

Short abstract
This article reviews how the targets of transcription factors have previously been isolated and outlines new technologies that are being developed to identify novel direct targets.
Abstract
Identifying the targets of transcription factors is important for understanding cellular processes. We review how targets have previously been isolated and outline new technologies that are being developed to identify novel direct targets, including chromatin immunoprecipitation combined with microarray screening and bioinformatic approaches.
The control of many cellular processes requires the coordinated activation or repression of genes in the correct spatial and temporal patterns. This regulation is carried out in large part by transcription factors, which bind to DNA sequences within chromatin and activate or repress the transcription of nearby genes. This binding is frequently sequence-specific, with sequence recognition being carried out by the transcription factor itself or by other proteins complexed to it. Identification of the targets of each transcription factor provides information about individual processes and how transcription factors interact in a transcriptional network. These networks can then be used to describe a particular cellular process, or even something as complicated as embryonic development [1,2].
The first step in identifying targets of a transcription factor usually involves overexpression or knockdown of the factor in question and analysis of the resulting changes in gene expression. The development of microarray technology has facilitated this kind of analysis, allowing identification of many more downstream genes than was previously feasible. But this method gives no information about whether targets are regulated directly by the transcription factor through binding to regulatory sequences within the gene or whether regulation is indirect, through the activation of intermediate genes. Other techniques, such as chromatin immunoprecipitation (ChIP) and Dam methylase identification (DamID), have therefore been developed. These reveal where in the genome the transcription factor is bound; these approaches allow identification of many direct target sequences, particularly when it is combined with microarrays of genomic DNA. This type of information, in combination with genomic sequences, is now being used to develop computational algorithms that scan genomic sequence with the aim of distinguishing functional binding sites and target genes of transcription factors.
Identification of downstream genes
Comparison of two cell populations in which a given transcription factor is differentially expressed, either by overexpression or knockdown, has been used to identify the target genes activated by transcription factors in a wide range of systems. The resulting mRNA populations may be analyzed in a number of ways, such as reverse-transcriptase-coupled (RT-)PCR of candidates, subtractive hybridization, differential display or serial analysis of gene expression (SAGE; see Figure 1). For instance, a large-scale screen to describe transcriptional networks in the development of sea urchins has recently been undertaken: genes involved in endomesoderm development - including those encoding transcription factors - were overexpressed or knocked down, and mRNA populations were compared using subtractive hybridization and RT-PCR of candidate genes [1].
Figure 1.
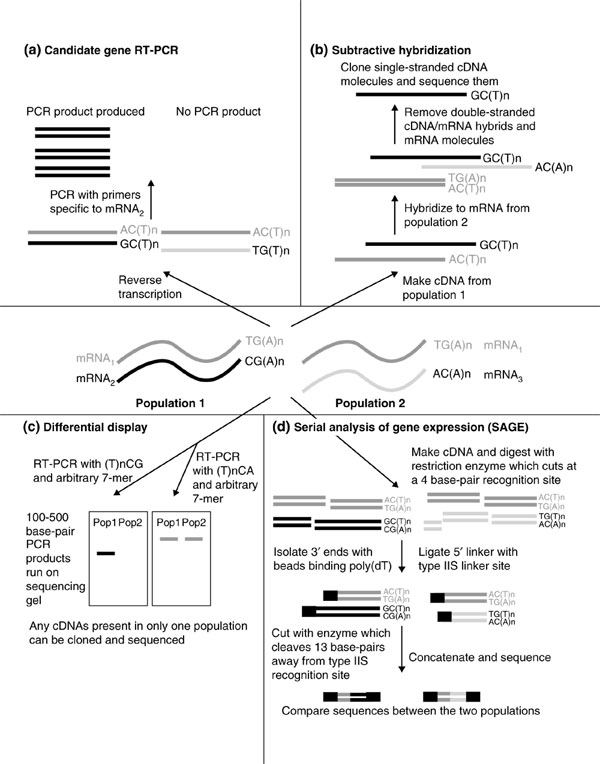
Four established techniques that are used to identify transcription-factor targets. These methods all compare mRNAs extracted from two populations of cells, one of which has the transcription factor in question overexpressed or knocked out. (a) Differences in the levels of specific candidate target genes in the two populations can be analyzed by reverse-transcriptase-coupled (RT-)PCR (for example, see [1,40]). (b) Any mRNAs that are equally expressed in both populations are subtracted, or removed, by cDNA-RNA hybridization. The remaining cDNAs are derived from mRNAs that are differentially expressed in one of the populations, and these can then be cloned and sequenced [3]. (c) With differential display, partial cDNA sequences are amplified from mRNA pools by RT-PCR. One primer - (T)nNN - binds to the polyadenylated tail of a subset of mRNAs that is defined by the two bases immediately 53 to the tail. The other binds to short sequences (6 or 7 base-pairs) that will occur with moderate frequency within the transcriptome. The products are radiolabeled and analyzed by polyacrylamide gel electrophoresis. Short cDNAs present in only one population can be isolated and sequenced [41,42]. (d) In serial analysis of gene expression (SAGE), cDNA is synthesized from mRNA and cleaved by a restriction enzyme that recognizes a 4 nucleotide sequence. The 33 end of the cleaved cDNA is isolated using beads that bind to oligo-dT, and 53 linkers are ligated to the restriction sites. These linkers contain type-IIS restriction sites, which are recognized by endonucleases that cleave a defined distance away (up to 20 base-pairs). This produces short DNA tags whose sequence and position are sufficient to identify the original transcript, provided cDNA sequences or expressed sequence tags (ESTs) are already known. The tags can be concatenated and sequenced, providing quantitative analysis of many transcripts simultaneously [43].
Such approaches have their limitations, however. Overexpression or misexpression of a transcription factor may not lead to up-regulation of its target genes if transcription is tightly controlled, or alternatively it may lead to indiscriminate activation of other genes that are not usually activated by the transcription factor under physiological conditions. On the other hand, knockdown of a transcription factor may cause embryonic or cellular lethality, or there may be redundancy with another factor so that bona fide target genes are not downregulated and therefore may not be identified. Nevertheless, these methods have been used successfully to identify transcription-factor target genes (see, for example, [3,4]).
Once putative target genes have been identified, they are often verified by examination of their expression pattern in tissues or whole organisms, since direct targets are expected to be activated in the regions where the transcription factor is expressed. Expression of putative target genes can also be compared between wild-type and mutant systems, as targets should not be expressed in the absence of the transcription factor (see [5] for example).
These methods identify only a limited number of targets, but more recently high-throughput techniques have allowed the identification of many more. Projects for sequencing both genomic DNA and expressed sequence tags (ESTs) have led to the development of expression microarrays, which enable simultaneous screening of most or all of the transcriptome and thus increase the number of targets that can be easily identified through comparisons of mRNA populations. In such experiments, RNA from each of the two cell populations, as described above and in Figure 1, is labeled with a different fluorescent dye. The RNA is then mixed and hybridized to microarrays, consisting of cDNAs or oligonucleotides arrayed on glass slides. The fluorescence intensity of each dot, which corresponds to one gene, can be measured and correlated to a change in expression of each gene [6]. For example, circadian gene expression in Drosophila, which is at least partially controlled by the Clock (Clk) transcription factor, was recently analyzed using microarrays [7]. Comparison of gene expression in wild-type and clk mutant flies led to the identification of 134 genes that require Clk for expression and whose expression levels cycle over 24 hours in wild-type flies [7].
In addition to giving increased numbers of potential transcription-factor targets, the ease with which large numbers of genes can now be investigated allows comparison of more than two different conditions, giving a clearer indication about which genes may be direct targets. For example, to identify targets of the sterol-regulatory-element binding protein (SREBP) genes in mice, Horton et al. [8] compared gene expression in the livers of one knockout strain and two transgenic strains of mice that overexpress different forms of SREBP. They applied stringent combinatorial criteria to identify direct targets, restricting themselves only to genes that were upregulated in both transgenic lines and downregulated in the knockout line. As a result, 33 genes were identified by this method, only 38% of the genes that would have been identified by comparing just two of the strains. Although this combinatorial method clearly increases the likelihood of predicting a direct target, other methods must be used to be more confident of a direct interaction of the transcription factor with the target gene.
Testing for direct activation of putative target genes
A variety of methods can be used to identify targets that are likely to be regulated directly by a transcription factor. Timing is one criterion: for example, immediate early genes, which are switched on shortly after the activation of a transcription factor, are more likely to be activated directly by that factor, because there has been little time for another gene to be activated and then for that to activate the target gene. This type of analysis is facilitated by the use of inducible gene expression, so the precise moment at which the transcription factor is activated and able to induce expression of downstream genes is known [9].
This technique can be further improved by the use of protein-synthesis inhibitors, such as cycloheximide. Transcription factors that are already present within the cell are able to activate the expression of their target genes, but in the presence of cycloheximide the target genes cannot be translated, and so cannot switch on further downstream genes as indirect targets. Thus, only those genes upregulated in the presence of cycloheximide are direct targets [10]. For instance, although microarray expression analysis identified 134 targets of Drosophila Clk, expression of a hormone-inducible form of Clk in cell culture in the presence of cycloheximide indicates that only nine of the genes are in fact direct targets [7].
These methods provide further evidence that a target is direct but do not show that the transcription factor binds directly to a regulatory sequence in the gene; this can be tested by other approaches, such as the electrophoretic mobility shift assay (EMSA). This technique identifies binding of specific proteins to DNA sequences, and so can demonstrate direct binding of a transcription factor to the promoter region of its target gene [11]. This in vitro method may not accurately reflect the situation in vivo, however, as binding is likely to be less tightly regulated in the assay.
To overcome this difficulty, two methods have been developed to demonstrate direct binding of a transcription factor to promoter regions of DNA in vivo: chromatin immunoprecipitation (ChIP) and Dam methylase identification (DamID; both are described in Figure 2). In addition to being used to ask whether a particular candidate gene is a direct target of a transcription factor, these techniques can also be adapted to identify new target genes. For example, regulatory DNA sequences enriched by ChIP can be used as probes to identify the coding regions of direct target genes [12-14]. Even these approaches have their limitations, however. In ChIP, protein-DNA interactions may not survive the procedure, and there is the risk of artifactual binding being introduced during the fixation process; similarly, expression of a fusion protein with DamID may not accurately replicate the situation in vivo. Nevertheless, these approaches prove to be very powerful and, as described below, can be scaled up to analyze the binding of transcription factors across the entire genome (so-called genome-wide location analysis).
Figure 2.
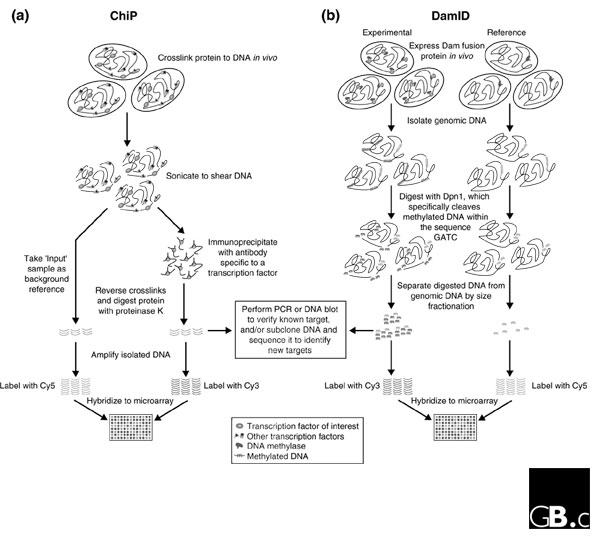
Experimental procedures for identifying transcription-factor targets in vivo by chromatin immunoprecipitation (ChIP) and Dam methylase identification (DamID), using microarrays. (a) In ChIP, formaldehyde is used to fix proteins bound to DNA in vivo. The DNA is then isolated and sheared by sonication into fragments of 200-700 base-pairs. An antibody against the transcription factor of interest is used to immunoprecipitate the factor and associated chromatin; or, if an epitope-tagged version of the protein is expressed in cells, an antibody can be used that is specific to the epitope. Protein is then removed from the DNA by reversal of the crosslinks and digestion with proteinase K. At this point, the isolated DNA can be used to verify targets by PCR or dot blot, or the DNA may be sub-cloned and sequenced to identify new targets [44]. For ChIP array analysis, the purified DNA is amplified by PCR and then labeled with a fluorophore, such as Cy3. As a reference for background binding, input DNA that is not enriched by immunoprecipitation is also amplified and labeled with another fluorophore, such as Cy5 [16,18]. Alternatively, non-enriched reference DNA is isolated after immunoprecipitation from cells that do not contain the transcription factor of interest [15]. The two populations of DNA are then hybridized to a microarray containing genomic sequences, and target sequences bound by the factor are identified according to the relative fluorescent intensity of each spot. (b) With DamID, the transcription factor of interest is fused to the Escherichia coli enzyme DNA adenine methylase (Dam). The fusion protein is expressed in vivo and Dam methylates DNA in the immediate vicinity of the binding site of the transcription factor, specifically acting on adenines in the sequence GATC. Dam alone is also expressed in cells as a reference, to identify background binding and methylation. Given that endogenous methylation of adenine does not occur in the DNA of most eukaryotes, methylated DNA can then be digested with Dpn1 (which cuts at the sequence GAmeTC) and isolated from uncut genomic DNA by size fractionation. The resulting DNA can then be analyzed by Southern blot to verify putative targets [14]. For genome-wide analysis, DNA from the experimental and reference samples is labeled with two different fluorophores (such as Cy3 and Cy5) and hybridized to a microarray [17,28,29].
Genome-wide location analysis
Several groups have recently developed techniques for high-throughput identification of genomic regions associated with transcription-factor binding [15-18], using ChIP and DamID approaches.
ChIP arrays
One approach, which was first described for Saccharomyces cerevisiae but has since been applied to human cell lines [15,16,18-21], has extended the ChIP protocol to the analysis of immunoprecipitated DNA with genomic microarrays (see Figure 2; reviewed in more detail in [22,23]).
The design of microarrays differs between different research groups and between organisms. For S. cerevisiae, which has a small and relatively simple genome containing approximately 6,200 genes, it is possible to design microarrays containing all yeast intergenic regions [15,16] in addition to coding regions [15,24]. Designing microarrays for human studies is more difficult, because higher eukaryotes have a more complex genome and more complex mechanisms of gene regulation. Unlike yeast, where the majority of transcription-factor-binding sites are found in upstream proximal promoter regions [15,24], higher eukaryotic gene expression is also controlled by factors binding at enhancer sequences located many kilobases from the gene. These enhancers may be situated 5' or 3' relative to the gene, in introns or even occasionally in exons (see below).
Initial studies of transcription-factor binding in human cells concentrated on E2Fs, a family of transcription factors that play a role in cell-cycle progression and proliferation [16,18]. Thus Ren and colleagues [19] designed arrays containing sequences upstream of 1,444 genes available from the human genome sequence, about 1,200 of which had previously been identified as cell-cycle-regulated. As more human genome sequence data and annotation has become available, however, the Ren and Young labs have now produced microarrays containing 6,000 and 13,000 sequences, again consisting mostly of 5' proximal sequences [21,25]. A different approach was taken by Weinmann and colleagues [18] who arrayed 7,776 human genomic fragments enriched for CpG islands, which are generally associated with upstream regulatory regions in vertebrates (reviewed in [26]).
Although such approaches are very powerful, one drawback of intergenic arrays is that they are biased by the design. In particular, 5' upstream sequence arrays will not detect interactions in introns, downstream sequences, non-annotated genomic regions, or exons. To overcome this bias, another group has designed a microarray containing the non-repetitive sequence of human chromosome 22 [27]. They then used this array in a ChIP assay to analyze binding of the p65 subunit of NF-κB when cells were stimulated with tumor necrosis factor (TNF) α. This approach not only identified new targets for p65 on chromosome 22, but also revealed binding sites in areas of the chromosome that are currently not annotated. Although costly, this technique could be extended to the other chromosomes as more completed human chromosome sequences become available, and in this way an unbiased view of genomic binding-site architecture can be built up.
DamID arrays
DNA isolated from DamID experiments has also been used to probe microarrays (Figure 2b). In the first reports of using this technique in Drosophila, cDNA arrays were used [17,28]. More recently, however, Sun and colleagues have used a microarray spotted with contiguous regions of Drosophila chromosomes 2 and 3 for this analysis [29], and it should not be long before genomic arrays are also commonplace when using this technique.
One interesting picture that is emerging from these genome-wide location analyses is the pattern of transcription-factor binding across the genome. Several studies have searched for consensus binding sites for a particular factor using bioinformatic approaches (see below), and such sites have been found scattered throughout the genome, in both intergenic and coding regions (see, for example, [15,24]). Genome-wide location analysis reveals, however, that only a subset of these sites is actually bound in vivo. This could be because binding-site recognition may be influenced by transcription-factor binding partners or by chromatin structure. For instance, when the binding of yeast transcription factors, Swi4 and Rap1, was analyzed using arrays containing both intergenic and coding regions of the genome, most binding sites were found to be in the proximal promoter regions of genes, and very few in coding sequence [15,24]. In human cells, when binding of p65 was analyzed across chromosome 22, 28% of binding sites were found within 5 kilobases upstream of the translation start codon, 40% were found in intronic regions, and less than 1% of sites (2/209) were found in exons [27]. To date, such observations have been made for only a small number of factors and it will be interesting to see how the results for other factors compare.
Bioinformatic approaches
Ideally, we would like to be able to predict the expression pattern of a gene from its regulatory sequences. Are we moving towards a time when bona fide regulatory sequences bound by transcription factors can be identified in silico? Databases of consensus transcription-factor-binding sites have been assembled over the last decade and computational algorithms that operate ab initio have been developed in an attempt to identify transcription-factor-binding sequences across the genome (see [30-32] for more detailed information). The programs exhibit different levels of stringency depending upon the algorithms used, but because they rely only on sequence data all are subject to false positives and false negatives. This is because transcription factors do not bind to all instances of their consensus binding site, as outlined above, and may also bind to other sequences that vary from the known consensus sequence (see below).
The development of computational algorithms has been improved by comparative genomics, or phylogenetic footprinting (for example [33], reviewed in [31]). This approach is based upon the fact that non-coding sequences that are highly conserved between species are much more likely to be involved in gene regulation. But difficulties arise in identification of organisms that are significantly closely related for regions to be conserved but sufficiently divergent for this conservation to be significant.
In order to improve the reliability of computational prediction of functional binding sites, other information, often derived from experimental studies, must be included in the analysis (see [31,32,34]). A common method involves comparing the promoters of genes co-regulated by a transcription factor to identify conserved motifs. Recently, targets of Dorsal, a transcription factor involved in specifying the dorsoventral axis in Drosophila, were identified using expression microarrays, and subsequent analysis identified up to 40 targets that have the expected restricted expression pattern in the embryo [35]. Examination of the genomic sequence around a subset of these target genes discovered that consensus Dorsal-binding sites generally cluster together, either upstream of the start codon ATG or within introns [35]. A computational algorithm was developed from this information and used to scan the rest of the Drosophila genome, identifying 3 known Dorsal target genes and 15 new putative targets [34]. Two of these targets have been tested and found to exhibit asymmetric expression patterns across the dorsoventral axis, as would be expected for Dorsal target genes ([34], reviewed in [36]).
Similarly, Kel et al. [37] were able to identify composite modules consisting of clusters of binding sites for E2F and other transcription factors that are involved in the regulation of known E2F targets. Examination of these regulatory sequences led to the identification of a range of characteristic motifs in addition to the known binding sites. Using this information, computational methods were then developed to search the promoter regions of cell-cycle-regulated genes. This led to the identification of 29 genes known to be regulated by E2F, plus an additional 313 putative E2F targets that contained the identified upstream regulatory modules. Some of these putative targets have now been confirmed as direct targets by ChIP analysis [37].
Interestingly, in those ChIP-array studies where it has been examined, a proportion of sequences bound to transcription factors did not contain the known consensus binding site for the transcription factor tested. For example, Iyer et al. [15] found that in S. cerevisiae about half of the targets of the transcription factors MBF and SBF do not contain the consensus binding sites for the factors. In human cells, Ren et al. [19] and Weinmann et al. [18] found that up to 25% of identified E2F targets did not contain the E2F consensus site. Further characterization revealed that some of these target genes are repressed rather than up-regulated by E2F [18]. Although no sequence that is common to these repressing regions has yet been described, applying computational techniques may reveal such a site. Thus, genome-wide location analysis combined with computational analysis may be useful in identifying previously unknown binding sequences for other transcription-factors.
Transcriptional networks
The development of high throughput methods for the identification of direct transcription-factor target genes has led to a large increase in our understanding of combinatorial networks of gene regulation. The combination of genome-wide expression data with genome-wide location analysis constitutes a powerful tool not only in verifying predicted interactions, but also in elucidating transcriptional networks. Simon et al. [38] performed genome-wide location analysis with the nine known cell-cycle activators in yeast and showed that cell-cycle transcriptional control is a connected network. For example, transcriptional regulators that act at one stage of the cycle to up-regulate genes promoting cell-cycle progression also up-regulate the transcription of factors that act during the next stage of the cycle. This group has since extended its analysis to (nearly) all yeast transcription-factors [20]. This has identified simple network motifs (the building blocks of a network) that have been used to describe networks controlling, for example, metabolism and the response to mating factor [20,39]. As these kinds of analyses become more commonplace, we can look forward to a time when each transcription factor can be placed in a network that describes a complex cellular process, such as those that led to the development of an embryo.
References
- Davidson EH, Rast JP, Oliveri P, Ransick A, Calestani C, Yuh CH, Minokawa T, Amore G, Hinman V, Arenas-Mena C, et al. A genomic regulatory network for development. Science. 2002;295:1669–1678. doi: 10.1126/science.1069883. [DOI] [PubMed] [Google Scholar]
- Wyrick JJ, Young RA. Deciphering gene expression regulatory networks. Curr Opin Genet Dev. 2002;12:130–136. doi: 10.1016/S0959-437X(02)00277-0. [DOI] [PubMed] [Google Scholar]
- Lee SW, Tomasetto C, Sager R. Positive selection of candidate tumor-suppressor genes by subtractive hybridization. Proc Natl Acad Sci USA. 1991;88:2825–2829. doi: 10.1073/pnas.88.7.2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menssen A, Hermeking H. Characterization of the c-MYC-regulated transcriptome by SAGE: identification and analysis of c-MYC target genes. Proc Natl Acad Sci USA. 2002;99:6274–6279. doi: 10.1073/pnas.082005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakin L, Reversade B, Virlon B, Rusniok C, Glaser P, Elalouf JM, Brulet P. Gene expression profiles in normal and Otx2-/-early gastrulating mouse embryos. Proc Natl Acad Sci USA. 2000;97:14388–14393. doi: 10.1073/pnas.011513398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- McDonald MJ, Rosbash M. Microarray analysis and organization of circadian gene expression in Drosophila. Cell. 2001;107:567–578. doi: 10.1016/s0092-8674(01)00545-1. [DOI] [PubMed] [Google Scholar]
- Horton JD, Shah NA, Warrington JA, Anderson NN, Park SW, Brown MS, Goldstein JL. Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci USA. 2003;100:12027–12032. doi: 10.1073/pnas.1534923100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature. 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- Rosa FM. Mix.1, a homeobox mRNA inducible by mesoderm inducers, is expressed mostly in the presumptive endodermal cells of Xenopus embryos. Cell. 1989;57:965–974. doi: 10.1016/0092-8674(89)90335-8. [DOI] [PubMed] [Google Scholar]
- Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047–3060. doi: 10.1093/nar/9.13.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RA, Brookman JJ, Gould AP, Meadows LA, Shashidhara LS, Strutt DI, Weaver TA. Targets of homeotic gene regulation in Drosophila. J Cell Sci Suppl. 1992;16:53–60. doi: 10.1242/jcs.1992.supplement_16.7. [DOI] [PubMed] [Google Scholar]
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem Sci. 2000;25:99–104. doi: 10.1016/S0968-0004(99)01535-2. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Henikoff S. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol. 2000;18:424–428. doi: 10.1038/74487. [DOI] [PubMed] [Google Scholar]
- Iyer VR, Horak CE, Scafe CS, Botstein D, Snyder M, Brown PO. Genomic binding sites of the yeast cell-cycle transcription-factors SBF and MBF. Nature. 2001;409:533–538. doi: 10.1038/35054095. [DOI] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, et al. Genome-wide location and function of DNA binding proteins. Science. 2000;290:2306–2309. doi: 10.1126/science.290.5500.2306. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Delrow J, Henikoff S. Chromatin profiling using targeted DNA adenine methyltransferase. Nat Genet. 2001;27:304–308. doi: 10.1038/85871. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Yan PS, Oberley MJ, Huang TH, Farnham PJ. Isolating human transcription-factor targets by coupling chromatin immunoprecipitation and CpG island microarray analysis. Genes Dev. 2002;16:235–244. doi: 10.1101/gad.943102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD. E2F integrates cell-cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002;16:245–256. doi: 10.1101/gad.949802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Rinaldi NJ, Robert F, Odom DT, Bar-Joseph Z, Gerber GK, Hannett NM, Harbison CT, Thompson CM, Simon I, et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci USA. 2003;100:8164–8169. doi: 10.1073/pnas.1332764100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nal B, Mohr E, Ferrier P. Location analysis of DNA-bound proteins at the whole-genome level: untangling transcriptional regulatory networks. BioEssays. 2001;23:473–476. doi: 10.1002/bies.1066. [DOI] [PubMed] [Google Scholar]
- Shannon MF, Rao S. Transcription. Of chips and ChIPs. Science. 2002;296:666–669. doi: 10.1126/science.1062936. [DOI] [PubMed] [Google Scholar]
- Lieb JD, Liu X, Botstein D, Brown PO. Promoter-specific binding of Rap1 revealed by genome-wide maps of protein-DNA association. Nat Genet. 2001;28:327–334. doi: 10.1038/ng569. [DOI] [PubMed] [Google Scholar]
- Odom DT, Zizlsperger N, Gordon D, Bell GW, Rinaldi NJ, Murray HL, Volkert TL, Schreiber J, Rolfe A, Gifford D, et al. Control of pancreas and liver gene expression by HNF transcription factors. Science. 2004;303:1378–1381. doi: 10.1126/science.1089769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr Biol. 1999;9:R661–R667. doi: 10.1016/S0960-9822(99)80418-7. [DOI] [PubMed] [Google Scholar]
- Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, et al. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci USA. 2003;100:12247–12252. doi: 10.1073/pnas.2135255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orian A, van Steensel B, Delrow J, Bussemaker HJ, Li L, Sawado T, Williams E, Loo LW, Cowley SM, Yost C, et al. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription-factor network. Genes Dev. 2003;17:1101–1114. doi: 10.1101/gad.1066903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LV, Chen L, Greil F, Negre N, Li TR, Cavalli G, Zhao H, Van Steensel B, White KP. Protein-DNA interaction mapping using genomic tiling path microarrays in Drosophila. Proc Natl Acad Sci USA. 2003;100:9428–9433. doi: 10.1073/pnas.1533393100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Ding W, Jiang Y, Greene JR, Wang L. Computational analysis of composite regulatory elements. Mamm Genome. 2002;13:327–332. doi: 10.1007/s00335-001-2141-8. [DOI] [PubMed] [Google Scholar]
- Pennacchio LA, Rubin EM. Comparative genomic tools and databases: providing insights into the human genome. J Clin Invest. 2003;111:1099–1106. doi: 10.1172/JCI200317842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohler U, Niemann H. Identification and analysis of eukaryotic promoters: recent computational approaches. Trends Genet. 2001;17:56–60. doi: 10.1016/S0168-9525(00)02174-0. [DOI] [PubMed] [Google Scholar]
- Lenhard B, Sandelin A, Mendoza L, Engstrom P, Jareborg N, Wasserman WW. Identification of conserved regulatory elements by comparative genome analysis. J Biol. 2003;2:13. doi: 10.1186/1475-4924-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markstein M, Markstein P, Markstein V, Levine MS. Genome-wide analysis of clustered Dorsal binding sites identifies putative target genes in the Drosophila embryo. Proc Natl Acad Sci USA. 2002;99:763–768. doi: 10.1073/pnas.012591199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopoulos A, Van Drenth M, Erives A, Markstein M, Levine M. Whole-genome analysis of dorsal-ventral patterning in the Drosophila embryo. Cell. 2002;111:687–701. doi: 10.1016/s0092-8674(02)01087-5. [DOI] [PubMed] [Google Scholar]
- Markstein M, Levine M. Decoding cis-regulatory DNAs in the Drosophila genome. Curr Opin Genet Dev. 2002;12:601–606. doi: 10.1016/S0959-437X(02)00345-3. [DOI] [PubMed] [Google Scholar]
- Kel AE, Kel-Margoulis OV, Farnham PJ, Bartley SM, Wingender E, Zhang MQ. Computer-assisted identification of cell-cycle-related genes: new targets for E2F transcription-factors. J Mol Biol. 2001;309:99–120. doi: 10.1006/jmbi.2001.4650. [DOI] [PubMed] [Google Scholar]
- Simon I, Barnett J, Hannett N, Harbison CT, Rinaldi NJ, Volkert TL, Wyrick JJ, Zeitlinger J, Gifford DK, Jaakkola TS, Young RA. Serial regulation of transcriptional regulators in the yeast cell-cycle. Cell. 2001;106:697–708. doi: 10.1016/s0092-8674(01)00494-9. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Simon I, Harbison CT, Hannett NM, Volkert TL, Fink GR, Young RA. Program-specific distribution of a transcription-factor dependent on partner transcription-factor and MAPK signaling. Cell. 2003;113:395–404. doi: 10.1016/s0092-8674(03)00301-5. [DOI] [PubMed] [Google Scholar]
- Endomesoderm Gene Network http://sugp.caltech.edu/endomes/
- Liang P, Pardee AB. Differential display of eukaryotic messenger RNA by means of the polymerase chain reaction. Science. 1992;257:967–971. doi: 10.1126/science.1354393. [DOI] [PubMed] [Google Scholar]
- Matz MV, Lukyanov SA. Different strategies of differential display: areas of application. Nucleic Acids Res. 1998;26:5537–5543. doi: 10.1093/nar/26.24.5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science. 1995;270:484–487. doi: 10.1126/science.270.5235.484. [DOI] [PubMed] [Google Scholar]
- Weinmann AS, Farnham PJ. Identification of unknown target genes of human transcription-factors using chromatin immunoprecipitation. Methods. 2002;26:37–47. doi: 10.1016/S1046-2023(02)00006-3. [DOI] [PubMed] [Google Scholar]