

Abstract
Segregation of mitochondrial DNA (mtDNA) is an important underlying pathogenic factor in mtDNA mutation accumulation in mitochondrial diseases and aging, but the molecular mechanisms of mtDNA segregation are elusive. Lack of high-throughput single-cell mutation load assays lies at the root of the paucity of studies in which, at the single-cell level, mitotic mtDNA segregation patterns have been analyzed. Here we describe development of a novel fluorescence-based, non-gel PCR restriction fragment length polymorphism method for single-cell A3243G mtDNA mutation load measurement. Results correlated very well with a quantitative in situ Padlock/rolling circle amplification—based genotyping method. In view of the throughput and accuracy of both methods for single-cell A3243G mtDNA mutation load determination, we conclude that they are well suited for segregation analysis.
Keywords: segregation, heteroplasmy, mitochondrial diseases, aging, A3243G mtDNA, mutation load, Padlock probing, PCR-RFLP
As a norm, all mitochondrial DNA (mtDNA) molecules in all cells of an individual have identical sequences, a situation referred to as homoplasmy (Enriquez 2003). Due to lack of sophisticated DNA repair systems in the mitochondrial compartment, mutation rates of mtDNA are five to ten times higher than those of nuclear DNA and deviation from the norm of homoplasmy will inevitably occur by acquisition of mtDNA mutations. Thus, invariably the situation arises in which two mtDNA sequence variants are present within cells of individuals, a condition referred to as heteroplasmy. Whether inherited or acquired, as a consequence of segregation, deleterious mtDNA mutations can accumulate to a level (the critical threshold) at which too little compensation from the wild-type molecules occurs, accounting for cell and tissue failure and hence disease.
Studies with mice that are heteroplasmic for neutral mtDNA haplotypes contributed significantly to the establishment of the germ line mitochondrial genetic bottleneck: reduction of the mtDNA copy number in early oogenesis in combination with random segregation, leading to a return to homoplasmy in one or only a few generations (Jenuth et al. 1996). Recently, however, it has been suggested that actual mtDNA copy numbers in early oogenesis are not compatible with the genetic bottleneck/random segregation mechanism (Cao et al. 2007). Also with heteroplasmic mice, it has been shown that postnatally, tissue-specific non-random or directional segregation occurs (Jenuth et al. 1997). Furthermore, in human postembryonic life, non-random segregation of inherited, pathogenic mtDNA mutations occurs (Chinnery et al. 1999; Chinnery 2002). Gaining further knowledge of segregation mechanisms will thus contribute to a better understanding of the nature of the mitochondrial genetic bottleneck and clonal accumulation of acquired mtDNA mutation with age, and of how differences in pathogenic mtDNA mutation load among different tissues and organs are generated.
So-called transmitochondrial cybrids are created by the fusion of enucleated cells carrying two different mtDNA sequence variants with a nucleated cell that has no mtDNA (ρ0 cells). Heteroplasmic cybrid cells have been a valuable resource for experimental mitotic segregation studies. In the human situation, they have been used particularly in studying segregation of the A3243G mtDNA sequence variants. This mutation causes a variety of disease phenotypes. It may be considered as exemplary for a central question in diseases with mtDNA mutation in their etiology: how can a single mutation cause variable phenotypes? Segregation is an obvious but elusive factor (Torroni et al. 2003).
By heteroplasmy analysis of bulk DNA of passages of cybrid clones carrying the A3243G pathogenic mutation, various patterns have been found: stable heteroplasmy and heteroplasmy shifts to either wild-type or mutant (for review, see Enriquez 2003). Of significance, stable heteroplasmy, as measured on bulk DNA of cells in passages of cultured cybrid clones, can be the result of random mitotic mtDNA segregation or non-segregation. Single-cell heteroplasmy determinations are needed to discriminate the two, but such studies are rare. Lehtinen et al. (2000) used limiting dilution to clone cells of late passages of continuously cultured A3243G cybrid founder cells. Following limited outgrowth, the mutation load of individual cells in the passages was assessed from bulk DNA using a PCR restriction fragment length polymorphism (PCR-RFLP) method with gel electrophoresis to quantify mutant fractions.
The workload associated with the gel-based PCR-RFLP method for A3243G single-cell heteroplasmy quantification after limited clonal expansion of cells in cybrid clone passages limits throughput. As an illustration, a total of six clones were analyzed after 30 weeks of continuous culture with, on average, ∼33 single cells analyzed (range 22–63) (Lehtinen et al. 2000; Turner et al. 2005). We therefore sought to develop high-throughput A3243G mutation load assays enabling analysis of hundreds of single cells in long-term serial passages of multiple cybrid clones. This will enable the analysis of heteroplasmy evolution longitudinally, which is essential in mechanistically sorting out segregation processes that determine heteroplasmy.
Here we used two strategies for single-cell A3243G mtDNA mutation load quantization: (1) physical isolation of individual cells by single-cell sorting, followed by closed-tube, real-time fluorescence PCR load measurement; and (2) bi-color in situ mtDNA genotyping by Padlock/rolling circle amplification (Padlock-RCA) (Larsson et al. 2004), in combination with image analysis for quantification purposes.
In preliminary experiments with Taqman and Molecular Beacon probes for closed-tube, real-time fluorescence readout, it was found, however, that the mutant probes reported both mutant and wild-type targets. Similar observations have been reported by Bai and Wong (2004). This poor discriminatory power is probably a consequence of the unfavorable thermodynamics associated with G—T mismatches. It complicates closed-tube assays anyhow, but is further exacerbated in this particular case, because the mutation locates in a GC-rich environment which, being in the D-loop of the mitochondrial tRNALeu(UUR), is additionally poised to form secondary structures.
PCR-RFLP is a sturdy and time-honored approach for A3243G mutant detection. We therefore resorted to non-gel—based PCR-RFLP fragment analyses employing melting temperature characteristics (PCR-RFMT) of the fragments using SYBR Green as reporter. The only sacrifice to a closed-tube assay is the one-time addition of the restriction enzyme. Here we describe the development and performance of the PCR-RFMT method.
Recently, we described qualitative in situ genotyping of the 3243 locus by Padlock-RCA (Larsson et al. 2004). Here we applied dedicated image analysis (Allalou et al. 2007) to quantitatively correlate the in situ assay with the PCR-RFMT. On the basis of their specifications in terms of throughput and accuracy, we conclude that both methods are suitable for A3243G mtDNA segregation analyses.
Materials and Methods
Cells, Cell Culture, and Cell Sorting
The A3243G transmitochondrial 143B cybrid clones were grown on DMEM containing 4.5 mg/ml glucose and 110 μg/ml pyruvate supplemented with 50 μg/ml uridine and 10% fetal bovine serum. Transmitochondrial cybrids used in this work were sampled from an ongoing mtDNA segregation study in which we cloned heteroplasmic cells and expanded them to 9-cm cultures following continuous culture with a 10% split twice a week. Cybrid cells were produced by fusing skin fibroblasts from two maternally inherited diabetes and deafness (MIDD) patients (coded V and GB) with 143 B ρ0 cells. For single-cell sorting, cells at near-confluence were harvested in 2 ml trypsin/EDTA. Five hundred μl was resuspended in a 5-ml Falcon tube containing 1500 μl complete medium. Single cells were sorted in the wells of an optical 96-well plate (Abgene; Epsom, UK) with the FACSVantage SE (BD Biosciences; Breda, The Netherlands) cell sorter using forward and side scatter gating. Plates were sealed and stored with no further processing at −20C for at least one night and up to several months.
For in situ genotyping, cells of a given clone and passage number were harvested by trypsin treatment and seeded in a culture plate containing microscopic object slides. Cells were allowed to adhere overnight, after which they were fixed for in situ genotyping with Padlock-RCA.
Primers, Probes, and PCR
The primers for the PCR-RFMT were designed with Primer Express version 1.0 and were manufactured at Eurogentec (Seraing, Belgium). The first forward primer, 5′-CGCCTTCC-CCCGAAATGAT, anneals from location 3162 to 3181 on the mtDNA. The second forward primer, 5′-CCCACACCCACCCAAGAACA, anneals from location 3204 to 3223; and the common reverse primer, 5′-TGGCCATGGGTATGTTGTTA, at 3300 to 3319. The first forward and the common reverse primers are referred to as primer pair A and the other pair as primer pair B. PCR was performed in an end volume of 20 μl, containing 10 μl SYBR Green Mastermix (Applied Biosystems; Nieuwerkerk aan de Ijssel, The Netherlands) and 250 nM of both primers. The PCR begins with 10 min hot-start at 95C, followed by 42 cycles alternating between 15 sec 95C and 1 min 63C using an ABI 7700 or 7900 spectrofluorometric Thermal Cycler (Applied Biosystems).
Restriction Enzyme Digestion and Melt Analysis
For restriction enzyme digestion of the amplicons, 5 μl ApaI restriction enzyme mix was added directly to the PCR product. The enzyme mix was composed of 0.5 μl BSA (10 mg/ml), 0.5 μl 10 × buffer A (Promega; Leiden, The Netherlands), 50 nl ApaI enzyme (80 U/μl; Promega) and 4 μl of water. Digestion was performed overnight at 37C and stored at 4C until further processing, up to a few weeks.
The melt curves were recorded with an ABI-Prism 7700 or 7500 spectrofluorimetric Thermal Cycler (Applied Biosystems) by gradually increasing the temperature over 20 min from 65C to 90C. The ABI-Prism software package did not include software for baseline correction and peak area calculations. To perform automated analysis and data plotting, melt data were exported in Excel file format and a macro was developed in-house, as described in Results.
Genotyping mtDNA at the 3243 Locus In Situ and Image Analysis
In situ mtDNA genotyping by Padlock-RCA was performed as described previously (Larsson et al. 2004), with slight modifications. In short, cells grown on microscopic object slides were fixed in 4% formaldehyde and 0.5% sucrose in PBS for 20 min at ambient temperature, washed with PBS, and stored in 70% ethanol at 4C. Following extensive rinses with PBS, they were treated with 0.05% pepsin (P7000; Sigma) in 0.01 M HCl for 5 min to gain access to the mtDNA for enzymes and Padlock probes. Next, the cells were dehydrated with graded ethanol series followed by a preincubation of 15 min in the enzyme reaction buffer supplemented with 0.2 μg/μl BSA. Then, MscI (0.5 U/μl) and T7 exonuclease (10 U/μl) (New England Biolabs; Leusden, The Netherlands) were added and incubated at 37C at the specified times. After digestion and exonuclease treatment, the Padlock probes (100 nM) were hybridized and circularized simultaneously. The hybridization/ligation was performed for 1 hr at 55C with Ampligase (Epicentre; Madison, WI) in the supplied buffer supplemented with 1 mM nicotinamide adenine di-nucleotide, 10% glycerol, and 125 mM KCl. Subsequently, the cells were washed with TBS supplemented with 0.05% Tween-20. RCA was performed for 1 hr at 37C after addition of 1 U/μl of Φ29 DNA polymerase (Fermentas; Ontario, Canada) and 0.2 mg/ml histone (calf thymus, type 2A; Sigma) to condense the RCA DNA product. For hybridization of the detection probes, we used 250 nM Lin16-FITC for the wild-type mtDNA and 250 nM Lin33-Cy5 for the A3243G mtDNA (Larsson et al. 2004). Incubation was for 30 min at 37C in 2× standard saline citrate supplemented with 20% formamide. Following three washes with TBS, 0.01% Tween-20, nuclear DNA was counterstained with 4′,6-diamidino-2-phenylindole.
For digital microscopy, a computer-controlled standard epifluorescence microscope (Leica DM; Leica Microsystems, Rijswijk, The Netherlands), equipped with a 100-W mercury arc lamp, a 100× 1.3-numerical aperture objective, and a scientific-grade 16-bit charge-coupled device camera, was used. The computer controls image acquisition through a motorized 8-position filter-block rotor and shutters.
The image analysis algorithm has as the key feature that it segments cells in the absence of a cytoplasmic counterstain. Instead, cytoplasm delineation is based on a fixed radial distance from each cell nucleus, a strategy made possible because of the absence of Padlock-RCA signals outside the cytoplasm (Allalou et al. 2007). The program runs as a plug-in for the VIS Image Analysis Software (Visiopharm; Horsholm, Denmark). The number of red (Cy5) over the number of red (Cy5) plus green (FITC) signals per cell is taken as the mutation load.
Results
As measures of the mutant and wild-type A3243G fractions in the single-cell PCR products, we used the areas under the peaks of the first derivative of the SYBR Green fluorescence melting curve of the ApaI-fragmented PCR product. Figure 1A presents relevant optimization experiments with two different primer pairs for amplification of the A3243G region and ∼100 pg genomic DNA from cybrid clones that were 100% wild-type and near 100% mutant. A heteroplasmic sample was also included in this experiment.
Figure 1.
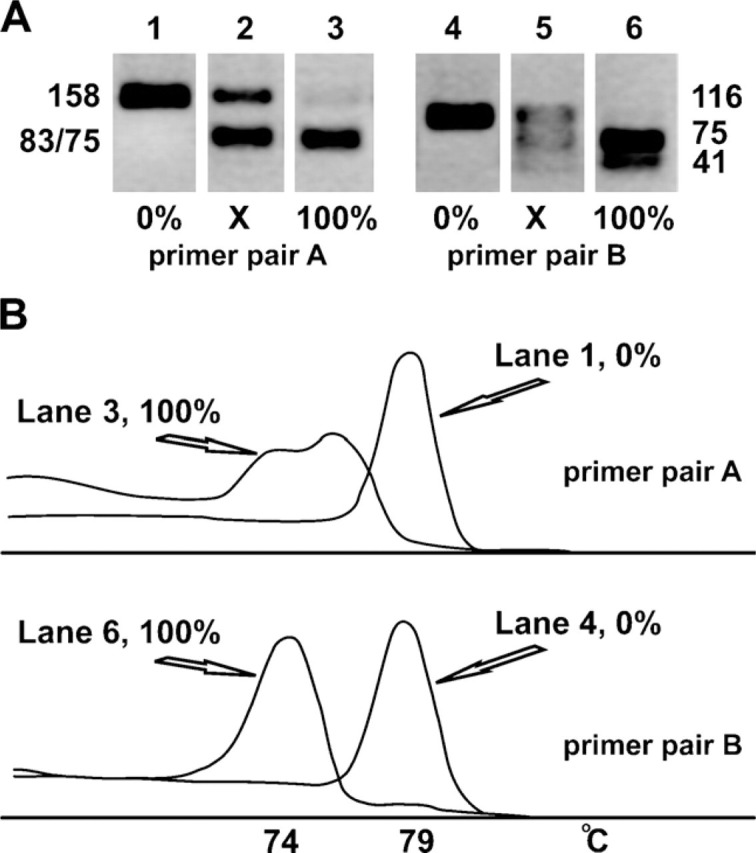
Electrophoretic and melt characteristics of the PCR restriction fragment-length polymorphism (PCR-RFLP) fragments. (A) Gel electrophoresis of the restriction enzyme fragments obtained after ApaI digestion of PCR products obtained with primer sets A and B. The amount of input DNA was ∼100 pg DNA, the equivalent of ∼16 cells. Products in Lanes 1 and 4 are from homoplasmic wild-type cybrid cells (0% mutant); in Lanes 3 and 6, from homoplasmic mutant cells (100% mutant); and in Lanes 2 and 5, from a heteroplasmic cybrid cell clone. (B) Derivatives of the melting curves of homoplasmic A3243G mtDNA. The top graph corresponds to Lanes 1 and 3 of A, the bottom graph to Lanes 4 and 6. The curves represent the derivative of the SYBR Green signal intensity (y-axis) as a function of temperature (x-axis). Note that the two mutant fragments generated with primer set A do not resolve electrophoretically (Lane 3 in A), whereas they do so in the melt analysis. The two mutant fragments generated with primer set B, in contrast, resolve electrophoretically (Lane 6 in A) but not in the melt analysis. The melting temperature (Tm) of both mutant fragments is ∼74C.
Primer pair A generates a 158-bp-long fragment with 43% GC and a melting temperature (Tm) of 79C. Digestion with the restriction enzyme ApaI cleaves only the mutant amplicon into a piece of 83 bp (46% GC, Tm = 76C) and a piece of 75 bp (40% GC, Tm = 74C).
Primer pair B generates a 116-bp-long fragment with 45% GC and also a Tm of 79C. Digestion with the restriction enzyme ApaI cleaves the mutant amplicon into a piece of 41 bp and 54% GC and a piece of 75 bp and 40% GC, both with a Tm of 74C. These Tms are measured in the PCR solution containing the restriction buffer and enzyme. In the absence of the restriction enzyme mix, the (undigested) amplicon melts at a temperature of 76C.
The electrophoretic behavior of the PCR-RFLP fragments generated from the genomic DNA of the three clones in a 3% agarose gel is shown in Figure 1A, and Figure 1B shows the derivatives of the fluorescent melting curves of the homoplasmic DNAs. The mutant restriction fragments of the amplicon from primer pair A do not resolve electrophoretically, but do so in the melt analysis. Conversely, the mutant fragments of primer pair B do resolve electrophoretically but do not in the melt analysis. Note also that with primer pair B, the wild-type fragment is much better separated from the mutant fragments in the melt analysis compared with primer pair A. The near-perfect coincidence of the melt peaks of the mutant fragments of primer pair B, the bell shapes of both peaks, and their near-complete separation provide an excellent starting point for baseline correction and estimation of the peak areas as measures for the mutant and wild-type fractions. Primer pair B was therefore chosen for further development.
To evaluate the performance of the PCR-RFMT at the single-cell level, we sorted single cells from various passages of heteroplasmic cybrid clones in 96-well optical plates and performed the PCR-RFMT procedure. In Figure 2, melt curves are shown for a heteroplasmic cell and two cells with different homoplasmic states (top panel), as well as nine derivative curves (lower panel). To exclude incomplete ApaI digestion under PCR-RFMT conditions, we mixed 100% wild-type and 100% mutant cell, single-cell sorted them, and performed PCR-RMFT. Only wild-type and mutant melt curves were obtained.
Figure 2.
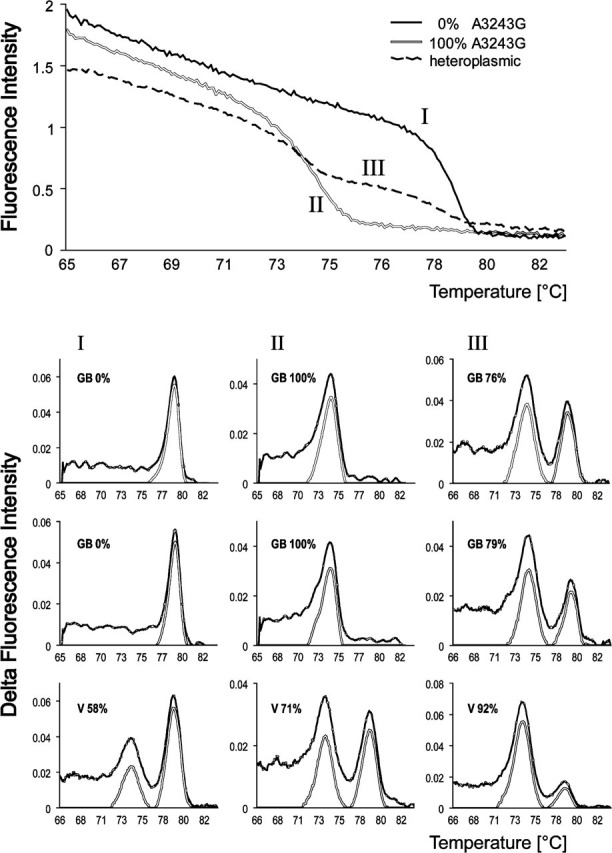
Single-cell PCR-RFLP fragment analyses employing Tm characteristics (PCR-RFMT) analysis. Upper panel: Melt curves of PCR-amplified ApaI-digested DNA from two homoplasmic (I, wild-type; II, A3243G) cells and one heteroplasmic (III) A3243G mtDNA cybrid cell. Lower panel: Examples of derivative melt curves and their analysis. The experimental data (black line) are processed by an Excel macro to generate baseline-corrected peaks (gray line) from which the mutant peak fraction is determined. Roman numerals refer to the melt curves in the upper panel. The nine plots represent the analysis of single cybrid cells derived from two patients, GB and V.
The results led us to conclude that also at the single-cell level, the PCR-RFMT principle works. To automatically measure the areas under the peaks and process the data into suitable plots, we next developed a macro in Excel. The macro searches for the two maxima and the minimum between the two peaks. It then employs the bell shapes of the peaks for baseline correction. The right-hand flank of the mutant peak (from mutant peak position to the minimum between the peaks) is mirrored to the left, and the line connecting the base of the bell is used as baseline. Similarly, the left-hand flank of the wild-type peak is mirrored to the right and baseline corrected. Baseline-corrected peaks for six A3243G cybrid cells are plotted in the graphs (Figure 2, lower panel). Finally, the macro calculates mutation load from the areas under the peaks. The data from a complete 96-well plate are plotted both as a histogram and as a scatter plot as a function of Ct (threshold cycle) value for easy evaluation of the results.
We next compared the single-cell PCR-RFMT approach with traditional gel-based imaging and quantification of fragments. For this we used single-cell PCR-RFLP fragments that by RFMT analysis showed a wide range of heteroplasmy. The correlation proved to be good (Figure 3).
Figure 3.

Correlation between A3243G mtDNA gel-based PCR-RFLP and PCR-RFMT analysis at the single-cell level. Individually sorted cells were subjected to PCR-RFMT analysis, followed by gel electrophoresis and image analysis of the same samples.
The correlation between the analysis methods may be good, but both methods probably suffer from underestimation of the mutant fraction due to heteroduplex formation and the inability of ApaI to digest heteroduplexes. To analyze the magnitude of this problem and to eventually compensate mathematically for this phenomenon, we cloned wild-type and mutant fragments, prepared mixtures of purified wild-type and mutant plasmids, and submitted them to PCR-RFMT analysis at various dilutions down to the sub-attomol range. If p represents the wild-type and q the mutant fraction in the sample, then at maximum heteroduplex formation, the relation between the wild-type homoduplex and heteroduplex, and mutant homoduplex fractions and the original input fractions are given by the equation: p2 + 2pq + q2 = 1. Thus, the mutation load q may be derived from the square root of the cleavable fraction q2, because both p2 and pq are not recognized by ApaI. Figure 4 plots the expected mutant fraction against the square root of the mutant peak fraction. The good linear relationship between expected mutation load and the square root of the mutant peak fraction shows that full heteroduplex formation occurs, enabling mathematical corrections to be made.
Figure 4.

Heteroduplex formation during A3243G PCR-RFMT analysis. mtDNA from a heteroplasmic cybrid cell line was cloned into a plasmid vector. Plasmids containing either wild-type or mutant mtDNA sequence clones were identified, purified, and mixed in predetermined ratios. Data are plotted as input mutation load (x-axis) versus the square root of the mutant peak fraction as measured by PCR-RFMT. Error bars are the standard deviation of three measurements per data point.
Previously, we developed an in situ A3243G mtDNA genotyping method. It is based on target-primed RCA of in situ—hybridized and —ligated Padlock probes (Larsson et al. 2004). The exquisite sensitivity of the ligase reaction to mismatches confers high specificity to A3243G sequence variant detection, not achievable by hybridization-based methods such as Taqman, and the signal generation capacity of RCA provides single molecule detection sensitivity with standard fluorescence microscopy and virtually no background signals. Results are primarily judged visually, but image analysis can confer quantitative information.
We therefore recently developed dedicated image analysis software to quantify Padlock-RCA mtDNA signals in cells (Allalou et al. 2007). Here we apply quantitative Padlock-RCA and correlate results with RFMT analysis. Figure 5A presents the mutation load histograms of cells from a given passage of clone GB_20 measured with the PCR-RFMT and the quantitative Padlock-RCA technique. For PCR-RFMT, six independently processed, single-cell-sorted 96-well plates were analyzed, resulting in the mutation load histogram of 541 cells shown in Figure 5A. The peak average was 92.2% with a standard deviation of 3.4%. For Padlock-RCA, 458 cells were analyzed from the same GB_20 clone passage. The peak average was 92.5% with a standard deviation of 4.3%. Images of Padlock-RCA of a coculture of homoplasmic wild-type and mutant cybrids, and GB_20 clone are shown in Figures 5B and 5C, respectively.
Figure 5.
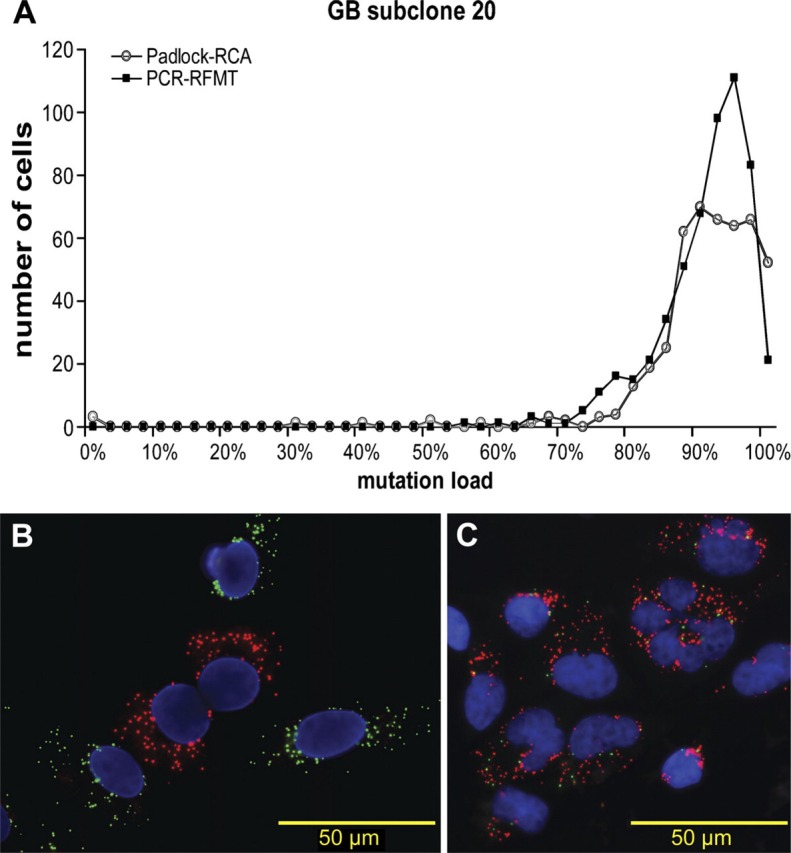
Correlating single-cell A3243G mtDNA PCR-RFMT with Padlock-RCA. (A) Individual cells of a heteroplasmic A3243G clone (GB_20) were analyzed by PCR-RFMT (n=541) and Padlock-RCA (n=458). According to computer simulations of random segregation, the very great majority of the cells in this late passage should have been genetically fixed, i.e., homoplasmic mutant. (B, C) Photomicrographs of cells from, respectively, a coculture of wild-type and mutant cybrids, and the heteroplasmic GB_20 clone genotyped in situ by Padlock-RCA. Red dots correspond to mutant and green dots to wild-type mtDNA.
Discussion
In mtDNA segregation studies, one needs to measure mtDNA point mutation levels in individual members of cell populations. Throughput and accuracy obviously are important parameters to get statistically sound data for biological interpretation. We describe here development of PCR-RFMT as a semi-high throughput single-cell A3243G mtDNA mutation load assay and correlate results with those obtained by image analysis of cells in situ—genotyped for the A and G variants at the 3243 locus by Padlock-RCA.
In their current versions, the two methods perform about equally in terms of throughput: hundreds of cells per person per day. Increase in throughput for both can be achieved by further automation of PCR-RFMT by robotized PCR and melt analysis. Use of 384-well plates would quadruple throughput of the PCR-RFMT, but single-cell sorting is limited to 96-well plates at present. Image acquisition has been identified as the rate-limiting factor for Padlock-RCA; thus automation of microscopy and image acquisition will increase throughput.
In terms of analytical accuracy, differences exist. PCR-RFMT is prone to heteroduplex formation. Mathematical correction allows the true mutation load to be determined, but the square root relationship between found and true mutation level values may lead to inaccuracies in the lower mutant range. Padlock-RCA is free of heteroduplex formation problems and will have the same accuracy over the full heteroplasmy range. The results shown in Figure 5 are interesting from an analytical and segregation biology point of view. They originate from cells of a high passage number. Computer simulations of random mtDNA segregation specified to this clone clearly indicated that at this passage, segregation should have been obvious by flattening of the peak and appearance of homoplasmic mutant cells (results not shown). It thus appears that this clone displays the phenomenon of stable heteroplasmy as reported by Lehtinen et al. (2000). If so, then variance in mutation load due to segregation would be absent and the variance observed in heteroplasmy would be due to assay variability only.
In an in vitro A3243G segregation study, we have successfully analyzed >10,000 cells from various passages of 10 A3243G cybrid clones with both methods, and results were concordant (unpublished observation). We conclude that the newly developed PCR-RFMT and the Padlock-RCA assay for quantifying single-cell A3243G mutation loads are both suitable for in vitro A3243G mtDNA segregation studies aimed at resolving enigmatic segregation mechanisms.
Acknowledgments
This work was supported by the Prinses Beatrix Fonds, the Stichting Spieren voor Spieren, and the EC-Strep Project ENLIGHT (ENhanced LIGation based Histochemical Techniques).
The authors thank Visiopharm, Denmark, for a free VIS license and help with integration of new functionality in VIS.
Literature Cited
- Allalou A, Van de Rijke FM, Jahangir, Tafrechi RS, Raap AK, Wahlby C. (2007) Image based measurement of single cell mtDNA mutation load. In Lecture Notes in Computer Science, Scandinavian Conference on Image Analysis 2007. Aalborg, Springer Publishing [Google Scholar]
- Bai RK, Wong LJ. (2004) Detection and quantification of heteroplasmic mutant mitochondrial DNA by real-time amplification refractory mutation system quantitative PCR analysis: a single-step approach. Clin Chem 50: 996–1001 [DOI] [PubMed] [Google Scholar]
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, et al. (2007) The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells. Nat Genet 39: 386–390 [DOI] [PubMed] [Google Scholar]
- Chinnery PF. (2002) Modulating heteroplasmy. Trends Genet 18: 173–176 [DOI] [PubMed] [Google Scholar]
- Chinnery PF, Zwijnenburg PJ, Walker M, Howell N, Taylor RW, Lightowlers RN, Bindoff L, et al. (1999) Nonrandom tissue distribution of mutant mtDNA. Am J Med Genet 85: 498–501 [PubMed] [Google Scholar]
- Enriquez JA. (2003) Segregation and dynamics of mitochondrial DNA in mammalian cells. In Holt IJ, ed. Genetics of Mitochondrial Diseases. Oxford, Oxford University Press, 279–294 [Google Scholar]
- Jenuth JP, Peterson AC, Fu K, Shoubridge EA. (1996) Random genetic drift in the female germline explains the rapid segregation of mammalian mitochondrial DNA. Nat Genet 14: 146–151 [DOI] [PubMed] [Google Scholar]
- Jenuth JP, Peterson AC, Shoubridge EA. (1997) Tissue-specific selection for different mtDNA genotypes in heteroplasmic mice. Nat Genet 16: 93–95 [DOI] [PubMed] [Google Scholar]
- Larsson C, Koch J, Nygren A, Janssen GMC, Raap AK, Landegren U, Nilsson M. (2004) In situ genotyping individual DNA molecules by target-primed rolling-circle amplification of Padlock probes. Nat Methods 1: 227–232 [DOI] [PubMed] [Google Scholar]
- Lehtinen SK, Hance N, El Meziane A, Juhola MK, Juhola KM, Karhu R, Spelbrink JN, et al. (2000) Genotypic stability, segregation and selection in heteroplasmic human cell lines containing np 3243 mutant mtDNA. Genetics 154: 363–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torroni A, Campos Y, Rengo C, Sellitto D, Achilli A, Magri C, Semino O, et al. (2003) Mitochondrial DNA haplogroups do not play a role in the variable phenotypic presentation of the A3243G mutation. Am J Hum Genet 72: 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CJ, Granycome C, Hurst R, Pohler E, Juhola MK, Juhola MI, Jacobs HT, et al. (2005) Systematic segregation to mutant mitochondrial DNA and accompanying loss of mitochondrial DNA in human NT2 teratocarcinoma cybrids. Genetics 170: 1879–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]