- Sigurdardottir S, Goodman BK, Rutberg J, Thomas GH, Jabs EW. (1999) Clinical, cytogenetic, and fluorescence in situ hybridization findings in two cases of “complete ring” syndrome. Am J Med Genet 87: 384–390 [DOI] [PubMed] [Google Scholar]
- Stankiewicz P, Brozek I, Hélias-Rodzewicz Z, Wierzba J, Pilch J, Bocian E, Balcerska A, et al. (2001) Clinical and molecularcytogenetics studies in seven patients with ring chromosome 18. Am J Med Genet 101: 226–239 [DOI] [PubMed] [Google Scholar]
- Starke H, Nietzel A, Weise A, Heller A, Mrasek K, Belitz B, Kelbova C, et al. (2003) Small supernumerary marker chromosomes (SMCs): genotype-phenotype correlation and classification. Hum Genet 114: 51–67 [DOI] [PubMed] [Google Scholar]
- Strenge S, Froster UG. (2004) Diaphragmatic hernia in 18p- syndrome. Am J Med Genet A 125: 97–99 [DOI] [PubMed] [Google Scholar]
- Sutcliffe MJ, Mueller OT, Koussef BG, Dumont DP, McFarland JA, Mawani F, Conforto D, et al. (2001) Three cell line mosaicism involving structural and numerical abnormalities of chromosome 18 in a 3.5-year-old girl: 47, XX, + 18/47, XX, + del(18)(q22)/46, XX. Am J Med Genet 102: 192–199 [DOI] [PubMed] [Google Scholar]
- Tümer Z, Harboe TL, Blennow E, Kalscheuer VM, Tommerup N, Brøndum-Nielsen K. (2004) Molecular cytogenetic characterization of ring chromosome 15 in three unrelated patients. Am J Med Genet A 130: 340–344 [DOI] [PubMed] [Google Scholar]
- Vermeesch JR, Baten E, Fryns J-P, Devriendt K. (2002) Ring syndrome caused by ring chromosome 7 without loss of subtelomeric sequences. Clin Genet 62: 415–417 [DOI] [PubMed] [Google Scholar]
- Yardin C, Esclaire F, Terro F, Baclet MC, Barthe D, Laroche C. (2001) First familial case of ring chromosome 18 and monosomy 18 mosaicism. Am J Med Genet 104: 257–259 [PubMed] [Google Scholar]