

Abstract
Patterns of microbial distribution represent the integrated effects of historical and biological processes and are thus a central issue in ecology. However, there is still active debate on whether dispersal limitation contributes to microbial diversification in strongly connected systems. In this study, sediment samples were collected along a transect representing a variety of seawater pollution levels in the East China Sea. We investigated whether changes in sediment bacterial community structures would indicate the effects of the pollution gradient and of dispersal limitation. Our results showed consistent shifts in bacterial communities in response to pollution. More geographically distant sites had more dissimilar communities (r = −0.886, P < 0.001) in this strongly connected sediment ecosystem. A variance analysis based on partitioning by principal coordinates of neighbor matrices (PCNM) showed that spatial distance (dispersal limitation) contributed more to bacterial community variation (8.2%) than any other factor, although the environmental factors explained more variance when combined (11.2%). In addition, potential indicator taxa (primarily affiliated with Deltaproteobacteria and Gammaproteobacteria) were identified; these taxa characterized the pollution gradient. This study provides direct evidence that dispersal limitation exists in a strongly connected marine sediment ecosystem and that candidate indicator taxa can be applied to evaluate coastal pollution levels.
INTRODUCTION
Atmospheric deposition and agricultural and industrial effluents transport a continuous supply of nitrogen (N) into the marine environment (1), producing a pollution gradient in coastal zones. It is probable that this trend will continue in the near future, with unknown ecological consequences (2). A high and unbalanced N supply is changing the nutrient stoichiometry (3), resulting in substrates that resist decomposition by microorganisms. Eventually, these substrates are deposited into the benthic sediment via the microbial carbon pump (4). Much of the marine environment is historically resource limited, especially in N (5). In particular, it is estimated that up to 80% of the nitrogen required by phytoplanktonic organisms is derived from sediment microbial remineralization in shallow seas (6). Thus, sediment microbes can potentially make marine sediment either a source or sink for the nutrient pool (7). It has been extensively documented that the magnitude and direction of remineralization depend on the local sediment characteristics and resident microbial communities (8, 9). Accordingly, identifying microbial community variation and its driving forces along N pollution gradients is important in predicting ecosystem consequences based on environmental changes.
There is good evidence from empirical research and theoretical ecology that N pollution exerts strong selective pressures on the variation of microbial communities. For instance, an excess N supply could select for nitrophilic microbes that require more N and decompose recalcitrant carbon substrates more efficiently (10). Accordingly, community composition may shift to favor organisms that are physiologically better adapted to such conditions (11). However, reciprocal transplantation experiments have provided extensive evidence that resident and immigrant microbial communities respond differently to the same disturbance (10, 12), indicating the apparent involvement of the microbial community in responses to environmental changes. Recently, increasing evidence has shown that the spatial distributions of microbes are nonrandom and that distance decay of similarity occurs for microbial compositional (13, 14) and functional (15) structures, although the results of certain studies are not consistent with these findings (16, 17). In particular, we have found that geographic distance imposed greater constraints on community variation than any of the individual sediment variables selected (18). Similarly, it has been shown that geographic distance (historical contingencies) is the dominant factor driving variation in bacterial diversity, in which geographic distance is applied as a proxy of past evolutionary and ecological events because of the spatial dissimilarity (19). Therefore, dispersal limitation might be more important than previously inferred (16, 17). Moreover, a comprehensive study revealed that sediment microbial assemblages show a low turnover rate (14). As mass effects would be greater in strongly connected systems (i.e., those with minor physical barriers), we therefore examined marine benthic sediments to test whether the distance decay of similarity of the microbial community in marine sediment mirrors that detected in physically isolated sites (18). This information is essential to include marine microorganisms on the biogeographic map on a global scale (13, 20).
The East China Sea is particularly important in fisheries, shipping, and environmental issues (21). However, increasingly serious eutrophication in this region, due to industrial and agricultural discharge, results in frequently harmful algal blooms (22). Nevertheless, how and to what extent the structure of the sediment bacterial community varies in response to N pollution in situ has not yet been investigated in the East China Sea. As a result, it is unclear whether microbial assemblages can be applied to indicate N pollution levels, mirroring the results obtained for contrasting cases, such as polycyclic aromatic hydrocarbons (23) and arsenic pollution (9). The present study was conceived because we observed native community effects and the uncertainty of dispersal limitation in interconnecting systems. Thus, we collected benthic sediment samples along a transect (corresponding to an array of seawater pollution levels) in the East China Sea (see Fig. S1 in the supplemental material) to determine the relative importance of local characteristics and spatial limitation in shaping the variation in the microbial community. We analyzed the bacterial community structure with 16S rRNA gene amplicon pyrosequencing and correlated it with environmental variables and spatial distances to achieve the following goals: (i) to evaluate how sediment bacterial communities respond to a pollution gradient, (ii) to determine the driving forces in shaping the bacterial community distributions, i.e., environmental heterogeneity, geographic distance, or both, and (iii) to explore taxa serving as indicators of such a pollution gradient.
MATERIALS AND METHODS
Sample collection.
Sediment samples were collected from the East China Sea during a cruise conducted by the Marine Environmental Monitoring Center of Ningbo on 25 October 2012. Five sites were chosen to cover a gradient of seawater pollution levels along a specified transect (see Fig. S1 in the supplemental material). At each site, five biological replicates were collected within a 50-m by 50-m area for a total of 25 samples. To avoid minor stochastic mass variation, surface sludge was discarded before homogenization. The samples were packaged in airtight sterile plastic bags with dry ice during the cruise and stored at −80°C after transport to the laboratory. The global positioning system (GPS) coordinates recorded at each sampling point (from 29°22′58″N 122°2′30″E to 29°22′43″N 122°34′48″E) were imported into the NOAA website (http://www.nhc.noaa.gov/gccalc.shtml) to calculate the pairwise geographic distance between samples.
Sediment geochemical analyses.
The pH values were measured using a probe (Starter 2100; Ohaus, USA) with a 1:2.5 (wt/vol) ratio of sediment to water. The sediments were immediately freeze-dried to analyze their geochemical characteristics. Sulfate (SO42−) was measured by barium chloride titration. The concentrations of total phosphorus (TP) were measured using inductively coupled plasma mass spectrometry (ICP-MS) (ELAN 9000/DRC-e; PerkinElmer, USA). Total organic carbon (TOC) and total nitrogen (TN) were measured with a CN analyzer (Vario Max CN; Elementar, Germany). Ammonium, nitrate, and nitrite were extracted by adding 50 ml of 1 M KCl to 10 g fresh sediment, shaking for 1 h, and filtering through filter paper and were determined with an automated procedure (Skalar SAN plus segmented flow analyzer). The dissolved inorganic nitrogen (DIN) content was calculated as the sum of NH4+, NO3−, and NO2−.
DNA extraction and purification.
Community DNA was extracted from 0.5-g amounts of sediments using a FastDNA spin kit (Bio 101, Carlsbad, CA, USA) according to the manufacturer's protocol. The genomic DNA (gDNA) extracts were quantified with a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA) and stored at −80°C until amplification.
Bacterial 16S rRNA amplification and 454 pyrosequencing.
An aliquot (50 ng) of purified DNA from each sample was used as the template for amplification. The V4 to V5 hypervariable regions of bacterial 16S rRNAs (Escherichia coli positions 515 to 907 [24]) were amplified using the primer set F515 (GTGCCAGCMGCCGCGG), with the Roche 454 “A” pyrosequencing adapter and a unique 11-bp barcode sequence, and R907 (CCGTCAATTCMTTTRAGTTT), with the Roche 454 “B” sequencing adapter at the 5′ end of each primer. Each sample was amplified in triplicate in a 50-ml reaction mixture under the following conditions: 30 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 30 s, with a final extension at 72°C for 10 min. The PCR products were pooled and purified with a PCR fragment purification kit (TaKaRa). The purified PCR products were quantified using the Quant-It Pico Green kit (Invitrogen, Carlsbad, CA).
Equimolar amounts of PCR products (assuming that amplicons of the same size had similar molar masses) for each sample were combined in a single tube and run on a Roche FLX 454 pyrosequencing machine (Roche Diagnostics Corporation, Branford, CT, USA), producing reads from the forward direction (F515 with barcode).
Processing of pyrosequencing data.
Sequencing reads were quality filtered, chimera checked, and PyroNoise analyzed using the Quantitative Insights Into Microbial Ecology (QIIME 1.6.0) workflow (25). Briefly, bacterial phylotypes were identified using uclust (7) and assigned to operational taxonomic units (OTUs, 97% similarity). Representative sequences from each phylotype were aligned using PyNAST (26), and the most abundant sequence in the OTU was selected as the representative sequence. The taxonomic identity of each phylotype was determined using the RDP database (27). The sequence data generated in this study were deposited in the DDBJ (http://www.ddbj.nig.ac.jp/) Sequence Read Archive and are available under the project number DRA001258. To correct for sequencing depth, we used a randomly selected subset of 4,800 sequences (corresponding to the smallest sequencing effort for any of the samples) per sample for further analysis.
Statistical analysis.
A SIMPER (similarity percentage) analysis was used to identify the taxa primarily responsible for the differences observed among the sample sites based on PAST (28). Several analyses were implemented in R version 3.0.2 (29). Specifically, nonmetric multidimensional scaling (NMDS) was used to evaluate the overall differences in microbial community structure (30). Partial Mantel tests were used to calculate the correlation between the distances separating the bacterial communities and the sediment characteristics or geographic distance in PASSaGE (31). To minimize the autocorrelation between spatial distance and environmental variables, a principle coordinates of neighbor matrices (PCNM) analysis was used to reduce the number of explanatory variables to retain only the most significant and important ones (32–34). The OTU abundance data contained many null abundances, and thus, a Hellinger transformation was made prior to the subsequent analysis (35). Before detrending, we tested whether the trend was significant, and then the bacterial community was detrended by regressing all variables on the x and y coordinates and retaining the residuals (34). Then, a forward selection procedure was employed to select the best PCNM variables (no spatial correlation). The forward selection was stopped if significance level alpha (P > 0.05) was reached (36). Variation partitioning enabled us to determine the various unique and combined fractions of variation explained in the bacterial community data by the environmental data, the broad-scale spatial pattern (trend surface analysis), and the fine-scale spatial patterns (PCNMs) (32).
RESULTS
Geochemical characteristics of sediments.
The concentrations of NO3−-N and DIN in the sediment decreased consistently (Fig. 1) from nearshore to offshore (see Fig. S1 in the supplemental material) as the salinity and depth increased (see Table S1), indicating N pollution due to coastal effluents. Conversely, the concentrations of TP and SO42− increased along the transect (Fig. 1). In contrast, the contents of PO43−, NH4+, and NO2− in the sediment did not vary linearly (see Table S1) but varied among sites. Moreover, pH and TOC were relatively stable along the pollution gradient.
FIG 1.
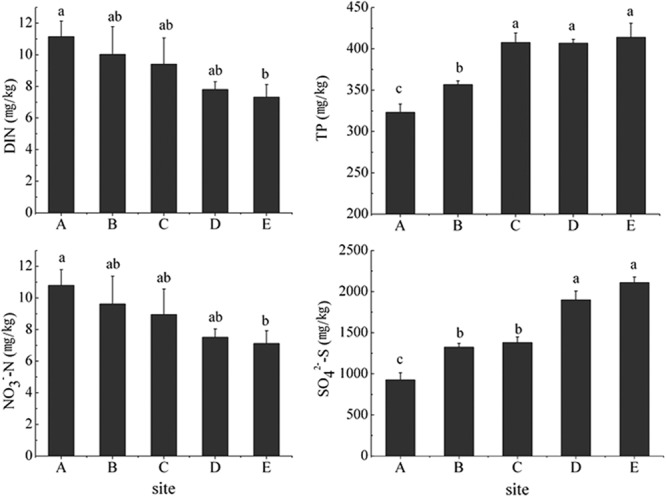
Linear changes in parameters along the pollution gradient. The data represent the means ± standard deviations (n = 5). Different small letters indicate significant differences between sites for that parameter. Means were compared using one-way ANOVA; P < 0.05.
Taxon-specific patterns.
Across all sediment samples, we obtained a total of 148,925 quality sequences, 4,873 to 6,869 sequences per sample (mean, 5,957). We were able to classify 88.4% of those sequences at the phylum level. The dominant phyla across all sediments were Gammaproteobacteria, Deltaproteobacteria, Planctomycetes, Bacteroidetes, and Chloroflexi (relative abundance, >5%) (see Fig. S2 in the supplemental material), representing more than 57% of the bacterial sequences. Consistent patterns (linear increases or decreases) were observed for the dominant phylum or class. For example, the relative abundances of Deltaproteobacteria and Bacteroidetes decreased, whereas those of Nitrospirae and Alphaproteobacteria increased along the pollution gradient (see Fig. S2). Regardless of the parameters, i.e., phylogenetic diversity, number of OTUs (phylotypes), and Shannon diversity, the alpha diversity was relatively stable, although it appeared to show a peak at the moderately polluted site C (see Fig. S3).
Bacterial community structure.
Based on the OTUs detected across the samples, an NMDS ordination analysis clearly revealed differentiation in the bacterial communities along our monitored pollution gradient (Fig. 2), although the community diversities did not change markedly (see Fig. S3 in the supplemental material). The patterns were further corroborated by an analysis of similarity (ANOSIM), which demonstrated that the sampling site was an important determinant of community composition (global r = 0.668, P = 0.001). Note that the community composition did not vary markedly between adjacent sites, such as site B versus C and site D versus E (Table 1). However, the communities separated well from nearshore to offshore (Fig. 2). In addition, the linear function showed a significant correlation (Pearson coefficient) between NMDS axis 1 (as a proxy for the bacterial community dissimilarity) and DIN (ρ = −0.504, P = 0.01), TP (ρ = 0.640, P = 0.001), and SO42− (ρ = 0.835, P < 0.001) (see Fig. S4). Taken together, the results clearly revealed the spatial dynamics of bacterial composition along this pollution gradient.
FIG 2.
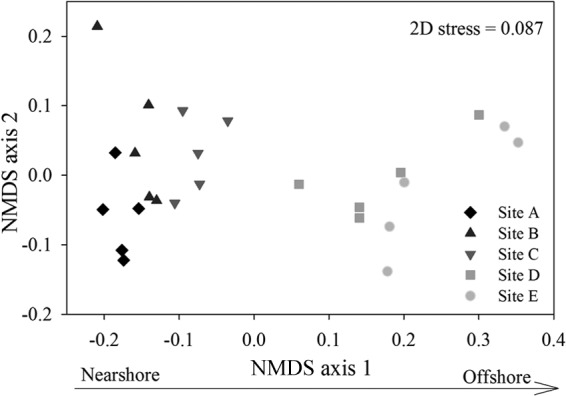
Nonmetric multidimensional scaling (NMDS) ordination of the dissimilarity (Bray-Curtis distance) in bacterial community composition (OTU level 0.03, NMDS stress = 0.09). Symbols are coded by sampling site. The shades of gray indicate the pollution gradient. 2D, 2 dimensional.
TABLE 1.
Community dissimilarity test based on analysis of similarity using Bray-Curtis distancea
| Site | A | B | C | D | E |
|---|---|---|---|---|---|
| A | |||||
| B | 0.0258 | ||||
| C | 0.0483 | 0.1410 | |||
| D | 0.0068 | 0.0084 | 0.0082 | ||
| E | 0.0071 | 0.0092 | 0.0068 | 0.1503 |
Values in boldface represent significant differences (P < 0.05) between bacterial communities in pairs of sampling sites.
Spatial distance decay of similarity for the bacterial community.
To test whether spatial distance was an important factor triggering the shifts in the bacterial community, a distance decay of similarity model was employed (14, 18). Due to minor distances among the biological samples within a given site, we calculated the average of community similarities between the samples within each pair of sites to represent the similarity between the two sites. The slope of the distance decay of similarity relationship was estimated by a linear regression between the similarity between the two sites and the corresponding geographic distance. As a result, a significant (r = −0.879, P = 0.002) distance decay of similarity relationship was observed, with a turnover of 0.002 (Fig. 3). Partial Mantel tests further supported the distance effect if the effect of the sediment environmental similarity matrix was held constant (P < 0.001). In addition, the overlaps of OTUs between sites were greater between adjacent sampling sites than between distant pairs and decreased consistently along the transect (see Table S2). Together, the results showed that spatial distance affects community structure, presumably due to dispersal limitation, even in this strongly interconnected ecosystem.
FIG 3.
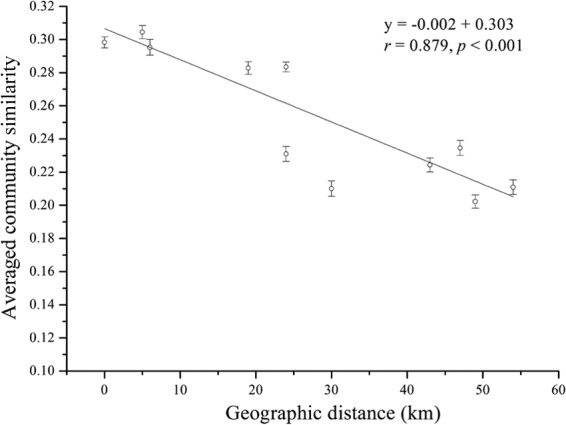
Correlation between community dissimilarities (Bray-Curtis distance) and geographic distances. Error bars represent means ± 1 standard error. Regression line, y = −0.002x + 0.313; r = 0.494; P < 0.001.
Linking bacterial community structure to sediment variables and geographic distances.
To better assess the effects of spatial distances (as a proxy for dispersal barriers) and sediment environmental parameters on the variations of the bacterial communities, we conducted a variance partitioning analysis of the combined PCNM output and illustrated our results using a modified variation partitioning diagram (Fig. 4). Two geochemical variables (no spatial correlation), sea depth (P < 0.001) and DIN (P = 0.005), and geographic distance (P < 0.001) were then selected for a subsequent variance partitioning analysis. Together, the whole set (environmental component, linear trend, and broad scale) of spatial and sediment environmental variables explained 19.4% of the variation in the undetrended (32) bacterial communities. The spatial variables (broad scale plus linear trend) alone explained 8.2% of the variations, and the sediment environmental variables explained 11.2% (among them, 5.7% of the variation was spatially structured) (Fig. 4). The interactions between environmental variables and spatial distance were moderate, indicating that the effects of spatial scales and environmental variables were to a certain extent dependent on each other. However, a large part (80.6%) of the variation was unexplained. Overall, both sediment properties and geographic distance are important factors shaping bacterial community structure.
FIG 4.
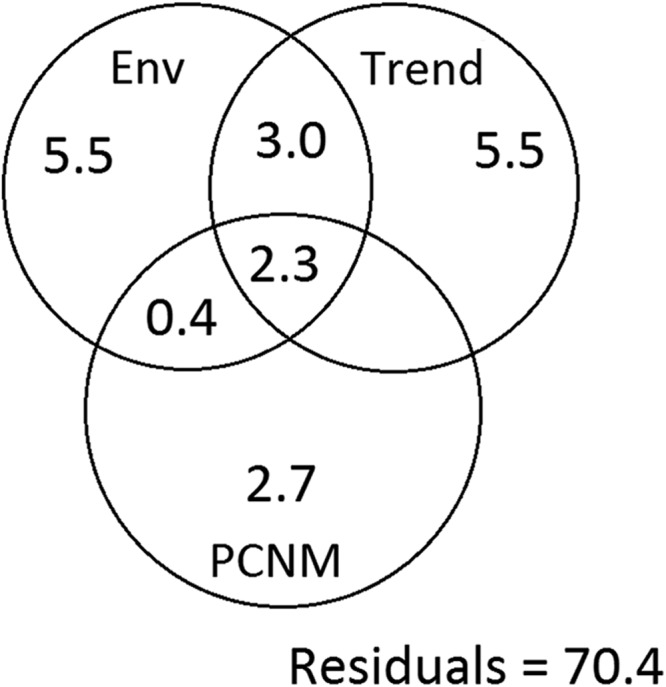
Variation partitioning by principal coordinates of neighbor matrices (PCNM)-based analysis for the phylogenetic structure of bacterial communities into an environmental component (top left), a linear trend (top right), and a broad-scale spatial pattern (bottom). A forward selection procedure was used to select the best PCNM variables (no spatial correlation) important to the bacterial community structure. Sediment environmental variables included dissolved inorganic nitrogen (DIN) and sea depth, which constrained 5.8% and 5.4% of the variation, respectively.
The abundances of taxa correlated with pollution.
At the highest level of taxonomic resolution, a total of 15 OTUs were detected in the sediment samples. These OTUs were primarily responsible for the differences (31.6% overall) in the bacterial communities among the sites (Fig. 5). These taxa were dominated by populations of the marine benthic group from the Deltaproteobacteria (members of Desulfobulbaceae and Desulfuromonadaceae) and Gammaproteobacteria (members of Xanthomonadales and Chromatiales). Cumulatively, these two groups of Proteobacteria contributed 14.2% and 12.8%, respectively, to the overall difference. A striking general pattern was that the abundance of members affiliated with the Deltaproteobacteria consistently increased along the pollution gradient, whereas those affiliated with the Gammaproteobacteria decreased (with the exception of OTUs 3200 and 37508) along the same gradient (Fig. 5). In addition, the abundances of these taxa were significantly correlated with the concentrations of DIN and TP along the pollution gradient (Table 2).
FIG 5.

Taxonomic identities of the 15 OTUs serving as indicator taxa along the pollution gradient, with their relative contributions to community dissimilarities. The diameters of the circles are proportional to the abundances of the OTUs, with the size of the circle indicating the average abundance (square root transformed) of each OTU at a given site.
TABLE 2.
Pearson correlation coefficients between the 15 indicator OTUs and pollution variablesa
| OTU | Taxa | DIN |
TP |
SO4 |
|||
|---|---|---|---|---|---|---|---|
| ρ | P | ρ | P | ρ | P | ||
| 36402 | Nitrospirae, Nitrospira, Thermodesulfovibrionaceae, GOUTA19 | −0.683 | <0.001 | −0.749 | <0.001 | ||
| 11599 | Chloroflexi, Anaerolineae, SB-34 | −0.645 | <0.001 | −0.750 | <0.001 | ||
| 20070 | Acidobacteria, OS-K | −0.507 | 0.009 | 0.397 | 0.049 | ||
| 28256 | Gemmatimonadetes, Gemm-2 | 0.429 | 0.032 | −0.457 | 0.022 | −0.665 | <0.001 |
| 20720 | Gammaproteobacteria, Xanthomonadales | 0.476 | 0.016 | −0.491 | 0.013 | −0.659 | <0.001 |
| 3200 | Gammaproteobacteria, Xanthomonadales | −0.518 | 0.008 | −0.616 | 0.001 | ||
| 37508 | Gammaproteobacteria, Xanthomonadales | −0.342 | 0.047 | 0.492 | 0.012 | 0.700 | <0.001 |
| 2574 | Gammaproteobacteria, Chromatiales | −0.485 | 0.014 | −0.561 | 0.004 | ||
| 41689 | Gammaproteobacteria, Chromatiales | 0.474 | 0.017 | ||||
| 2106 | Gammaproteobacteria | 0.477 | 0.016 | 0.620 | 0.001 | ||
| 19635 | Deltaproteobacteria, Syntrophobacterales, Desulfococcus | −0.562 | 0.003 | 0.473 | 0.017 | 0.664 | <0.001 |
| 35580 | Deltaproteobacteria, Syntrophobacterales | −0.645 | <0.001 | 0.504 | 0.010 | 0.674 | <0.001 |
| 28637 | Deltaproteobacteria, Desulfuromonadales, Desulfuromonadaceae | −0.422 | 0.036 | 0.545 | 0.005 | 0.806 | <0.001 |
| 41623 | Deltaproteobacteria, Desulfobacterales, Desulfobulbaceae | −0.500 | 0.011 | 0.611 | 0.001 | 0.874 | <0.001 |
| 24733 | Deltaproteobacteria, Desulfobacterales, Desulfobulbaceae | −0.481 | 0.015 | 0.566 | 0.003 | 0.744 | <0.001 |
Only the significant coefficients (P < 0.05) are listed.
DISCUSSION
The effects of heavy metal contamination gradients on microbial community structure and function are well established (9, 37). In contrast, far less attention has been paid to nutrient pollution gradients in marine coastal ecosystems, especially in terms of the apparent community effects. In this study, we analyze the relative effects of a pollution gradient and geographic distance (for historical contingencies, see references 19 and 18) on the fluctuations of the sediment bacterial community, with particular emphasis on the identification of bacterial taxa as indicators of such a pollution gradient.
Important factors influencing sediment bacterial community structure.
Anthropogenic effluents have produced a pollution gradient in this coastal zone; e.g., DIN increased consistently along the transect (Fig. 1). However, bacterial communities can respond rapidly to pollution (9, 37, 38). A high level of diversity is considered an important aspect of ecosystem health (39). However, the diversity did not change linearly along the pollution gradient studied, which had its peak diversity at the moderately polluted site C (see Fig. S3 in the supplemental material). This pattern is in line with the intermediate disturbance hypothesis, which states that a maximum of diversity is found in communities experiencing intermediate disturbance (40). It has been reported that diversity can recover due to the divergence and proliferation of tolerant species over time (37). Thus, diversity might be a poor indicator of ecosystem stress in long-term and chronically polluted ecosystems, whereas the bacterial community might serve as a good indicator.
The bacterial communities investigated in this study were dominated by Gammaproteobacteria, Deltaproteobacteria, and Planctomycetes. This dominance pattern is distinct from those found in lake sediments (18) and marine coastal waters (41). However, this pattern is similar to those found in other marine sediments worldwide (23, 37, 42), suggesting that a deterministic process (habitat specialization) has shaped the sediment bacterial community (14). We consistently observed robust dynamics of bacterial assemblages among sites (Fig. 2 and 3), and such variations were significantly correlated with DIN and sea depth (Fig. 4). Sea depth, which is most likely related to unmeasured variables, such as oxygen availability and pressure, explained 5.4% of the variation of the community, similar to the amount explained by the pollution parameter DIN (5.8%) (Fig. 4). Although it is widely accepted that depth is an important factor in controlling the vertical distribution of bacteria in the soil and in the water column (38, 43), our emphasis in this study is on the role of depth in a landscape, rather than along a vertical column.
Dispersal limitation in a strongly connected sediment ecosystem.
An issue relevant to our study and to many other biogeographic studies is whether spatial distance creates genetic variation (18, 34, 44). This information is essential to put microorganisms on the biogeographic map and, subsequently, to predict the associated functional relationships on the global scale (13, 20). We detected a close correlation between variation in the bacterial community and geographic distances (Fig. 3), even when a local geochemical role was excluded (P < 0.001, partial Mantel test). The overlaps of microbial assemblages were linearly decreased along the transect, resulting in relatively low similarities. Therefore, dispersal limitation could exist in a strongly connected marine sediment ecosystem such as this. In addition, the geographic distance explained more variation in community structure than any other individual factor (Fig. 4). The proportion of unexplained variation is comparable with that in a comprehensive biogeographic study (45). This result may be due to additional factors not measured in this study, such as the redox state of the sediment (46). However, some of the unexplained variation could also be due to ecologically neutral processes of diversification (i.e., evolutionary noise produced through random ecological drift) (39).
Many studies have shown that dispersal between distant environments is limited (14, 18). In contrast, water flow and tidal activity primarily govern the dispersal of bacteria and their transportation to the benthic zone (47). For these reasons, high dispersal rates (mass effects) in the marine sediment investigated in this study may produce a random distribution of bacterial composition (48). Thus, geographic distance would have minor or negligible effects on taxon diversification. However, this is not the case in the current study. One potential explanation of this outcome is that the dispersal capabilities of bacterial groups vary greatly (49), resulting in a legacy effect associated with geographic distance. Alternatively, the observed patterns may reflect the interactive effects of biotic and abiotic pressures, which impose variations in reproductive success across individuals and species (14, 50). Indeed, the importance of geographic distance in structuring bacterial communities does not erase the effect of local features in driving compositional responses. Consistently, we observed that local geochemical factors have a stronger effect on variation than geographic distance (Fig. 4), accompanied with a low spatial turnover rate (w = 0.002) (Fig. 3). This is in concert with the notion that dispersal limitation alone, however, is not enough to cause spatial turnover in community composition (50). In such a case, dispersed bacteria are filtered by new habitats (species sorting) or by competitive interactions with resident taxa (50, 51), resulting in the diversification of physiologically better adapted organisms (11). Overall, our results showed that the spatial distribution of bacterial communities is controlled not only indirectly, through complex trophic interactions, but also directly, by dispersal limitation. Thus, the study of global patterns in microbial communities should integrate the effects of dispersal limitation, even in highly connected marine ecosystems on a regional scale, to improve prediction models.
Bacterial taxa indicating the pollution gradient.
The samples from a range of levels of seawater pollution collected in this study allowed us to obtain bacterial taxa that showed sensitivity to the pollution gradient. As a result, we identified 15 abundant taxa that cumulatively contributed 31.7% of the variation among sample sites (Fig. 5). Thus, these taxa may be good indicator species for environmental monitoring; their abundances changed linearly along the pollution gradient and were significantly correlated with the pollution parameters measured (Table 2). The key indicator taxa displayed markedly different ecological strategies across environments (52). Thus, changes in their abundance may be associated with the ecological consequences of anthropogenic contamination. For instance, the candidate indicator taxa belonging to the Syntrophobacterales/Desulfuromonadales are strictly anaerobic genera belonging to the known sulfate-reducing bacteria (SRB) (53). In sulfate-rich marine sediments, SRB typically mineralize organic matter, coupled with sulfate reduction, which contributes up to 50% of the total organic mineralization in coastal ecosystems (53, 54). The abundances of these SRB are positively correlated with the concentrations of SO42−. This positive correlation implies that the SRB are, most likely, active there. Thus, the abundant and active SRB potentially play a substantial role in N remineralization in this region (6, 55). In contrast, the abundances of Chromatiales and Xanthomonadales (OTU 20720) (affiliated to Gammaproteobacteria) decreased consistently along the pollution gradient (Fig. 5). This pattern is in concert with the notion that the heterotrophic Gammaproteobacteria grow rapidly at high levels of marine nutrients (55). Although individual classes contain a range of phylogenetic diversity, the members from certain classes appear to share a consistent set of attributes that respond to pollution. In combination, these taxa might be applied to evaluate coastal pollution levels, at least in this case.
Supplementary Material
ACKNOWLEDGMENTS
This work was financially supported by the National High Technology Research and Development Program of China (863 Program, grant 2012AA092000), the Natural Science Foundation of Ningbo City (grant 2013A610169), Research Fund from 2011 Center of Modern Marine Aquaculture of East China Sea, and the KC Wong Magna Fund of Ningbo University.
Footnotes
Published ahead of print 10 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.03731-13.
REFERENCES
- 1.Galloway JN, Townsend AR, Erisman JW, Bekunda M, Cai Z, Freney JR, Martinelli LA, Seitzinger SP, Sutton MA. 2008. Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320:889–892. 10.1126/science.1136674 [DOI] [PubMed] [Google Scholar]
- 2.Mackenzie FT, Vera LM, Lerman A. 2002. Century-scale nitrogen and phosphorus controls of the carbon cycle. Chem. Geol. 190:13–32. 10.1016/S0009-2541(02)00108-0 [DOI] [Google Scholar]
- 3.Lunau M, Maren Voss M, Erickson M, Dziallas C, Casciotti K, Ducklow H. 2013. Excess nitrate loads to coastal waters reduces nitrate removal efficiency: mechanism and implications for coastal eutrophication. Environ. Microbiol. 15:1492–1504. 10.1111/j.1462-2920.2012.02773.x [DOI] [PubMed] [Google Scholar]
- 4.Jiao N, Herndl GJ, Hansell DA, Benner R, Kattner G, Wilhelm SW, Kirchman DL, Weinbauer MG, Luo T, Chen F, Azam F. 2010. Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat. Rev. Microbiol. 8:593–599. 10.1038/nrmicro2386 [DOI] [PubMed] [Google Scholar]
- 5.Grzymski JJ, Dussaq AM. 2012. The significance of nitrogen cost minimization in proteomes of marine microorganisms. ISME J. 6:71–80. 10.1038/ismej.2011.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dale AW, Prego R. 2002. Physico-biogeochemical controls on benthic-pelagic coupling of nutrient fluxes and recycling in a coastal upwelling system. Mar. Ecol. Prog. Ser. 235:15–28. 10.3354/meps235015 [DOI] [Google Scholar]
- 7.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. 10.1093/bioinformatics/btq461 [DOI] [PubMed] [Google Scholar]
- 8.Comte J, del Giorgio PA. 2011. Composition influences the pathway but not the outcome of the metabolic response of bacterioplankton to resource shifts. PLoS One 6:e25266. 10.1371/journal.pone.0025266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiong J, Wu L, Tu S, Van Nostrand JD, He Z, Zhou J, Wang G. 2010. Microbial communities and functional genes associated with soil arsenic contamination and rhizosphere of the arsenic hyper-accumulating plant Pteris vittata L. Appl. Environ. Microbiol. 76:7277–7284. 10.1128/AEM.00500-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Treseder KK, Kivlin SN, Hawkes CV. 2011. Evolutionary trade-offs among decomposers determine responses to nitrogen enrichment. Ecol. Lett. 14:933–938. 10.1111/j.1461-0248.2011.01650.x [DOI] [PubMed] [Google Scholar]
- 11.Allison SD, Lu Y, Weihe C, Goulden ML, Martiny AC, Treseder KK, Martiny JB. 2013. Microbial abundance and composition influence litter decomposition response to environmental change. Ecology 94:714–725. 10.1890/12-1243.1 [DOI] [PubMed] [Google Scholar]
- 12.Christiaen B, McDonald A, Cebrian J, Ortmann AC. 2013. Response of the microbial community to environmental change during seagrass transplantation. Aquat. Bot. 109:31–38. 10.1016/j.aquabot.2013.03.008 [DOI] [Google Scholar]
- 13.Martiny JB, Bohannan BJ, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, Krumins JA, Kuske CR, Morin PJ, Naeem S, Ovreås L, Reysenbach AL, Smith VH, Staley JT. 2006. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 4:102–112. 10.1038/nrmicro1341 [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, He J, Liu X, Zhang L, Zhang E. 2013. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J. 7:1310–1321. 10.1038/ismej.2013.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Kang S, Schadt CW, Garten CT. 2008. Spatial scaling of functional gene diversity across various microbial taxa. Proc. Natl. Acad. Sci. U. S. A. 105:7768–7773. 10.1073/pnas.0709016105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Finlay BJ. 2002. Global dispersal of free-living microbial eukaryote species. Science 296:1061–1063. 10.1126/science.1070710 [DOI] [PubMed] [Google Scholar]
- 17.Lozupone CA, Knight R. 2007. Global patterns in bacterial diversity. Proc. Natl. Acad. Sci. U. S. A. 104:11436–11440. 10.1073/pnas.0611525104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xiong J, Liu Y, Lin X, Zhang H, Zeng J, Hou J, Yang Y, Yao T, Knight R, Chu H. 2012. Geographic distance and pH drive bacterial distribution in alkaline lake sediments across Tibetan Plateau. Environ. Microbiol. 4:2457–2466. 10.1111/j.1462-2920.2012.02799.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ge Y, He JZ, Zhu YG, Zhang JB, Xu Z, Zhang LM, Zheng YM. 2008. Differences in soil bacterial diversity: driven by contemporary disturbances or historical contingencies? ISME J. 2:254–264. 10.1038/ismej.2008.2 [DOI] [PubMed] [Google Scholar]
- 20.Larsen PE, Field D, Gilbert JA. 2012. Predicting bacterial community assemblages using an artificial neural network approach. Nat. Methods 9:621–625. 10.1038/nmeth.1975 [DOI] [PubMed] [Google Scholar]
- 21.Li D, Daler D. 2004. Ocean pollution from land-based sources: East China Sea, China. Ambio 33:107–113. 10.1579/0044-7447-33.1.107 [DOI] [PubMed] [Google Scholar]
- 22.Wang JH, Wu JY. 2009. Occurrence and potential risks of harmful algal blooms in the East China Sea. Sci. Total Environ. 407:4012–4021. 10.1016/j.scitotenv.2009.02.040 [DOI] [PubMed] [Google Scholar]
- 23.Sun MY, Dafforn KA, Johnston EL, Brown MV. 2013. Core sediment bacteria drive community response to anthropogenic contamination over multiple environmental gradients. Environ. Microbiol. 15:2517–2531. 10.1111/1462-2920.12133 [DOI] [PubMed] [Google Scholar]
- 24.Biddle JF, Fitz-Gibbon S, Schuster SC, Brenchley JE, House CH. 2008. Metagenomic signatures of the Peru Margin subseafloor biosphere show a genetically distinct environment. Proc. Natl. Acad. Sci. U. S. A. 105:10583–10588. 10.1073/pnas.0709942105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. QIIME allows integration and analysis of high-throughput community sequencing data. Nat. Methods 7:335–336. 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267. 10.1093/bioinformatics/btp636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM. 2005. The Ribosomal Database Project (RDP-II): sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294–D296. 10.1093/nar/gki038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke KR. 1993. Non-parametric multivariate analysis of changes in community structure. Aust. J. Ecol. 18:117–143. 10.1111/j.1442-9993.1993.tb00438.x [DOI] [Google Scholar]
- 29.R Development Core Team 2013. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://cran.r-project.org/ [Google Scholar]
- 30.Legendre P, Legendre L. 1998. Numerical ecology. Elsevier, New York, NY [Google Scholar]
- 31.Rosenberg MS, Anderson CD. 2011. PASSaGE: pattern analysis, spatial statistics and geographic exegesis. Methods Ecol. Evol. 2:229–232. 10.1111/j.2041-210X.2010.00081.x [DOI] [Google Scholar]
- 32.Borcard D, Legendre P. 2002. All-scale spatial analysis of ecological data by means of principal coordinates of neighbour matrices. Ecol. Modell. 153:51–68. 10.1016/S0304-3800(01)00501-4 [DOI] [Google Scholar]
- 33.Dray S, Legendre P, Peres-Neto PR. 2006. Spatial modelling: a comprehensive framework for principal coordinate analysis of neighbour matrices (PCNM). Ecol. Modell. 196:483–493. 10.1016/j.ecolmodel.2006.02.015 [DOI] [Google Scholar]
- 34.Ramette A, Tiedje JM. 2007. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc. Natl. Acad. Sci. U. S. A. 104:2761–2766. 10.1073/pnas.0610671104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Legendre P, Gallagher ED. 2001. Ecologically meaningful transformations for ordination of species data. Oecologia 129:271–280. 10.1007/s004420100716 [DOI] [PubMed] [Google Scholar]
- 36.Blanchet FG, Legendre P, Borcard D. 2008. Forward selection of explanatory variables. Ecology 89:2623–2632. 10.1890/07-0986.1 [DOI] [PubMed] [Google Scholar]
- 37.Gillan DC, Danis B, Pernet P, Joly G, Dubois P. 2005. Structure of sediment-associated microbial communities along a heavy metal contamination gradient in the marine environment. Appl. Environ. Microbiol. 71:679–690. 10.1128/AEM.71.2.679-690.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiong J, He Z, Van Nostrand JD, Luo G, Tu S, Zhou J, Wang G. 2012. Assessing the microbial community and functional genes in a vertical soil profile with long-term arsenic contamination. PLoS One 7:e50507. 10.1371/journal.pone.0050507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Allison SD, Martiny JBH. 2008. Resistance, resilience, and redundancy in microbial communities. Proc. Natl. Acad. Sci. U. S. A. 105:11512–11519. 10.1073/pnas.0801925105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson JB. 1990. Mechanisms of species coexistence: twelve explanations for Hutchinson's ‘Paradox of the Plankton': evidence from New Zealand plant communities. N. Z. J. Ecol. 13:17–42 [Google Scholar]
- 41.Fortunato CS, Eiler A, Herfort L, Needoba JA, Peterson TD, Crump B. 2013. Determining indicator taxa across spatial and seasonal gradients in the Columbia River coastal margin. ISME J. 7:1899–1911. 10.1038/ismej.2013.79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gobet A, Böer SI, Huse SM, van Beusekom JEE, Quince C, Sogin ML, Boetius A, Ramette A. 2012. Diversity and dynamics of rare and of resident bacterial populations in coastal sands. ISME J. 6:542–553. 10.1038/ismej.2011.132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson CE, Alldredge AL, McCliment EA, Amaral-Zettler LA, Carlson CA. 2011. Depleted dissolved organic carbon and distinct bacterial communities in the water column of a rapid-flushing coral reef ecosystem. ISME J. 5:1374–1387. 10.1038/ismej.2011.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martiny JBH, Eisen JA, Penn K, Allison SD, Horner-Devine MC. 2011. Drivers of bacterial β-diversity depend on spatial scale. Proc. Natl. Acad. Sci. U. S. A. 108:7850–7854. 10.1073/pnas.1016308108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Van der Gucht K, Cottenie K, Muylaert K, Vloemans N, Cousin S, Declerck S, Jeppesen E, Conde-Porcuna JM, Schwenk K, Zwart G, Degans H, Vyverman W, De Meester L. 2007. The power of species sorting: local factors drive bacterial community composition over a wide range of spatial scales. Proc. Natl. Acad. Sci. U. S. A. 104:20404–20409. 10.1073/pnas.0707200104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liang Y, Van Nostrand JD, N′Guessan LA, Peacock AD, Deng Y, Long PE, Resch CT, Wu L, He Z, Li G, Hazen TC, Lovley DR, Zhou J. 2012. Microbial functional gene diversity with a shift of subsurface redox conditions during in situ uranium reduction. Appl. Environ. Microbiol. 78:2966–2972. 10.1128/AEM.06528-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Augspurger C, Karwautz C, Mußmann M, Daims H, Battin TJ. 2010. Drivers of bacterial colonization patterns in stream biofilms. FEMS Microbiol. Ecol. 72:47–57. 10.1111/j.1574-6941.2009.00830.x [DOI] [PubMed] [Google Scholar]
- 48.Mouquet N, Loreau M. 2003. Community patterns in source-sink metacommunities. Am. Nat. 162:544–557. 10.1086/378857 [DOI] [PubMed] [Google Scholar]
- 49.Verreydt D, De Meester L, Decaestecker E, Villena MJ, Van Der Gucht K, Vannormelingen P, Vyverman W, Declerck SA. 2012. Dispersal-mediated trophic interactions can generate apparent patterns of dispersal limitation in aquatic metacommunities. Ecol. Lett. 15:218–226. 10.1111/j.1461-0248.2011.01728.x [DOI] [PubMed] [Google Scholar]
- 50.Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, Rockhold ML, Konopka A. 2013. Quantifying community assembly processes and identifying features that impose them. ISME J. 7:2069–2079. 10.1038/ismej.2013.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Székely AJ, Berga M, Langenheder S. 2013. Mechanisms determining the fate of dispersed bacterial communities in new environments. ISME J. 7:61–71. 10.1038/ismej.2012.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fierer N, Bradford MA, Jackson RB. 2007. Toward an ecological classification of soil bacteria. Ecology 88:1354–1364. 10.1890/05-1839 [DOI] [PubMed] [Google Scholar]
- 53.Plugge CM, Zhang W, Scholten JC, Stams AJ. 2011. Metabolic flexibility of sulfate-reducing bacteria. Front. Microbiol. 2:81. 10.3389/fmicb.2011.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muyzer G, Stams AM. 2008. The ecology and biotechnology of sulphate-reducing bacteria. Nat. Microbiol. Rev. 6:441–454. 10.1038/nrmicro1892 [DOI] [PubMed] [Google Scholar]
- 55.Zhou J, He Q, Hemme CL, Mukhopadhyay A, Hillesland K, Zhou A, He Z, Van Nostrand JD, Hazen TC, Stahl DA, Wall JD, Arkin AP. 2011. How sulphate-reducing microorganisms cope with stress: lessons from systems biology. Nat. Rev. Microbiol. 9:452–466. 10.1038/nrmicro2575 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.