

Abstract
The qseBC-encoded quorum-sensing system regulates the motility of Escherichia coli O157:H7 in response to bacterial autoinducer 3 (AI-3) and the mammalian stress hormones epinephrine (E) and norepinephrine (NE). The qseC gene encodes a sensory kinase that autophosphorylates in response to AI-3, E, or NE and subsequently phosphorylates its cognate response regulator QseB. In the absence of QseC, QseB downregulates bacterial motility and virulence in animal models. In this study, we found that 8- to 10-month-old calves orally inoculated with a mixture of E. coli O157:H7 and its isogenic qseBC mutant showed significantly higher fecal shedding of the qseBC mutant. In vitro analysis revealed similar growth profiles and motilities of the qseBC mutant and the parental strain in the presence or absence of NE. The magnitudes of the response to NE and expression of flagellar genes flhD and fliC were also similar for the qseBC mutant and the parental strain. The expression of ler (a positive regulator of the locus of enterocyte effacement [LEE]), the ler-regulated espA gene, and the csgA gene (encoding curli fimbriae) was increased in the qseBC mutant compared to the parental strain. On the other hand, growth, motility, and transcription of flhD, fliC, ler, espA, and csgA were significantly reduced in the qseBC mutant complemented with a plasmid-cloned copy of the qseBC genes. Thus, in vitro motility and gene expression data indicate that the near-parental level of motility, ability to respond to NE, and enhanced expression of LEE and curli genes might in part be responsible for increased colonization and fecal shedding of the qseBC mutant in calves.
INTRODUCTION
Enterohemorrhagic Escherichia coli (EHEC) O157:H7 causes a broad spectrum of diarrheal illnesses, including uncomplicated diarrhea, hemorrhagic colitis, and hemolytic uremic syndrome (1). Cattle, the major reservoir for EHEC O157:H7, are colonized at the terminal portion of the large intestine, especially the recto-anal junction (RAJ) (2, 3). Intestinal colonization results in the fecal shedding of EHEC O157:H7, a major risk factor in the contamination of beef and other meat products (4–6). EHEC O157:H7 colonization of the large intestine produces characteristic histopathology called attaching and effacing (A/E) lesions (7).
The genes for the production of A/E lesions are carried on a 43-kb pathogenicity island termed the locus of enterocyte effacement (LEE) (8). The LEE contains 41 open reading frames (ORFs) that are organized into five major operons (LEE1 through LEE5). Several genes contained in LEE1, LEE2, LEE3, and LEE4 encode proteins for the synthesis of a type III secretion system, which secrets various LEE and non-LEE-encoded proteins required for the adherence of EHEC O157:H7 (9). The expression of LEE is activated by Ler, encoded by the ler gene of the LEE1 operon (10).
Besides the requirement for LEE, bacterial motility has been suggested to be important in the ability of EHEC O157:H7 to colonize cattle (11). Quorum-sensing signaling systems have been implicated in the regulation of LEE and bacterial motility (12, 13). Specifically, the qseBC-encoded two-component quorum-sensing pathway regulates bacterial motility in response to bacterially produced quorum-sensing signals called autoinducers (AI) and the mammalian stress hormones epinephrine and norepinephrine (NE) (14, 15). Homologues of EHEC O157:H7 QseBC are present in other pathogenic bacteria, including uropathogenic E. coli (UPEC), enteropathogenic E. coli (EPEC), Salmonella enterica serovar Typhimurium, Salmonella enterica serovar Typhi, Pasteurella multocida, Francisella tularensis, and Haemophilus influenzae, as well as nonpathogenic E. coli K-12 strains (16).
QseC is a transmembrane protein that interacts through its periplasmic sensory domain with AI-3 and the stress hormones epinephrine and norepinephrine to undergo autophosphorylation at its cytoplasmic domain (14, 15). The phosphorylated QseC transfers the phosphoryl group to its cognate response regulator QseB, which activates transcription of FlhDC, the master transcriptional regulator of the flagellar gene complex, and autoregulates its own transcription by binding to the flhDC and qseBC promoters, respectively (16, 17). However, constitutive expression of QseB, which occurs in qseC deletion mutants, has been shown to attenuate virulence of EHEC O157:H7, UPEC, S. Typhimurium, and F. tularensis (18–21).
The mechanism for virulence attenuation in qseC deletion mutants appears to be the presence of constitutively phosphorylated QseB, which causes metabolic perturbations leading to dysregulation of virulence gene expression (19). On the other hand, virulence of a qseB deletion mutant of Aeromonas hydrophila is reportedly attenuated due to changes in the expression of cyclic diGMP levels resulting in reduced motility, biofilm formation, and protease production (22). According to another study, qseBC double deletion mutants of S. Typhimurium colonized swine intestine at the parental levels but exhibited increased motility compared to that of the parent strain in response to norepinephrine, lending support to the conclusion that additional genes might be involved in norepinephrine-mediated regulation of bacterial motility (23). These conclusions seem reasonable considering that the qseBC-encoded quorum-sensing system cross talks with other two-component bacterial quorum-sensing systems to enhance LEE expression, thereby enhancing EHEC O157:H7 adherence to target host tissues (15). Thus, one could suggest that there are other two-component systems to compensate for the loss of the QseBC system in some bacterial pathogens, enabling them to express genes in response to host or bacterial signals and to ascertain that they have reached the right site in the host intestine to establish colonization.
EHEC O157:H7 and many sorbitol-fermenting, nonmotile EHEC O157 serotypes recovered from patients with the hemolytic uremic syndrome produce a variety of fimbrial adhesins, including curli fimbriae, which play direct or indirect roles in adherence of these bacteria to cultured epithelial cells and colonization of animal intestines (24–27). In earlier reports, we demonstrated that the hha deletion mutant of EHEC O157:H7 expresses LEE and the curli-encoding gene csgA at higher levels, exhibits increased adherence to cultured HEp-2 cells, and produces increased amounts of biofilm biomass (28–30). However, neither the production of curli fimbriae in animal intestines nor their role in promoting EHEC O157:H7 adherence to and colonization of intestinal tissues is fully understood. Considering that QseBC enhances bacterial motility (13) and knowing that there is an inverse relationship between bacterial motility and curli expression (31), EHEC O157:H7 lacking QseBC might show an aberrant relationship between the expression of curli and bacterial motility.
In this study, we assessed the impact of the qseBC deletion on the colonization of EHEC O157:H7 in cattle intestine by comparing the duration and magnitude of fecal shedding of the qseBC mutant with those of its isogenic parental strain. In addition, we determined if the effects of qseBC deletion on EHEC O157:H7 colonization of bovine intestine would differ from those reported for intestinal colonization of S. enterica serovar Typhimurium in swine (18). We also correlated the relative effects of the qseBC deletion on intestinal colonization with in vitro expression of virulence genes, especially those that promote bacterial motility and enhance bacterial adherence to tissues of the large intestine, a prerequisite for increased intestinal colonization and fecal shedding of EHEC O157:H7 in cattle.
MATERIALS AND METHODS
Bacterial strains, culture media, and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. EHEC O157:H7 strain NADC 5570, a streptomycin-resistant isolate of EHEC O157:H7 strain 86-24 (32, 33), was used as a parental strain for constructing an isogenic qseBC deletion mutant (NADC 6557). Bacterial strains were cultivated in Luria-Bertani broth (LB) or LB agar (Sigma-Aldrich, St. Louis, MO) supplemented with antibiotics (Sigma-Aldrich) as needed (streptomycin, 100 mg liter−1; kanamycin, 50 mg liter−1; and carbenicillin, 100 mg liter−1).
TABLE 1.
Bacterial strains and plasmidsa
| Strain or plasmid | Genotype and description | Source or references |
|---|---|---|
| E. coli strains | ||
| NADC 5570 | stx2+ and streptomycin-resistant strain 86-24 | 32, 49 |
| NADC 6557 | ΔqseBC mutant of NADC 5570 | This study |
| TOP 10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara-leu)7697 galU galK rpsL (Strr) endA1 nupG λ | Invitrogen |
| Plasmids | ||
| pSMART-HC | Cloning vector | Lucigen |
| pAM450 | Plasmid with a temp-sensitive origin of replication | 30, 33 |
| pSM558 | pCR2.1 carrying a cloned kan (neo) cassette encoding kanamycin resistance | This study |
| pSM644 | pSMART-LC containing in tandem 750-bp US and 750-bp DS regions of qseBC ORFs | This study |
| pSM646 | pSM644 with a 1.07-kb fragment encoding kanamycin resistance cloned between US and DS fragments | This study |
| pSM648 | 2.57-kb fragment of pSM646 cloned at the XbaI site of pAM450 | This study |
| pSM639 | 3.51-kb fragment containing qseBC ORFs and 750 bp US and DS sequences cloned in pSMART-LCamp | This study |
Detailed descriptions of bacterial strains and plasmids are provided in Materials and Methods.
Determination of bacterial growth in pure and mixed cultures.
Overnight bacterial cultures grown in LB-carbenicillin broth at 37°C with shaking (200 rpm) were diluted 1:100 in low-glucose Dulbecco modified Eagle medium (DMEM) (Life Technologies, Grand Island, NY) containing carbenicillin, and 300 μl of diluted cultures was added to the wells of a 100-well Honeycomb 2 plate. The plate was incubated at 37°C in an automated growth curve reader programmed for continuous shaking and collecting readings of optical density at 600 nm (OD600) every 30 m over a 24-h period (Growth Curves USA, Piscataway, NJ). The optical density data were analyzed by using the GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA). The bacterial growth rate was calculated by using the equation μ = 2.303(log OD2 − log OD1)/(t2 − t1). For determining bacterial growth in mixed cultures, the overnight cultures of the parental strain (streptomycin resistant) and its isogenic qseBC mutant (resistant to streptomycin and kanamycin) were standardized to an OD600 of 0.10 in low-glucose DMEM and incubated at 37°C with shaking (200 rpm). The 10-fold serial dilutions of the culture aliquots taken hourly were plated on LB agar containing streptomycin (100 μg ml−1). After overnight incubation, the colonies that grew on these plates were counted (representing the parental and the qseBC mutant colony counts) and then transferred by replica plating to LB agar containing kanamycin (50 μg ml−1). The number of colonies that grew on LB agar-kanamycin plates (representing the qseBC mutant colony count) after overnight incubation at 37°C were subtracted from the total colony counts obtained on LB-streptomycin agar plates to compute the colony count of the parental strain.
Recombinant DNA procedures.
For constructing an in-frame qseBC deletion mutant of EHEC O157:H7 strain NADC 5570, a 0.75-kb DNA fragment upstream (US) and a 0.75 kb DNA fragment downstream (DS) of the qseBC ORFs were PCR amplified using primer pairs qseBC-USF/qseBC-USR and qseBC-DSF/qseBC-DSR, respectively (Table 2). XbaI restriction sites were built into the primers qseBC-USF and qseBC-DSR, and BglII restriction sites were built into the primers qseBC-USR and qseBC-DSF. The US and DS fragments were cloned in the order 5′ US-DS 3′ into pSMART-HC (Lucigen Corp., Middleton, WI) and electroporated into E. coli TOP10 according to the manufacturer's instructions (Life Technologies) to generate recombinant plasmid pSM644. A 1.07-kb fragment encoding kanamycin resistance (kan) was isolated by PCR from a kan (neo) cassette (28, 34) using the primers kanF and kanR containing the BglII restriction sites (Table 2). The 1.07-kb kan fragment was purified by using the QIAquick PCR purification kit (Qiagen, Valencia, CA) and ligated into the pCR2.1TOPO TA cloning vector, and the ligated DNA was electroporated into E. coli TOP10 cells according to the manufacturer's instructions (Life Technologies). The pCR2.1-kan recombinant plasmid (pSM558) was treated with the Change-IT multiple-mutation site-directed mutagenesis kit according to manufacturer's instructions (USB Corp., Cleveland, OH). This mutagenesis treatment allowed removal of the XbaI restriction sites, one located in the flippase recognition target (FRT) sequence upstream and the other in the FRT sequence downstream of the kan gene of the kan cassette (28, 34). Plasmid pSM558 was digested with BglII, the digested DNA was resolved using standard agarose gel electrophoresis, and the 1.07-kb kan fragment was recovered from gel slices using a gel extraction kit (Qiagen). The kan fragment was cloned at the BglII restriction site located at the junction of the 5′ US and 3′ DS fragments of plasmid pSM644 to generate recombinant plasmid pSM646. The 2.57-kb US-kan-DS fragment isolated by the XbaI restriction digestion of pSM646 was cloned at the XbaI site of a plasmid (pAM450) encoding ampicillin resistance and temperature sensitive for its replication (30, 33). The recombinant plasmid (pSM648) was electroporated into EHEC O157:H7 strain NADC 5570 for deleting the qseBC genes by using a previously described allelic replacement method (30). The genomic DNA from kanamycin-resistant and ampicillin-sensitive mutants recovered by the allelic replacement method was screened by PCR using the primers qseBCF and qseBCR (Table 2) to confirm that the qseBC genes were replaced by the 1.07-kb kan fragment. For complementation experiments, a 3.51-kb DNA fragment containing the qseB and qseC genes was isolated from strain NADC 5570 by PCR using the primers qseBC-USF and qseBC-DSR and cloned into the low-copy vector pSMART-LC (Lucigen Corp.). The recombinant qseBC-pSMART-LC (pSM639) or the empty-vector pSMART-LC (Lucigen Corp.) was electroporated into the qseBC deletion mutant strain (NADC 6557) to generate a qseBC-complemented qseBC mutant (qseBC+/ΔqseBC) and the noncomplemented qseBC mutant (ΔqseBC) containing the vector pSMART-LC, respectively.
TABLE 2.
Primers used for PCR and qRT-PCR
| Primera | Nucleotide sequenceb | Location or referencec |
|---|---|---|
| qseBC-USF | GCGTCTAGATCTGGCTTTGGTTAACAGGAG | 3975815–3975834 |
| qseBC-USR | GATCAGATCTTTTTTCATCCCTGCGATAACCG | 3976571–3976549 |
| qseBC-DSF | GCGAGATCTAGATCTAGGGTAAGACTTTTGCTAAATC | 3978576–39978598 |
| qseBC-DSR | GATCTCTAGAGAGTACGGATTTCTGCGATTAC | 3979328–3979307 |
| kanF | GATCAGATCTCGATAGCTGAATGAGTGACGTGC | 18 |
| kanR | GATCAGATCTGCATAGAGCAGTGACGTAGTCGC | 18 |
| qseBCF | CAGGGCCATTACTGCGATTAC | 3976381–3976401 |
| qseBCR | TCGTTCAGTTGACCATTGGAG | 3978750–3978729 |
| fliCF (qRT-PCR) | TTAGCTGCCACCCTTCATG | 2700535–2700517 |
| fliCR (qRT-PCR) | TCGTCAAGTTGCCTGCATC | 2700385–2700403 |
| fliCP (qRT-PCR) | TCTACGTATGCCTGGCTTCCACCG | 2700405–2700428 |
| csgAF (qRT-PCR) | TACTATTACCCAGCATGGTGG | 1548878–1548898 |
| csgAR (qRT-PCR) | CAAGAGTGGCGCTGTTACC | 1548984–1548966 |
| csgAP (qRT-PCR) | CCACGTTGGGTCAGATCGATTGAG | 1548962–1548938 |
| flhDCF (qRT-PCR) | ACAACATTAGCGGCACTGAC | 2655207–2655188 |
| flhDCR (qRT-PCR) | AGAGTAATCGTCTGGTGGCTG | 2655107–2655124 |
| flhDCP (qRT-PCR) | AAACGGAAGTGACAAACCAGCTGATTG | 2655131–2655158 |
| rpoAF (qRT-PCR) | GGCTTGACGATTTCGACATC | 4242887–4242906 |
| rpoAR (qRT-PCR) | GGTGAGAGTTCAGGGCAAAG | 4242997–4242978 |
| rpoAP (qRT-PCR) | TGAAGTTATTCTTACCTTGAATAAATCTGGCATTG | 4242976–4242942 |
Subscripts F, R, and P denote forward primer, reverse primer, and TaqMan probe, respectively.
Nucleotide sequences of the primers used in this study were selected from the published genome sequence of E. coli O157:H7 strain EDL933 (50) (accession number AE005174.2).
Position of primer sequence in the genome of EDL933 (50).
qRT-PCR.
Bacterial strains were grown to an OD600 of 1.1 to 1.2 in low-glucose DMEM lacking or containing 50 μM norepinephrine (18, 35). One milliliter of each bacterial culture was mixed with 2 ml of RNA-Protect reagent, and total RNA was isolated by using the RNeasy kit according to the manufacturer's instructions (Qiagen, Valencia, CA). RNA was subjected to DNase treatment using the Ambion Turbo DNA-free kit to remove DNA contamination according to the manufacturer's instructions (Invitrogen). Quantitative reverse transcription-PCR (qRT-PCR) was performed by adding 25 ng of DNase-treated RNA, 0.75 μM (each) antisense and sense primers, 0.25 μM TaqMan probe (labeled at the 5′ end with 6-carboxyfluorescein [FAM] reporter and at 3′ end with 6-carboxytetramethylrhodamine [TAMRA] quencher) (IDT, Coralville, IA) to a qRT-PCR master mix (Agilent Technologies, Santa Clara, CA). The nucleotide sequences of the primers and probes used in qRT-PCR are listed in Table 2. qRT-PCR was carried out in Mx3005P (50°C for 30 min for cDNA synthesis; 95°C for 5 min; and 35 cycles of 95°C for 30 s, 55°C for 60 s, 72°C for 30 s for cDNA amplification and detection of amplification products). The relative expression of the target genes was determined by comparing the base-corrected and normalized (housekeeping gene rpoA) fluorescence generated for the target gene in the mutant and the complemented mutant strains to that in the parent strain.
Animal experiments.
The animals were housed in climate-controlled BL-2 barns per the guidelines of the American Association for Laboratory Animal Care. The animals were fed twice daily with pelleted feed and alfalfa hay cubes in amounts equal to 1% of their body weight, and water was offered ad libitum. All animal protocols were approved by the National Animal Disease Center Animal Care and Use Committee. Two animal trials were conducted to study the effect of qseBC deletion on fecal shedding of EHEC O157:H7. In each trial, 8- to 10-month-old male calves (n = 8) were orally inoculated with 50 ml of phosphate-buffered saline containing 1 × 1010 CFU of the parental strain (streptomycin-resistant strain NADC 5570) and 1 × 1010 CFU of the isogenic qseBC mutant (streptomycin and kanamycin resistant NADC 6557). Ten grams of feces, collected by rectal palpation, was resuspended in 90 ml of an enrichment broth prepared by adding novobiocin (10 μg ml−1) and potassium tellurite (0.75 μg ml−1) to tryptic soy broth. Ten-fold serial dilutions of fecal suspensions were spread-plated on sorbitol-MacConkey agar plates containing 0.75 μg ml−1 potassium tellurite (SMACT) and 100 μg ml−1 of streptomycin. After incubation at 37°C for 24 h, sorbitol-negative colonies produced on SMACT agar plates were counted and confirmed as O157 by agglutination with anti-O157 antiserum (Thermo Fisher Scientific Inc., Waltham, MA). About 30 colonies confirmed as EHEC O157:H7 were picked from the streptomycin-SMACT medium to kanamycin-SMACT medium to determine the relative proportion of the kanamycin-resistant qseBC mutant strain. In order to detect very low levels of the inoculated strains in feces, fecal suspensions prepared in the enrichment broth were incubated overnight at 37°C before plating on SMACT agar plates. The proportion of kan-resistant colonies was determined as described above. A competitive index (CI) was determined using the formula CI = (mutant CFU per g feces/parent CFU per g feces)/(mutant CFU input/parent CFU input). To determine competitive index, fecal counts were first normalized by using Y = Y + K, where K = 20. This normalization allowed conversion of any fecal shedding value of zero to nonzero value that could then be log transformed. A CI of <1.0 means that the parent strain colonized at a higher level and showed higher fecal shedding, and a CI of >1 indicates increased colonization and fecal shedding of the mutant strain. The CI values for the fecal shedding were evaluated by using a single-sample t test with a P value of <0.05 indicating the significance of the CI for each sampling day.
Determination of bacterial motility.
For swimming motility assays, bacterial strains were grown in LB containing carbenicillin (100 μg ml−1) at 37°C with shaking (175 rpm) for 18 to 24 h. The overnight-grown bacterial cultures were standardized to an OD600 of 4.00, and 2 μl of these cultures was spotted on soft-agar motility plates (DMEM containing 0.3% Noble agar and 100 μg ml−1 carbenicillin with or without 50 μM norepinephrine). The diameter of motility halos produced on motility plates was measured after 18 to 24 h of incubation at 37°C.
Statistical analyses.
A two-sample, nonparametric Mann-Whitney test was used to determine the significance of the difference in motilities and expression of selected genes in the mutant and the complemented mutants relative to the parent strain. The differences were considered significant at a P value of <0.05. The significance of the CI values for fecal shedding on each sampling day was evaluated by using a single-sample t test with a P value of <0.05 indicating the significance of the CI.
RESULTS
The qseBC deletion enhances fecal shedding of EHEC O157:H7 in cattle.
The qseBC deletion mutant (ΔqseBC) generated by the allelic replacement procedure as described in Materials and Methods was confirmed by PCR using primers qseBCF and qseBCR (Table 2) for the replacement of the qseBC genes by the kanamycin gene (data not shown). Oral inoculations of calves (n = 8 per trial) with a bacterial inoculum containing equal numbers of the streptomycin-resistant parental strain (NADC 5570) and its isogenic streptomycin- and kanamycin-resistant qseBC deletion mutant (NADC 6557) showed that the mutant strain was always the predominant type, representing 77 to 99% of the total recoverable CFU per gram of feces, during the course of a 4-week study in the first trial (Fig. 1A and B). For example, in the first animal trial, the mutant strain was recovered at about 5 × 103 CFU g−1 of feces from day 1 through day 10, compared to the recovery of about 4 × 102 CFU g−1 of feces for the parent strain on these days. The mutant strain continued to be excreted at >102 CFU g−1 of feces from day 13 until day 28, compared to the shedding of <102 CFU g−1 of feces of the parent strain during these days. In a second independent trial performed using 8- to 10-month-old calves (n = 8), the fecal shedding of the qseBC mutant strain during the 4 weeks of sampling represented about 85 to 99% of the total recoverable bacterial counts in the feces of the animals inoculated with the equal amounts of the mutant and the parental strain (Fig. 1C). The competitive indices (CIs) computed by using the results shown in Fig. 1A and C are presented in Fig. 1B and D. The CI for each sampling day was determined to be significant (P < 0.0001) based on a single-sample t test. The results shown in these graphs represent bacterial counts of the two inoculated strains obtained by the direct plating of fecal samples on SMACT agar plates. However, the fecal counts of the parental strain for day 17 (Fig. 1A) and days 13, 15, 17, and 20 (Fig. 1C), which are shown as gray circles on these graphs, could be obtained only by plating of fecal suspensions grown overnight in the enrichment broth.
FIG 1.

Magnitude of fecal shedding of the qseBC mutant relative to that of the parental strain. Eight- to 10-month-old calves (n = 8 per trial) inoculated with a mixture of the parent (NADC 5570) and qseBC mutant (NADC 6557) strains were monitored over 28 days for fecal shedding of the inoculated strains in two independent trials. (A and C) Bacterial counts are plotted as percentage of the average CFU per gram of feces recovered for the mutant and the parent strain on each of the 13 fecal sampling days during the two animal trials. (B and D) Competitive indices (CIs), defined as the ratio of the mutant to the parent recovered in the feces of calves, were calculated from the data presented in panels A and C, respectively. CIs of >1 are indicative that the mutant strain colonized at levels significantly higher than the parental strain (P < 0.0001).
The qseBC deletion does not affect the growth of the qseBC mutant in a pure culture or in a mixed culture with the parental strain.
In order to determine if an altered growth profile of the qseBC mutant compared to the parental strain could have contributed to the enhanced colonization by the qseBC mutant in the calf intestine, we compared the growth of the mutant to that of the parental strain in low-glucose DMEM with or without norepinephrine. The growth rate for the qseBC deletion mutant (0.23 h−1) was not significantly different (P > 0.05) from that of the parental strain (0.27 h−1) (Fig. 2A). The qseBC-complemented mutant showed a significantly (P < 0.0001) lower growth rate (0.12 h−1) and an early onset of the stationary phase, suggesting that the increased expression of QseB and QseC exerts a negative effect on bacterial growth (Fig. 2A). The examination of the growth of the qseBC mutant in mixed cultures with the parental strain showed equivalent recovery of the viable counts of the mutant and the parental strains, indicating that the qseBC deletion does not affect the growth and the viability of the mutant strain (Fig. 2B).
FIG 2.
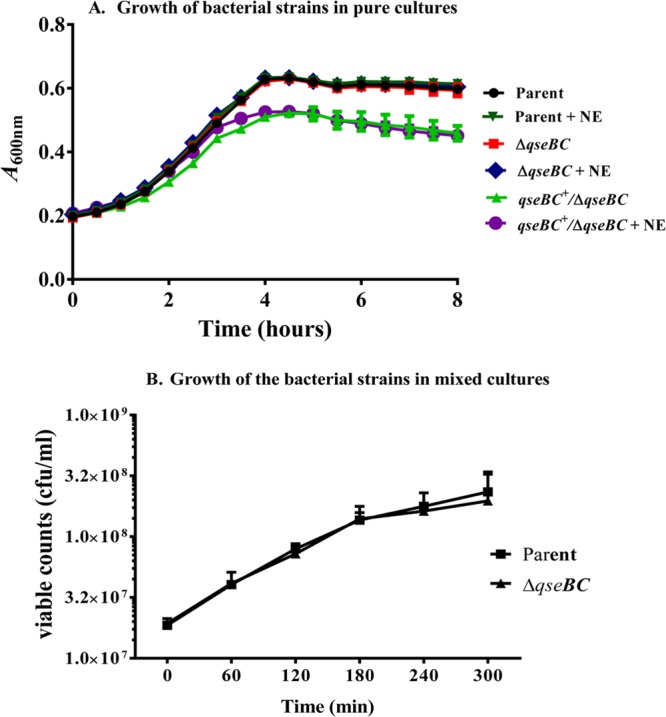
Comparison of growth curves of bacterial strains grown in independent or mixed cultures. (A) The growth curves of the parent, qseBC mutant, and the complemented qseBC mutant strains grown as independent cultures were deduced by measuring the optical density every 30 min over a period of 8 h. Three independent cultures were tested in triplicate, and each time point on the growth curve represents an average of nine readings. (B) The parent and the qseBC mutant strains were also grown in mixed cultures at 37°C, and 10-fold serial dilutions were plated on LB agar containing streptomycin to obtain total viable colony counts representing both strains. The colonies from LB-streptomycin plates were screened for kanamycin resistance to determine relative proportions of the qseBC mutant and the parental strain. The viable colony counts for each strain represent the mean CFU ml−1 ± standard error of the mean (SEM) for the three independent mixed cultures of the parental and the mutant strains.
Motility and NE signaling are not significantly impacted in the qseBC mutant strain.
Since QseBC-mediated quorum-sensing signaling in EHEC O157:H7 enhances flagellar gene expression and motility (13, 15), which have indirectly been shown to be important for colonization (36, 37), we compared the motility of the qseBC deletion mutant to those of the parental and the qseBC-complemented mutant strains in the presence or absence of the QseBC-activating signaling molecule norepinephrine (NE). The phenotypic analysis of motility on soft-agar plates revealed that a deletion of the qseBC genes did not compromise bacterial motility. As shown in Fig. 3A, the sizes of the motility zones produced by the qseBC deletion mutant (19.0 mm ± 0.63 mm; P = 0.82) on DMEM lacking NE were similar to those for the parental strain (19.0 mm ± 0.43 mm). Similarly, the motility zones produced by the qseBC mutant (20.0 mm ± 0.49 mm; P = 0.048) in the presence of NE were only slightly smaller than those produced by the parental strain (22.0 ± 0.77 mm) on DMEM containing NE (Fig. 3A). The qseBC mutant complemented with the qseBC-carrying plasmid (pSM639) produced motility zones that were significantly smaller on medium lacking (13.0 mm ± 0.52 mm; P = 0.002) or containing (16.0 ± 0.20 mm; P < 0.0001) NE in comparison to the parental and qseBC mutant strains (Fig. 3A). Figure 3B shows a graphic presentation of the motility data generated by determining the sizes of the motility zones produced by the spot inoculation of soft-agar motility plates (containing or lacking NE) from three independently grown cultures of the parent, qseBC mutant, and qseBC-complemented mutant strains. As shown in Fig. 3B, the mean diameter of the motility zones produced by the qseBC deletion mutant on medium containing NE was significantly greater (20.0 mm ± 0.47 mm; P = 0.031) than those on medium without NE (19.0 mm ± 0.43 mm). The parental strain showed the highest increase in the size of its motility zones on medium with NE (22.0 mm ± 0.77 mm; P = 0.003) compared to that in the absence of NE (19.0 mm ± 0.63 mm) (Fig. 3B). The next-highest increase in motility in the presence of NE was observed for the qseBC-complemented mutant, in which motility zones increased from 13.0 mm ± 0.32 mm in the absence of NE to 16.0 mm ± 0.20 mm (P = 0.0002) in the presence of NE (Fig. 3B). Overall, the qseBC-complemented mutant strain produced the smallest motility zones but showed a positive response to NE. The production of smaller motility zones by the complemented mutant could presumably be attributed to the reduced growth of the complemented strain (Fig. 2A) due to the increased expression of the QseBC genes from the complementing plasmid (pSM639).
FIG 3.
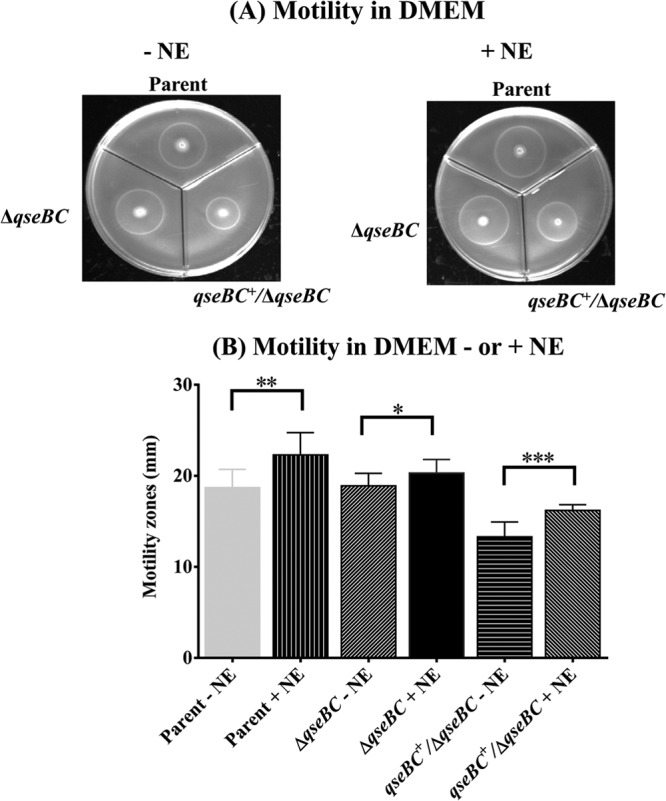
Determination of the relative motility of the parent, qseBC, and qseBC-complemented mutant strains. Motilities of the parent, qseBC mutant (ΔqseBC), and qseBC-complemented mutant (qseBC+/ΔqseBC mutant) strains were determined by spotting overnight bacterial cultures grown in LB-carbenicillin broth on motility agar plates containing or lacking NE. The diameters of motility zones produced around the point of bacterial inoculations (A) were measured after 24 h of incubation at 37°C. The mean diameters (mm) of swimming or motility zones were computed from three independent bacterial cultures and 9 or 10 replicates spotted on motility plates containing or lacking NE (B). Statistical significance of the differences in the motilities of the qseBC and qseBC-complemented mutants relative to parental strain is indicated (*, P = 0.0309; **, P = 0.0029; ***, P = 0.0002).
Transcription of flhD and fliC correlates with motility of the parental, qseBC mutant, and complemented mutant strains.
It has been shown in previous studies that the reduced motility of EHEC O157:H7 deleted of the sensor kinase-encoding qseC results from the reduced transcription of flhD and fliC (13). The effects of deleting both qseC and its cognate response regulator-encoding gene qseB were not reported previously. Since we did not observe a significant difference in the motility phenotypes produced by the qseBC mutant and the parental strain but observed a significant reduction in the motility of the qseBC-complemented mutant, we examined if the motility phenotypes produced by these three strains would correlate with the transcriptional levels of flhD, the master regulator of the flagellar gene expression, and the fliC gene, which encodes a major flagellar protein. qRT-PCR analysis of RNAs prepared from bacterial strains grown in the presence or absence of NE showed no significant (P > 0.05) differences in the transcriptional levels of flhD and fliC between the qseBC mutant and the parental strain (Fig. 4). For example, as shown in Fig. 4A, the transcriptional levels of flhD in the qseBC mutant were 0.95 ± 0.13 and 0.97 ± 0.09, compared to the transcriptional levels of 1.03 ± 0.01 and 1.01 ± 0.05 in the parental strain, in the absence or presence of NE, respectively. Similarly, the transcriptional levels of fliC in the qseBC mutant were 0.97 ± 0.09 and 1.02 ± 0.08, compared to the transcriptional levels of 1.0 ± 0.05 and 1.00 ± 0.04 in the parental strain, in the absence or presence of NE, respectively (Fig. 4B). However, the transcriptional levels of flhD (0.49 ± 0.11 without NE and 0.41 ± 0.06 with NE) and fliC (0.32 ± 0.08 without NE and 0.56 ± 0.15 with NE) were significantly reduced (P < 0.05) in the qseBC-complemented mutant relative to the parental and qseBC mutant strains (Fig. 4A and B).
FIG 4.

Quantification of transcriptional levels of flhD and fliC. qRT-PCR was performed using DNA-free RNA prepared from bacterial strains grown in DMEM with or without NE for 5 h to an OD600 of 1.1 to 1.2. (A) Relative expression of flhD in the parent, qseBC mutant (ΔqseBC), and qseBC-complemented mutant (qseBC+/ΔqseBC mutant) strains grown in the absence or presence of NE. (B) Transcriptional levels of fliC in the parent, qseBC mutant, and qseBC-complemented mutant strains grown in medium lacking NE or containing NE. Bars represent the average of the relative expression, expressed as means ± SEM for three independent bacterial cultures and with each culture tested in triplicate. Significant (P < 0.05) differences in motility between the parent and mutant strains were determined using the Mann-Whitney test (*, P = 0.0306; **, P = 0.003; ***, P = 0.0002).
Deletion of qseBC enhances LEE expression.
The QseBC-mediated quorum-sensing pathway has been implicated in cross talk with the other two-component signal transduction systems, such as those that enhance expression of LEE (15). Since LEE encodes a type III secretion system for secreting both the LEE- and non-LEE-encoded virulence factors required for the adherence of EHEC O157:H7 to the bovine intestine (33, 38, 39), we assessed the expression of LEE-encoded ler and ler-regulated espA in the qseBC mutant relative to the parental strain. qRT-PCR analysis revealed 1.77-fold (P < 0.0001)- and 1.8-fold (P < 0.0001)-higher expression of ler in the qseBC mutant than in the parental strain in the absence or presence of NE, respectively (Fig. 5A). The transcriptional levels of espA, which is positively regulated by Ler, were significantly upregulated, as indicated by 2.47-fold (P < 0.0001) and 2.4-fold (P = 0.002) increases in the espA expression in the qseBC mutant compared to the parental strain in medium lacking or containing NE, respectively (Fig. 5B). However, the transcriptional levels of ler (0.83 ± 0.17 [P = 0.037] without NE and 0.69 ± 0.14 [P = 0.22] with NE) and espA (0.59 ± 0.16 [P = 0.015] without NE and 0.56 ± 0.08 [P = 0.002] with NE) were variably but significantly lower in the qseBC-complemented mutant than in the parental and qseBC mutant strains (Fig. 5A and B). The reduced transcription of ler and espA could at least partly be attributed to reduced growth of the qseBC-complemented mutant strain (Fig. 2A). This reduced growth is presumably caused by an imbalanced stoichiometry of the sensory kinase and response regulator due to the increased expression of QseC and QseB, respectively, from the complementing plasmid.
FIG 5.

Effects of qseBC deletion on transcriptional levels of ler and espA. The transcriptional levels of ler and espA in DNA-free RNA prepared from bacterial strains grown in DMEM with or without NE for 5 h (OD600 of 1.1 to 1.2) were determined by qRT-PCR. (A) Relative expression of ler in the parent, qseBC mutant, and qseBC-complemented mutant (ΔqseBC/qseBC+) strains grown in the absence or presence of NE. (B) Transcriptional levels of espA in the parent, qseBC, and qseBC-complemented mutant strains grown in medium lacking NE or containing NE. Bars represent the average of the relative expression, expressed as means ± SEM for three independent bacterial cultures and with each culture tested in triplicate. Significance (P < 0.05) of the difference in the expression of genes was evaluated by using the Mann-Whitney test (*, P < 0.05; **, P = 0.0083; *** P = 0.002; ****, P < 0.0001).
The qseBC deletion enhances curli gene expression.
Curli fimbriae are expressed by many pathogenic as well as nonpathogenic E. coli strains, and production of curli promotes adherence and biofilm formation (24). Since several reports have also implicated curli fimbriae in the virulence of many pathogenic E. coli serotypes (26, 27), we determined the effect of the qseBC deletion on the curli gene expression in EHEC O157:H7. The transcriptional levels of csgA, which encodes the curlin protein of curli fimbriae, were 2.67-fold higher in the qseBC mutant (2.67 ± 0.55; P = 0.026) than in the parental strain (1.02 ± 0.12) when these strains were grown without NE (Fig. 6A). In medium containing NE, the expression of csgA in the qseBC mutant appeared to be higher (7.36 ± 4.4) than that in the parental strain (1.0 ± 0.00), but this increase was not significant (P = 0.20) perhaps due to a large standard error of the mean (Fig. 6B). The qseBC-complemented mutant showed significantly lower transcriptional levels of csgA in medium lacking (0.18 ± 0.11; P = 0.026) (Fig. 6A) or supplemented with (0.002 ± 0.001; P = 0.028) (Fig. 6B) NE than the parental strain (1.02 ± 0.12 without NE and 1.14 ± 0.34 with NE). Significantly higher csgA transcription in the qseBC mutant than in the parental strain also correlated with increased Congo red binding by the mutant relative to the parental and the qseBC-complemented mutant strains (data not shown).
FIG 6.
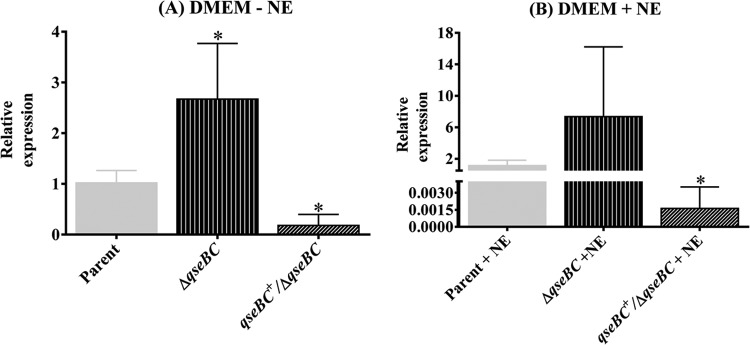
Effects of qseBC deletion on transcriptional levels of csgA and Congo red binding by bacterial cells. The transcription of csgA, the gene encoding curlin of curli fimbriae, was determined by qRT-PCR using DNA-free RNA prepared from the parent, qseBC mutant, and complemented qseBC mutant strains. Bars represent the average of the relative expression of csgA determined using RNA from three independent bacterial cultures grown at 37°C in the absence (A) or presence (B) of NE. Bars represent the mean relative expression ± SEM. The significance of the difference in the expression of csgA for the qseBC and qseBC-complemented mutants relative to the parent strain was determined by using the nonparametric Mann-Whitney test (*, P = 0.0286).
DISCUSSION
In this study, we show for the first time that a qseBC deletion mutant of EHEC O157:H7 gained a competitive advantage over the parent strain in colonizing cattle intestines. There was a significant increase in the magnitude of fecal shedding of the qseBC mutant by the calves inoculated with a mixture of the isogenic mutant and the parent strain during the course of a 4-week study. That the mutant strain was better in colonizing the animals inoculated with the mixed inoculum was also indicated by competitive indices (CIs) of >1 for the mutant strain on each fecal sampling day. These results are unexpected compared to the reported effects on bacterial virulence resulting from a qseC or qseB deletion alone. For example, a qseC deletion reportedly attenuated virulence and colonization of EHEC O157:H7, UPEC, and Salmonella Typhimurium, and Francisella tularensis in rabbit, mouse, and porcine animal models (19, 21, 23). These negative effects of qseC deletion on bacterial virulence have been attributed to the accumulation of phosphorylated QseB due to a constitutive positive feedback loop causing dysregulation of virulence and impairment of bacterial metabolism (19). The appropriate balance between QseB and its phosphorylated counterpart appears to be maintained by the phosphatase activity of QseC, which dephosphorylates QseB to regulate virulence gene expression in response to quorum sensing (20). Augmentation of colonization with the qseBC deletion mutant, as indicated by the increased fecal shedding, suggests that the complete removal of the qseBC-encoded quorum-sensing system overcomes the negative effects on the virulence gene expression that would otherwise result from the elevated levels of phosphorylated QseB in qseC deletion mutants. The increased colonization of the qseBC deletion mutant in calves, as reported in this study, also deviates from no effects of qseBC or qseB deletion on virulence of S. Typhimurium and UPEC in porcine and mouse challenge models, respectively (20, 23). Additionally, in a clinical isolate of Aeromonas hydrophila, qseB is essential for virulence in a septicemic mouse model in conjunction with qseC and cyclic diGMP-mediated regulation of gene expression (22). These results therefore may suggest that the qseBC deletion affects different genetic networks in EHEC O157:H7 than in the other bacterial pathogens. It may also be valid to conclude, based on the reported varied effects of qseBC on the virulence of bacterial pathogens, that there are different mechanisms of pathogenicity in different animals.
In our attempts to correlate the competitive edge of the qseBC deletion mutant of EHEC O157:H7 in colonization of cattle intestine over the parent strain, we found that expression of specific sets of genes that are directly or indirectly linked to virulence and colonization was upregulated in the qseBC deletion mutant relative to the parental strain. Furthermore, we observed that the ability of the qseBC mutant to respond to NE was not significantly altered, as indicated by only a slight reduction in its motility compared to that of the parent strain. Correlated with almost similar motilities of the qseBC mutant and parent strains were the similar transcriptional levels of flagellar genes flhD and fliC irrespective of the presence or absence of NE in the culture medium. These findings, showing virtually unaltered motility of the qseBC mutant and its ability to respond to NE in a manner that was similar to that of the parent strain, were comparable to those reported for a Salmonella Typhimurium qseBC deletion mutant, which also showed parental motility and was unaffected in responding to NE (23). However, the same study showed that pigs inoculated with the Salmonella Typhimurium qseBC mutant shed the mutant strain at the same magnitude as the parent strain (23). Only the qseC mutant was shed poorly in these pigs, implying the importance of the QseC sensor kinase in regulating the phosphorylation status of QseB, which is essential for the virulence of Salmonella in pigs. However, our finding that the parental strain was outcompeted by the qseBC mutant suggests, as has been postulated for the qseBC deletion mutants of S. Typhimurium (18), that the activation of other, yet-unknown genetic systems could allow the expression of near-parental motility in response to the presence of NE and bacterial autoinducers in bovine intestines. For example, it is possible that one or more of the 32 response regulators identified in EHEC O157:H7 (40) could, through cross talk (15), affect the expression of a gene(s) that would allow the qseBC mutant to respond to NE to upregulate motility.
In addition to a postulated role of motility in colonization of bovine intestines by EHEC O157:H7 (36, 37), the type III secretion system-mediated secretion of LEE- as well as non-LEE-encoded effectors are critical for EHEC O157:H7 adherence to and colonization of cattle intestines (29, 37, 38, 41). Thus, significant but low levels of increases in the transcriptional levels of LEE-carried ler and espA, observed in the qseBC mutant irrespective of the presence or lack thereof of NE, could be instrumental in imparting a competitive edge to the mutant relative to the parent strain in intestinal colonization of calves. The transcriptional levels of ler, which encodes Ler for the positive regulation of LEE, are controlled by several known negative and positive transcriptional factors, including QseA and QseD, whose expression is also modulated in response to quorum-sensing signaling (30, 42–48). Thus, it is possible that either a derepression of one or more of these positive regulators or a repression of a negative regulator could enhance LEE expression in the qseBC mutant, allowing it to outcompete the parental strain in calves inoculated with a mixture of these two strains.
Curli fimbriae are important adhesive structures that enable E. coli to bind to various host proteins, colonize animal tissues, modulate immune functions, and promote biofilm formation on a variety of matrices (24). Both EHEC O157:H7 and nonmotile EHEC O157 serotypes are capable of producing curli, which have been implicated in adherence of these bacterial strains to epithelial cells (26, 27). Thus, higher transcription of csgA and increased Congo red binding, which are indicative of increased curli production (51), could promote adherence of the qseBC mutant to intestinal epithelial cells even in the absence of NE. This increased adherence due to enhanced curli production might be one of the factors contributing to higher fecal shedding of the qseBC mutant than of the parental strain.
In summary, the near-parental-type motility, ability to respond to NE, and increased espA and csgA transcription observed under in vitro growth conditions are suggestive that similar in vivo increases in the expression of LEE and the genes encoding bacterial motility and adhesive curli confer a competitive advantage to the qseBC mutant in colonizing cattle intestines. In a recent study, we demonstrated the importance of motility and LEE for increased colonization of bovine intestines by EHEC O157:H7, since mutants expressing LEE at very high levels but compromised in bacterial motility were unable to colonize and persist in cattle intestines (37). Thus, regulatory pathways that allow increased expression of LEE and other adhesins, such as curli, without causing significant reductions in bacterial motility might explain the increased colonization of cattle intestines by the qseBC mutant of EHEC O157:H7.
ACKNOWLEDGMENTS
We thank Lindsay Andersen, Matt Inbody, and Kaitlin Johnson for technical assistance in this study and Dalene Whitney, Daniel Jenkins, and Brian Conrad for animal caretaking and sample collection work. We also thank Randy Sacco and Thad Stanton for their critical reviews of the manuscript.
Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
Published ahead of print 10 January 2014
REFERENCES
- 1.Karmali MA. 2004. Infection by Shiga toxin-producing Escherichia coli: an overview. Mol. Biotechnol. 26:117–122. 10.1385/MB:26:2:117 [DOI] [PubMed] [Google Scholar]
- 2.Dean-Nystrom EA, Stoffregen WC, Bosworth BT, Moon HW, Pohlenz JF. 2008. Early attachment sites for Shiga-toxigenic Escherichia coli O157:H7 in experimentally inoculated weaned calves. Appl. Environ. Microbiol. 74:6378–6384. 10.1128/AEM.00636-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naylor SW, Low JC, Besser TE, Mahajan A, Gunn GJ, Pearce MC, McKendrick IJ, Smith DG, Gally DL. 2003. Lymphoid follicle-dense mucosa at the terminal rectum is the principal site of colonization of enterohemorrhagic Escherichia coli O157:H7 in the bovine host. Infect. Immun. 71:1505–1512. 10.1128/IAI.71.3.1505-1512.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arthur TM, Brichta-Harhay DM, Bosilevac JM, Kalchayanand N, Shackelford SD, Wheeler TL, Koohmaraie M. 2010. Super shedding of Escherichia coli O157:H7 by cattle and the impact on beef carcass contamination. Meat Sci. 86:32–37. 10.1016/j.meatsci.2010.04.019 [DOI] [PubMed] [Google Scholar]
- 5.Elder RO, Keen JE, Siragusa GR, Barkocy-Gallagher GA, Koohmaraie M, Laegreid WW. 2000. Correlation of enterohemorrhagic Escherichia coli O157 prevalence in feces, hides, and carcasses of beef cattle during processing. Proc. Natl. Acad. Sci. U. S. A. 97:2999–3003. 10.1073/pnas.97.7.2999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soon JM, Chadd SA, Baines RN. 2011. Escherichia coli O157:H7 in beef cattle: on farm contamination and pre-slaughter control methods. Anim. Health Res. Rev. 12:197–211. 10.1017/S1466252311000132 [DOI] [PubMed] [Google Scholar]
- 7.Moon HW, Whipp SC, Argenzio RA, Levine MM, Giannella RA. 1983. Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41:1340–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Elliott SJ, Yu J, Kaper JB. 1999. The cloned locus of enterocyte effacement from enterohemorrhagic Escherichia coli O157:H7 is unable to confer the attaching and effacing phenotype upon E. coli K-12. Infect. Immun. 67:4260–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarvis KG, Kaper JB. 1996. Secretion of extracellular proteins by enterohemorrhagic Escherichia coli via a putative type III secretion system. Infect. Immun. 64:4826–4829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elliott SJ, Sperandio V, Giron JA, Shin S, Mellies JL, Wainwright L, Hutcheson SW, McDaniel TK, Kaper JB. 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE- and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infect. Immun. 68:6115–6126. 10.1128/IAI.68.11.6115-6126.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McNeilly TN, Naylor SW, Mahajan A, Mitchell MC, McAteer S, Deane D, Smith DG, Low JC, Gally DL, Huntley JF. 2008. Escherichia coli O157:H7 colonization in cattle following systemic and mucosal immunization with purified H7 flagellin. Infect. Immun. 76:2594–2602. 10.1128/IAI.01452-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. 1999. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 96:15196–15201. 10.1073/pnas.96.26.15196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sperandio V, Torres AG, Kaper JB. 2002. Quorum sensing Escherichia coli regulators B and C (QseBC): a novel two-component regulatory system involved in the regulation of flagella and motility by quorum sensing in E. coli. Mol. Microbiol. 43:809–821. 10.1046/j.1365-2958.2002.02803.x [DOI] [PubMed] [Google Scholar]
- 14.Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. 2006. The QseC sensor kinase: a bacterial adrenergic receptor. Proc. Natl. Acad. Sci. U. S. A. 103:10420–10425. 10.1073/pnas.0604343103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hughes DT, Clarke MB, Yamamoto K, Rasko DA, Sperandio V. 2009. The QseC adrenergic signaling cascade in enterohemorrhagic E. coli (EHEC). PLoS Pathog. 5:e1000553. 10.1371/journal.ppat.1000553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke MB, Sperandio V. 2005. Transcriptional regulation of flhDC by QseBC and sigma (FliA) in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 57:1734–1749. 10.1111/j.1365-2958.2005.04792.x [DOI] [PubMed] [Google Scholar]
- 17.Clarke MB, Sperandio V. 2005. Transcriptional autoregulation by quorum sensing Escherichia coli regulators B and C (QseBC) in enterohaemorrhagic E. coli (EHEC). Mol. Microbiol. 58:441–455. 10.1111/j.1365-2958.2005.04819.x [DOI] [PubMed] [Google Scholar]
- 18.Bearson BL, Bearson SM. 2008. The role of the QseC quorum-sensing sensor kinase in colonization and norepinephrine-enhanced motility of Salmonella enterica serovar Typhimurium. Microb. Pathog. 44:271–278. 10.1016/j.micpath.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 19.Hadjifrangiskou M, Kostakioti M, Chen SL, Henderson JP, Greene SE, Hultgren SJ. 2011. A central metabolic circuit controlled by QseC in pathogenic Escherichia coli. Mol. Microbiol. 80:1516–1529. 10.1111/j.1365-2958.2011.07660.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kostakioti M, Hadjifrangiskou M, Pinkner JS, Hultgren SJ. 2009. QseC-mediated dephosphorylation of QseB is required for expression of genes associated with virulence in uropathogenic Escherichia coli. Mol. Microbiol. 73:1020–1031. 10.1111/j.1365-2958.2009.06826.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rasko DA, Moreira CG, Li de R, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. 2008. Targeting QseC signaling and virulence for antibiotic development. Science 321:1078–1080. 10.1126/science.1160354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kozlova EV, Khajanchi BK, Popov VL, Wen J, Chopra AK. 2012. Impact of QseBC system in c-di-GMP-dependent quorum sensing regulatory network in a clinical isolate SSU of Aeromonas hydrophila. Microb. Pathog. 53:115–124. 10.1016/j.micpath.2012.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bearson BL, Bearson SM, Lee IS, Brunelle BW. 2010. The Salmonella enterica serovar Typhimurium QseB response regulator negatively regulates bacterial motility and swine colonization in the absence of the QseC sensor kinase. Microb. Pathog. 48:214–219. 10.1016/j.micpath.2010.03.005 [DOI] [PubMed] [Google Scholar]
- 24.Barnhart MM, Chapman MR. 2006. Curli biogenesis and function. Annu. Rev. Microbiol. 60:131–147. 10.1146/annurev.micro.60.080805.142106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lloyd SJ, Ritchie JM, Torres AG. 2012. Fimbriation and curliation in Escherichia coli O157:H7: a paradigm of intestinal and environmental colonization. Gut Microbes 3:272–276. 10.4161/gmic.20661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosser T, Dransfield T, Allison L, Hanson M, Holden N, Evans J, Naylor S, La Ragione R, Low JC, Gally DL. 2008. Pathogenic potential of emergent sorbitol-fermenting Escherichia coli O157:NM. Infect. Immun. 76:5598–5607. 10.1128/IAI.01180-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saldana Z, Xicohtencatl-Cortes J, Avelino F, Phillips AD, Kaper JB, Puente JL, Giron JA. 2009. Synergistic role of curli and cellulose in cell adherence and biofilm formation of attaching and effacing Escherichia coli and identification of Fis as a negative regulator of curli. Environ. Microbiol. 11:992–1006. 10.1111/j.1462-2920.2008.01824.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma VK, Bearson BL. 2013. Hha controls Escherichia coli O157:H7 biofilm formation by differential regulation of global transcriptional regulators FlhDC and CsgD. Appl. Environ. Microbiol. 79:2384–2396. 10.1128/AEM.02998-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharma VK, Carlson SA, Casey TA. 2005. Hyperadherence of an hha mutant of Escherichia coli O157:H7 is correlated with enhanced expression of LEE-encoded adherence genes. FEMS Microbiol. Lett. 243:189–196. 10.1016/j.femsle.2004.12.003 [DOI] [PubMed] [Google Scholar]
- 30.Sharma VK, Zuerner RL. 2004. Role of hha and ler in transcriptional regulation of the esp operon of enterohemorrhagic Escherichia coli O157:H7. J. Bacteriol. 186:7290–7301. 10.1128/JB.186.21.7290-7301.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pesavento C, Becker G, Sommerfeldt N, Possling A, Tschowri N, Mehlis A, Hengge R. 2008. Inverse regulatory coordination of motility and curli-mediated adhesion in Escherichia coli. Genes Dev. 22:2434–2446. 10.1101/gad.475808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffin PM, Ostroff SM, Tauxe RV, Greene KD, Wells JG, Lewis JH, Blake PA. 1988. Illnesses associated with Escherichia coli O157:H7 infections. A broad clinical spectrum. Ann. Intern. Med. 109:705–712 [DOI] [PubMed] [Google Scholar]
- 33.McKee ML, Melton-Celsa AR, Moxley RA, Francis DH, O'Brien AD. 1995. Enterohemorrhagic Escherichia coli O157:H7 requires intimin to colonize the gnotobiotic pig intestine and to adhere to HEp-2 cells. Infect. Immun. 63:3739–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bearson BL, Bearson SM, Uthe JJ, Dowd SE, Houghton JO, Lee I, Toscano MJ, Lay DC., Jr 2008. Iron regulated genes of Salmonella enterica serovar Typhimurium in response to norepinephrine and the requirement of fepDGC for norepinephrine-enhanced growth. Microbes Infect. 10:807–816. 10.1016/j.micinf.2008.04.011 [DOI] [PubMed] [Google Scholar]
- 35.Kendall MM, Rasko DA, Sperandio V. 2007. Global effects of the cell-to-cell signaling molecules autoinducer-2, autoinducer-3, and epinephrine in a luxS mutant of enterohemorrhagic Escherichia coli. Infect. Immun. 75:4875–4884. 10.1128/IAI.00550-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mahajan A, Currie CG, Mackie S, Tree J, McAteer S, McKendrick I, McNeilly TN, Roe A, La Ragione RM, Woodward MJ, Gally DL, Smith DG. 2009. An investigation of the expression and adhesin function of H7 flagella in the interaction of Escherichia coli O157 : H7 with bovine intestinal epithelium. Cell. Microbiol. 11:121–137. 10.1111/j.1462-5822.2008.01244.x [DOI] [PubMed] [Google Scholar]
- 37.Sharma VK, Sacco RE, Kunkle RA, Bearson SM, Palmquist DE. 2012. Correlating levels of type III secretion and secreted proteins with fecal shedding of Escherichia coli O157:H7 in cattle. Infect. Immun. 80:1333–1342. 10.1128/IAI.05869-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dean-Nystrom EA, Bosworth BT, Moon HW, O'Brien AD. 1998. Escherichia coli O157:H7 requires intimin for enteropathogenicity in calves. Infect. Immun. 66:4560–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naylor SW, Roe AJ, Nart P, Spears K, Smith DG, Low JC, Gally DL. 2005. Escherichia coli O157 : H7 forms attaching and effacing lesions at the terminal rectum of cattle and colonization requires the LEE4 operon. Microbiology 151:2773–2781. 10.1099/mic.0.28060-0 [DOI] [PubMed] [Google Scholar]
- 40.Mizuno T. 1997. Compilation of all genes encoding two-component phosphotransfer signal transducers in the genome of Escherichia coli. DNA Res. 4:161–168. 10.1093/dnares/4.2.161 [DOI] [PubMed] [Google Scholar]
- 41.Gally DL, Naylor SW, Low JC, Gunn GJ, Synge BA, Pearce MC, Donachie W, Besser TE. 2003. Colonisation site of E. coli O157 in cattle. Vet. Rec. 152:307. [PubMed] [Google Scholar]
- 42.Habdas BJ, Smart J, Kaper JB, Sperandio V. 2010. The LysR-type transcriptional regulator QseD alters type three secretion in enterohemorrhagic Escherichia coli and motility in K-12 Escherichia coli. J. Bacteriol. 192:3699–3712. 10.1128/JB.00382-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansen AM, Kaper JB. 2009. Hfq affects the expression of the LEE pathogenicity island in enterohaemorrhagic Escherichia coli. Mol. Microbiol. 73:446–465. 10.1111/j.1365-2958.2009.06781.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laaberki MH, Janabi N, Oswald E, Repoila F. 2006. Concert of regulators to switch on LEE expression in enterohemorrhagic Escherichia coli O157:H7: interplay between Ler, GrlA, HNS and RpoS. Int. J. Med. Microbiol. 296:197–210. 10.1016/j.ijmm.2006.02.017 [DOI] [PubMed] [Google Scholar]
- 45.Njoroge JW, Gruber C, Sperandio V. 2013. The interacting Cra and KdpE regulators are involved in the expression of multiple virulence factors in enterohemorrhagic Escherichia coli. J. Bacteriol. 195:2499–2508. 10.1128/JB.02252-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sharp FC, Sperandio V. 2007. QseA directly activates transcription of LEE1 in enterohemorrhagic Escherichia coli. Infect. Immun. 75:2432–2440. 10.1128/IAI.02003-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Torres AG, Lopez-Sanchez GN, Milflores-Flores L, Patel SD, Rojas-Lopez M, Martinez de la Pena CF, Arenas-Hernandez MM, Martinez-Laguna Y. 2007. Ler and H-NS, regulators controlling expression of the long polar fimbriae of Escherichia coli O157:H7. J. Bacteriol. 189:5916–5928. 10.1128/JB.00245-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Chaudhuri RR, Constantinidou C, Hobman JL, Patel MD, Jones AC, Sarti D, Roe AJ, Vlisidou I, Shaw RK, Falciani F, Stevens MP, Gally DL, Knutton S, Frankel G, Penn CW, Pallen MJ. 2004. Regulators encoded in the Escherichia coli type III secretion system 2 gene cluster influence expression of genes within the locus for enterocyte effacement in enterohemorrhagic E. coli O157:H7. Infect. Immun. 72:7282–7293. 10.1128/IAI.72.12.7282-7293.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melton-Celsa AR, Rogers JE, Schmitt CK, Darnell SC, O'Brien AD. 1998. Virulence of Shiga toxin-producing Escherichia coli (STEC) in orally-infected mice correlates with the type of toxin produced by the infecting strain. Jpn. J. Med. Sci. Biol. 51(Suppl):S108–S114 [DOI] [PubMed] [Google Scholar]
- 50.Perna NT, Plunkett G, III, Burland V, Mau B, Glasner JD, Rose DJ, Mayhew GF, Evans PS, Gregor J, Kirkpatrick HA, Posfai G, Hackett J, Klink S, Boutin A, Shao Y, Miller L, Grotbeck EJ, Davis NW, Lim A, Dimalanta ET, Potamousis KD, Apodaca J, Anantharaman TS, Lin J, Yen G, Schwartz DC, Welch RA, Blattner FR. 2001. Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409:529–533. 10.1038/35054089 [DOI] [PubMed] [Google Scholar]
- 51.Uhlich GA, Cooke PH, Solomon EB. 2006. Analyses of the red-dry-rough phenotype of an Escherichia coli O157:H7 strain and its role in biofilm formation and resistance to antibacterial agents. Appl. Environ. Microbiol. 72:2564–2572. 10.1128/AEM.72.4.2564-2572.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]