

Abstract
Coxsackievirus B3 (CVB3) is the most common pathogen that induces acute and chronic viral myocarditis in children. The cytopathic effect (CPE)-based neutralization test (Nt-CPE) and the plaque reduction neutralization test (PRNT) are the most common methods for measuring neutralizing antibody titers against CVB3 in blood serum samples. However, these two methods are inefficient for CVB3 vaccine clinical trials, which require the testing of a large number of serum specimens. In this study, we developed an efficient neutralization test based on the enzyme-linked immunospot (Nt-ELISPOT) assay for measuring CVB3-neutralizing antibodies. This modified ELISPOT assay was based on the use of a monoclonal antibody against the viral capsid protein VP1 to detect the cells that are infected with CVB3, which, after immunoperoxidase staining, are counted as spots using an automated ELISPOT analyzer. Using the modified ELISPOT assay, we characterized the infection kinetics of CVB3 and divided the infection process of CVB3 on a cluster of cells into four phases. The stability of the Nt-ELISPOT was then evaluated. We found that over a wide range of infectious doses (102 to 106.5× 50% tissue culture infectious dose [TCID50] per well), the neutralizing titers of the sera were steady as long as they were tested during the log phase or the first half of the stationary phase of growth of the spots. We successfully shortened the testing period from 7 days to approximately 20 h. We also found that there was a good correlation (R2 = 0.9462) between the Nt-ELISPOT and the Nt-CPE assays. Overall, the Nt-ELISPOT assay is a reliable and efficient method for measuring neutralizing antibodies in serum.
INTRODUCTION
Coxsackievirus B3 (CVB3) is a small nonenveloped single-stranded positive-sense RNA icosahedral virus that belongs to the Enterovirus genus within the Picornaviridae family. The genome of picornaviruses consists of a single open reading frame, which is expressed as a large polyprotein that is considered to have three general regions, P1 to P3. The P1 region encodes four structural proteins: VP1, VP2, VP3, and VP4. VP1 is the major antigenic determinant of CVB3 and is the most external and immunodominant of its capsid proteins (1). CVB3 can cause meningoencephalitis (2), acute pancreatitis (3), and childhood-onset diabetes (4, 5). It is the most common pathogen that induces acute and chronic viral myocarditis in children (6). The development of a vaccine to prevent CVB3-induced myocarditis was started >26 years ago (7). Using different murine model systems, it has been demonstrated that classic and newly developed vaccination procedures are quite successful for the prevention of CVB3 infections (8). However, no vaccines or therapeutic reagents have been approved for clinical use.
Humoral immunity plays an important role in the defense against virus infection, particularly infections from enteroviruses (9). Thus, the assessment of the humoral immunity response is indispensable in the development of vaccines against enteroviruses. A neutralization test is a commonly used method for in vitro detection of neutralizing antibodies. The cytopathic effect (CPE)-based neutralization test (Nt-CPE) and the plaque reduction neutralization test (PRNT) are the standard neutralization tests used for many types of viruses (10–13). However, these two traditional neutralization tests are time-consuming (taking approximately 7 days) and labor-intensive; thus, these tests can hardly meet the demands of vaccine clinical trials, which require the screening of a large number of samples. Therefore, an efficient neutralization test needs to be developed.
The enzyme-linked immunospot (ELISPOT) assay is a powerful tool for detecting and enumerating individual cells that secrete a particular biomarker of interest ex vivo (14). By employing high-affinity capture and detection antibodies, each individual cell producing the biomarker of interest (such as antibodies, cytokines, and proteins) is visualized as a spot after an enzyme-catalyzed color reaction. The plates are then scanned and analyzed using an automated ELISPOT analyzer to determine the number of antigen-specific cells. Because it is highly sensitive, quantitative, easy to use, and amenable to high throughput, the ELISPOT assay has been widely used in many fields of biomedical research, including vaccine development, transplantation studies, and research on HIV, cancer, and allergies (14). In the field of vaccine development, the ELISPOT assay is often used for the monitoring of specific cellular immune responses. However, the ELISPOT assay is seldom used to assess humoral immunity, even though it was originally developed to analyze antibody-secreting cells (15, 16). Exploiting the highly sensitive and high-throughput properties of the ELISPOT assay, some studies in the last few years have attempted to apply it to measure neutralizing antibodies (17, 18). This type of ELISPOT-based neutralization test (Nt-ELISPOT) has properties similar to those of the ELISPOT assay, which makes Nt-ELISPOT an ideal alternative method for the measurement of the neutralizing capacities of serum on a large scale. In this study, the Nt-ELISPOT was successfully applied for the measurement of neutralizing antibody titers against CVB3 in blood serum.
MATERIALS AND METHODS
Viruses and cells.
The CVB3 XM08-2035 strain (GenBank accession no. JQ042700) was isolated from a throat swab of a hand-foot-and-mouth disease (HFMD) patient (two-year-old girl, mild case, not hospitalized) from the Centers for Disease Control and Prevention (CDC) of Xiamen, China, in 2008. The other two virus strains used in this study were enterovirus 71 (EV71) strain 10-123 (GenBank accession no. JQ042703, isolated from the Xiamen CDC in 2008) and coxsackievirus A16 (CA16) strain TW2007-00190 (GenBank accession no. JF420555, a gift from National Taiwan University). Rhabdomyosarcoma (RD) cells were obtained from the American Type Culture Collection (ATCC) and maintained in minimal essential medium (MEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) plus 2 mM l-glutamine, 100 IU of penicillin, and 100 μg/ml streptomycin. To estimate the viral infectivity titers, serially diluted virus samples (from 10−1 to 10−10) were added to the RD cells (5,000 cells per well) in 96-well plates, and eight wells were used for each dilution. The 96-well plates were incubated for five days prior to the observation of the presence of a cytopathic effect. The 50% tissue culture infectious doses (TCID50s) of CVB3 were calculated by the method described by Reed and Muench (19). To calculate the multiplicities of infection (MOIs), the titers were converted from TCID50s to PFU by multiplying by 0.69, and this number was then divided by the number of cells.
Blood serum samples.
Human blood serum specimens were collected from HFMD patients, who provided informed consent, at the Xiamen CDC during the outbreak of HFMD in 2010. All of the serum samples were stored at −20°C. Ethical approval was obtained from the Research Ethics Review Committee of Xiamen University. IgG was purified from the serum samples through ammonium sulfate precipitation and protein G affinity chromatography (20).
Monoclonal antibody.
Anti-CVB3 specific monoclonal antibodies (MAbs) were produced in our laboratory using the method described by Chen et al. (21). Four 6-week-old female BALB/c mice were immunized subcutaneously with the XM08-2035 strain of CVB3, which was inactivated by heating at 56°C for 1 h and emulsified in Freund's adjuvant. Blood samples were obtained from the tails of the immunized mice after two booster immunizations. The use of mice was approved by the Institutional Animal Care and Use Committee at Xiamen University. All of the MAbs were conjugated to horseradish peroxidase (HRP) using the method reported by Nakane and Kawaoi (22) and stored at −20°C.
Western blot analysis.
The RD cells were prepared in MEM supplemented with 2% FBS and seeded at 20,000 cells per well into a 96-well plate ≥4 h prior to the experiment. The infectious dose was set to 30,000× TCID50 per well. Twelve hours later, the cells were harvested and subjected to three freeze-thaw cycles. The samples were then separated by 12% SDS-PAGE and transferred onto a nitrocellulose membrane (Whatman). Subsequently, the membrane was blocked with 5% nonfat milk in phosphate-buffered saline (PBS) (pH 7.45) for 1 h, incubated with HRP-labeled MAb A13H11 (1:3,000 dilution) for 1 h, and washed three times with PBS containing 0.1% Tween 20. The 3,3′,5,5′-tetramethylbenzidine (TMB) substrate solution (Sigma) was added to visualize the immunoreactive protein bands.
Nt-CPE.
The RD cells were prepared in MEM supplemented with 2% FBS and seeded at 5,000 cells per well into 96-well plates ≥4 h prior to the experiment. The sera were first diluted 16-fold with MEM and then heat inactivated at 56°C for 30 min. Two-fold serial dilutions from 1:32 to 1:16,384 (10 dilutions, four wells for each dilution) were prepared in MEM. The serially diluted sera were challenged with CVB3 at 100× TCID50 per well. The serum and virus mixtures were incubated at 37°C for 1 h and then added to the cell plates. The cell plates were incubated at 37°C and 5% CO2 for 5 to 7 days. The neutralizing titers were read as the highest dilution that completely inhibited CPE in >50% of the wells.
Nt-ELISPOT.
The RD cells were prepared in MEM supplemented with 2% FBS and seeded at 20,000 cells per well into a 96-well plate ≥4 h prior to the experiment. The sera were first diluted 16-fold with MEM and then heat inactivated at 56°C for 30 min. Two-fold serial dilutions from 1:32 to 1:16,384 (10 dilutions) were prepared in MEM. Next, 50 μl of the serially diluted sera was challenged with an equal volume of CVB3 (30,000× TCID50/well). The serum-virus mixtures were incubated at 37°C for 1 h and then added to the plates. The plates were incubated at 37°C and 5% CO2 for 12 h. After incubation, the plates were fixed with 100 μl of 0.2% glutaraldehyde in phosphate-buffered saline (PBS) for 1 h at room temperature and permeated with 100 μl of 1% Triton X-100 in PBS for 30 min. HRP-labeled MAbs against CVB3 were diluted in PBS with 2% gelatin and 5% casein, added to the wells of the 96-well plates, and incubated at 37°C for 30 min. The plates were washed three times with PBS containing 0.05% Tween 20 (PBST). The TMB substrate solution (Sigma) was added at 37°C for 10 min. The stained plates were patted dry and then scanned and counted by the ImmunoSpot image analyzer 3.2 (Cellular Technology, Ltd.) using the blue color system. The resolution of the captured picture for each well was 512 by 512 pixels. In most situations, the counting parameters were set as follows: the optical sensitivity ranged from 175 to 210, the diffuse spot process and the background balance were 0, and the spot size threshold was 0.0001 mm2 to 8.7781 mm2. The other counting parameters were maintained at the default settings. The wells in the last two columns of the 96-well plate are the virus control wells and the cell control wells, respectively. The virus control wells were infected with the same amount of virus as the test wells, and the cell control wells contained an equal volume of MEM. The inhibition rate of the serum on the spots can be calculated using the equation P = [1 − (Ntest − Ncell control)/(Nvirus control − Ncell control)] × 100%. In this equation, P is the inhibition rate of the serum on the spots, Ntest is the number of spots in the test well, Ncell control is the average number of spots in the cell control wells, and Nvirus control is the average number of spots in the virus control wells.
Data analysis.
The neutralizing titers were read as the highest dilution that completely neutralized >50% of viruses (Nt50). If the MOI is ≤1, the Nt50 can be read as the highest approximate dilution that completely inhibits >50% of the spots. However, it is worth noting that the neutralization of 50% of the viruses is no longer manifested as a 50% reduction in the number of spots when the MOI is >1. A Poisson distribution can be used to predict the fraction of cells in a population infected with a given number of particles at different MOIs (23). Assuming that one viral unit can efficiently infect one cell, the percent reduction of the number of spots that corresponds to the Nt50 at a given MOI can easily be deduced: Preduction = [1 − (1 − e−m/2)/(1 − e−m)] × 100%. In this equation, Preduction is the percent reduction of the number of spots that corresponds to Nt50, m represents the MOI, and e is the base of the natural logarithm. Similarly, the percent reduction of the number of spots that corresponds to the neutralization of 90% of the viruses (Nt90) at a given MOI is Preduction = [1 − (1 − e−m/10)/(1 − e−m)] × 100%. The neutralization of 50% of the viruses corresponds to a 37.7% reduction in the number of spots when the MOI is 1. If the MOI is >4.5, the reduction in the number of spots is <10% and the Nt50 is not stable due to the normal fluctuation in the number of spots. In this case, the Nt90 can be determined and multiplied by 2 to calculate the Nt50, because the Nt50 value is typically 2-fold higher than the Nt90 value. The determination of the neutralizing titer becomes difficult for MOI values of >23. If the Nt50 of a serum sample is <32, it is defined as negative; in contrast, if the Nt50 of a serum sample is ≥32, it is defined as positive.
The results obtained under different conditions were compared through a repeated measures analysis of variance (ANOVA). The null hypothesis states that the mean Nt50 values obtained under different conditions are the same. If the means are significantly different, Bonferroni's multiple comparison test was used to compare two of the means. The results of the two assay methods were compared through a paired-sample t test, and the correspondence between the two methods was analyzed by linear regression using the GraphPad Prism 5.04 software. A P value of 0.05 was considered statistically significant for all parameters, and the confidence interval (CI) was 95%.
RESULTS
Establishment of a modified ELISPOT assay for the detection of cells infected with CVB3.
The modified ELISPOT assay was based on the use of a high-affinity MAb against viral protein to label the cells infected with CVB3, and the MAb was conjugated to horseradish peroxidase (HRP). The addition of 3,3′,5,5′-tetramethylbenzidine (TMB) substrate stained the labeled cells that were infected with CVB3 blue. Thus, under a microscope, the infected cells appeared as blue spots. Therefore, the selection of a high-affinity MAb is a key step in the establishment of the modified ELISPOT assay. A total of 29 MAbs against CVB3 were produced in our laboratory. The use of the 29 MAbs as the detection antibody in the modified ELISPOT assay revealed that 10 MAbs showed good reactivities with CVB3-infected cells (see Fig. S1 in the supplemental material). Lastly, a high-affinity and specific MAb, A13H11, was selected as the detection antibody from these 10 MAbs. The A13H11 MAb reacted with the viral protein VP1 of CVB3 (Fig. 1A). The HRP-conjugated A13H11 can label CVB3-infected cells specifically and does not react with uninfected cells or cells infected with EV71 or CA16 (Fig. 1B). The background is commonly <10 spots and fluctuates in practice.
FIG 1.
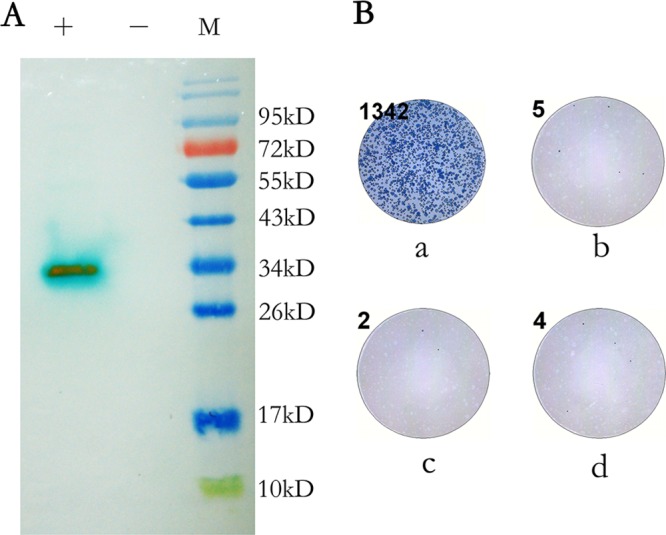
(A) Western blot analysis of the reactivity of A13H11 to VP1. +, lysis of RD cells infected with CVB3 at 30,000× TCID50 per well; −, lysis of normal RD cells; M, molecular weight marker (catalog no. SM0671; Fermentas). (B) Specificity of the detection antibody A13H11. The number in the upper left corner of each well indicates the number of spots calculated by the ImmunoSpot image analyzer. Shown are the wells infected with the XM08-2035 strain of CVB3 (a), the 10-123 strain of EV71 (b), the 2007-00190 strain of CA16 (c), and the control well, which contains an equal volume of minimal essential medium (MEM) (d).
Infection kinetics of CVB3.
Because this new ELISPOT assay can quantify the number of infected cells, we used it to characterize the infection kinetics of CVB3. RD cells were infected with CVB3 at 40× TCID50 per well. The number of spots was detected every 6 h for a total of 72 h. As shown in Fig. 2A, the growth curve of the number of spots was S-shaped and was divided into four phases.
FIG 2.

Infection kinetics of CVB3. (A) The growth curve of the number of spots for an infectious dose of CVB3 of 40× TCID50 per well. The data points indicate the average numbers of spots from six wells, and the error bars show the standard deviations. (B) Contour plot of the number of spots versus the infectious dose and incubation time. RD cells were seeded onto 96-well plates at 20,000 cells per well and infected with CVB3 at different doses. The number of spots was detected over a total of 48 h. Every 6 h, six wells were fixed and stained to evaluate the number of spots. ●, sampling points. The numbers of spots are distinguished by labeled contour lines. (C) Relationship between the infectious dose and the incubation time. A series of infectious dose and incubation time combinations that yield a specific number of spots (400, 800, and 1,200) were chosen from Fig. 2B.
(i) Lag phase. During the first few hours, the viruses started to infect the cells. However, there was little or no increase in the number of spots during this phase. The possible reasons are that the viral proteins were not yet synthesized during this period and that the infectious dose was so low that only a small proportion of cells was infected.
(ii) Log phase. In this phase, the viral protein VP1 was synthesized and accumulated to a detectable level or the viruses multiplied and continuously infected new cells. The number of spots increased rapidly in a logarithmic manner during this phase. This phase lasted approximately 18 h.
(iii) Stationary phase. Almost all of the cells were infected, and the number of spots did not continue to increase. This phase lasted approximately 12 h.
(iv) Death phase. All of the cells were infected. Some displayed complete CPE and became detached from the culture dish. Thus, the number of spots began to decline.
Moreover, we also characterized the infection kinetics of CVB3 at other infectious doses (Fig. 2B). The growth curves of the number of spots obtained for different infectious doses were similar, with the exception of the length of the lag phase (Fig. 2B, lower left corner). Increases in the infectious doses decreased the length of the lag phase. At an infectious dose of 105× TCID50 per well, many spots were detected 6 h postinfection.
We selected a set of infectious dose and incubation time combinations that yielded a certain number of spots (e.g., 400, 800, and 1,200) from Fig. 2B and found that there was a negative linear correlation between the relative infectious dose and the incubation time (Fig. 2C). The average slope was approximately −9, which means that if the infectious dose is increased 10-fold, the incubation time required to yield the same number of spots decreases by 9 h. Due to the correspondence between the number of spots and the actual amount of virus, the slope can also be explained by the following: if the original infectious dose is increased 10-fold, the time required to multiply the same amount of virus will be reduced by 9 h.
Feasibility of the Nt-ELISPOT.
To assess the feasibility of the Nt-ELISPOT for the measurement of neutralizing antibodies against CVB3, we designed the following experiments.
First, a neutralizing antibody-positive human serum sample and a neutralizing antibody-negative human serum sample were chosen. The neutralizing titers of the two serum samples were 512 and <32, respectively, as determined with the Nt-CPE. The sera were then tested using the Nt-ELISPOT. The incubation time was set to 12 h, and the infectious dose was set to 30,000× TCID50 per well. As shown in Fig. 3A, the number of the spots in the neutralizing antibody-positive human serum sample decreased, whereas the number of the spots in the neutralizing antibody-negative human serum sample did not. An increase in the dilution of the neutralizing antibody-positive human serum sample decreased the inhibition percentage. However, the inhibition effect may be due to factors other than antibodies in the human sera. To rule out this possibility, immunoglobulin G (IgG) was purified from the neutralizing antibody-positive serum sample, and the experiment was repeated. The purified antibodies still inhibited the number of spots (Fig. 3A).
FIG 3.

Feasibility of the Nt-ELISPOT. Three serum samples were used in this assessment. Their neutralizing capacities were measured by the Nt-CPE and the Nt-ELISPOT in parallel. For the Nt-ELISPOT, the incubation time was set to 12 h and the infectious dose was set to 30,000× TCID50 per well. (A) Number of spots obtained from different dilutions of the sera. Data for the neutralizing antibody-positive human serum, the neutralizing antibody-negative human serum, IgG purified from the neutralizing antibody-positive human serum, and serum from a mouse immunized with CVB3 are shown. (B) The neutralization titers of the three samples were measured by the Nt-CPE and Nt-ELISPOT. For the Nt-ELISPOT, the neutralizing titers of the sera were read as the highest dilutions that inhibited >50% of the spots. (C) The neutralizing antibody-positive serum was 2-fold serially prediluted to obtain multiple test samples with decreasing neutralizing capacities. The neutralizing capacities of the samples were then assayed with the Nt-ELISPOT.
If the neutralizing titers of sera are read as the highest dilutions that inhibit >50% of the spots, the neutralizing titers of the neutralizing antibody-positive serum sample and the neutralizing antibody-negative serum sample were 1,024 and <32, respectively. In addition, using the Nt-ELISPOT and the Nt-CPE, we measured the neutralizing capacity of a serum sample from a mouse immunized with CVB3. The results obtained with the Nt-ELISPOT were almost the same as those obtained with the Nt-CPE (Fig. 3B). This finding indicates that the Nt-ELISPOT can be used to measure neutralizing antibodies against CVB3 in both human and mouse serum.
Furthermore, the neutralizing antibody-positive serum sample was 2-fold serially prediluted with the neutralizing antibody-negative serum sample to obtain multiple test samples with decreasing neutralizing capacities. The neutralizing capacities of the samples were then assayed using the Nt-ELISPOT. The neutralizing titers that were measured were identical to the expected neutralizing titers (Fig. 3C), which proves that the neutralizing titers measured by the Nt-ELISPOT accurately reflect the neutralizing capacity of the serum.
Influence of the infectious dose and incubation time on the Nt-ELISPOT.
In their studies of the neutralization reaction between phages and neutralizing antibodies, Andrewes and Elford (24) summarized their findings using the “percentage law”: over a very wide range of virus concentrations, a given concentration of antibody neutralizes the same relative proportion of viral infectivity as long as the antibody is in considerable excess over the virus. Additional studies have shown that the percentage law can apply to the virus-antibody interaction in general (24–28). However, the traditional neutralization tests cannot directly reflect the percentage law due to their limited sensitivities or the methods that are used with these tests to determine the neutralizing titer of serum. The Nt-ELISPOT is sensitive and can quantify infected cells. Therefore, we speculate that the Nt-ELISPOT can directly reflect the percentage law and that the infectious dose of the virus will not influence the neutralizing titer of the serum in the Nt-ELISPOT.
In the first set of experiments that were used to test this hypothesis, the neutralizing capacities of seven serum samples (Table 1) were measured with the Nt-ELISPOT using different infectious doses and a fixed incubation time of 12 h. The mean logarithm of the Nt50 was plotted against the logarithm of the virus concentration (Fig. 4A). As shown, the infectious dose did not significantly influence the neutralizing titers of the sera when it was approximately <106× TCID50 per well (P > 0.05).
TABLE 1.
Background of human blood serum samples
| Serum sample no. | Donor characteristics |
||||
|---|---|---|---|---|---|
| Sex | Age (yr) | Diagnosisa |
|||
| EV71 | CA16 | CVB3 | |||
| 6832b | Male | 1 | − | − | + |
| 6851b | Male | 2.3 | − | + | − |
| 6852b,c | Female | 6.6 | + | − | − |
| 6859b,c | Male | 3.3 | − | + | − |
| 6865b,c | Male | 3.6 | − | + | − |
| 6866b,c | Female | 3 | − | + | − |
| 6891b,c | Female | 2.8 | + | + | − |
| 6839c | Female | 0.4 | − | + | + |
| 6854c | Male | 5.4 | + | − | − |
| 6862c | Male | 2.8 | + | − | − |
| 6876c | Male | 2.3 | − | − | + |
| 6895c | Male | 2.9 | − | + | − |
| 6914c | Female | 3.3 | + | − | − |
| 6947c | Female | 2.5 | − | + | − |
| 6964c | Female | 3 | − | + | − |
| 7027c | Male | 4 | − | + | − |
| 7046c | Male | 5 | − | + | + |
| 7047c | Female | 3 | − | + | − |
| 7060c | Female | 4.5 | − | + | − |
The diagnosis of the donor was based on detection of the viral RNA by RT-PCR (45).
Sera used in the evaluation of the influence of the infectious dose and incubation time on the Nt-ELISPOT.
Sera used in the experiment for comparison of the Nt-CPE and Nt-ELISPOT. +, viral RNA is detected; −, viral RNA is not detected.
FIG 4.

Influence of the infectious dose and the incubation time on the Nt-ELISPOT. The neutralizing capacities of seven sera were measured under different experimental conditions. All of the neutralizing titers of the sera were log2-transformed. ●, average Nt50 of the seven serum samples; ○, average of the Nt50 values that were calculated by multiplying 2 by the Nt90 of the seven serum samples. The error bars show the standard deviations. The dotted curve represents the number of spots under different experimental conditions. (A) The incubation time was fixed at 12 h, and the infectious dose per well was changed. (B) The infectious dose was fixed at 30,000× TCID50 per well, and the incubation time was changed. (C) Several incubation time and infectious dose combinations that yield 1,500 spots were selected to measure the neutralizing capacities of the sera.
In the next set of experiments, the infectious dose was fixed at 30,000× TCID50 per well and the incubation time was changed to evaluate its influence on the neutralizing titers of the sera. The neutralizing capacities of the same seven serum samples were tested every 2 h starting at 6 h postinfection. The mean logarithm of the Nt50 was plotted against the incubation time (Fig. 4B). As shown, the mean Nt50 was found to be constant between 8 and 16 h; however, the Nt50 value significantly decreased when the sera were tested at 18 h postinfection or later (P < 0.01). When the infectious dose was fixed at 30,000× TCID50 per well, the log and stationary phases of growth of the spots occurred at 8 to 12 h and 12 to 20 h postinfection, respectively (Fig. 2B). Therefore, we can speculate that the incubation time may not influence the neutralizing titers of sera as long as the sera are tested during the log phase or the first half of the stationary phase of growth in the number of spots after infection.
The next set of experiments evaluated the combined influence of the infectious dose and the incubation time on the neutralizing titers of sera. A set of infectious dose and incubation time combinations that yield the same amount of spots were chosen. Concretely, the number of spots was fixed to 1,500, and the infectious dose and incubation time combinations were chosen from Fig. 2B. The combined conditions were then used to test the neutralizing titers of the seven serum samples. As shown in Fig. 4C, the neutralizing titers of the sera were almost identical under the different conditions.
In summary, we can conclude that the Nt-ELISPOT complies with the percentage law: over a wide range, the infectious dose does not significantly influence the neutralizing titers of the sera. In addition, if the sera are tested during the log phase or the first half of the stationary phase of the growth of spots, the incubation time has little influence on the neutralizing titers of sera.
Comparison of the Nt-CPE and Nt-ELISPOT.
The Nt-CPE is the standard method that is recommended by the World Health Organization (WHO) for the measurement of neutralizing antibodies against polioviruses (10) and has been widely applied to determine neutralizing antibody titers against other enteroviruses, including CVB3. Therefore, we evaluated the concordance between the Nt-ELISPOT and Nt-CPE. First, using the Nt-ELISPOT, we screened hundreds of serum samples collected from children with characteristic symptoms of hand-foot-and-mouth disease in 2010. In addition, 17 serum samples (Table 1) with different neutralizing titers were selected. The neutralizing titers of the 17 samples were tested four separate times using the Nt-CPE and Nt-ELISPOT. In the Nt-ELISPOT, the neutralizing titers of the sera were calculated in two ways, with the highest dilutions that were able to neutralize >50% and >90% of the viral infectivity (Nt50 and Nt90, respectively). As shown in Fig. 5A, the Nt50 values measured by the Nt-ELISPOT were mainly 2- to 4-fold higher than the corresponding neutralizing titers measured by the Nt-CPE. In contrast, the Nt90 values that were measured by the Nt-ELISPOT were almost equal to the neutralizing titers that were measured by the Nt-CPE (P = 0.1544); in fact, a linear regression analysis indicated that there was a high degree of correlation (R2 = 0.9462, Fig. 5B) between these two measurements. The infectious dose that is commonly used in the Nt-CPE is 100× TCID50, and the neutralizing titers of the sera tested with the Nt-CPE are read as the highest dilution that can neutralize a virus to 1× TCID50. Consequently, the judgment standard of the neutralizing titer used in the Nt-CPE is 99% neutralization (Nt99). Thus, the Nt90 of the Nt-ELISPOT is almost equal to the Nt99 of the Nt-CPE. However, the number of spots is more sensitive to the dilution of serum at the 50% neutralization condition. Therefore, we chose Nt50 as the judgment standard of the neutralizing titer in the Nt-ELISPOT.
FIG 5.

Comparison of the Nt-CPE and Nt-ELISPOT. The neutralizing titers of 17 human serum samples (with different neutralizing titers) were independently tested four times using the Nt-CPE and the Nt-ELISPOT. The titers were then log2-transformed, and the average neutralizing titer of each serum was calculated. (A) The sera were aligned by their neutralizing titers. ● and ○, average Nt50 and Nt90 of each serum sample, respectively; these values were determined using the Nt-ELISPOT assay. ■, the average neutralizing titer of each serum sample, which was determined using the Nt-CPE. The error bars show the standard deviations of the quadruplicate neutralizing titers. (B) The average Nt90 values of the sera that were determined by the Nt-ELISPOT were plotted against the average neutralizing titers of the corresponding sera that were determined by the Nt-CPE.
DISCUSSION
In this study, we developed an ELISPOT assay to measure serum-neutralizing antibodies against CVB3. Because the traditional neutralization tests are time-consuming and labor-intensive, several different neutralization tests have been developed (reviewed by Zha et al. [29]). The common cell-based neutralization methods are the following. The first is common staining. In traditional neutralization tests, the experimenters have to examine the cytopathic effect under a microscope, which is subjective and not easily discerned. To visualize the plaques, the monolayer is commonly stained with dyes, such as crystal violet or neutral red. Zielinska et al. (30) used an ELISPOT analyzer to automatically count the stained plaques. Although this automatic counting method makes the PRNT more efficient and objective, it still takes a long time (3 days) to obtain the results. The second cell-based neutralization method uses the reporter gene. Due to their high infectivity and mutation efficiency, pseudoviruses or infectious clones of many viruses have been constructed. A number of neutralization tests based on a reporter gene of recombinant viruses have been reported (31–34). These methods are easy to use, rapid, and objective. However, these tests rely on the existence of a recombinant virus and therefore cannot be used to reflect epidemic strains in a serological survey. The third cell-based neutralization method is the immunological method. Immunological methods have been widely used in neutralization tests, such as ELISA (35–37) and the immunofluorescence assay (38, 39). These tests rely on antibodies to recognize the infected cells that express viral proteins. The expression of viral proteins always occurs before the cytopathic effect; consequently, enzyme amplification can enhance both the speed and sensitivities of these neutralization tests. In addition, neutralization tests based on flow cytometry (40–42), which can detect infected cells at the single-cell level, have been reported. However, this method is not suitable for high-throughput screening. Abai et al. (17) developed a fast neutralization assay for human cytomegalovirus based on the ELISPOT assay. This method quantifies the infected cells based on the expression of the immediate-early 1 (IE1) viral protein through immunoperoxidase staining and the use of an automated ELISPOT analyzer. Because it was performed in 96-well cell culture plates and the data were automatically collected and analyzed, this test is very suitable for high-throughput screening.
Although multiple new neutralization tests have been developed, all of these assays measure the neutralizing titers of sera through the detection of the residual infectivity after the virus and antibody reaction. Most neutralization tests with different infectious doses read the highest dilution that protects 50% of the cells from infection as the neutralizing titer of the sera. However, according to a Poisson distribution, the percentage of the reduction of infected cells does not equal the percentage of neutralized viruses at high infectious doses. Therefore, this method of calculating serum neutralizing antibody titers is not exact and should take the infectious dose into account.
Because the Nt-CPE is the standard neutralization test used for enteroviruses, we evaluated the concordance between the Nt-ELISPOT and Nt-CPE. If the neutralizing titer is read as the highest dilution that neutralizes >50% of the virus, the results obtained with the Nt-ELISPOT are 2- to 4-fold higher than those obtained with the Nt-CPE. However, if the neutralizing titer is read as the highest dilution that neutralizes >90% of the virus, the two tests produce almost identical results. This is because the judgment standard of the neutralizing titer used in the Nt-CPE can be considered 99% neutralization. Thus, if the neutralizing titers of sera are read as the highest dilutions that neutralize >50% of the virus in a newly established neutralization test, it should be noted that the results obtained with the newly established method may be different from those obtained with the Nt-CPE.
The percentage law states that “over a wide range of viral concentrations, a given concentration of antibody neutralizes the same relative proportion of virus infectivity” (24). This law, however, has to be qualified: the concentration of antibody has to be in considerable excess compared with the concentration of virus. In addition, this law is actually the expected effect of the excess of antibody compared with antigen, which makes the fraction of the complexed antibody negligible (43). Using poliovirus as an example, Brioen and Boxyé (44) studied the validity range of the percentage law and found that it is valid when the virus concentration is approximately <1 × 108 PFU/ml. At higher concentrations, the result was determined by the ratio of antibody to virus. Using the Nt-ELISPOT, we proved that the law is valid when the virus concentration is <1 × 107× TCID50/ml. However, the actual validity range may be wider because it was difficult to accurately evaluate the percentage of neutralized viruses through the number of spots for an MOI of >23, which is beyond the suitable range of the Nt-ELISPOT. In addition, we also proved that the incubation time of the Nt-ELISPOT has little influence on the neutralizing titers of the sera when the spots are counted during the log phase or the first half of the stationary phase of growth of the spots. In fact, this finding is simply the indirect reflection of the percentage law in the Nt-ELISPOT.
Compared with the Nt-CPE, the Nt-ELISPOT is an efficient neutralization test that has many advantages: it is rapid, in that the incubation time is optional and can be shortened to <12 h postinfection (compared to 4 to 7 days for the Nt-CPE); it is stable, in that the infectious dose has little influence on the test results over a wide range (102 to 106.5× TCID50 per well), whereas the infectious dose used in the Nt-CPE has to be limited to approximately 100× TCID50 per well; there is low serum sample consumption, as there is no need to use repeated wells in the Nt-ELISPOT, and only 5 μl of the serum specimen is needed if the dilution starts at 32-fold; and it is high throughput, in that at least eight samples can be tested on a 96-well plate, and hundreds of samples can be tested in a single test. Because an automatic spot analyzer is used with the Nt-ELISPOT, the result is more quantitative and objective.
Unlike the neutralization tests that are based on a reporter gene, the virus used in the Nt-ELISPOT is a clinical isolate without any modifications. The Nt-ELISPOT is applicable to other viruses and is especially helpful for viruses that cannot form plaques. To develop this assay for a new virus, it is only necessary to select a susceptible cell and screen a high-affinity antibody. We have applied the Nt-ELISPOT to the detection of many viruses in our laboratory, such as enterovirus 71, coxsackievirus 16, echovirus 30, and rotavirus (data not shown; our unpublished data). Our approach to the measurement of infected cells using an ELISPOT analyzer can be adapted to a variety of applications, such as susceptible cell screening, virus titration, and virus identification. However, there are some drawbacks associated with the validation of our method, such as the limited sample size and the use of only a single strain of CVB3. Further studies are still needed to support a wider use of the modified ELISPOT assay described herein.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Science and Technology Major Project of the Ministry of Science and Technology of China (no. 2010ZX09401-403), the National Natural Science Fund for Distinguished Young Scholar (no. 30925030), and the National High Technology Research and Development Program of China (no. 2012AA02A408). The sponsors had no role in the study design, data collection or analysis, decision to publish, or preparation of the manuscript.
Footnotes
Published ahead of print 3 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/CVI.00359-13.
REFERENCES
- 1.Haarmann CM, Schwimmbeck PL, Mertens T, Schultheiss HP, Strauer BE. 1994. Identification of serotype-specific and nonserotype-specific B-cell epitopes of coxsackie B virus using synthetic peptides. Virology 200:381–389. 10.1006/viro.1994.1202 [DOI] [PubMed] [Google Scholar]
- 2.Melnick JL. 1996. Enteroviruses: polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses, p 655–712 In Fields BN, Howley PM, Chanock RM, Melnick JL, Monath TP, Roizman B, Straus SE. (ed), Fields virology. Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 3.Imrie C, Ferguson J, Sommerville R. 1977. Coxsackie and mumpsvirus infection in a prospective study of acute pancreatitis. Gut 18:53–56. 10.1136/gut.18.1.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoon JW, Austin M, Onodera T, Notkins AL. 1979. Virus-induced diabetes mellitus—isolation of a virus from the pancreas of a child with diabetic ketoacidosis. N. Engl. J. Med. 300:1173–1179. 10.1056/NEJM197905243002102 [DOI] [PubMed] [Google Scholar]
- 5.Clements GB, Galbraith DN, Taylor KW. 1995. Coxsackie B virus infection and onset of childhood diabetes. Lancet 346:221. 10.1016/S0140-6736(95)91270-3 [DOI] [PubMed] [Google Scholar]
- 6.Kawai C. 1999. From myocarditis to cardiomyopathy: mechanisms of inflammation and cell death: learning from the past for the future. Circulation 99:1091–1100. 10.1161/01.CIR.99.8.1091 [DOI] [PubMed] [Google Scholar]
- 7.Godney EK, Arizpe HM, Gaunti CJ. 1987. Characterization of the antibody response in vaccinated mice protected against coxsackievirus B3-induced myocarditis. Viral Immunol. 1:305–314. 10.1089/vim.1987.1.305 [DOI] [PubMed] [Google Scholar]
- 8.Henke A, Jarasch N, Wutzler P. 2003. Vaccination procedures against coxsackievirus-induced heart disease. Expert Rev. Vaccines 2:805–815. 10.1586/14760584.2.6.805 [DOI] [PubMed] [Google Scholar]
- 9.McKinney RE, Jr, Katz SL, Wilfert CM. 1987. Chronic enteroviral meningoencephalitis in agammaglobulinemic patients. Rev. Infect. Dis. 9:334–356. 10.1093/clinids/9.2.334 [DOI] [PubMed] [Google Scholar]
- 10.WHO 1997. Manual for the virological investigation of polio. Global Programme for Vaccines and Immunization, Expanded Programme on Immunization, World Health Organization, Geneva, Switzerland. http://whqlibdoc.who.int/hq/1997/WHO_EPI_GEN_97.01.pdf [Google Scholar]
- 11.Plummer G, Benyesh-Melnick M. 1964. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine, New York, NY: [DOI] [PubMed] [Google Scholar]
- 12.Roehrig JT, Hombach J, Barrett AD. 2008. Guidelines for plaque-reduction neutralization testing of human antibodies to dengue viruses. Viral Immunol. 21:123–132. 10.1089/vim.2008.0007 [DOI] [PubMed] [Google Scholar]
- 13.Coates HV, Alling DW, Chanock RM. 1966. An antigenic analysis of respiratory syncytial virus isolates by a plaque reduction neutralization test. Am. J. Epidemiol. 83:299–313 [DOI] [PubMed] [Google Scholar]
- 14.Kalyuzhny AE. 2005. Handbook of ELISPOT: methods and protocols, vol 302 Humana Press, Totowa, NJ [Google Scholar]
- 15.Czerkinsky CC, Nilsson LA, Nygren H, Ouchterlony O, Tarkowski A. 1983. A solid-phase enzyme-linked immunospot (ELISPOT) assay for enumeration of specific antibody-secreting cells. J. Immunol. Methods 65:109–121. 10.1016/0022-1759(83)90308-3 [DOI] [PubMed] [Google Scholar]
- 16.Sedgwick JD, Holt PG. 1983. A solid-phase immunoenzymatic technique for the enumeration of specific antibody-secreting cells. J. Immunol. Methods. 57:301–309. 10.1016/0022-1759(83)90091-1 [DOI] [PubMed] [Google Scholar]
- 17.Abai AM, Smith LR, Wloch MK. 2007. Novel microneutralization assay for HCMV using automated data collection and analysis. J. Immunol. Methods. 322:82–93. 10.1016/j.jim.2007.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodrigo WW, Alcena DC, Rose RC, Jin X, Schlesinger JJ. 2009. An automated Dengue virus microneutralization plaque assay performed in human Fc{gamma} receptor-expressing CV-1 cells. Am. J. Trop. Med. Hyg. 80:61–65 [PubMed] [Google Scholar]
- 19.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 20.Ma JK, Hikmat BY, Wycoff K, Vine ND, Chargelegue D, Yu L, Hein MB, Lehner T. 1998. Characterization of a recombinant plant monoclonal secretory antibody and preventive immunotherapy in humans. Nat. Med. 4:601–606. 10.1038/nm0598-601 [DOI] [PubMed] [Google Scholar]
- 21.Chen Y, Li C, He D, Cheng T, Ge S, Shih JW, Zhao Q, Chen PJ, Zhang J, Xia N. 2012. Antigenic analysis of divergent genotypes human enterovirus 71 viruses by a panel of neutralizing monoclonal antibodies: current genotyping of EV71 does not reflect their antigenicity. Vaccine 32:425–430. 10.1016/j.vaccine.2012.10.032 [DOI] [PubMed] [Google Scholar]
- 22.Nakane P, Kawaoi A. 1974. Peroxidase-labeled antibody. A new method of conjugation. J. Histochem. Cytochem. 22:1084–1091. 10.1177/22.12.1084 [DOI] [PubMed] [Google Scholar]
- 23.Dulbecco R, Vogt M. 1954. One-step growth curve of Western equine encephalomyelitis virus on chicken embryo cells grown in vitro and analysis of virus yields from single cells. J. Exp. Med. 99:183–199. 10.1084/jem.99.2.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andrewes CH, Elford WJ. 1933. Observations on anti-phage sera. I: “The percentage law”. Br. J. Exp. Pathol. 14:367–376 [Google Scholar]
- 25.Dulbecco R, Vogt M, Strickland AG. 1956. A study of the basic aspects of neutralization of two animal viruses, western equine encephalitis virus and poliomyelitis virus. Virology 2:162–205. 10.1016/0042-6822(56)90017-4 [DOI] [PubMed] [Google Scholar]
- 26.Dimmock NJ. 1993. Neutralization of animal viruses. Curr. Top. Microbiol. Immunol. 183:1–149 [DOI] [PubMed] [Google Scholar]
- 27.Mandel B. 1978. Neutralization of animal viruses. Adv. Virus Res. 23:205–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nara PL, Hatch WC, Dunlop NM, Robey WG, Arthur LO, Gonda MA, Fischinger PJ. 1987. Simple, rapid, quantitative, syncytium-forming microassay for the detection of human immunodeficiency virus neutralizing antibody. AIDS Res. Hum. Retroviruses 3:283–302. 10.1089/aid.1987.3.283 [DOI] [PubMed] [Google Scholar]
- 29.Zha JF, Xiao CY, Zuo MZ. 2008. Research progress in vitro detection of vaccine neutralizing antibody. Chinese Medicinal Biotechnology 3:374–377 [Google Scholar]
- 30.Zielinska E, Liu D, Wu HY, Quiroz J, Rappaport R, Yang DP. 2005. Development of an improved microneutralization assay for respiratory syncytial virus by automated plaque counting using imaging analysis. Virol. J. 2:84. 10.1186/1743-422X-2-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z, Mo C, Kemble G, Duke G. 2004. Development of an efficient fluorescence-based microneutralization assay using recombinant human cytomegalovirus strains expressing green fluorescent protein. J. Virol. Methods 120:207–215. 10.1016/j.jviromet.2004.05.010 [DOI] [PubMed] [Google Scholar]
- 32.Kolokoltsov AA, Davey RA. 2004. Rapid and sensitive detection of retrovirus entry by using a novel luciferase-based content-mixing assay. J. Virol. 78:5124–5132. 10.1128/JVI.78.10.5124-5132.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arya SC, Agarwal N. 2012. Poliovirus neutralization test with poliovirus pseudovirus to measure neutralizing antibody in humans. Clin. Vaccine Immunol. 19:458. 10.1128/CVI.05568-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pierson TC, Sánchez MD, Puffer BA, Ahmed AA, Geiss BJ, Valentine LE, Altamura LA, Diamond MS, Doms RW. 2006. A rapid and quantitative assay for measuring antibody-mediated neutralization of West Nile virus infection. Virology 346:53–65. 10.1016/j.virol.2005.10.030 [DOI] [PubMed] [Google Scholar]
- 35.Huang ML, Chiang PS, Luo ST, Liou GY, Lee MS. 2010. Development of a high-throughput assay for measuring serum neutralizing antibody against enterovirus 71. J. Virol. Methods 165:42–45. 10.1016/j.jviromet.2009.12.015 [DOI] [PubMed] [Google Scholar]
- 36.Ivanov AP, Dragunsky EM. 2005. ELISA as a possible alternative to the neutralization test for evaluating the immune response to poliovirus vaccines. Expert Rev. Vaccines 4:167–172. 10.1586/14760584.4.2.167 [DOI] [PubMed] [Google Scholar]
- 37.Muhamuda K, Madhusudana SN, Ravi V. 2007. Development and evaluation of a competitive ELISA for estimation of rabies neutralizing antibodies after post-exposure rabies vaccination in humans. Int. J. Infect. Dis. 11:441–445. 10.1016/j.ijid.2006.09.013 [DOI] [PubMed] [Google Scholar]
- 38.Smith JS, Yager PA, Baer GM. 1973. A rapid reproducible test for determining rabies neutralizing antibody. Bull. World Health Organ. 48:535–541 [PMC free article] [PubMed] [Google Scholar]
- 39.Rey F, Barré-Sinoussi F, Schmidtmayerova H, Chermann JC. 1987. Detection and titration of neutralizing antibodies to HIV using an inhibition of the cytopathic effect of the virus on MT4 cells. J. Virol. Methods 16:239–249. 10.1016/0166-0934(87)90008-5 [DOI] [PubMed] [Google Scholar]
- 40.Earl PL, Americo JL, Moss B. 2003. Development and use of a vaccinia virus neutralization assay based on flow cytometric detection of green fluorescent protein. J. Virol. 77:10684–10688. 10.1128/JVI.77.19.10684-10688.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kraus AA, Messer W, Haymore LB, de Silva AM. 2007. Comparison of plaque-and flow cytometry-based methods for measuring dengue virus neutralization. J. Clin. Microbiol. 45:3777–3780. 10.1128/JCM.00827-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sashihara J, Burbelo PD, Savoldo B, Pierson TC, Cohen JI. 2009. Human antibody titers to Epstein-Barr Virus (EBV) gp350 correlate with neutralization of infectivity better than antibody titers to EBV gp42 using a rapid flow cytometry-based EBV neutralization assay. Virology 391:249–256. 10.1016/j.virol.2009.06.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burnet F, Keogh EV, Lush D. 1937. The immunological reactions of the filterable viruses. Immunology Cell Biol. 15:227–368. 10.1038/icb.1937.23 [DOI] [Google Scholar]
- 44.Brioen P, Boxyé A. 1985. Poliovirus neutralization and the percentage law. Arch. Virol. 83:105–111. 10.1007/BF01310968 [DOI] [PubMed] [Google Scholar]
- 45.Ge S, Yan Q, He S, Zhuang S, Niu J, Xia N. 2013. Specific primer amplification of the VP1 region directed by 5′ UTR sequence analysis: enterovirus testing and identification in clinical samples from hand-foot-and-mouth disease patients. J. Virol. Methods 193:463–469. 10.1016/j.jviromet.2013.06.009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.