

Abstract
Acetoin reductase is an important enzyme for the fermentative production of 2,3-butanediol, a chemical compound with a very broad industrial use. Here, we report on the discovery and characterization of an acetoin reductase from Clostridium beijerinckii NCIMB 8052. An in silico screen of the C. beijerinckii genome revealed eight potential acetoin reductases. One of them (CBEI_1464) showed substantial acetoin reductase activity after expression in Escherichia coli. The purified enzyme (C. beijerinckii acetoin reductase [Cb-ACR]) was found to exist predominantly as a homodimer. In addition to acetoin (or 2,3-butanediol), other secondary alcohols and corresponding ketones were converted as well, provided that another electronegative group was attached to the adjacent C-3 carbon. Optimal activity was at pH 6.5 (reduction) and 9.5 (oxidation) and around 68°C. Cb-ACR accepts both NADH and NADPH as electron donors; however, unlike closely related enzymes, NADPH is preferred (Km, 32 μM). Cb-ACR was compared to characterized close homologs, all belonging to the “threonine dehydrogenase and related Zn-dependent dehydrogenases” (COG1063). Metal analysis confirmed the presence of 2 Zn2+ atoms. To gain insight into the substrate and cofactor specificity, a structural model was constructed. The catalytic zinc atom is likely coordinated by Cys37, His70, and Glu71, while the structural zinc site is probably composed of Cys100, Cys103, Cys106, and Cys114. Residues determining NADP specificity were predicted as well. The physiological role of Cb-ACR in C. beijerinckii is discussed.
INTRODUCTION
Clostridia are well known for their capacity to produce various acids and alcohols as end products of the anaerobic breakdown of different polysaccharides (1). These fermentation products are of interest as building blocks in the chemical industry or as biofuels. A renowned example, which has been studied extensively in the past, is the production of acetone, 1-butanol, and ethanol (ABE) by solventogenic clostridia. This process, also known as ABE fermentation, has been studied extensively in the past (2). In addition to ABE, some clostridial species, like Clostridium beijerinckii or Clostridium acetobutylicum, are able to produce isopropanol or acetoin, respectively (3). Acetoin can be converted to 2,3-butanediol via a single reduction step, catalyzed by acetoin reductase (ACR) (or 2,3-butanediol dehydrogenase [BDH]; EC 1.1.1.4). Acetoin and 2,3-butanediol are well-known metabolic end products of various microorganisms, and their biosynthesis in most cases involves pyruvate and α-acetolactate as intermediates (4, 5). Alternatively, both molecules may also be degraded by certain microorganisms (6). Although many microorganisms, including eukaryotes, are known to interconvert acetoin and 2,3-butanediol, only a few enzymes have actually been characterized in detail (7, 8). 2,3-Butanediol and especially the pure enantiomeric forms are very valuable for the synthesis of chiral high-value pharmaceuticals. Alternatively, 2,3-butanediol may be enzymatically converted into 2-butanol, a potential biofuel with the same energetic value as 1-butanol but with less toxicity to the fermenting organism (1).
Although wild-type C. acetobutylicum is known to produce acetoin (3), it is not able to produce 2,3-butanediol (9). However, heterologous expression of an acetoin reductase from C. beijerinckii enabled production of 2,3-butanediol, as has recently been shown by Siemerink et al. (10). Acetoin reductases are alcohol dehydrogenases (ADHs; EC 1.1.1), a large group of enzymes which are found in all three domains of life and which are capable of catalyzing the reversible reaction between aldehydes and ketones and their corresponding alcohols (11, 12). Clostridia harbor many putative ADH gene sequences, but the specificity of the corresponding enzymes can often not be concluded from the primary structure. Most characterized ACRs/BDHs belong to the superfamily of medium-chain dehydrogenases/reductases (MDRs), although some belong to the short-chain dehydrogenases (SDHs) (13).
This work reports on the discovery and detailed characterization of the acetoin reductase from C. beijerinckii NCIMB 8052, which was previously used to enable 2,3-butanediol production in C. acetobutylicum (10). The enzyme was selected from a set of eight putative acetoin reductases, six of which were heterologously expressed in Escherichia coli and analyzed for the desired activity. The selected enzyme was purified to homogeneity and characterized with respect to substrate specificity, temperature and pH optima, kinetics, and structural modeling. Its characteristics are compared to those of related characterized enzymes of the MDR superfamily.
MATERIALS AND METHODS
Materials used.
All chemicals were analytical grade and purchased from Sigma-Aldrich (unless stated otherwise). Primers were obtained from Biolegio (Nijmegen, The Netherlands). The restriction enzymes were obtained from Invitrogen (Breda, The Netherlands). For heterologous expression, the vectors pET24d and pET26d (kanamycin resistance; Merck Millipore, Darmstadt, Germany) were used.
Organisms and growth conditions.
E. coli DH5α (New England BioLabs, Ipswich, MA, USA) was used as a host for the cloning vectors, and E. coli BL21(DE3) was used as an overproduction strain to obtain recombinant protein for purification. All E. coli strains were grown at 37°C, 200 rpm, in Luria-Bertani (LB) medium supplemented with MgSO4 (1 mM), kanamycin (50 μg/ml), and ZnSO4 (250 μM) (unless stated otherwise). Additional zinc was added to ensure full occupancy of the zinc binding sites of the enzyme (14), and additional magnesium was added to increase E. coli biomass (15).
Identification and cloning of potential acetoin reductase-encoding genes.
The genome of C. beijerinckii NCIMB 8052 was screened for potential acetoin reductases by performing BLAST-P searches with the sequences from a set of characterized acetoin reductase/2,3-butanediol dehydrogenases from different organisms (Bacillus subtilis subsp. subtilis GI:16077691, Saccharomyces cerevisiae GI:330443361, Thermoanaerobacter brockii GI:1771790, and Klebsiella pneumoniae GI:1468939), including the closely related Gram-positive Bacillus cereus YUF-4 (GI:14475601) (http://www.ncbi.nlm.nih.gov/blast) (16, 17). The putative acetoin reductase genes, CBEI_0223, CBEI_0685, CBEI_1464 (C. beijerinckii ACR [Cb-ACR]), CBEI_2243, CBEI_3864, and CBEI_3890, were amplified by PCR on C. beijerinckii genomic DNA using the indicated primers (see Table S1 in the supplemental material). All PCR products were ligated into their corresponding pET24d or pET26b vector. The cloned genes did not contain a stop codon, resulting in a hexahistidine tag at the C terminus of the enzymes after expression. Chemically competent E. coli DH5α cells were transformed with the ligation mixtures according to the manufacturer's protocol and selected for kanamycin resistance. Colonies were checked by colony PCR, after which restriction analysis and sequence analysis confirmed the correct constructs. Chemically competent E. coli BL21(DE3) cells were transformed with the resulting pWUR plasmids (see Table S2 in the supplemental material).
Production and purification of heterologously expressed Cb-ACR.
For screening the candidate acetoin reductases, E. coli BL21(DE3), containing the different pWUR plasmids, was used to inoculate a 5-ml overnight LB medium culture with the mentioned supplements. This preculture was used to inoculate 50 ml LB medium in conical flasks and incubated for 8 h (37°C, 120 rpm). After 8 h of growth, the cultures were placed on ice for 1 h to induce host chaperones (18) and subsequently induced by adding 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG). E. coli cells were harvested after 15 h of growth at 20°C and resuspended in 20 mM Tris-HCl buffer, pH 7.5, containing 1 mM tris(2-carboxyethyl)phosphine (TCEP) as a reducing agent (19). After three cycles of sonication (20 W for 20 s; intermittent cooling on ice for 30 s), the total cell extract was centrifuged (30 min; 16,000 × g; 4°C), resulting in a clear cell extract. The crude cell extracts were used for screening purposes.
To overproduce Cb-ACR, a 2-liter culture of E. coli BL21(DE3) harboring pWUR450 was grown and induced as described above. The collected cell suspension was treated twice with a French pressure cell (110 MPa), centrifuged for 20 min (16,500 × g; 4°C), and passed through a 0.45-μm filter (Sartorius Stedim Biotech). The resulting cell extract was applied to a 20-ml Matrex Gel Red A affinity column (Amicon) equilibrated with 20 mM Tris-HCl buffer (pH 7.5) (buffer A). Unbound protein was removed by washing with 2 column volumes of buffer A, and the recombinant protein was eluted using a linear gradient of 0 to 2 M NaCl in the same buffer. The fractions containing Cb-ACR activity were pooled and applied to a Ni-chelating column (20 ml) equilibrated in buffer A, containing 300 mM NaCl with a flow rate of 2 ml min−1. Proteins were eluted with a linear gradient of 20 to 500 mM imidazole, and fractions (10 ml) were collected. The fractions with highest acetoin reductase activity were pooled and applied at a flow rate of 10 ml min−1 to a HiPrep desalting column (53 ml) (GE Healthcare Europe GmbH, Belgium), equilibrated in buffer A. The desalted fractions were supplemented with 1 mM TCEP and frozen at −20°C under anaerobic conditions.
Screening for acetoin reductase activity.
An initial screening for d/l-acetoin reduction was performed with crude cell extracts. Each reduction reaction was performed in a degassed reaction mixture containing 100 mM NaPi buffer (pH 6.5), 50 mM d/l-acetoin, and 0.28 mM NAD(P)H as described below. The following were included as controls: (i) crude cell extract of E. coli BL21(DE3) harboring an empty pET24d vector and (ii) a blank with no substrate. A correction was made for the temperature-dependent spontaneous degradation of NAD(P)H. The reactions in this screening were initiated by adding d/l-acetoin.
Acetoin reductase activity assays.
Alcohol oxidations and aldehyde/ketone reductions were determined by following either the reduction of NAD(P)+ or the oxidation of NAD(P)H at 340 nm, using a Hitachi U2010 spectrophotometer equipped with a temperature-controlled cuvette holder. Unless stated otherwise, all reactions were performed at 37°C under standard reaction conditions as described below. Each oxidation reaction was performed in a degassed reaction mixture containing 100 mM glycine-NaOH buffer (pH 9.5), 50 mM alcohol, and 0.28 mM NAD(P)+. Each reduction reaction was performed in a degassed reaction mixture containing 100 mM sodium phosphate buffer (pH 6.5), 50 mM aldehyde or ketone, and 0.28 mM NAD(P)H.
In all assays, the reaction was initiated by the addition of an appropriate amount of purified enzyme. One unit of Cb-ACR was defined as the amount of enzyme that oxidized or reduced 1 micromole of NAD(P)H or NAD(P)+ per minute, respectively. A correction was made for the (temperature-dependent) spontaneous degradation of NAD(P)H. The protein concentration was determined using the Bradford reagent (Bio-Rad) with bovine serum albumin as a standard (20).
Optimal pH and temperature.
The optimal pH of the reduction reaction was determined using a 100 mM NaPi buffer (pH 5.5 to 7.5). The oxidation reaction was determined using a 100 mM glycine-NaOH buffer (pH 8.0 to 10.0). Both reactions were done at 37°C. The optimal temperature of the oxidation and reduction reaction was determined (range, 20°C to 85°C) using the 100 mM NaPi buffer (pH 6.5) or the 100 mM glycine-NaOH buffer (pH 9.5), respectively. The pH of the buffers was set at 25°C, and temperature corrections were made using their temperature coefficients (±0.0028 pH/°C for sodium phosphate buffer and ±0.025 pH/°C for the glycine buffer).
Substrate specificity and kinetics.
The activity of purified Cb-ACR was tested on various potential substrates, in either the reductive or the oxidative direction, as described above for the general assay. Kinetic parameters were determined using acetoin as the substrate. Multiple measurements with d/l-acetoin were done (0.0244 mM, 0.0488 mM, 0.098 mM, 0.19 mM, 0.391 mM, 0.781 mM, 1.563 mM, 3.125 mM, 12.5 mM, and 50.0 mM) in the presence of 0.28 mM NAD(P)H. In addition, multiple measurements were performed with the cofactors NADH and NADPH (0.014 mM, 0.028 mM, 0.035 mM, 0.042 mM, 0.056 mM, 0.070 mM, 0.14 mM, and 0.28 mM). The Km and Vmax values were calculated by a computer-aided direct fit to the Michaelis-Menten equation. Variability is expressed as standard error of the mean. All reactions followed Michaelis-Menten-type kinetics.
Size exclusion chromatography.
The molecular mass of the native enzyme was determined by size exclusion chromatography on a Superdex 200 high-resolution 10/30 column (24 ml; GE Healthcare Europe GmbH, Belgium) equilibrated in 50 mM Tris-HCl containing 100 mM NaCl (pH 7.8). In total, 250 μl purified enzyme in 50 mM Tris-HCl buffer (pH 7.8) was applied to the column. Proteins used for calibration were Blue dextran 2000 (>2,000 kDa), aldolase (158 kDa), bovine serum albumin (67 kDa), ovalbumin (43 kDa), chymotrypsinogen (25 kDa), and RNase A (13.7 kDa).
Metal content.
The metal content of purified Cb-ACR was determined by inductively coupled plasma atomic emission spectroscopy (ICP-AES) using buffer A as a blank. The analyses included the metal ions Ca2+, Cd2+, Cu2+, Mg2+, Mn2+, Ni2+, and Zn2+. The effect of metal ions on acetoin reductase activity was determined using different metal salts (CaCl2, CoCl2, FeCl3, FeSO4, NiCl2, MgCl2, MnCl2, and ZnCl2) at a final concentration of 1 mM. To remove any divalent metals, the enzyme preparation was first preincubated in 1 mM EDTA at 37°C for 60 min, after which the EDTA was removed by a desalting column. Subsequently, the residual activity of the acetoin reductase was measured as described above. The activity of Cb-ACR without pretreatment with EDTA or addition of metal ions was defined as 100%.
Sequence and structure analysis.
The secondary structure of Cb-ACR was predicted using Psipred (21). Phylogenetic analysis was performed by aligning the amino acid sequence of Cb-ACR with the amino acid sequences of known characterized reductases (Table 1) using T-Coffee (22). A bootstrapped phylogenetic tree was constructed using Clustal W (23) and displayed using the neighbor-joining method with NJPlot 1.0 (24).
TABLE 1.
Overview of characterized acetoin reductases/2,3-butanediol dehydrogenases and threonine dehydrogenases
| Organism | GI (gene identifier) | COG | Molecular mass, kDa (no. of residues) | Quaternary structure | Activity for ACR/BDHa | Activity for TDHb | Coenzyme | PDB accession no. | Reference(s) |
|---|---|---|---|---|---|---|---|---|---|
| C. beijerinckii (Cb-ACR) | 149902809 | 1063 | 38.84 (360) | α2 | 100/0/99 | 0 | NADP | This paper | |
| B. cereus YUF-4 | 14475601 | 1063 | 38.10 (349) | α2 | +/+/− | NDd | NAD | 17 | |
| P. polymyxa | 317183731 | 1063 | 37.84 (350) | α2 | 100/0/72 | ND | NAD | 37 | |
| B. subtilis subsp. subtilis 168 | 16077691 | 1063 | 37.34 (346) | ND | 100/1/ND | ND | NAD | 52, 53 | |
| Mycobacterium strain B-009 | 327442548 | 1063 | 36.78 (350) | α2 | 100/4/61 | ND | NAD | 54 | |
| S. cerevisiae | 330443361 | 1063 | 41.45 (382) | α2 | 100/0/43 | NAD | 55 | ||
| T. kodakarensis | 57640851 | 1063 | 38.10 (350) | α4 | ND | + | NAD | 3GFB | 39, 56 |
| P. furiosus | 18893044 | 1063 | 37.82 (348) | α4 | + | + | NAD | 42 | |
| E. coli | 135560 | 1063 | 37.24 (341) | α4 | ND | + | NAD | 57 | |
| T. guaymasensis | 317411316 | 1063 | 39.53 (365) | α4 | 98/17/100 | − | NADP | 36 | |
| T. brockii | 1771790 | 1063 | 37.78 (353) | α4 | 100/1/ND | ND | NADP | 1YKF/1BXZ | 41, 53, 58 |
| C. beijerinckii (CbADH)c | 60592974 | 1063 | 37.72 (351) | α4 | 100/1/0 | ND | NADP | 1KEV | 41, 53, 58 |
| K. pneumoniae | 1468939 | 1028 | 26.64 (256) | α4 | 0/0/98 | ND | NAD | 1GEG | 13 |
Specificity of the 2,3-butanediol dehydrogenase activity for the d/l/meso stereoisomer is given in percent.
Threonine dehydrogenase activity.
This ADH from C. beijerinckii NRRL B593 has no homolog in C. beijerinckii NCIMB 8052, from which Cb-ACR was derived.
ND, not determined.
Structural modeling.
A structural model of Cb-ACR was built using the automatic protein structure prediction server of Phyre V2.0 with the modeling mode set to intensive (25). Intensive performs complete modeling of the entire protein using multiple templates and ab initio techniques. Ab initio techniques for structure prediction are generally unreliable, but they can provide valuable clues to structural features. Model reliability and accuracy were analyzed by QMEAN (26, 27). The QMEAN score ranges between 0 and 1 with higher values for better models. The QMEAN Z-score provides an estimate of the absolute quality of a model by relating it to reference structures solved by X-ray crystallography. Predicted models of low quality are expected to have strongly negative QMEAN Z-scores. The quality of the model was further analyzed using Ramachandran plot analysis (Procheck) (28) and WhatCheck (a subset of the WhatIf program) (29) (http://swift.cmbi.ru.nl/servers/html/index.html).
RESULTS AND DISCUSSION
Identification and cloning of potential acetoin reductase-encoding genes.
The well-studied solvent-producing C. acetobutylicum is also known to produce acetoin but not 2,3-butanediol (9). In an effort to have C. acetobutylicum produce 2,3-butanediol, a metabolic engineering approach was done previously, requiring the introduction of a heterologous acetoin reductase gene (10). The search for the desired ACR and the further characterization are described here. BLAST-P searches on the genome of another solventogenic clostridium, C. beijerinckii NCIMB 8052, using available ACR/BDH amino acid sequences, yielded a set of putative ACR/BDH genes. The highest scores were obtained with the acetylacetoin reductase/2,3-butanediol dehydrogenase gene from Bacillus cereus YUF-4 (17), which resulted in eight putative acetoin reductase-encoding genes, with identities ranging between 24% and 54%. These eight predicted acetoin reductases encoded by CBEI_0223, CBEI_0528, CBEI_0544, CBEI_0685, CBEI_1464, CBEI_2243, CBEI_3864, and CBEI_3890 are all annotated in NCBI (http://www.ncbi.nlm.nih.gov/) as alcohol dehydrogenase. CBEI_1464 showed the highest similarity (53%) to the B. cereus query and thus was the most likely candidate to possess acetoin reductase activity.
Six of the candidate acetoin reductase genes were successfully expressed in E. coli; the gene constructs of CBEI_0528 and CBEI_0544 failed to produce transformants in E. coli and were therefore no longer considered here. The initial screening in crude cell extracts showed that only CBEI_1464 (Cb-ACR) exhibited significant acetoin reductase activity with racemic acetoin as the substrate (Fig. 1). Minor acetoin reductase activity was also found with CBEI_0685, CBEI_2243, and CBEI_3864, although these activities hardly exceeded the background.
FIG 1.

Acetoin reductase activity of six potential acetoin reductases from C. beijerinckii NCIMB 8052. The genes were heterologously expressed in E. coli BL21(DE3). Acetoin reductase activity was measured in crude cell extracts.
In silico analysis of C. beijerinckii CBEI_1464.
C. beijerinckii CBEI_1464 encodes a protein of 360 amino acids and has a calculated molecular mass of approximately 38.8 kDa with a theoretical pI of 6.42 (http://img.jgi.doe.gov/cgi-bin/w/main.cgi). The protein belongs to COG1063 (cluster of orthologous groups of proteins), which comprises l-threonine dehydrogenase and related Zn-dependent dehydrogenases (http://www.ncbi.nlm.nih.gov/COG/). A BLAST-P analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) discloses many putative GroES-containing, Zn-binding alcohol dehydrogenases from the genera Clostridium and Bacillus. The best hits with characterized enzymes concern those from the Gram-positive species B. cereus YUF-4 (56% identity), Paenibacillus polymyxa (50% identity), B. subtilis subsp. subtilis (53% identity), and Mycobacterium strain B-009 (41% identity). Identification of the protein family and protein domains was performed using an Interpro scan from EMBL-EBI (http://www.ebi.ac.uk/interpro/). This scan confirmed that the protein is a member of the zinc-containing alcohol dehydrogenase superfamily (IPR002085), containing an N-terminal GroES-like alcohol dehydrogenase domain (IPR011032 and IPR013154) (amino acids [aa] 1 to 183), a conserved GroES-related zinc-binding domain (IPR002328) (aa 69 to 83), and an NAD(P)-binding alcohol dehydrogenase domain (IPR016040) (aa 175 to 295).
Purification of recombinant Cb-ACR.
Cb-ACR was successfully purified to homogeneity from cell extract of E. coli BL21(DE3) containing pWUR450 by using three subsequent chromatographic columns, namely, a Matrex Gel Red A affinity column, a Ni-chelating affinity column, and a HiPrep desalting column. SDS-PAGE analysis indicated an estimated molecular subunit mass of approximately 40 kDa (not shown). Size exclusion chromatography on a calibrated S200 column suggested that the quaternary structure of Cb-ACR is predominantly a homodimer, although a larger protein fraction, presumably representing tetramers, was also observed.
Optimal temperature and pH.
The effect of temperature on Cb-ACR activity was studied for both the reduction and the oxidation reaction. The activity increased from 20°C to 80°C (Fig. 2A), with a temperature optimum for the reduction and oxidation reaction of around 68°C. This is remarkable, since the original host, C. beijerinckii NCIMB 8052, is a mesophilic microorganism. However, it has been observed more often that ADHs from C. beijerinckii are relatively stable at moderate temperatures (30–32). Also, closely related thermophilic Clostridium species are known to possess proteins with activity optima at higher temperatures (33). An Arrhenius analysis resulted in a linear plot in the temperature range of 20°C to 60°C (Fig. 2B), with a calculated activation energy for the formation of the enzyme/substrate complex of 96 kJ/mol. The linearity of the plot indicates that the conformation of Cb-ACR does not change throughout this temperature range.
FIG 2.

Effect of temperature and pH on acetoin reductase activity. (A) Effect of temperature; 20°C to 85°C for the reduction reaction (◆) and 40°C to 80°C for the oxidation reaction (○). (B) Arrhenius plot of the temperature dependence of the reduction reaction from 20°C to 60°C. (C) Effect of pH; pH 5.5 to 7.5 for the reduction reaction (◆) and 8.0 to 10.0 for the oxidation reaction (○).
To determine the optimal pH for both the reduction of acetoin and the oxidation of 2,3-butanediol, the activity of Cb-ACR was measured in a pH range of 5.5 to 10.0 (Fig. 2C). Cb-ACR displayed >70% of its maximal activity in the pH range of 6 to 7, with an optimal pH at approximately 6.5 for the reduction of acetoin. For the oxidation of 2,3-butanediol, >90% of its maximal activity was in the pH range of 9 to 9.5, with an optimal pH at approximately 9.5.
Substrate specificity and kinetics.
Purified Cb-ACR was tested for multiple potential substrates. The specific reductase activity of Cb-ACR was found to be highest on d/l-acetoin (Table 2). Also, the conversions of diacetyl, 3-acetoxy-2-butanone, and acetol were shown to proceed with a relatively high activity (92%, 66%, and 46%, respectively). 2-Butanone showed a very low activity (2%). In the oxidative direction, the highest activities were found with diols with 2 adjacent hydroxyl groups, with d-2,3-butanediol and meso-2,3-butanediol performing best. The l-isomer of 2,3-butanediol did not show any activity. Some activity was also observed for 1,2-propanediol (24%), but compounds with more distant hydroxyl groups (1,3-propanediol and ethylene glycol) or a single hydroxyl group (2-butanol, 1-propanol, 2-propanol, and ethanol) showed no activity. Also, d- or l-threonine could not be used as a substrate. Altogether, these results indicate that enzyme activity requires a hydroxyl, keto, or acetoxy group, adjacent to the redox-active keto/hydroxyl group (Table 2 shows the structural formulas). The electronegative nature of the indicated groups suggests that they are probably involved in hydrogen bonding.
TABLE 2.
Substrate specificity of Cb-ACRa
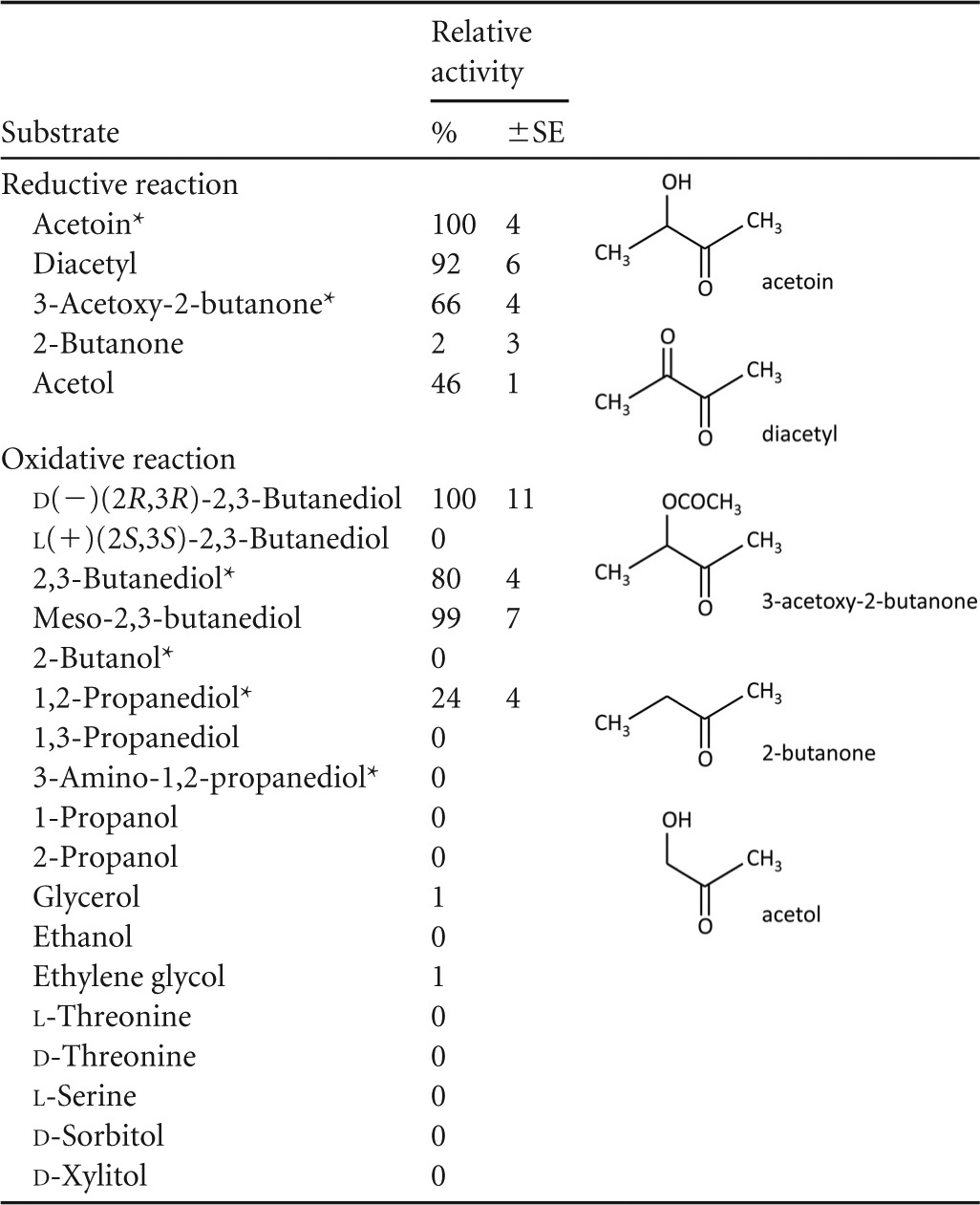
The specific activity of Cb-ACR in the reductive and oxidative direction is shown. Activities are expressed as percentages of the most active substrate. Standard errors based on 3 or more replicates are shown. Structural formulas of the most active substrates in the reductive direction are shown. An asterisk indicates a racemic mixture.
The specific activity of the d-2,3-butanediol dehydrogenation was 5.4-fold lower than the acetoin reduction (1.7 U/mg and 9.1 U/mg, respectively). With respect to the cofactor usage, Cb-ACR showed the highest activities with NADPH as the electron donor. A reasonable activity was, nevertheless, also obtained on NADH. The specific activity of the reaction with NADPH is about 2-fold higher and the apparent Km value for NADPH was ∼10-fold lower than those found for NADH (0.032 mM and 0.304 mM, respectively). This evidently results in ∼20-fold-higher catalytic efficiency for the NADPH-dependent reaction (Table 3). Using racemic acetoin and NADPH, specific activities amounted to ∼11 U/mg. The Km for acetoin (0.4 to 0.5 mM) is not significantly different when NADH or NADPH is used as the electron donor.
TABLE 3.
Kinetic data of purified Cb-ACR
| Reaction | Vmax (U · mg−1) |
Km (mM) |
kcat/Km NAD(P)H (s−1 · mM−1) | |
|---|---|---|---|---|
| Acetoin | NAD(P)H | |||
| Acetoin + NADPH | 10.6 ± 0.6 | 0.39 ± 0.11 | 0.032 ± 0.003 | 214 ± 23 |
| Acetoin + NADH | 5.41 ± 0.1 | 0.48 ± 0.05 | 0.304 ± 0.024 | 11.5 ± 0.94 |
Effect of metal ions and oxygen.
Based on the in silico analysis, Cb-ACR is expected to be a member of the zinc-containing alcohol dehydrogenase superfamily. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) confirmed the presence of approximately two zinc atoms and one calcium per monomer of Cb-ACR. Remarkably, the residual activity in the control after preincubation in the presence of EDTA remained 100%. This suggests that the zinc molecules are tightly bound to the Cb-ACR or that they do not have a catalytic role. Addition of most salts and metals caused no significant inhibition or activation of Cb-ACR. Further research is needed to explain the presence of the calcium atom. Addition of extra calcium did not result in differences in specific activities.
Furthermore, Cb-ACR was found to be oxygen sensitive, with a half-life of approximately 6 h (aerobic at 0°C). However, addition of common reducing agents like dithiothreitol (DTT) or β-mercaptoethanol caused a strong inhibition. Possibly, these compounds (respectively, C4 and C2) mimic the substrate acetoin and as such occupy the active site, resulting in inhibition. Moreover, both mercaptoethanol and DTT contain an electronegative group (S) adjacent to the carbon containing the hydroxyl group (see below), as was also seen for the diols, discussed above. Inhibition by β-mercaptoethanol has been described also for the meso-BDH of K. pneumoniae, where the crystal structure revealed that mercaptoethanol specifically binds to the active site, forming hydrogen bonds to Gln140 and Gly183 (34). Also, for DTT and other thiols, strong inhibition has been reported for a sheep liver sorbitol dehydrogenase, where DTT forms a tight ternary complex with the enzyme-NAD complex (35). When tris(2-carboxyethyl)phosphine (TCEP) was used as a reducing agent, inhibition did not occur (19). In the presence of TCEP, the oxygen sensitivity was severely diminished, especially when stored at −20°C.
The oxygen-sensitive nature of the Cb-ACR might be caused by one or more of the 8 cysteines present in the primary sequence. However, all known ACRs contain several cysteine residues (ranging from 3 to 11), while a sensitivity to oxygen has never been reported, except for the ACR/BDH from the archaeon Thermococcus guaymasensis (36). Moreover, as discussed above, several of the cysteine residues are proposed to be involved in binding of the zinc ions. These residues are therefore conserved in ACRs, including those which are not oxygen sensitive. Cys123 and Cys191 are, however, unique to Cb-ACR (Fig. 3) and may therefore be responsible for the observed oxygen sensitivity. Also, the ACR/BDH from T. guaymasensis contains a cysteine in an unusual position, possibly explaining its oxygen sensitivity (36).
FIG 3.

Multiple sequence alignment of Cb-ACR and other characterized close homologs. Gene identifiers of the genes can be found in Table 1. The sequence of the ketose reductase of Bemisia argentifolii (GI:4106363) was included because a three-dimensional structure was available (PDB 1E3J), which was used for the modeling of Cb-ACR. The sequences were aligned using the web-based T-Coffee program (22). Conserved residues involved in metal and NAD(P) binding are shaded in black. Asterisks indicate residues involved in binding of the catalytic and structural zinc. The secondary structure prediction of Cb-ACR (using the psipred program [59]) is shown above the alignment.
Cb-ACR in comparison to other characterized ACRs/BDHs.
Although the currently available genome sequences harbor hundreds of closely related representatives of the zinc-containing alcohol dehydrogenase superfamily, only a limited number of ACRs/BDHs has been characterized in more detail. All closely related ACRs/BDHs are highly specific for the d-isomer (2R,3R) of 2,3-butanediol (Table 1). The more distantly related and smaller ACRs/BDHs from K. pneumoniae and homologs (not discussed in detail here) have been shown to be specific for meso-2,3-butanediol or the l-isomer (2S,3S) (13). As discussed recently by Yu et al., these clear differences in specificity can be associated with different superfamilies, viz., the medium-chain dehydrogenase/reductase and the short-chain dehydrogenase/reductase superfamily (37). All of the medium-chain dehydrogenase/reductase representatives discussed here belong to COG1063 (Table 1), covering “threonine dehydrogenase and related Zn-dependent dehydrogenases.” According to the classification of Riveros-Rosas et al., COG1063 represents one of the families (polyol dehydrogenase) within macrofamily I and one which can be subdivided into 12 subfamilies, involved in various metabolic roles (38).
The alignment (Fig. 3) encompasses the closest orthologs, including a set of archaeal threonine dehydrogenases which belong to the same COG (1063). Conserved residues (shown with asterisks in the alignment) are those involved in binding of a catalytic zinc atom (Cys37, His70, and Glu71) and those involved in NAD(P)+ binding (G180XG182XXG185), as suggested by the identified Interpro domains. In addition to the residues that bind the catalytic zinc, a set of 4 conserved cysteines (Cys100, Cys103, Cys106, and Cys114) is present in most sequences, which has been suggested to constitute the binding site of a structural zinc atom (12). This structural zinc binding site is, however, not present in the ACR/BDH (ydjL) of B. subtilis subsp. subtilis 168, which is closely related to the Cb-ACR. This observation is remarkable, as its primary sequence nicely aligns, except for the four missing cysteines. As yet, we have no explanation for this. Also, the BDHs of T. brockii, T. guaymasensis, and another C. beijerinckii strain (NRRL B593) do not contain these cysteine residues. The latter BDHs constitute a more distantly related group, as can be seen in the phylogenetic comparison (Fig. 4). In addition to the absence of the structural zinc, this group has also been shown to prefer NADP over NAD (Table 1). Structure analysis has indicated that the NADP specificity is determined by the residues Gly198, Ser199, Arg200, and Tyr218 (T. brockii numbering) (39–41). Indeed, most other sequences shown here contain other residues at these positions, e.g., the Gly is usually replaced by a Glu (Fig. 3). In this respect, it is remarkable that the Cb-ACR has a preference for NADP as well but does not contain the indicated residues. The gene sequence of Cb-ACR contains a Gln instead of the Gly or Glu.
FIG 4.

Phylogenetic tree of characterized ACRs/BDHs. Amino acid sequences used for the multiple alignment were also used to construct a phylogenetic tree, except that sequences of ACRs/BDHs that belong to a different superfamily, viz., K. pneumoniae and Corynebacterium glutamicum, were added. The tree includes sequences of archaeal, bacterial, and eukaryotic origin, revealing the broad phylogenetic spreading of this group of enzymes. The different clades of NAD- and NADP-dependent enzymes and threonine dehydrogenases are indicated.
Based on the alignment (Fig. 3), the phylogenetic tree (Fig. 4), and the data in Table 1, we show that the medium-chain ACRs/BDHs (COG1063) can be subdivided into three clades, which differ in cofactor specificity, quaternary structure, and catalytic activity. T. guaymasensis, T. brockii, and C. beijerinckii (CbADH) require NADP(H) and have an α4 tetrameric structure. The archaeal enzymes from Pyrococcus furiosus and Thermococcus kodakarensis and the one from E. coli also have an α4 structure but use NAD(H) and are able to convert threonine. These might be true threonine dehydrogenases, although the enzyme from P. furiosus also shows high activity on 2,3-butanediol (42). A third clade contains the Cb-ACR and related enzymes. All have a dimeric α2 structure, and all are specific for NAD(H), except for the Cb-ACR, which can use NADP(H) as well (Table 1). Cb-ACR shows no activity on threonine. For the other members of this clade, threonine has not been tested. Thus, whether the absence of threonine dehydrogenase activity also holds for the other clade members remains to be investigated.
Structural modeling.
Until now, no structure has been determined for any acetoin reductase of this superfamily. Hence, a structural model of Cb-ACR was built using the automatic protein structure prediction server of Phyre V2.0. Multiple fits with structural similarities were found, and based on heuristics to maximize confidence, percent identity, and alignment coverage, six templates (PDB 1e3j, 1pl6, 1kol, 4a10, 2dph, and 4a2c) were selected and used to model Cb-ACR. In addition, 4 residues were modeled ab initio. In the final model, 99% of residues were modeled at >90% confidence. Model reliability and accuracy were checked by QMEAN, resulting in the following scores: QMEAN score, 0.691; Z-score, −0.59. The QMEAN analysis included an image of the structure showing the regions with high error values. The majority of the model had low-error values, but three loops showed higher error values: Gly47-Pro65, Arg205-Gln210, and Pro224-Ala230. The Ramachandran plot (not shown) indicated that 96.3% of the residues are in the most favored regions and additionally allowed regions. WhatCheck analysis showed that bond lengths and angles were found to deviate normally from the standard bond lengths and angles. Torsion angle evaluation showed only a few unusual residues. The backbone torsion angle evaluation, however, showed unusual conformations in loop Gly47-Pro65. Apparently, this loop is difficult to model and decreases the quality of the model. Although there are some concerns about the details of the structural model, we consider the obtained model useful to gain insight into the three-dimensional structure of Cb-ACR.
The Cb-ACR model structure has an overall fold very similar to that of other ADHs, despite sharing less than 35% protein sequence identity with those ADHs for which structures are known. As already concluded from the in silico analysis, the Cb-ACR structural model (Fig. 5) is comprised of two domains, a cofactor binding domain (residues 160 to 297) and a catalytic domain (residues 1 to 159 and 298 to 360), which are separated by a deep cleft. The cofactor binding domain has the characteristic α/β NAD(P) binding Rossmann fold composed of 6 β-strands and 6 accompanying α-helices (Fig. 5A) (43). The catalytic domain also has an α/β-fold similar to that observed in other ADHs. By analogy to the Bemisia argentifolii ketose reductase (Ba-KR) (PDB 1e3j) (44), which was one of the templates used for the modeling, we can propose that Cb-ACR contains a catalytic zinc atom in the putative active site, which is located in the catalytic domain at the bottom of the cleft, and a structural zinc atom coordinated by four cysteine residues (Cys100, Cys103, Cys106, and Cys114). Furthermore, as in the Ba-KR, the Cb-ACR catalytic zinc is bound by the side chains of Cys37, His70, Glu71, and a water molecule, in a variation of the zinc site generally found in ADHs. In addition, the proposed adenyl phosphate moiety (of NADP) binding site in Ba-KR is also conserved in the Cb-ACR structure (Fig. 5B) and consists of Arg205 and Arg209, which is in agreement with the observed preference of Cb-ACR for NADP. Also, the residues of the conserved NAD(P) binding motif GxGxxG (Fig. 3), composed of Gly180, Gly182, and Gly185, are accessible in the model and enable binding of the nucleotide.
FIG 5.

Three-dimensional model of Cb-ACR. (A) Overall structure of Cb-ACR. Residues Cys37, His70, and Glu71 of the catalytic site are indicated. The catalytic and structural zinc molecules (purple spheres) were modeled into the structure using a superposition with B. argentifolii Ba-KR ADH (PDB 1e3j) (44). The four cysteines (Cys100, Cys103, Cys106, and Cys114) binding the structural zinc and the conserved arginines (Arg205 and Arg209) involved in binding the adenyl phosphate moiety of NADP are indicated. (B) Surface representation of Cb-ACR model. An NADP molecule was modeled into the structure using a superposition with T. brockii TBADH (PDB 1ykf) (41), giving an indication of how NADP is bound. These images were generated using PyMOL (PyMOL Molecular Graphics System, Version 1.2r3pre; Schrödinger, LLC).
Cofactor specificity.
With respect to the cofactor usage, Cb-ACR showed the highest activities with NADPH as electron donor. The preference for NADP(H) is rather unexpected. All close homologs prefer NAD(H) over NADP(H) (Table 1), and NADP(H) dependence has been shown only for the more distantly related ACRs/BDHs from T. guaymasensis, T. brockii, and C. beijerinckii (Table 1; Fig. 4). In B. cereus YUF-4, two acetylacetoin reductases (AACRI and AACRII) have been described, with AACRI showing a preference for NADP(H) and AACRII showing a preference for NAD(H) (45). However, for AACRI no amino acid sequence is known, making it impossible to assess its relatedness to Cb-ACR. But according to the properties described by Hosaka et al. (45), AACRI is likely to belong to a different ACR/BDH family. NAD(H)-dependent AACRII, evidently, resembles the Cb-ACR, as it was used as a query to screen the C. beijerinckii genome. The unusual preference of Cb-ACR for NADP(H) might be a reflection of a different physiological role, e.g., the NADPH dependence suggests that Cb-ACR might fulfill an anabolic rather than a catabolic role.
The structural basis for the preference of NADP over NAD has been discussed by Baker et al. (46). The primary determinant is the residue at position 204 (CBEI_1464 numbering). A small and neutral residue at this position (G, A, or S) leaves room for the phosphate group of NADP, whereas dehydrogenases specific for NAD have a larger negatively charged residue (E or D) at this position. Indeed, the alignment shown in Fig. 3 appears to confirm this theory. All NAD(H)-dependent ACRs/BDHs have an E or a D at the indicated position, while the NAD(P)-dependent enzymes have a G. Cb-ACR is somewhat unusual in this respect, as it has a bulky glutamine (Q) at position 204. Apparently, the absence of a negative charge is sufficient to accommodate NADP(H). It has been suggested that the nearby presence of one or two basic residues is also important for NADP(H) specificity (47). This is indeed the case for the Cb-ACR, where the Gln204 is followed by two arginines, Arg205 and Arg209. Also, the NADP(H)-dependent Ba-KR from B. argentifolii, whose structure was used to construct the Cb-ACR structural model, contains a neutral alanine at the determining position and also two arginines (Arg200 and Arg204) in the vicinity.
Possible physiological and industrial role of Cb-ACR.
As to the physiological role of Cb-ACR, it is interesting that CBEI_1464 clusters with a putative oxidoreductase (CBEI_1466) and a sigma54-specific transcriptional regulator from the Fis family (CBEI_1463). Several gene pairs that are homologous to the genes encoding CBEI_1463 and Cb-ACR are conserved within several species of the Firmicutes phylum. A BLAST-P search indicated that CBEI_1463 is similar to the aco operon expression regulatory protein (AcoR) of Alcaligenes eutrophus H16 and Bacillus subtilis (48, 49). The aco operon is known to code for subunits of an acetoin dehydrogenase, involved in the catabolism of acetoin as a carbon source. B. subtilis secretes acetoin in the medium, but after depletion of an external C source, it is able to use acetoin as a carbon source again by expressing its aco operon (48). The expression of this aco operon is regulated by acoR, which is highly similar to CBEI_1463. In Clostridium magnum, an acetoin dehydrogenase enzyme system, resembling the one encoded by the aco operon of B. subtilis, is described, which has also a similar AcoR-like protein (50). The presence of CBEI_1463 suggests that Cb-ACR of C. beijerinckii might be involved in the degradation of 2,3-butanediol to acetoin, which is then catabolized as in C. magnum and B. subtilis. However, an acetoin dehydrogenase complex as found in C. magnum and B. subtilis is not present in C. beijerinckii. A recent genome-wide transcriptional analysis of C. beijerinckii by whole-transcriptome shotgun sequencing (RNA-Seq) technology revealed that CBEI_1464 is significantly expressed, especially during the solventogenic phase (51). This suggests that Cb-ACR might have a role in the formation of an alcohol. Alternatively, as discussed above, the NADP-dependence points to a more anabolic role of Cb-ACR. C. beijerinckii is, however, not able to produce acetoin, which agrees with the fact that a gene for an acetolactate decarboxylase is missing. Altogether, this leaves the role of Cb-ACR in C. beijerinckii enigmatic.
Unlike C. beijerinckii, C. acetobutylicum does have the capacity to produce acetoin, but not 2,3-butanediol (2). By expression of CBEI_1464 in C. acetobutylicum, it has recently been shown that Cb-ACR indeed accomplishes in vivo the stoichiometric conversion of acetoin to 2,3-butanediol (10). Future research continues on the use of the engineered C. acetobutylicum (Cb-ACR) strain. Its capacity to produce 2,3-butanediol is a first step toward a 2-butanol-producing strain. 2-Butanol is a potential biofuel, with an energy content similar to that of 1-butanol but with less toxicity to the producing strain (1).
Supplementary Material
ACKNOWLEDGMENTS
This project is financially supported by the Netherlands Ministry of Economic Affairs and the B-Basic partner organizations (www.b-basic.nl) through B-Basic, a public-private NWO-ACTS (Advanced Chemical Technologies for Sustainability) program.
Footnotes
Published ahead of print 17 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.04007-13.
REFERENCES
- 1.Lopez Contreras AM, Kuit W, Siemerink MAJ, Kengen SWM, Springer J, Claassen PAM. 2010. Production of longer-chain alcohols from biomass—butanol, isopropanol and 2,3-butanediol, p 415–460 In Waldron K. (ed), Bioalcohol production. Woodhead Publishing Ltd, Cambridge, United Kingdom [Google Scholar]
- 2.Jones DT, Woods DR. 1986. Acetone-butanol fermentation revisited. Microbiol. Rev. 50:484–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doremus MG, Linden JC, Moreira AR. 1985. Agitation and pressure effects on acetone-butanol fermentation. Biotechnol. Bioeng. 27:852–860. 10.1002/bit.260270615 [DOI] [PubMed] [Google Scholar]
- 4.Ji XJ, Huang H, Ouyang PK. 2011. Microbial 2,3-butanediol production: a state-of-the-art review. Biotechnol. Adv. 29:351–364. 10.1016/j.biotechadv.2011.01.007 [DOI] [PubMed] [Google Scholar]
- 5.Xiao Z, Xu P. 2007. Acetoin metabolism in bacteria. Crit. Rev. Microbiol. 33:127–140. 10.1080/10408410701364604 [DOI] [PubMed] [Google Scholar]
- 6.Schink B. 1984. Fermentation of 2,3-butanediol by Pelobacter carbinolicus sp-nov and Pelobacter propionicus sp-nov, and evidence for propionate formation from C-2 compounds. Arch. Microbiol. 137:33–41. 10.1007/BF00425804 [DOI] [Google Scholar]
- 7.Hohn-Bentz H, Radler F. 1978. Bacterial 2,3-butanediol dehydrogenases. Arch. Microbiol. 116:197–203. 10.1007/BF00406037 [DOI] [PubMed] [Google Scholar]
- 8.Syu MJ. 2001. Biological production of 2,3-butanediol. Appl. Microbiol. Biotechnol. 55:10–18. 10.1007/s002530000486 [DOI] [PubMed] [Google Scholar]
- 9.Wardwell SA, Yang YT, Chang HY, San KY, Rudolph FB, Bennett GN. 2001. Expression of the Klebsiella pneumoniae CG21 acetoin reductase gene in Clostridium acetobutylicum ATCC 824. J. Ind. Microbiol. Biotechnol. 27:220–227. 10.1038/sj.jim.7000179 [DOI] [PubMed] [Google Scholar]
- 10.Siemerink MAJ, Kuit W, Lopez Contreras AM, Eggink G, van der Oost J, Kengen SWM. 2011. d-2,3-Butanediol production due to heterologous expression of an acetoin reductase in Clostridium acetobutylicum. Appl. Environ. Microbiol. 77:2582–2588. 10.1128/AEM.01616-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radianingtyas H, Wright PC. 2003. Alcohol dehydrogenases from thermophilic and hyperthermophilic archaea and bacteria. FEMS Microbiol. Rev. 27:593–616. 10.1016/S0168-6445(03)00068-8 [DOI] [PubMed] [Google Scholar]
- 12.Reid MF, Fewson CA. 1994. Molecular characterization of microbial alcohol dehydrogenases. Crit. Rev. Microbiol. 20:13–56. 10.3109/10408419409113545 [DOI] [PubMed] [Google Scholar]
- 13.Ui S, Okajima Y, Mimura A, Kanai H, Kobayashi T, Kudo T. 1997. Sequence analysis of the gene for and characterization of D-acetoin forming meso-2,3-butanediol dehydrogenase of Klebsiella pneumoniae expressed in Escherichia coli. J. Ferment. Bioeng. 83:32–37. 10.1016/S0922-338X(97)87323-0 [DOI] [Google Scholar]
- 14.Brouns SJ, Turnbull AP, Willemen HL, Akerboom J, van der Oost J. 2007. Crystal structure and biochemical properties of the D-arabinose dehydrogenase from Sulfolobus solfataricus. J. Mol. Biol. 371:1249–1260. 10.1016/j.jmb.2007.05.097 [DOI] [PubMed] [Google Scholar]
- 15.Shiloach J, Fass R. 2005. Growing E. coli to high cell density—a historical perspective on method development. Biotechnol. Adv. 23:345–357. 10.1016/j.biotechadv.2005.04.004 [DOI] [PubMed] [Google Scholar]
- 16.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hosaka T, Ui S, Ohtsuki T, Mimura A, Ohkuma M, Kudo T. 2001. Characterization of the NADH-linked acetylacetoin reductase/2,3-butanediol dehydrogenase gene from Bacillus cereus YUF-4. J. Biosci. Bioeng. 91:539–544. 10.1016/S1389-1723(01)80170-5 [DOI] [PubMed] [Google Scholar]
- 18.Brouns SJ, Smits N, Wu H, Snijders AP, Wright PC, de Vos WM, van der Oost J. 2006. Identification of a novel alpha-galactosidase from the hyperthermophilic archaeon Sulfolobus solfataricus. J. Bacteriol. 188:2392–2399. 10.1128/JB.188.7.2392-2399.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Getz EB, Xiao M, Chakrabarty T, Cooke R, Selvin PR. 1999. A comparison between the sulfhydryl reductants tris(2-carboxyethyl)phosphine and dithiothreitol for use in protein biochemistry. Anal. Biochem. 273:73–80. 10.1006/abio.1999.4203 [DOI] [PubMed] [Google Scholar]
- 20.Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248–254. 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 21.McGuffin LJ, Bryson K, Jones DT. 2000. The PSIPRED protein structure prediction server. Bioinformatics 16:404–405. 10.1093/bioinformatics/16.4.404 [DOI] [PubMed] [Google Scholar]
- 22.Notredame C, Higgins DG, Heringa J. 2000. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302:205–217. 10.1006/jmbi.2000.4042 [DOI] [PubMed] [Google Scholar]
- 23.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 24.Perriere G, Gouy M. 1996. WWW-query: an on-line retrieval system for biological sequence banks. Biochimie 78:364–369. 10.1016/0300-9084(96)84768-7 [DOI] [PubMed] [Google Scholar]
- 25.Kelley LA, Sternberg MJE. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 26.Benkert P, Kunzli M, Schwede T. 2009. QMEAN server for protein model quality estimation. Nucleic Acids Res. 37:W510–W514. 10.1093/nar/gkp322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benkert P, Tosatto SCE, Schomburg D. 2008. QMEAN: a comprehensive scoring function for model quality assessment. Proteins Struct. Funct. Bioinformatics 71:261–277. 10.1002/prot.21715 [DOI] [PubMed] [Google Scholar]
- 28.Laskowski RA, Macarthur MW, Moss DS, Thornton JM. 1993. Procheck—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26:283–291. 10.1107/S0021889892009944 [DOI] [Google Scholar]
- 29.Hooft RWW, Vriend G, Sander C, Abola EE. 1996. Errors in protein structures. Nature 381:272. 10.1038/381272a0 [DOI] [PubMed] [Google Scholar]
- 30.Bogin O, Peretz M, Hacham Y, Korkhin Y, Frolow F, Kalb AJ, Burstein Y. 1998. Enhanced thermal stability of Clostridium beijerinckii alcohol dehydrogenase after strategic substitution of amino acid residues with prolines from the homologous thermophilic Thermoanaerobacter brockii alcohol dehydrogenase. Protein Sci. 7:1156–1163. 10.1002/pro.5560070509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ismaiel AA, Zhu CX, Colby GD, Chen JS. 1993. Purification and characterization of a primary-secondary alcohol dehydrogenase from two strains of Clostridium beijerinckii. J. Bacteriol. 175:5097–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li C, Heatwole J, Soelaiman S, Shoham M. 1999. Crystal structure of a thermophilic alcohol dehydrogenase substrate complex suggests determinants of substrate specificity and thermostability. Proteins 37:619–627. [DOI] [PubMed] [Google Scholar]
- 33.Demain AL, Newcomb M, Wu JH. 2005. Cellulase, clostridia, and ethanol. Microbiol. Mol. Biol. Rev. 69:124–154. 10.1128/MMBR.69.1.124-154.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otagiri M, Kurisu G, Ui S, Takusagawa Y, Ohkuma M, Kudo T, Kusunoki M. 2001. Crystal structure of meso-2,3-butanediol dehydrogenase in a complex with NAD+ and inhibitor mercaptoethanol at 1.7 A resolution for understanding of chiral substrate recognition mechanisms. J. Biochem. 129:205–208. 10.1093/oxfordjournals.jbchem.a002845 [DOI] [PubMed] [Google Scholar]
- 35.Lindstad RI, McKinley-McKee JS. 1996. Reversible inhibition of sheep liver sorbitol dehydrogenase by thiol compounds. Eur. J. Biochem. 241:142–148. 10.1111/j.1432-1033.1996.0142t.x [DOI] [PubMed] [Google Scholar]
- 36.Ying X, Ma K. 2011. Characterization of a zinc-containing alcohol dehydrogenase with stereoselectivity from the hyperthermophilic archaeon Thermococcus guaymasensis. J. Bacteriol. 193:3009–3019. 10.1128/JB.01433-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu B, Sun J, Bommareddy RR, Song L, Zeng AP. 2011. Novel (2R,3R)-2,3-butanediol dehydrogenase from potential industrial strain Paenibacillus polymyxa ATCC 12321. Appl. Environ. Microbiol. 77:4230–4233. 10.1128/AEM.02998-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riveros-Rosas H, Julian-Sanchez A, Villalobos-Molina R, Pardo JP, Pina E. 2003. Diversity, taxonomy and evolution of medium-chain dehydrogenase/reductase superfamily. Eur. J. Biochem. 270:3309–3334. 10.1046/j.1432-1033.2003.03704.x [DOI] [PubMed] [Google Scholar]
- 39.Bowyer A, Mikolajek H, Stuart JW, Wood SP, Jamil F, Rashid N, Akhtar M, Cooper JB. 2009. Structure and function of the l-threonine dehydrogenase (TkTDH) from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Struct. Biol. 168:294–304. 10.1016/j.jsb.2009.07.011 [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez E, Fernandez MR, Larroy C, Pares X, Biosca JA. 2001. Characterization and functional role of Saccharomyces cerevisiae 2,3-butanediol dehydrogenase. Chem. Biol. Interact. 130–132:425–434. 10.1016/S0009-2797(00)00282-9 [DOI] [PubMed] [Google Scholar]
- 41.Korkhin Y, Kalb AJ, Peretz M, Bogin O, Burstein Y, Frolow F. 1998. NADP-dependent bacterial alcohol dehydrogenases: crystal structure, cofactor-binding and cofactor specificity of the ADHs of Clostridium beijerinckii and Thermoanaerobacter brockii. J. Mol. Biol. 278:967–981. 10.1006/jmbi.1998.1750 [DOI] [PubMed] [Google Scholar]
- 42.Machielsen R, van der Oost J. 2006. Production and characterization of a thermostable L-threonine dehydrogenase from the hyperthermophilic archaeon Pyrococcus furiosus. FEBS J. 273:2722–2729. 10.1111/j.1742-4658.2006.05290.x [DOI] [PubMed] [Google Scholar]
- 43.Rossmann MG, Moras D, Olsen KW. 1974. Chemical and biological evolution of nucleotide-binding protein. Nature 250:194–199. 10.1038/250194a0 [DOI] [PubMed] [Google Scholar]
- 44.Banfield MJ, Salvucci ME, Baker EN, Smith CA. 2001. Crystal structure of the NADP(H)-dependent ketose reductase from Bemisia argentifolii at 2.3 A resolution. J. Mol. Biol. 306:239–250. 10.1006/jmbi.2000.4381 [DOI] [PubMed] [Google Scholar]
- 45.Hosaka T, Ui S, Mimura A. 1999. Separation and properties of two acetylacetoin reductases from Bacillus cereus YUF-4. Biosci. Biotechnol. Biochem. 63:199–201. 10.1271/bbb.63.199 [DOI] [PubMed] [Google Scholar]
- 46.Baker PJ, Britton KL, Rice DW, Rob A, Stillman TJ. 1992. Structural consequences of sequence patterns in the fingerprint region of the nucleotide binding fold. Implications for nucleotide specificity. J. Mol. Biol. 228:662–671 [DOI] [PubMed] [Google Scholar]
- 47.Carugo O, Argos P. 1997. NADP-dependent enzymes. I. Conserved stereochemistry of cofactor binding. Proteins 28:10–28 [DOI] [PubMed] [Google Scholar]
- 48.Ali NO, Bignon J, Rapoport G, Debarbouille M. 2001. Regulation of the acetoin catabolic pathway is controlled by sigma L in Bacillus subtilis. J. Bacteriol. 183:2497–2504. 10.1128/JB.183.8.2497-2504.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kruger N, Steinbuchel A. 1992. Identification of acoR, a regulatory gene for the expression of genes essential for acetoin catabolism in Alcaligenes eutrophus H16. J. Bacteriol. 174:4391–4400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kruger N, Oppermann FB, Lorenzl H, Steinbuchel A. 1994. Biochemical and molecular characterization of the Clostridium magnum acetoin dehydrogenase enzyme system. J. Bacteriol. 176:3614–3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Y, Li X, Mao Y, Blaschek HP. 2012. Genome-wide dynamic transcriptional profiling in Clostridium beijerinckii NCIMB 8052 using single-nucleotide resolution RNA-Seq. BMC Genomics 13:102. 10.1186/1471-2164-13-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nicholson WL. 2008. The Bacillus subtilis ydjL (bdhA) gene encodes acetoin reductase/2,3-butanediol dehydrogenase. Appl. Environ. Microbiol. 74:6832–6838. 10.1128/AEM.00881-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan Y, Lee CC, Liao JC. 2009. Enantioselective synthesis of pure (R,R)-2,3-butanediol in Escherichia coli with stereospecific secondary alcohol dehydrogenases. Org. Biomol. Chem. 7:3914–3917. 10.1039/b913501d [DOI] [PubMed] [Google Scholar]
- 54.Takeda M, Muranushi T, Inagaki S, Nakao T, Motomatsu S, Suzuki I, Koizumi J. 2011. Identification and characterization of a mycobacterial (2R,3R)-2,3-butanediol dehydrogenase. Biosci. Biotechnol. Biochem. 75:2384–2389. 10.1271/bbb.110607 [DOI] [PubMed] [Google Scholar]
- 55.Gonzalez E, Fernandez MR, Larroy C, Sola L, Pericas MA, Pares X, Biosca JA. 2000. Characterization of a (2R,3R)-2,3-butanediol dehydrogenase as the Saccharomyces cerevisiae YAL060W gene product. Disruption and induction of the gene. J. Biol. Chem. 275:35876–35885. 10.1074/jbc.M003035200 [DOI] [PubMed] [Google Scholar]
- 56.Bashir Q, Rashid N, Jamil F, Imanaka T, Akhtar M. 2009. Highly thermostable L-threonine dehydrogenase from the hyperthermophilic archaeon Thermococcus kodakaraensis. J. Biochem. 146:95–102. 10.1093/jb/mvp051 [DOI] [PubMed] [Google Scholar]
- 57.Boylan SA, Dekker EE. 1978. L-threonine dehydrogenase of Escherichia coli K-12. Biochem. Biophys. Res. Commun. 85:190–197. 10.1016/S0006-291X(78)80028-X [DOI] [PubMed] [Google Scholar]
- 58.Peretz M, Bogin O, Tel-Or S, Cohen A, Li G, Chen JS, Burstein Y. 1997. Molecular cloning, nucleotide sequencing, and expression of genes encoding alcohol dehydrogenases from the thermophile Thermoanaerobacter brockii and the mesophile Clostridium beijerinckii. Anaerobe 3:259–270. 10.1006/anae.1997.0083 [DOI] [PubMed] [Google Scholar]
- 59.Jones DT. 1999. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292:195–202. 10.1006/jmbi.1999.3091 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.