

Abstract
Bacterial colonization in the gastrointestinal tracts (GIT) of preweaned calves is very important, since it can influence early development and postweaning performance and health. This study investigated the composition of the bacteria along the GIT (rumen, jejunum, ileum, cecum, and colon) of preweaned bull calves (3 weeks old) using pyrosequencing to understand the segregation of bacteria between the mucosal surface and digesta. Phylogenetic analysis revealed that a total of 83 genera belonging to 13 phyla were distributed throughout the GIT of preweaned calves, with the Firmicutes, Bacteroidetes, and Proteobacteria predominating. Quantitative PCR (qPCR) analysis of selected abundant bacterial genera (Prevotella, Bacteroides, Lactobacillus, and Faecalibacterium) revealed that their prevalence was significantly different among the GIT regions and between mucosa- and digesta-associated communities. Rumens contained the most diverse bacterial population, consisting of 47 genera, including 16 rumen-specific genera, followed by the large intestine and then the small intestine. Bacterial species richness was higher at the mucosal surface than in the local digesta, with the exception of the rumen. The majority of bacteria found on the rumen epithelial surface and within the small intestine could not be identified due to a lack of known genus-level information. Thus, future studies will be required to fully characterize the microbiome during the development of the rumens and the mucosal immune systems of newborn calves. This is the first study to analyze in depth the bacterial composition of the GIT microbiome in preweaned calves, which extends previous findings regarding early rumen colonization and bacterial segregation between mucosa- and digesta-associated microbial communities.
INTRODUCTION
The gastrointestinal tracts (GIT) of newborns contain a less diverse microbiome than those of adults, and progressive colonization over time increases this diversity (1). Based on the fecal microbiomes of infants, the pioneer gut bacteria are comprised of facultative anaerobes such as Staphylococcus, Streptococcus, Enterococcus, and Enterobacteriaceae spp. during the first few days of life (2). These species create the reduced environment that is required for obligate anaerobic gut microbes (3). Besides the facultative anaerobes, anaerobic Bifidobacterium species are also present at high levels in the guts of 1-week-old neonates (3). An increasing number of studies are investigating microbial establishment and the factors influencing this process in newborn livestock, such as dairy calves. A recent study reported a link between the prevalence of Faecalibacterium during the first week of life and body weight gain as well as diarrhea incidences in 4-week-old calves (4). This suggests a potential role for gut bacteria in both animal health and production. Evidence is also emerging that the initial acquisition of and continuous exposure to microbes result in a host-specific gut microbiome, which plays a vital role in the maturation of the mucosal immune system (5–7). Hence, knowledge regarding the pioneer bacterial community of calves provides an opportunity to understand how perturbations in microbial composition and diversifications may alter health and production in cattle.
The fecal bacterial composition of dairy calves undergoes dynamic changes during the first 12 weeks of life (8). These changes include the appearance of new species such as Ruminococcus flavefaciens and Fibrobacter species and the disappearance of Bifidobacterium, Enterobacteriaceae, Streptococcus, and Lactobacillus species (8), suggesting that both diet and gut development may drive changes in the bacterial composition during early life. A study comparing mucosa- and digesta-associated bacterial phylotypes in preweaned calves revealed a much greater richness in mucosa-associated bacterial phylotypes throughout the GIT than in those in the regional digesta (9). Moreover, mucosa-associated bacteria in the murine distal colon not only differed significantly from fecal bacteria but also correlated with Toll-like receptor 2 (TLR2) and TLR4 gene expression in colon epithelial cells (10). These observations indicate the importance of studying bacterial segregation between mucosal surfaces and digesta throughout the GIT, to better understand host-microbe interactions. Currently, our understanding of the gut microbiome and its segregation between mucosal surfaces and digesta in the neonatal calves is very limited. Therefore, the present study used pyrosequencing of 16S rRNA genes to provide a more complete analysis of the taxonomic segregation of bacteria throughout the GIT, including the rumen, of preweaned calves.
MATERIALS AND METHODS
Sample collection.
This was a companion study conducted using samples collected from 3-week-old Holstein bull calves used in our previous study (9). One-week-old bull calves (n = 8) were purchased from a commercial dairy farm and housed at the Vaccine and Infectious Disease Organization (VIDO), University of Saskatchewan. All experimental protocols were approved by the University of Saskatchewan Committee on Animal Care (animal protocol 20020105), and all the procedures were performed following the Canadian Council on Animal Care guidelines. Calves were fed fresh, nonpasteurized whole milk and Blue Medallion calf supplement (20% crude protein, 3% crude fat, 5.7% crude fiber, 1% Ca, 0.6% P; Ridley Inc., Fed-Rite, Manitoba, Canada). When calves were 3 weeks old, mucosal tissue and digesta samples were collected from the rumen, jejunum, ileum, cecum, and colon within 20 min after euthanization. Mucosal tissue samples were rinsed 3 times with sterile phosphate-buffered saline (PBS) (pH 7.0) to remove digesta, cut into 4- to 5-mm2 pieces, and immersed in RNAlater (Life Technologies, Carlsbad, CA, USA). Digesta was collected from each mucosal tissue collection site, and 200 μl of digesta was mixed with 1 ml RNAlater. Samples were stored at −80°C until further analysis.
DNA extraction and amplicon preparation for pyrosequencing.
Total DNA was extracted from all the samples using the bead-beating method as described by Li et al. (11). DNA quality and quantity was measured using NanoDrop 1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). Total DNA was then diluted to final concentrations of 25 ng/μl and 50 ng/μl for mucosal tissue and digesta, respectively, and used in amplicon preparation for pyrosequencing.
Conventional PCR was performed for all the samples to amplify the V1-V3 region of 16S rRNA gene using primer-containing Titanium A and B adaptors (27F [primer B], 5′CCTATCCCCTGTGTGCCTTGGCAGTCTCAGAGAGGTTTGATCCTGGCTCAG3′; 338R [primer A], 5′CCATCTCATCCCTGCGTGTCTCCGACTCAGRLTGCTGCCTCCCGTAGGAGT3′, where RL is the rapid library adaptor; boldface type indicates letters that do not represent primer bases) (12). The reverse primer contained 10 different multiplex identifiers (RL1 to RL8, RL10, and RL11; 454 Life Sciences, Branford, CT, USA) and was used to amplify samples from 5 different GIT regions for mucosal tissue and digesta separately using the following program; 94°C for 5 min, 35 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 30 s followed by 72°C for 7 min. The PCR was first performed for individual animals separately, using primer A with the same assigned RL based on the gut region and sample type (e.g., rumen tissue samples with primer A containing RL1; rumen content samples with primer A containing RL2 and so on), and then the PCR products were pooled by the RL index. DNA fragments (∼400 bp) were extracted from 1% agarose gel using QIAEX II gel extraction kit (Qiagen Science, MD, USA) following the manufacturer's instructions. The purified PCR products were then quantified using NanoDrop 1000, and amplicons from each sample were pooled in equal amounts to a final concentration of 40 ng/μl and subjected to pyrosequencing.
Pyrosequencing and data analysis.
The pooled PCR amplicons were subjected to pyrosequencing using Roche GS-FLX System with Titanium chemistry at Genome Quebec (Montreal, QC, Canada). Data were analyzed using the QIIME (Quantitative Insight into Microbial Ecology) tool kit (13). The raw sequences obtained from pyrosequencing were filtered through a quality control pipeline, and bases with quality scores higher than 25 were retained for further analysis. The high-quality reads were then assigned to operational taxonomic units (OTUs) at a 97% identity threshold using the Uclust algorithm (14), and taxonomy was assigned using the latest Greengenes database (Greengenes May 2013 release). Taxonomy assignment was done separately for mucosal tissue and digesta from each gut region (rumen, jejunum, ileum, cecum, and colon). The taxonomic identification and comparisons were performed at the genus level. Alpha diversity of mucosa- and digesta-associated bacterial communities of each gut region was obtained using various diversity indices (Chao1 index, Shannon index, and observed species) using the alpha rarefaction pipeline. The hierarchical clustering of microbial communities (jackknifed beta diversity) was performed using weighted UniFrac distance and unweighted pair group method with arithmetic mean (UPGMA) clustering.
Quantification of abundant bacterial genera along the GIT.
The copy numbers of 16S rRNA genes of 5 selected bacterial genera (Prevotella, Bacteroides, Lactobacillus, Clostridium cluster XIV, and Faecalibacterium) were estimated using genus-specific primers (see Table S1 in the supplemental material) and SYBR green chemistry (fast SYBR green master mix; Applied Biosystems) with a StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA, USA). The standard curves for each bacterial genus were prepared using purified PCR products amplified with each primer pair. The copy number of the 16S rRNA gene per gram of fresh tissue or digesta was then calculated using the equation described in reference 11. The prevalence of bacterial genera was calculated by dividing the 16S rRNA gene copy number for each genus by the 16S rRNA gene copy number for total bacteria.
Statistical analysis.
The data obtained from the qPCR were analyzed using the MIXED procedure in SAS (version 9.2; SAS Institute Inc., Cary, NC). The effects of gut region and sample type (mucosal tissue versus digesta) on the bacterial prevalence were determined using a two-way analysis of variance (ANOVA), and the statistical significances were declared at P < 0.05. Prevalence was determined as follows: Yijk = μ + Gi + Sj + GSij + eijk, where Y is bacterial prevalence, μ is mean, G is gut region effect, S is sample type effect, GS is gut region by sample type effect, e is residual error, i is the number of gut regions, j is the number of sample types, and k is the number of observations.
Nucleotide sequence accession numbers.
The identified sequences from this study were deposited in the NCBI sequence read archive (SRA) under the accession numbers SRR1036467 to SRR1036476.
RESULTS
Bacterial diversity along the GIT of preweaned calves.
Pyrosequencing analysis of the GIT samples of preweaned calves generated a total of 565,854 sequences and 15,771 operational taxonomic units (OTUs), with Good's coverage ranging from 0.77 to 0.96. However, the distribution of these OTUs varied among individual GIT regions. The highest number of OTUs was observed in the rumen, followed by the large intestine (cecum and colon) and then the small intestine (jejunum and ileum) (Table 1). The alpha diversity indices (Chao1 and Shannon indices) were higher in the rumen bacterial community (mucosal tissue and digesta together) than in the large- and small-intestinal communities (Table 1). When the alpha diversity indices were compared between mucosal tissue and digesta of each gut region, higher values were observed in digesta than mucosal tissue only in the rumen. In contrast, higher Chao1 and Shannon index values were observed in the mucosa-associated communities of the jejunum, ileum, and cecum than in the respective digesta-associated communities (Table 1). The Chao1 index was higher for colonic digesta than colonic mucosal tissue, whereas the Shannon index was higher for colonic mucosal tissue than colonic digesta (Table 1).
TABLE 1.
Generated sequences, identified operational taxonomic units (OTUs), bacterial diversity, and species richness along the GIT of preweaned dairy calves
| GIT region | Totala |
Mucosa |
Digesta |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No. of sequences | No. of OTUs | Chao1 indexb | Shannon indexc | No. of sequences | No. of OTUs | Chao1 index | Shannon index | No. of sequences | No. of OTUs | Chao1 index | Shannon index | |
| Rumen | 94,116 | 5,693 | 6,046 | 8.6 | 48,639 | 6,051 | 3,908 | 8.2 | 46,110 | 7,374 | 4,608 | 8.8 |
| Jejunum | 96,086 | 2,632 | 2,522 | 6.2 | 75,960 | 3,394 | 2,970 | 6.3 | 20,796 | 2,362 | 2,041 | 6.1 |
| Ileum | 112,404 | 2,685 | 3044 | 6.1 | 57,783 | 3,744 | 3,865 | 6.9 | 55,416 | 2,571 | 2,036 | 5.2 |
| Cecum | 81,567 | 2,893 | 4,372 | 7.2 | 49,232 | 4,196 | 3,836 | 7.1 | 33,292 | 3,251 | 3,348 | 6.9 |
| Colon | 177,978 | 4,694 | 4,644 | 7.1 | 49,119 | 3,950 | 3,335 | 6.9 | 129,507 | 7,264 | 3,968 | 6.9 |
Values are for mucosa and digesta samples together for each gut region.
The Chao1 index is an estimation of species richness.
The Shannon index is an an estimation of diversity.
Gut bacterial composition of preweaned calves: distribution of the predominant bacterial phyla and genera along the GIT.
In total, 13 bacterial phyla were identified within the preweaned calf gut microbiota, and these were dominated by Firmicutes (42.7%), Bacteroidetes (36.3%), and Proteobacteria (11.9%). However, the relative abundance of these major phyla varied within the mucosa- and digesta-associated communities. Firmicutes (57.6%) were predominant in the digesta, while Bacteroidetes (42.3%) were predominant in the mucosa-associated community, when these two communities were compared collectively without considering regional variations. However, the relative abundance of these predominant phyla varied markedly among GIT regions as well as between mucosal tissue and digesta (Fig. 1). When the bacterial composition was compared regionally, Bacteroidetes dominated all digesta-associated communities along the GIT except for the small intestine, where Firmicutes (∼97%) was predominant. Bacteroidetes also dominated all the mucosa-associated communities with the exception of the jejunal tissue, where there was a mix of Firmicutes and Proteobacteria rather than Bacteroidetes.
FIG 1.
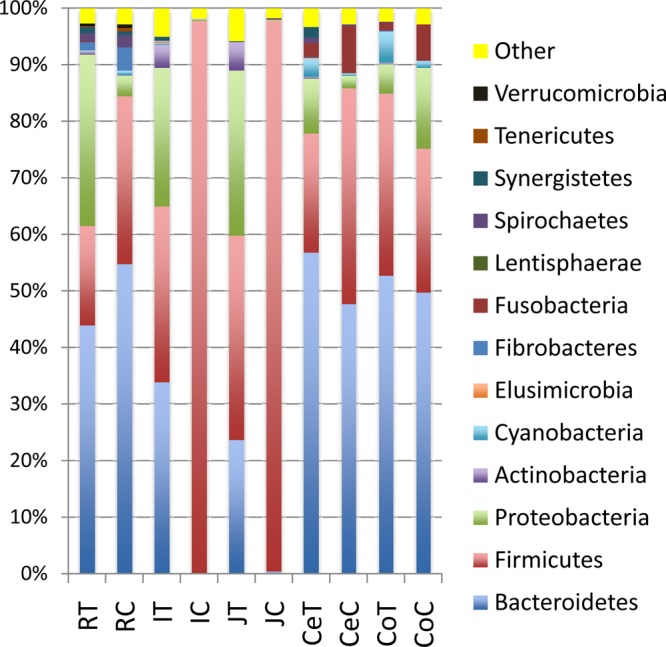
Phylum-level composition of gut bacteria in mucosa- and digesta-associated communities throughout the gastrointestinal tracts of dairy calves. RT, rumen tissue; RC, rumen contents; IT, ileum tissue; IC, ileum contents; JT, jejunum tissue; JC, jejunum contents; CeT, cecum tissue; CeC, cecum contents; CoT, colon tissue; CoC, colon contents.
A total of 83 genera belonging to the 13 phyla were observed throughout the GIT of preweaned calves; however, in the present study, 32.5% of all sequences were not identified at the genus level. The distribution of predominant bacterial genera (relative abundance ≥ 10% in at least one sample) was also different between mucosa- and digesta-associated communities throughout the GIT (Fig. 2). Bacteroides, Prevotella, Faecalibacterium, and Burkholderia were predominant in the mucosa-associated community, while Bacteroides, Prevotella, Lactobacillus, Clostridium, and Sharpea were predominant in the digesta-associated community (Fig. 2). Among the 83 bacterial genera identified, only 7 genera (Prevotella, Fibrobacter, Lactobacillus, Clostridium, Ruminococcus, Sharpea, and Corynebacterium, the least abundant genus) were detected in both the mucosa- and digesta-associated communities in all GIT regions.
FIG 2.

Relative abundance and distribution of 20 abundant (≥1% in at least one GIT region) bacterial genera detected throughout the GIT of 3-week-old preweaned calves. (a) Distribution of abundant genera in mucosa-associated tissue. (b) Distribution of abundant genera in digesta.
When the bacterial composition among the different GIT regions was compared using UniFrac distance and UPGMA clustering analysis for all OTUs as well as the abundant bacterial genera (20 genera; relative abundance > 1%), the rumen and large-intestinal bacterial communities clustered separately from the small intestine (Fig. 3a and b). Among the small-intestinal communities, jejunal and ileal mucosa-associated bacterial communities clustered more closely than the digesta-associated communities (Fig. 3a and b). The rumen digesta- and mucosa-associated communities clustered more closely to each other than to other members of the combined rumen and large-intestinal communities (Fig. 3a and b). The distribution of bacterial genera further revealed 10 shared genera throughout all GIT regions, 10 shared genera between the rumen and large intestine, 8 shared genera between the small intestine and large intestine, and finally only 4 shared genera between the rumen and small intestine (Fig. 3c).
FIG 3.

Clustering of bacterial genera and OTUs present in the GIT of 3-week-old preweaned calves. (a) Heat map generated using the relative abundance (percent) of 20 abundant bacterial genera. The heat map was generated using the gplots package in R by clustering GIT regions based on the distribution and relative abundance of bacterial genera. The heat map scale displays the row Z score (Z score = [actual relative abundances of a species in a specific GIT region − mean relative abundance of the same species along the GIT]/standard deviation). (b) Clustering of all OTUs obtained from pyrosequencing of GIT samples. Unweighted pair group method with arithmetic mean (UPGMA) clustering was based on all available data with jackknifed beta diversity, which compares the diversity among the samples. (c) Comparison of the number of shared bacterial genera among the rumen, small intestine, and large intestine. The number of bacterial genera represents only the unique genera in each region, shared genera between two GIT sections, and shared genera in all the gut regions.
Regional segregation of gut bacteria between mucosa- and digesta-associated communities.
The phylum Bacteroidetes dominated the rumen digesta- and tissue-associated communities of preweaned calves (Fig. 1). The majority of digesta-associated Bacteroidetes (54.8%) were comprised of Bacteroides (15.8%), Prevotella (15.1%), and Paludibacter (4.1%), while Prevotella (15.8%) and Bacteroides (10.5%) dominated the rumen tissue-associated community (see Fig. S1 in the supplemental material). Firmicutes, the second most abundant phylum in the rumen digesta and third most abundant phylum in the rumen tissue, consisted of 18 different bacterial genera (see Fig. S1 in the supplemental material). Among these 18 genera, Megamonas and p-75-a5 were not present in the rumen digesta-associated community, while Streptococcus and Sarcina were absent in the rumen tissue-associated community. Proteobacteria was the second most abundant phylum associated with the rumen tissue (30.2%), which was 10 times greater than that in the rumen digesta. However, 27.8% of Proteobacteria sequences were not assigned to any genus. In addition, the majority of sequences from ruminal tissue-associated community (58.3%) were not assigned to any genus. In contrast, only 33.1% of sequences from the rumen digesta-associated community were not assigned to any genus. The present study revealed 16 rumen-specific bacterial genera (Actinomyces, Porphyromonas, Butyricimonas, Elusimicrobium, p-75-a5, Megamonas, Desulfobulbus, Comamonas, Hylemonella, Sphaerochaeta, Anaeroplasma, Synergistes, Roseburia, BF311, TG5, and RFN20) in the preweaned calves. The genera Desulfobulbus, Comamonas, Hylemonella, Megamonas, TG5, and p-75-a5 were observed only in the rumen tissue-associated community, while Elusimicrobium and Anaeroplasma were present only in the rumen digesta-associated community.
The large-intestinal digesta- and mucosa-associated bacterial communities were primarily comprised of Bacteroidetes (cecal tissue, 56.8%; cecal digesta, 47.6%; colonic tissue, 52.8%; colonic digesta, 49.8%) (Fig. 1). The prevalence of Bacteroides was higher in cecal digesta (15.9%) than in cecal tissue (5.9%) (see Fig. S1 in the supplemental material). In contrast, Prevotella accounted for more Bacteroidetes in cecal tissue (21.7%) than digesta (9.1%). The prevalence of Prevotella was higher in colon digesta- and mucosa-associated communities (19.3%) than that of Bacteroides (colonic digesta, 15.4%; colon tissue, 11%). Firmicutes was the second most abundant bacterial phylum in the large-intestinal communities (cecal tissue, 21.1%; cecal digesta, 38.2%; colonic tissue, 32.1%; colonic digesta, 25.4%). A higher prevalence of Faecalibacterium than other Firmicutes genera was observed in all large-intestinal communities (cecal tissue, 9%; cecal digesta, 9.3%; colonic tissue, 10.8%; colonic digesta, 13.5%) (see Fig. S1 in the supplemental material). Besides Faecalibacterium, Clostridium was also abundant in cecum (12%) and colon (5.8%) digesta-associated communities. A higher prevalence of Lactobacillus was observed only in the cecal digesta (6.4%), in contrast to other large-intestinal communities. The bacterial genera Escherichia, Succinivibrio, Flavobacterium, Blautia, and L7AE11 were observed only in the large intestine. The present study also revealed 10 bacterial genera (Paludibacter, Parabacteroides, Oscillospira, Shuttleworthia, Acidaminococcus, Anaerovibrio, Fusobacterium, Ruminobacter, Gallibacterium, and Treponema) that were shared by the rumen and the large-intestinal communities but not present in the small intestine.
The small-intestinal (jejunum and ileum) bacterial communities displayed a unique segregation between mucosa- and digesta-associated communities. The ileal digesta of preweaned calves contained primarily Firmicutes (97.7%), consisting of Lactobacillus (44.5%), Clostridium (16.7%), and Sharpea (8.9%) (see Fig. S2 in the supplemental material). The ileal tissue was comprised of Bacteroidetes (33.7%), followed by Firmicutes (31.8%) and Proteobacteria (24.4%). Prevotella (8.2%) dominated the ileal mucosa-associated Bacteroidetes, while a high proportion (24.9%) was not identified at the genus level. In addition, 49.9% of ileal-tissue sequences could not be assigned to any genus. The jejunal digesta-associated community was also dominated by Firmicutes (97.4%), which consisted of genera Sharpea (31.8%), Butyrivibrio (4.2%), Ruminococcus (3.2%), and Lactobacillus (2.4%) (see Fig. S2 in the supplemental material). However, 53.2% of digesta-associated Firmicutes could not be assigned to any genus. The jejunal mucosa-associated bacteria contained Firmicutes (36.2%), Proteobacteria (29.2%), and Bacteroidetes (23.6%). However, 61.7% of total sequences from the jejunal mucosa-associated community could not be assigned to any genus. The prevalence of Burkholderia, which belongs to the Proteobacteria, was high only in the small-intestinal mucosa-associated communities (ileal issue, 11.8%; jejunal tissue, 10.3%). Moreover, 17 bacterial genera were detected only in the small-intestinal mucosa-associated bacterial community (Caulobacter, Variovorax, Acinetobacter, Lautropia, Diaphorobacter, Janthinobacterium, Staphylococcus, Microbacterium, Nocardiopsis, Thermoactinomyces, Facklamia, Enterococcus, Phascolarctobacterium, Atopobium, Bradyrhizobium, Jeotgalicoccus, and Pelomonas).
Prevalence of abundant bacterial genera throughout the GIT.
The analysis of bacterial prevalence by qPCR confirmed the results observed in the pyrosequencing of pooled samples from preweaned calves (Table 2). The prevalence of bacterial genera was significantly different among GIT regions with the exception of Clostridium cluster XIV. The highest prevalence of Prevotella was observed in colonic digesta, in contrast to other digesta-associated communities, while its prevalence was higher in the small-intestinal mucosa-associated communities than in the respective digesta-associated communities. A higher Bacteroides prevalence was observed in the rumen communities, while Lactobacillus prevalence was highest in the small intestine, relative to other GIT regions. The highest prevalence of Faecalibacterium was detected in the large-intestinal communities.
TABLE 2.
Prevalence of abundant bacterial genera along the GIT of 3-week-old calves
| Genus | Prevalence (%)a in: |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rumen |
Jejunum |
Ileum |
Cecum |
Colon |
||||||
| Tissue | Digesta | Tissue | Digesta | Tissue | Digesta | Tissue | Digesta | Tissue | Digesta | |
| Prevotella | 9.9 ± 0.8 | 15.1 ± 0.4 | 10.8 ± 6.8 | 0.5 ± 0.2 | 10.8 ± 2.2 | 0.2 ± 0.1 | 18.4 ± 4.6 | 16.9 ± 0.8 | 16.9 ± 2.2 | 21.6 ± 1.4 |
| Bacteroides | 29.0 ± 19.6 | 15.2 ± 0.7 | 0.01 ± 0.002 | ND | ND | ND | 5.5 ± 0.3 | 10.4 ± 0.3 | 10.4 ± 0.4 | 15.3 ± 1.0 |
| Lactobacillus | 0.1 ± 0.04 | 5.9 ± 0.3 | 5.5 ± 0.6 | 2.1 ± 0.1 | 6.4 ± 2.8 | 45.7 ± 2.9 | 0.02 ± 0.01 | 0.03 ± 0.3 | 0.03 ± 0.02 | 3.0 ± 0.5 |
| Faecalibacterium | 0.2 ± 0.03 | 0.2 ± 0.1 | ND | ND | ND | ND | 9.8 ± 0.6 | 14.2 ± 0.6 | 14.2 ± 0.5 | 10.3 ± 0.3 |
| Clostridium cluster XIV | 6.7 ± 6.2 | 1.3 ± 0.6 | 0.2 ± 0.1 | 0.5 ± 0.3 | 0.2 ± 0.1 | 1.0 ± 0.4 | 0.7 ± 0.3 | 0.9 ± 0.6 | 0.9 ± 0.1 | 1.6 ± 0.1 |
Prevalence = (16S rRNA gene copy number of each genus/total bacterial 16S rRNA gene copy number) × 100. Prevotella, PG*S = 0.02; Bacteroides, PG = 0.01; Lactobacillus, PG*S < 0.01; Faecalibacterium, PG*S < 0.01 (G, gut region; S, sample type [tissue versus digesta]). ND, not detected (represents a prevalence of <0.001%).
DISCUSSION
The distribution of OTUs obtained via pyrosequencing of mucosal tissue and digesta samples from the length of the GIT of 3-week-old preweaned calves in the present study was similar to that of bacterial phylotypes obtained via PCR-DGGE (PCR-denaturing gradient gel electrophoresis) profiling of the same samples in our previous study (9). The taxonomy analysis conducted in the present study was based on pooled samples from 8 animals. Therefore, to confirm that pooling of samples did not bias the description of microbiome composition in the GIT of preweaned calves, qPCR was used to compare the prevalence of 5 selected bacterial genera that belonged to the abundant category (relative abundance ≥ 10% in at least one sample). The bacterial prevalence estimated by qPCR was in agreement with the relative abundance revealed by the pyrosequencing of pooled samples. The analysis of qPCR data also confirmed the observed differences in bacterial prevalence among GIT regions as well as between mucosa- and digesta-associated communities. Although, pooling of samples prevents exploring individual animal variations, the present study succeeded in describing the composition and distribution of the gut microbiota of preweaned calves.
Analysis of the digesta-associated bacterial community along the GIT of a 2-year-old beef steer revealed a high prevalence of Bacteroidetes in the rumen and a high prevalence of Firmicutes in the small and large intestines (15). Similarly, Bacteroidetes dominated the rumen contents of preweaned calves, while Firmicutes dominated the small intestines. In contrast to the adult beef steer, Bacteroidetes dominated the large intestines of preweaned calves. Despite the differences observed in the bacterial composition due to the host age, diet, and breed (15), both studies revealed a region-dependent bacterial community throughout the GIT of cattle. Moreover, de Oliveira and colleagues (15) reported more diverse rumen and large-intestinal bacterial communities than small-intestinal communities, and these communities clustered separately. Our current data for preweaned calves are consistent with these previous observations. The present study also revealed that all mucosa-associated gut communities, with the exception of those in jejunal tissue, contained more Bacteroidetes than Firmicutes, implying that Bacteroidetes tend to more readily colonize mucosal surfaces during early life.
The observed regional variations in the bacterial composition of preweaned calves between small intestine and large intestine further revealed that fecal-sample-based studies may fail to detect the true gut microbiome. However, the regional perturbation of the microbial composition may be associated with enteric diseases. For example, the biopsy samples from Crohn's disease (CD) patients revealed the association of Faecalibacterium prausnitzii with CD as a decrease in bacterial numbers in ileal CD patients (16) and as an increase in bacterial numbers in colonic CD patients (17) compared to the healthy individuals. These observations demonstrate the importance of proper sampling when biomarkers for the associations between microbiota and enteric diseases are being identified. Therefore, the results of previous studies together with the present findings suggest that sampling of local mucosa and digesta is essential to understand enteric infections and diseases caused by the dysbiosis of the gut microbiome.
The higher number of OTUs and higher diversity in the digesta of the rumen and colon imply that a more complex microbial population has been established in those two GIT regions than the small intestines of preweaned calves. The rumen and colon are known to function as sites for microbial fermentation of indigestible dietary substrates and retain digesta for a longer interval than the small intestine (18). The increased retention time of digesta may facilitate the growth of a more complex and dense bacterial community. The milk-fed calves in the present study had access to supplemental calf starter, which may have accelerated the colonization of rumen with a more diverse bacterial population.
The rumen bacterial population of 2-week-old calves fed only milk replacer was reported to contain 45 bacterial genera belonging to 15 different phyla (19). Similarly, the present study reported 47 bacterial genera belonging to 13 phyla in the rumens of 3-week-old calves. In addition, Jami and colleagues (20) reported a diverse rumen bacterial community in 1- to 3-day-old calves, which was dominated by Streptococcus species. The Bacteroidetes population present in the rumen contents of adult cattle fed a grain-based diet was reported to contain more Prevotella than Bacteroides (21), while that in young calves fed whole milk or milk replacer mainly contained Bacteroides (19, 20). The rumen contents of 3-week-old calves used in the present study contained similar levels of Prevotella (15.1%) and Bacteroides (15.8%), suggesting that calf starter may have driven rumen microbiome development toward that of the adult cattle. This transition suggests an early development of the rumen microbiome to ferment a plant polysaccharide-based diet.
Previous studies conducted in preweaned calves relied on the collection of rumen contents. However, a comparison between the rumen digesta- and mucosa-associated communities in the present study revealed differences in the bacterial composition and the relative abundance of shared bacterial genera. Moreover, the observed rumen tissue-specific bacterial genera indicate that studies based only on rumen contents fail to adequately describe the rumen microbiome. Bacteroidetes dominated the rumen tissue-associated bacteria of preweaned calves. However, the rumen tissue-associated bacterial community in beef steers fed a grain-based diet and beef heifers fed hay- or grain-based diets consisted of more Firmicutes than Bacteroidetes (21, 22). The difference observed in the rumen epimural bacterial community might be due to the differences in age, diet of the host, and the breed of cattle (dairy versus beef) (23). The high prevalence of the phylum Proteobacteria, class Betaproteobacteria (25.1%), in the tissue-associated microbiome was another distinct feature in the rumens of preweaned calves. The Proteobacteria species associated with rumen epithelium of dairy cows were different from the species identified from ruminal fluid (24). Those species isolated from the rumen epithelium clustered along with other Betaproteobacteria, which are capable of oxidizing ammonia (24). Therefore, we speculate that the high prevalence of Proteobacteria in the rumen tissue-associated community may play an important role in scavenging the oxygen that diffuses from the blood to the epithelium and oxidizing ammonia produced in the rumen. This may facilitate rumen bacterial colonization and its functions by maintaining the anaerobic conditions required for microbial fermentation. However, the large proportion of unclassified tissue-specific Proteobacteria indicates that there are many unknown bacteria attached to the rumen tissue. Thus, future studies are necessary to understand epimural bacteria and their dynamics in response to rumen development and dietary changes in dairy calves.
A high abundance of genus Sharpea was observed in the small intestine of preweaned calves compared to other gut regions. Sharpea belongs to the order Erysipelotrichales and the predicted family Coprobacillaceae (by QIIME [13]), and this is the first time Sharpea has been detected in the GIT of preweaned calves. Sharpea azabuensis belongs to clostridial cluster XVII and has been detected in the feces of thoroughbred horses (25). S. azabuensis ferments a wide variety of sugars, including glucose, galactose, lactose, and fructose (25). Thus, the observed high abundance of Sharpea in the jejunal digesta of preweaned calves may facilitate the fermentation of milk sugars. Since the genus Sharpea is abundant in the GIT of preweaned calves, future studies may provide a better understanding of its role in host metabolism.
In contrast to the rumen, the small and large intestines revealed higher bacterial diversity (Shannon index) in the mucosa-associated community than in the adjacent digesta. In addition, the species richness (Chao1 index) was also higher in the mucosa-associated communities of the jejunum, ileum, and cecum than in the respective digesta. The higher species richness observed for the mucosa-associated communities is in agreement with our previous study conducted with the same samples but using PCR-DGGE (9). The association of mucosal bacteria with the expression of TLR2 and TLR4 in the colons of mice (10) suggests that mucosa-associated bacteria may play an important role in stimulating host immune responses through pattern recognition receptors. The present study revealed small-intestinal mucosa-specific bacteria that were not identified in other GIT regions, suggesting that such mucosa-specific bacteria may survive mucosal immune defense mechanisms and may be crucial for priming the host mucosal immune system. A previous study investigating the mucosal expression of TLRs along the GIT of 3-week-old calves, using the same mucosal tissue samples, revealed that TLR1, -9, and -10 gene expression was highest in the ileum, followed by the jejunum and then other GIT regions (26). Even though there are regional variations in immune cell distribution throughout the gut (27, 28), we speculate that regional differences in the gut bacteria may also contribute to the observed TLR expression pattern. Moreover, the small-intestinal bacterial density was negatively correlated with the mucosal expression of TLRs in the preweaned calves. For example, digesta-associated total bacterial density was negatively correlated with the expression of TLR2 and TLR6 in the jejunum and ileum, respectively, while tissue-associated total bacteria and lactic acid bacteria (LAB) density was negatively correlated with the expression of TLR2 in the jejunum (26). This hyporesponsiveness of mucosal TLRs to the predominant Gram-positive bacterial population (Firmicutes) present in the small-intestine communities may be a host mechanism to avoid immune responses to commensal microbes.
The preweaned-calf large-intestinal regions contained similar bacterial compositions in the mucosa- and digesta-associated communities which differed only in their relative abundance. Unlike the small-intestinal and ruminal tissue-associated communities, most of the sequences obtained from the large intestine were assigned to a genus, with the exception of the cecal tissue (51% unassigned sequences). Cecum and colon shared nearly 55% of the bacterial genera observed in the large intestine. However, in comparison to bacteria shared between the ileum and jejunum (∼85%), these two large-intestinal bacterial communities showed less similarity to each other. The high proportion of assigned genus information observed in the large-intestinal communities may be due to their resemblance to the fecal bacterial community, which has been studied more extensively than the small intestine community. Uyeno and colleagues reported that the fecal microbiomes of 3-week-old dairy calves consist of 39.2% Bacteroides-Prevotella group, 17.7% Clostridium coccoides-Eubacterium rectale group, and 11% Faecalibacterium (8). Colon samples from the 3-week-old calves used in the present study revealed similar levels for the Bacteroides-Prevotella group (32.5%) and Faecalibacterium (12.1%). However, the relative abundance of Clostridium was only 3.2%, and there were no members of the genus Eubacterium observed in any of the gut regions. These observations once again indicate that fecal samples best represent the composition of the large-intestinal bacterial community but not that of the entire GIT. The present study revealed a significantly higher prevalence of Faecalibacterium in large-intestinal communities than in communites in other GIT regions. A high abundance of F. prausnitzii in the feces was associated with increased weight gain and a decreased incidence of diarrhea in dairy calves (4). The fermentation of F. prausnitzii produces butyrate, which is the major energy source for colonic epithelial cells, and it also stimulates anti-inflammatory responses (4). Therefore, the observed higher prevalence of Faecalibacterium species in the large intestine of preweaned calves may be important for maintaining proper body weight and reducing enteric infections during early life.
Conclusion.
In conclusion, the present study revealed that the bacterial composition throughout the GIT of 3-week-old calves varied between mucosa and digesta communities as well as among GIT regions. The observed mucosa-specific bacterial genera further suggest that fecal samples do not adequately represent the complexity of the gut microbiome and potential relationships with the host. Moreover, the colonization of calf rumen starts early in life with a distinct segregation of bacteria between digesta and epithelial surfaces. This segregation suggests that future studies should focus on both communities. The large number of sequences throughout the GIT for which genus level information was not obtained suggests that our knowledge of the ruminant gut microbiome is not adequate to explain its complexity and functions in the host. In particular, analysis of tissue-attached bacteria may provide a better understanding of early rumen/gut development and functions. Previous studies of bacteria in digesta of preweaned calves reported large variation among individual animals, and it will be important to determine if a similar level of variation is present for tissue-attached bacteria. Understanding the full extent of gut microbiome diversity among individual animals will be critical for determining how it shapes the individual animal performances in terms of health and performance.
Supplementary Material
ACKNOWLEDGMENTS
This project was supported by Alberta Livestock and Meat Agency Ltd. (ALMA 2011F129R) and National Science Engineering Research council. P. J. Griebel is the holder of a Tier I CRC in Neonatal Mucosal Immunology, which is funded by the Canadian Institutes of Health Research (CIHR).
We thank P. Stothard and B. Ghoshal for their assistance in pyrosequencing data analysis.
Footnotes
Published ahead of print 17 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03864-13.
REFERENCES
- 1.Turroni F, Peano C, Pass DA, Foroni E, Severgnini M, Claesson MJ, Kerr C, Hourihane J, Murray D, Fuilgni F, Gueimonde M, Margolles A, De Bellis G, O'Toole PW, van Sinderen D, Marcheis JR, Ventura M. 2012. Diversity of Bifidobacteria within the infant gut microbiota. PLoS One 7:e36957. 10.1371/journal.pone.0036957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fanaro S, Chierici R, Guerrini O, Vigi V. 2003. Intestinal microflora in early infancy: composition and development. Acta Paediatr. Suppl. 91:48–55. 10.1111/j.1651-2227.2003.tb00646.x [DOI] [PubMed] [Google Scholar]
- 3.Jost T, Lacroix C, Braegger CP, Chassard C. 2012. New insights in gut microbiota establishment in healthy breast-fed neonates. PLoS One 7:e44595. 10.1371/journal.pone.0044595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oikonomou G, Teixeira AGV, Foditsch C, Bicalho ML, Machado VS, Bicalho RC. 2013. Fecal microbial diversity in pre-weaned dairy calves as described by pyrosequencing of metagenomic 16S rDNA. Associations of Faecalibacterium species with health and growth. PLoS One 8:e63157. 10.1371/journal.pone.0063157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mulder IE, Schmidt B, Stokes CR, Lewis M, Bailey M, Aminov RI, Prosser JI, Gill BP, Pluske JR, Mayer CD, Musk CC, Kelly D. 2009. Environmentally acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol. 7:79. 10.1186/1741-7007-7-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulder IE, Schmidt B, Lewis M, Delday M, Stokes CR, Bailey M, Aminov RI, Gill BP, Pluske JR, Mayer CD, Musk C, Kelly D. 2011. Restricting microbial exposure in early life negates the immune benefits associated with gut colonization in environments of high microbial diversity. PLoS One 6:e28279. 10.1371/journal.pone.0028279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chung H, Pamp SJ, Hill JA, Surana NJ, Edelman SM. 2012. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149:1578–1593. 10.1016/j.cell.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uyeno Y, Sekiguchi Y, Kamagata Y. 2010. rRNA-based analysis to monitor succession of faecal bacterial communities in Holstein calves. Appl. Microbiol. 51:570–577. 10.1111/j.1472-765X.2010.02937.x [DOI] [PubMed] [Google Scholar]
- 9.Malmuthuge N, Chen Y, Li M, Fries P, Griebel PJ, Baurhoo B, Zhao X, Guan LL. 2012. Distinct commensal bacteria associate with ingesta and mucosal epithelium throughout the gastrointestinal tract. FEMS Microbiol. Ecol. 79:337–347. 10.1111/j.1574-6941.2011.01220.x [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Devkota S, Musch MW, Jabri B, Nagler C, Antonopoulos DA, Chervonsky A, Chang EB. 2010. Regional mucosa-associated microbiota determine physiological expression of TLR2 and TLR4 in murine colon. PLoS One 5:e13607. 10.1371/journal.pone.0013607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li M, Penner GB, Hernandez-Sanabria E, Oba M, Guan LL. 2009. Effects of sampling location and time, and host animal on assessment of bacterial diversity and fermentation parameters in the bovine rumen. J. Appl. Microbiol. 107:1924–1934. 10.1111/j.1365-2672.2009.04376.x [DOI] [PubMed] [Google Scholar]
- 12.Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. 2008. Error-correcting barcoded primers allow hundreds of samples to be pyrosequenced in multiplex. Nat. Methods 5:235–237. 10.1038/nmeth.1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 15.de Oliveira MNV, Jewell KA, Freitas FS, Benjamin LA, Totola MR, Borges AC, Moraes CA, Suen G. 2013. Characterizing the microbiota across the gastrointestinal tract of a Brazilian Nelore steer. Vet. Microbiol. 164:307–314. 10.1016/j.vetmic.2013.02.013 [DOI] [PubMed] [Google Scholar]
- 16.Willing BP, Dicksved J, Halfvarson J, Andersson AF, Luccio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstarnd L. 2010. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139:1844–1854. 10.1053/j.gastro.2010.08.049 [DOI] [PubMed] [Google Scholar]
- 17.Hansen R, Russell RK, Reiff C, Louis P, Mclntosh F, Berry SH, Mukhopadhya I, Bisset WM, Barclay AR, Bishop J, Flynn DM, McGrogan P, Loganathan S, Mahdi G, Flint HJ, El-Omar EM, Hold GL. 2012. Microbiota of de-novo paediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn's but not in ulcerative colitis. Am. J. Gastroenterol. 107:1913–1922. 10.1038/ajg.2012.335 [DOI] [PubMed] [Google Scholar]
- 18.Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. 2011. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol. Rev. 35:681–704. 10.1111/j.1574-6976.2011.00270.x [DOI] [PubMed] [Google Scholar]
- 19.Li RW, Connor EE, Li C, Baldwin RL, VI, Sparks ME. 2012. Characterization of the rumen microbiota of pre-ruminant calves using metagenomics tools. Environ. Microbiol. 14:129–139. 10.1111/j.1462-2920.2011.02543.x [DOI] [PubMed] [Google Scholar]
- 20.Jami E, Israel A, Kotser A, Mizrahi I. 2013. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7:1069–1079. 10.1038/ismej.2013.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li M, Zhou M, Adamowicz E, Basarab JA, Guan LL. 2012. Characterization of bovine ruminal epithelial bacterial communities using 16S rRNA sequencing, PCR-DGGE and qRT-PCR analysis. Vet. Microbiol. 155:72–80. 10.1016/j.vetmic.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 22.Chen Y, Penner GB, Li M, Oba M, Guan LL. 2011. Changes in bacterial diversity associated with epithelial tissue in the beef cow rumen during the transition to a high-grain diet. Appl. Environ. Microbiol. 77:5770–5781. 10.1128/AEM.00375-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meng Q, Preston RL. 1998. Factors influencing bacterial attachment to the ruminal epithelium in vitro. J. Gen. Appl. Microbiol. 44:35–41. 10.2323/jgam.44.35 [DOI] [PubMed] [Google Scholar]
- 24.Misumori M, Ajisaka N, Tajima K, Kajikawa H, Kurihara M. 2002. Detection of Proteobacteria from the rumen by PCR using methanotroph-specific primers. Appl. Microbiol. 35:251–255. 10.1046/j.1472-765X.2002.01172.x [DOI] [PubMed] [Google Scholar]
- 25.Morita H, Shiratori C, Murakami M, Takami H, Toh H, Kato Y, Nakajima F, Takagi M, Akita H, Masaoka T, Hattori M. 2008. Sharpea azabuensis gen. nov., sp. nov., a Gram-positive, strictly anaerobic bacterium isolated from the faeces of thoroughbred horses. Int. J. Syst. Evol. Microbiol. 58:2682–2686. 10.1099/ijs.0.65543-0 [DOI] [PubMed] [Google Scholar]
- 26.Malmuthuge N, Li M, Fries P, Griebel PJ, Guan LL. 2012. Regional and age dependent changes in gene expression of toll-like receptors and key antimicrobial defense molecules throughout the gastrointestinal tract of dairy calves. Vet. Immunol. Immunopathol. 146:18–26. 10.1016/j.vetimm.2012.01.010 [DOI] [PubMed] [Google Scholar]
- 27.Menezies M, Ingham A. 2006. Identification and expression of toll-like receptors 1–10 in selected bovine and ovine tissues. Vet. Immunol. Immunopathol. 109:23–30. 10.1016/j.vetimm.2005.06.014 [DOI] [PubMed] [Google Scholar]
- 28.Abreu MT. 2010. Toll-like receptors signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 10:131–142. 10.1038/nri2707 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.