

Abstract
Standard cytomorphological examination of bone marrow (BM) aspirates does not appear to be sensitive enough to detect single neuroblastoma cells. The SIOPEN Neuroblastoma Bone Marrow Committee developed a sensitive and reproducible anti-GD2 immunocytochemical assay and introduced morphological and immunocytological criteria for the interpretation of results. Fixed cytospins were incubated with a commercially available anti-GD2 monoclonal antibody and an APAAP kit. Cells fulfilling all morphological and immunocytological criteria were called criteria-positive cells (CPCs). Not convincingly interpretable cells fulfilled some, but not all, criteria, and negative cells displayed only exclusion criteria. The genetic profile of doubtful cells was checked by fluorescence in situ hybridization. Ideally, 3 × 106 cells were analyzed to reach a 95% probability of detecting one tumor cell in 1 × 106 mononuclear cells. Four quality control rounds were organized to validate the method. A total of 111 quality control samples were analyzed. Two main improvements were achieved: in discordant cases, the range between the lowest and highest reported result was reduced by half, and discordant results were only found in samples with less than 10 CPCs per 1 × 106. This article describes the first internationally standardized protocol to detect and quantify rare neuroblastoma cells by immunocytochemistry. This method is an indispensable tool for multicenter studies evaluating the clinical significance of minimal residual disease in neuroblastoma.
Keywords: immunocytochemistry, minimal residual disease, neuroblastoma, bone marrow
Neuroblastoma (NB), a tumor originating from the sympathetic nervous tissue, is the most common extracranial solid tumor in children with a yearly incidence of 7–10 per million. The tumor consists of sympathetic neuronal elements of variable immaturity and shows a diverse clinical behavior. Approximately 40% of NB patients suffer from high-risk stage 4 disease with bone marrow (BM) involvement (Moss et al. 1991). These children have a poor clinical outcome despite intensive multimodal therapy (Brodeur and Castleberry 1997).
The demonstration of disseminated tumor cells in BM is important for clinical staging and risk assessment at diagnosis and for monitoring therapeutic response during treatment. In addition, screening of autologous stem cell preparations is crucial because the reinfusion of contaminated stem cell products could lead to systemic recurrence (Brenner et al. 1993; Deisseroth et al. 1994; Rill et al. 1994).
According to the International Neuroblastoma Staging System, conventional cytology of BM smears is still the only accepted technique for the detection of disseminated NB cells (Brodeur et al. 1988). However, the sensitivity of this approach is limited because a tumor cell number lower than 0.1% is virtually not detectable by conventional cytomorphology (Cheung et al. 1997; Mehes et al. 2003). Therefore, the development of more sensitive and specific detection methods is indispensable.
During the last few decades, several assays based on immunocytology (Cheung et al. 1986; Berthold et al. 1989; Nagai et al. 1994), automatic immunofluo-rescence plus fluorescence in situ hybridization (FISH) (AIPF) (Ambros et al. 2003), RT-PCR (Burchill et al. 1995; Miyajima et al. 1995; Cheung et al. 1998; Lo Piccolo et al. 2001), or flow cytometry were evaluated (Komada et al. 1998; Nagai et al. 2000; Warzynski et al. 2002; Swerts et al. 2004). However, the reliability of tumor cell detection and quantification by these methods remains controversial.
A Neuroblastoma Bone Marrow Committee (NBMC) was established by the European Neuroblastoma Study Group to evaluate and standardize procedures for the detection of minimal residual disease (MRD) in NB patients. In connection with the evaluation of a new high-risk protocol by the SIOP European Neuroblastoma (SIOPEN) group, the NBMC developed, optimized, and standardized an immunocytochemical assay based on the detection of the neuroblastoma-specific GD2 disialoganglioside. In addition, morphological and immunocytological criteria for the interpretation of results were introduced and standardized. Four multicenter quality control (QC) rounds were organized among nine European research groups to evaluate the technique and assess the interobserver concordance. The latter improved markedly after the adoption of the standardized protocol.
This article describes a standardized immunocytochemical staining method and minimal morphological and immunocytological criteria for the evaluation of stained BM samples. The application of this protocol will lead to a more reproducible and reliable assessment of MRD in NB. We believe that a standardized method is needed to generate comparable results in multicenter studies evaluating the clinical significance of MRD.
Materials and Methods
Sample Collection
After informed consent from patients, bilateral BM aspirates from the iliac crest were performed following previously published guidelines (Brodeur et al. 1988; Ambros and Ambros 2001). The second aspiration from each puncture site was used for immunocytology and FISH. Bilateral BM samples were not pooled and were transferred to the laboratory at room temperature as fast as possible.
Control Samples
Slides containing cells from a NB cell line (e.g., IMR32) were included in every experiment as positive controls. For negative control, the primary anti-GD2 antibody was replaced with an antibody of the same IgG2a isotype (Dako Corporation; Glostrup, Denmark). This allowed us to evaluate the background staining caused by the interaction of the anti-GD2 antibody with Fc-receptor-bearing leukocytes.
Isolation, Processing, and Storage of Mononuclear Cells
Mononuclear BM cells were isolated by density gradient centrifugation using Lymphoprep (Nycomed; Oslo, Norway) following the instructions of the manufacturer. Aspirates from different sites were processed separately. After separation, the mononuclear cells were collected from the interphase layer and washed twice in PBS (Gibco; Paisley, UK).
Large-diameter cytospins (17 mm) containing ∼5 × 105 mononuclear cells were prepared. These slides should not be overcrowded and the mononuclear cells must lie well separated from one another. This can be achieved by centrifuging no more than 7 × 105 cells down on precoated (e.g., poly-l-lysine) glass slides in a Hettich centrifuge (Hettich Zentrifugen; Tuttlingen, Germany).
The slides were air-dried overnight and stored in airtight plastic boxes or wrapped in aluminum foil at −24C until immunocytology was performed. Before staining, the slides were thawed in closed boxes to avoid formation of condensation water, because this could destroy the morphology of the cells.
Standardized Immunocytochemical Staining Protocol
The NBMC decided to standardize the fixation, the immunocytochemical staining procedure and the evaluation of immunocytological results to improve the sensitivity, specificity, and reproducibility of MRD detection in NB.
Fixation
The cytospins must be fixed in 4% buffered paraformaldehyde for 10 min. Commercially available formaldehyde can also be used, provided that it is free from methanol. To avoid artificial tumor cell contamination, slides must be incubated individually. After fixation, the cytospins were washed three times with PBS to remove remainders of the fixative.
Immunocytochemical Staining
The staining procedure comprised 10 subsequent steps. All incubations were performed in a humidifier at room temperature.
-
1.
Incubation of cytospins for 30 min with 30 μl of an unlabeled monoclonal mouse anti-human GD2 disialoganglioside antibody (clone 14.G2a; BD Biosystems, Erembodegem, Belgium) diluted 1/100 in 1% PBS/BSA (Gibco; Paisley, UK).
-
2.
Washed twice with PBS for 5 min.
-
3.
Incubation for 30 min with 30 μl of an unlabeled rabbit anti-mouse antibody (Dako Corporation) and diluted 1/20 in 1% PBS/BSA.
-
4.
Repetition of step two.
-
5.
Incubation with 30 μl of the APAAP complex (Dako Corporation) and diluted 1/20 in 1% PBS/BSA.
-
6.
Repetition of step two.
-
7.
Incubation with Dako Fuchsin+ Substrate Chromogene System (Dako Corporation), prepared as indicated by the manufacturer, for no longer than 10 min.
-
8.
Washed in running tap water for at least 5 min.
-
9.
Counterstained with hematoxylin (Sigma; St Louis, MO) until an appropriate blue nuclear stain was obtained.
-
10.
Mounted in aqueous mounting medium (e.g., Glycergel mounting medium; Dako Corporation) with cover slip.
Evaluation of Immunocytochemically
Stained Samples
The characteristics of GD2-positive disseminated NB cells and false-positive hematopoietic cells were examined in detail. The observations led to the following morphological and immunocytological guidelines for the identification of positive cells.
Morphological Criteria
Cells with a round nucleus, often, but not always, larger than that of small lymphocytes, displaying a granular chromatin structure and a scarce amount of cytoplasm were considered positive. Cells showing a low nuclear/cytoplasmic ratio or typical morphological features of hematopoietic cells were considered negative. Cells found outside or on the border of the cytospin field were excluded.
Immunocytological Criteria
Cells must display a strong, deep red staining localized to the entire cell membrane and cytoplasm. A weak staining and a staining restricted to a subcellular compartment or covering the nucleus was considered negative. In addition, cells surrounded by positively stained amorphous material were excluded.
Based on the morphological and immunocytological criteria, cells were classified into three groups:
-
1.
CPCs (criteria-positive cells): cells fulfilling all morphological and immunocytological inclusion criteria (Figures 1A-D).
-
2.
NCICs (not convincingly interpretable cells): cells fulfilling some but not all inclusion criteria (Figure 1E).
-
3.
NCs (negative cells) (Figures 1F and 1G): cells displaying only exclusion criteria.
Figure 1.

Immunocytochemical analysis of bone marrow (BM) slides from neuroblastoma (NB) patients according to the standardized protocol. (A) CPCs (criteria-positive cells) forming a clump. NB cells in clumps do not always display a round nucleus because they adjust their form to the clump. The entrapped erythroblast (arrow) is smaller than the NB cells and displays a different chromatin structure. (B) The two CPCs mold in a different way compared with the surrounding hematopoietic cells. (C) Two CPCs showing the typical nuclear size, chromatin structure, and nuclear/cytoplasmic ratio that clearly differs from hematopoietic cells. The myeloid cells (arrow) are passively stained because of the shedding of the GD2 antigen by the NB cells. (D) If the morphological and immunological criteria are fulfilled, even a single cell can be identified as CPC. (E) The staining intensity of this cell is comparable to that of a CPC but the nuclear shape, size, chromatin structure, and the nuclear cytoplasmic ratio do not fulfill the criteria. Therefore, the cell is classified as not convincingly interpretable cell. (F) Neither the nuclear/cytoplasmic ratio nor the vesicular structure of the cytoplasm fulfills the criteria. The cell has the cytological features of a histiocyte. (G) The cell on the left side has the same size and displays the same staining intensity as the CPC on the right. However, the nuclear shape and the nuclear/cytoplasmic ratio do not fulfill the criteria. Moreover, the cell contains GD2-positive material in the cytoplasm (arrow). This is a typical feature of a macrophage. (H) Strongly positive material (right) without a visible nucleus is not reported. CPC on the left.
Single cells as well as cells being part of a Homer Wright rosette or cell clump are evaluated, classified, and counted. Clumps or rosettes consisting of too many cells to evaluate or count are reported separately. In addition, the estimated number of evaluated mononuclear cells must be reported.
When evaluating or reporting immunocytochemical results, the work flow depicted in Figure 2 should be followed. No further review is needed when no GD2-positive cells or more than 10 CPCs are present. For samples with 1-10 CPCs or samples containing NCICs, central review by the members of the NBMC is obligatory. When no consensus is reached, the genetic profile of the doubtful cells should be checked by FISH to find out whether it corresponds to the cytogenetic aberrations found in the primary tumor. The sequential immunocytological staining and molecular cytogenetic characterization can be done using an automated scanning and relocation system (e.g., Metafer4/RCDetect; MetaSystems, Altlussheim, Germany) (Ambros et al. 2003). GD2-positive cells showing genetic aberrations (e.g., gain of whole chromosomes or 17q and MYCN amplification) are called FPCs (FISH-positive cells) (Figure 3).
Figure 2.
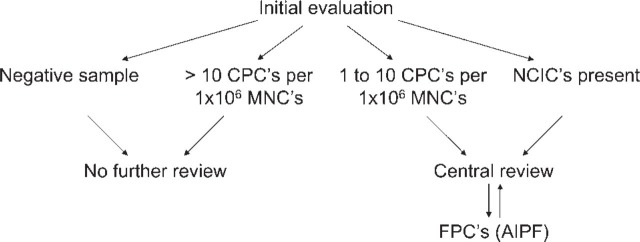
Work flow. (CPCs, criteria-positive cells; NCICs, not convincingly interpretable cells; MNCs, mononuclear cells; AIPF, automatic immunofluorescence plus FISH; FPCs, FISH-positive cells.)
Figure 3.
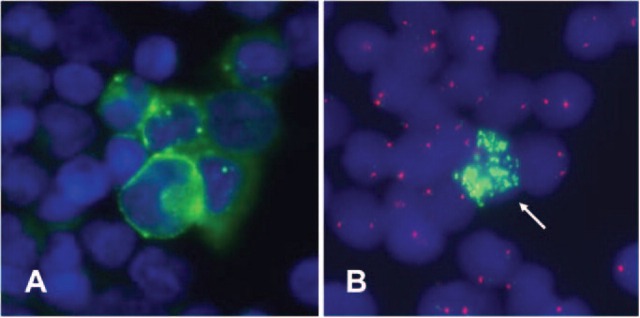
Automatic immunofluorescence plus FISH allows the sequential immunological staining and molecular cytogenetic characterization of disseminated neuroblastoma cells. (A) GD2-positive cells (green fluorescence). The nucleus is stained with DAPI (blue fluorescence). (B) The genetic makeup of the GD2-positive cells is visualized by FISH. Only one GD2-positive cell displays MYCN amplification (green fluorescence signals). GD2-positive cells displaying the same genetic aberrations as the primary tumor are called FISH-positive cells.
Results
Sensitivity
The sensitivity of the immunocytochemical assay is not limited by the technique itself. On the contrary, the number of analyzed cells defines the sensitivity. When enough cells are analyzed, a high sensitivity can be reached. The Poisson distribution f(X) = μX × e−μ/X! can be used to calculate the statistics of tumor cell detection (Cheung et al. 1986). The parameter μ = n × p (where n is the total number of cells analyzed and p is the true frequency of tumor cells) represents the total number of tumor cells present. The variable × denotes the number of tumor cells actually detected. When × = 0, f(X) is the possibility of missing a tumor cell. When only one tumor cell is present in the midst of 999,999 normal cells, the probability of missing the tumor cell after counting 3 × 106 cells is less than 5%. This means at least 3 × 106 cells should be analyzed to reach a 95% chance of detecting one tumor cell in 1 × 106 normal mononuclear cells. Therefore, six cytospins, each containing 5 × 105 cells, should be analyzed to secure the analysis of 3 × 106 mononuclear cells.
Quality Control
Between 2001 and 2003, four multicenter QC rounds were organized among the nine members of the NBMC to develop and validate the staining protocol, the morphological and immunocytological criteria, and the work flow. A total of 111 QC samples were analyzed. Every research group sent preferably three slides from at least two different BM samples to every other member. The participants fixed and stained the cytospins independently of each other. They evaluated the samples unaware of any clinical information. Individual screening results were disclosed and the level of interobserver concordance was assessed during subsequent QC meetings. In addition, samples with discordant results were reviewed by all participants, resulting in an optimized staining protocol and refined morphological and immunocytological criteria. The results of quality control round 1 and 4 are shown in Table 1. Only samples analyzed by at least four participants were included in the study. Participants 1-8 used the immunocytochemical staining assay to evaluate the quality control samples. Participant 9 detected residual NB cells using automatic immunofluorescence plus FISH.
Table 1.
Results of quality control rounds 1 and 4
| Results of Quality Control Round 1 | |||||||||
| Cytospin Label | Lab 1 | Lab 2 | Lab 3 | Lab 4 | Lab 5 | Lab 6 | Lab 7 | Lab 8 | Lab 9 |
| QC1 A1 | NEa | 0 | 0 | 1 | NE | NE | 0 | ND | 0 |
| QC1 A2 | NE | 0 | 0 | 0 | NDb | NE | 0 | ND | 0 |
| QC1 A3 | NE | 0 | 0 | 0 | ND | NE | 0 | ND | 0 |
| QC1 A4 | 0 | 0 | 0 | 0 | ND | 1 | 0 | ND | 0 |
| QC1 B1 | 0 | 6 | 9 | 5 | >100c | 53 | 42 | ND | 10 |
| QC1 B2 | 0 | 0 | 0 | 0 | ND | 3 | 15 | ND | 0 |
| QC1 B3 | 0 | 0 | 0 | 4 | ND | 3 | 1 | ND | 0 |
| QC1 C1 | >100 | >100 | NE | >100 | >100 | >100 | >100 | ND | >100 |
| QC1 C2 | 50 | >100 | NE | >100 | >100 | >100 | >100 | ND | >100 |
| QC1 C3 | NE | 0 | 0 | 2 | 1 | 0 | 4 | ND | 0 |
| QC1 G1 | >100 | >100 | >100 | >100 | NE | >100 | >100 | ND | >100 |
| QC1 G2 | Cd | 1 | 2 | 0 | NE | 0 | 15 | ND | 0 |
| QC1 G3 | NE | 0 | 0 | 0 | NE | 0 | 18 | ND | 0 |
| QC1 G4 | 0 | 1 | 0 | 0 | NE | 0 | 21 | ND | 0 |
| QC1 I1 | >100 | >100 | NE | NE | >100 | NE | >100 | ND | ND |
| QC1 I2 | 1 | 1 | 0 | NE | 0 | NE | ND | ND | ND |
| QC1 I3 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | ND | 0 |
| QC1 I4 | 11 | C | 5 | 2 | 4 | 3 | 32 | ND | 7 |
| QC1 I5 | 0 | 0 | 0 | 0 | ND | 0 | 0 | ND | 0 |
| QC1 I6 | 40 | >100 | >100 | >100 | ND | >100 | >100 | ND | >100 |
| QC1 I7 | 0 | 2 | ND | 0 | ND | 0 | 0 | ND | 0 |
| QC1 I8 | 0 | 0 | ND | 2 | ND | 0 | 8 | ND | 0 |
| QC1 N1 | 0 | >100 | C | C | ND | 23 | 92 | ND | 80 |
| QC1 N2 | 0 | 0 | 0 | NE | ND | 0 | 4 | ND | 2 |
| QC1 N3 | 1 | 1 | 2 | 0 | ND | 7 | 14 | ND | 7 |
| QC1 S1 | 0 | ND | 0 | 2 | 1 | NE | 16 | ND | 0 |
| QC1 S2 | 0 | ND | 0 | 0 | 0 | NE | 18 | ND | 0 |
| QC1 S3 | 0 | ND | 0 | 0 | 0 | 0 | 0 | ND | ND |
| QC1 S4 | 0 | ND | 0 | 0 | 0 | 0 | 0 | ND | ND |
| QC1 S5 | 0 | 1 | 3 | 0 | ND | 1 | 22 | ND | 0 |
| QC1 S6 | C | 3 | 2 | 0 | ND | 0 | 27 | ND | 0 |
| Results of Quality Control Round 4 | |||||||||
| Cytospin Label | Lab 1 | Lab 2 | Lab 3 | Lab 4 | Lab 5 | Lab 6 | Lab 7 | Lab 8 | Lab 9 |
| QC4 A1 | 0 | 0 | NE | NE | 0 | 0 | 0 | 0 | 0 |
| QC4 A2 | 0 | 0 | 0 | 0 | 0 | 0 | 8 | 0 | 0 |
| QC4 B1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 B2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 C1 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | ND | >100 |
| QC4 C2 | 0 | C | ND | ND | 0 | 0 | 1 | ND | 4 |
| QC4 F1 | 0 | 3 | 0 | 0 | 0 | 1 | 2 | 0 | 10 |
| QC4 F2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 G1 | 48 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| QC4 N1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 N2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 S1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| QC4 S2 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Not evaluable.
Not done.
More than 100 positive cells or more than 2 clusters.
One or two positive clusters.
Participants 1-8 used an immunocytological staining assay to evaluate the quality control samples. Participant 9 detected residual neuroblastoma cells using an automatic immunofluorescence plus FISH device (AIPF). The number of GD2-positive cells (quality control round 1) or the number of criteria-positive cells (quality control round 4) is reported.
Quality control round 1 was organized before the immunocytochemical staining protocol was standardized and the morphological and immunocytological criteria were formulated. A total of 33 QC samples were analyzed. Two samples were excluded because they were analyzed by only three participants. Considerable differences were found both in the number of positively scored samples and in the number of GD2-positive cells per individual sample. Six samples were scored positive by all participating centers. In five samples, no positive cells were found. Discordant results were found in 20 samples (65%). In these samples, the average difference between the highest and the lowest reported number of GD2-positive cells was 19.
Quality control round 4 was organized after the standardization of the staining method, the formulation of the criteria, and the design of the work flow. Thirteen samples were fixed, stained, and evaluated by each participant in a blinded way and, in accordance with the work flow, samples containing less than 10 CPCs or samples with NCICs were reviewed during a QC meeting. Because we noticed a remarkable improvement in the sensitivity and specificity of the method and in the reproducibility of the results after analyzing 13 samples, QC round 4 was terminated at that point. After central review, the results were concordant in 10 samples. Only in three samples (23%) were discordant results found. In these samples, the average difference between the highest and the lowest reported number of CPCs was 9. Discordant results were only found in samples with less than 10 CPCs per 1 × 106. The standardization of the assay led to a significant decrease in the number of discordant results (X2 = 4.91, p = 0.027, DF = 1). The range between the highest and the lowest reported number of positive cells decreased from 19 to 9.
The immunocytochemical results (participants 1-8) were also compared with those obtained with AIPF (participant 9). In QC round 1, 15 samples scored positive for the immunocytochemical assay, whereas no neuroblastoma cells were detected by AIPF. The discrepancies in four of these samples were probably from sample variability because only one or two immunocytochemistry positive cells were found. All other samples (11) were most likely false positive. In QC round 4, only one discordant result was found (8%). Participant 7 reported 8 CPCs, whereas no FPCs were detected by participant 9. These results prove that the standardization of the staining and evaluation procedures reduced the number of false positive results dramatically.
Discussion
The detection of occult NB cells in BM has important therapeutic and prognostic implications because BM disease is associated with an unfavorable outcome for most children (Hartmann et al. 1999; Cotterill et al. 2000). On the other hand, children with stage 4S disease have a good prognosis, although they may present with BM metastases. Cytomorphological screening of BM smears is still the only accepted method for the detection of disseminated NB cells. However, Mehes et al. reported that a tumor cell infiltrate lower than 0.1% can be overlooked by conventional cytomorphology because the unspecific morphological appearance of NB cells limits the sensitivity of this method (Mehes et al. 2003). In recent years, numerous alternative approaches using immunological and molecular biological techniques were developed to improve the detection of residual NB cells in BM (Cheung et al. 1986; Berthold et al. 1989; Nagai et al. 1994; Burchill et al. 1995; Miyajima et al. 1995; Cheung et al. 1998; Komada et al. 1998; Nagai et al. 2000; Lo Piccolo et al. 2001; Warzynski et al. 2002; Ambros et al. 2003; Swerts et al. 2004). However, the sensitivity and specificity of these assays vary markedly and hamper studies evaluating the clinical significance of MRD. Therefore, the standardization of detection techniques is urgently needed.
In connection with a phase III study organized by the SIOPEN group, the NBMC developed, optimized, and standardized an immunocytochemical assay based on the detection of the NB-specific GD2 disialoganglioside. This antigen is highly and consistently expressed in neuroectodermal tumors and is not found in normal BM or peripheral blood cells (Cheung et al. 1986; Wu et al. 1986; Sariola et al. 1991). Because all NB are believed to express GD2, the possibility of a false-negative result can usually be excluded. However, the uneven distribution of NB cells in the body may hamper the detection of disseminated tumor cells. To avoid false-negative results caused by sampling error, a sufficient number of cells must be analyzed. The Poisson distribution f(X) = μX × −μ/X! can be used to calculate the statistics of tumor cell detection (Cheung et al. 1986). At least 3 × 106 cells must be analyzed to reach a 95% probability of detecting one tumor cell in 1 × 106 normal mononuclear cells.
Immunocytochemical results can also be obscured by false-positive events (Pantel et al. 1994; Borgen et al. 1998; Mehes et al. 2001). These can be caused by the active or passive uptake of the tumor cell-derived GD2 ganglioside by hematopoietic cells. Furthermore, a very small subset of mature plasma cells producing antibodies against alkaline phosphatase can be false positive because they react directly with the enzyme (Borgen et al. 1998). Finally, the illegitimate expression of the targeted antigen, the cross-reactivity of the applied monoclonal antibody, and interactions between antibodies and Fc-bearing leukocytes can give rise to false-positive results.
When the members of the NBMC stained their slides using different immunocytochemical staining methods and analyzed their results according to individual morphological criteria, considerable discrepancies were observed. The evaluation of stained samples by the whole group using a multiheaded microscope clearly demonstrated the urgent need for developing a standardized immunocytochemical staining protocol and introducing morphological and immunocytological criteria. Consequently, the NBMC agreed on one staining method and formulated morphological and immunocytological criteria for the interpretation of the results. Only cells with a round nucleus, often, but not always larger than that of small lymphocytes, a granular chromatin, and a limited amount of cytoplasm are considered positive. In addition, a strong, deep red staining localized to the entire cell membrane and cytoplasm must be present. To our knowledge, this is the first time that a standardized protocol including morphological and immunological criteria for the detection of NB cells in BM has been designed.
However, when applying these criteria, the NBMC discovered that a small proportion of immunocytochemically stained cells could not unequivocally be classified as positive (i.e., NB cells) because they did not fulfill all postulated morphological and immunocytological criteria. Therefore, it was decided to categorize all immunocytochemically stained cells into three groups: CPCs fulfilling all postulated criteria; NCICs displaying some but not all inclusion criteria; and NCs, which, in spite of their staining, were identified as nonmalignant hematopoietic cells.
Borgen et al. published a similar approach for the analysis of circulating carcinoma cells by applying the anti-cytokeratin antibodies AE1/AE3 and an alkaline phosphatase-based detection method on cytospins prepared from mononuclear BM cells (Borgen et al. 1999). They also categorized immunologically stained cells into three groups which they called tumor cells, probable tumor cells (?), and hematopoietic cells. To discriminate among these groups, they presented a catalog of pictures illustrating a large number of morphological and immunological variants of these categories. However, regarding the detection of NB cells using an anti-GD2 antibody, the members of the NBMC do not believe that it is feasible to cover all possible variants of immunocytochemically stained BM cells by means of illustrations. Therefore, the NBMC decided to introduce a workflow including two additional analytical steps (Figure 2). First, samples with 1-10 CPCs or samples containing NCICs are simultaneously reviewed by the members of the NBMC. Second, if no consensus is reached, the genetic profile of the doubtful cells is checked by automatic immunofluorescence plus FISH to disclose the identity of these cells. If the genetic aberrations in the doubtful cells correspond to those found in the primary tumor, the cells are called FPCs. Finally, the morphological and immunocytological features of these FPCs are carefully studied to refine the standardized evaluation criteria.
The standardized staining protocol, the morphological and immunocytological criteria, and the work flow were evaluated during four multicenter QC rounds, organized among the nine members of the NBMC. A total of 111 QC samples was analyzed. The concordance between the different observers, with regard to the staining and the evaluation of the immunocytochemical results, was assessed. After standardization, a significant decrease in the number of discordant results was reported. In addition, the range between the highest and the lowest reported result was reduced by half, and discordant results were only found in samples with less than 10 CPCs per 1 × 106 mononuclear cells.
Immunocytology has been used in the clinical practice of hematology and oncology for many years and has many advantages compared with flow cytometry or RT-PCR. In contrast to the latter, immunocytology allows the reliable quantification of tumor cells. This is important when the number of disseminated tumor cells appears to be prognostically important and not purely the presence or the absence of disease. The immunocytochemical technique is cost-effective and simple, and, because no expensive equipment is needed, immunocytological stainings can be performed in virtually every routine laboratory around the world.
This article describes the first international standardization of an immunocytochemical staining and evaluation method developed to detect and quantify small numbers of neuroblastoma cells in BM. The results of our QC rounds show that the standardization of the staining method, the formulation of morphological and immunocytological criteria and the design of the work flow resulted in a higher reproducibility, sensitivity, and specificity. Methodological standardization is indispensable and must be agreed on before multicenter studies, designed to assess the clinical importance of minimal residual disease, can be initiated.
Acknowledgments
This work was supported by the SIOPEN-R-NET project (EC grant QLRI-CT-2002-01768). Part of this work was supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders (I.W.T).
Literature Cited
- Ambros PF, Ambros IM. (2001) Pathology and biology guidelines for resectable and unresectable neuroblastic tumors and bone marrow examination guidelines. Med Pediatr Oncol 37: 492–504 [DOI] [PubMed] [Google Scholar]
- Ambros PF, Mehes G, Ambros IM, Ladenstein R. (2003) Disseminated tumor cells in the bone marrow—chances and consequences of microscopical detection methods. Cancer Lett 197: 29–34 [DOI] [PubMed] [Google Scholar]
- Berthold F, Schneider A, Schumacher A, Bosslet K. (1989) Detection of minimal disease in bone-marrow of neuroblastoma patients by immunofluorescence. Pediatr Hematol Oncol 6: 73–83 [DOI] [PubMed] [Google Scholar]
- Borgen E, Beiske K, Trachsel S, Nesland JM, Kvalheim G, Herstad TK, Schlichting E, et al. (1998) Immunocytochemical detection of isolated epithelial cells in bone marrow: non-specific staining and contribution by plasma cells directly reactive to alkaline phosphatase. J Pathol 185: 427–434 [DOI] [PubMed] [Google Scholar]
- Borgen E, Naume B, Nesland JM, Kvalheim G, Beiske K, Fodstad Ø, Diel I, et al. (1999) Standardization of the immunocytochemical detection of cancer cells in BM and blood: I. Establishment of objective criteria for the evaluation of immunostained cells. Cytotherapy 1: 377–388 [DOI] [PubMed] [Google Scholar]
- Brenner MK, Rill DR, Moen RC, Krance RA, Mirro J, Anderson WF, Ihle JN. (1993) Gene-marking to trace origin of relapse after autologous bone marrow transplantation. Lancet 341: 85–86 [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Castleberry RP. (1997) Neuroblastoma. In Pizzo PA, Poplack DG. eds. Principles and practice of pediatric oncology. Philadelphia, Lippincott-Raven Publishers, 761–797 [Google Scholar]
- Brodeur GM, Seeger RC, Barrett A, Berthold F, Castleberry RP, D'Angio GJ, De Bernardi B, et al. (1988) International criteria for diagnosis, staging, and response to treatment in patients with neuroblastoma. J Clin Oncol 6: 1874–1881 [DOI] [PubMed] [Google Scholar]
- Burchill SA, Bradbury FM, Selby P, Lewis IJ. (1995) Early clinical evaluation of neuroblastoma cell detection by reverse transcriptase-polymerase chain reaction (RT-PCR) for tyrosine hydroxylase mRNA. Eur J Cancer 31A:553–556 [DOI] [PubMed] [Google Scholar]
- Cheung IY, Barber D, Cheung NKV. (1998) Detection of microscopic neuroblastoma in marrow by histology, immunocytology, and reverse transcription-PCR of multiple molecular markers. Clin Cancer Res 4: 2801–2805 [PubMed] [Google Scholar]
- Cheung NKV, Heller G, Kushner BH, Liu CY, Cheung IY. (1997) Detection of metastatic neuroblastoma in bone marrow: when is routine marrow histology insensitive? J Clin Oncol 15: 2807–2817 [DOI] [PubMed] [Google Scholar]
- Cheung NKV, Van Hoff DD, Strandjord SE, Coccia PF. (1986) Detection of neuroblastoma cells in bone marrow using GD2 specific monoclonal antibodies. J Clin Oncol 4: 363–369 [DOI] [PubMed] [Google Scholar]
- Cotterill SJ, Pearson ADJ, Pritchard J, Foot ABM, Roald B, Kohler JA, Imeson J. (2000) Clinical prognostic factors in 1277 patients with neuroblastoma: results of The European Neuroblastoma Study Group “Survey' 1982-1992. Eur J Cancer 36: 901–908 [DOI] [PubMed] [Google Scholar]
- Deisseroth AB, Zu ZF, Claxton D, Hanania EG, Fu SQ, Ellerson D, Goldberg L, et al. (1994) Genetic marking shows that Ph+ cells present in autologous transplants of chronic myelogenous leukemia (CML) contribute to relapse after autologous bone marrow in CML. Blood 83: 3068–3076 [PubMed] [Google Scholar]
- Hartmann O, Valteau-Couanet D, Vassal G, Lapierre V, Brugières L, Delgado R, Couanet D, et al. (1999) Prognostic factors in metastatic neuroblastoma in patients over 1 year of age treated with high-dose chemotherapy and stem cell transplantation: a multivariate analysis in 218 patients treated in a single institution. Bone Marrow Transplant 23: 789–795 [DOI] [PubMed] [Google Scholar]
- Komada Y, Zhang XL, Zhou YW, Inaba H, Deguchi T, Azuma E, Sakurai M. (1998) Flow cytometric analysis of peripheral blood and bone marrow for tumor cells in patients with neuroblastoma. Cancer 82: 591–599 [PubMed] [Google Scholar]
- Lo Piccolo MS, Cheung NKV, Cheung IY. (2001) GD2: a new molecular marker for detecting neuroblastoma. Cancer 92: 924–931 [DOI] [PubMed] [Google Scholar]
- Mehes G, Luegmayr A, Ambros IM, Ladenstein R, Ambros PF. (2001) Combined automatic immunological and molecular cytogenetic analysis allows exact identification and quantification of tumor cells in the bone marrow. Clin Cancer Res 7: 1969–1975 [PubMed] [Google Scholar]
- Mehes G, Luegmayr A, Kornmuller R, Ambros IM, Ladenstein R, Gadner H, Ambros PF. (2003) Detection of disseminated tumor cells in neuroblastoma: 3 log improvement in sensitivity by automatic immunofluorescence plus FISH (AIPF) analysis compared with classical bone marrow cytology. Am J Pathol 163: 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyajima Y, Kato K, Numata S, Kudo K, Horibe K. (1995) Detection of neuroblastoma cells in bone marrow and peripheral blood at diagnosis by the reverse transcriptase-polymerase chain reaction for tyrosine hydroxylase mRNA. Cancer 75: 2757–2761 [DOI] [PubMed] [Google Scholar]
- Moss TJ, Reynolds CP, Sather HN, Romansky SG, Hammond GD, Seeger RC. (1991) Prognostic value of immunocytologic detection of bone marrow metastases in neuroblastoma. N Engl J Med 324: 219–226 [DOI] [PubMed] [Google Scholar]
- Nagai J, Ishida Y, Koga N, Tanaka Y, Ohnuma K, Toyoda Y, Katoh A, et al. (2000) A new sensitive and specific combination of CD81/CD56/CD45 monoclonal antibodies for detecting circulating neuroblastoma cells in peripheral blood using flow cytometry. J Pediatr Hematol Oncol 22: 20–26 [DOI] [PubMed] [Google Scholar]
- Nagai J, Kigasawa H, Tomioka K, Koga N, Nishihira H, Nagao T. (1994) Immunocytochemical detection of bone marrow-invasive neuroblastoma cells. Eur J Haematol 53: 74–77 [DOI] [PubMed] [Google Scholar]
- Pantel K, Schlimok G, Angstwurm M, Weckermann D, Schmaus W, Gath H, Passlick B, et al. (1994) Methodological analysis of immunocytochemical screening for disseminated epithelial tumor cells in bone marrow. J Hematother 3: 165–173 [DOI] [PubMed] [Google Scholar]
- Rill DR, Santana VM, Roberts WM, Nilson T, Bowman LC, Krance RA, Heslop HE, et al. (1994) Direct demonstration that autologous bone-marrow transplantation for solid tumors can return a multiplicity of tumorigenic cells. Blood 84: 380–383 [PubMed] [Google Scholar]
- Sariola H, Terava H, Rapola J, Saarinen UM. (1991) Cell-surface ganglioside GD2 in the immunohistochemical detection and differential diagnosis of neuroblastoma. Am J Clin Pathol 96: 248–252 [DOI] [PubMed] [Google Scholar]
- Swerts K, De Moerloose B, Dhooge C, Brichard B, Benoit Y, Laureys G, Philippé J. (2004) Detection of residual neuroblastoma cells in bone marrow: comparison of flow cytometry with immunocytochemistry. Cytometry 61B:9–19 [DOI] [PubMed] [Google Scholar]
- Warzynski MJ, Graham DM, Axtell RA, Higgins JV, Hammers YA. (2002) Flow cytometric immunophenotyping test for staging/monitoring neuroblastoma patients. Cytometry 50: 298–304 [DOI] [PubMed] [Google Scholar]
- Wu Z, Schwartz E, Seeger RC, Ladisch S. (1986) Expression of GD2 ganglioside by untreated primary human neuroblastomas. Cancer Res 46: 440–443 [PubMed] [Google Scholar]