

Abstract
Salmonella enterica serovar Typhimurium (S. Typhimurium) pathogenicity island 1 (SPI-1) encodes a type III secretion system required for invasion of host gut epithelial cells. Expression of SPI-1 virulence genes is controlled by a complex hierarchy of transcription factors encoded within and outside SPI-1. The master regulator of SPI-1, HilA, is itself regulated by three homologous transcription factors, HilD, HilC, and RtsA. HilD activates transcription of hilA and other target genes in response to environmental conditions associated with the intestinal microenvironment of the host. We have mapped the binding of HilD across the S. Typhimurium genome using chromatin immunoprecipitation-sequencing (ChIP-seq). Thus, we have identified 17 regions bound by HilD, including 11 novel targets. The majority of HilD targets are located outside SPI-1. We demonstrate transcription activation of 8 genes by HilD; four of these genes have not been previously described as being regulated by HilD, including lpxR, which encodes a lipid A deacylase important for immune evasion. We also show that HilD-activated genes are frequently activated by HilC and RtsA, indicating extensive overlap of the HilD, HilC, and RtsA regulons.
INTRODUCTION
Salmonella enterica subsp. enterica serovar Typhimurium (S. Typhimurium) is a Gram-negative enteric pathogen that is a leading cause of bacterial food-borne illness worldwide. Capable of infecting a broad range of animal species, S. Typhimurium infection in humans typically manifests as a self-limiting gastroenteritis but in approximately 5% of cases progresses to a severe, potentially fatal bacteremia (1). In addition to its relevance as a pathogen, S. Typhimurium serves as an important model for host-pathogen interactions during infection (2). Successful S. Typhimurium pathogenesis is the consequence of complex temporal regulation of sets of virulence genes, each specific to a particular microenvironment encountered within the host.
An essential early step in the progression of S. Typhimurium infection is bacterial invasion of host intestinal epithelial cells. Invasion is facilitated by genes in a 40-kb region of horizontally acquired DNA known as Salmonella pathogenicity island 1 (SPI-1), one of several pathogenicity islands on the Salmonella chromosome (3). SPI-1 encodes a type III secretion system (T3SS) that is required for invasion of host cells and intestinal disease (4). The SPI-1 T3SS is a multiprotein, needle-like complex that delivers effector proteins to the host cell cytoplasm; these effectors directly induce rearrangements of the actin cytoskeleton, leading to membrane ruffling and uptake of invading Salmonella by these otherwise nonphagocytic cells (5).
Appropriate expression of the SPI-1 T3SS is controlled by a combination of regulatory proteins encoded both inside and outside the pathogenicity island. The OmpR/ToxR family transcription factor HilA is considered to be the master regulator of SPI-1, as it is capable of activating expression of all SPI-1 genes required for the assembly of a functional T3SS (6). However, HilA activation is dependent upon a coupled, positive-feedback loop comprising the AraC/XylS family transcription factors HilD, HilC, and RtsA, which drive HilA expression above the SPI-1 activation threshold (7, 8). HilD, HilC, and RtsA are capable of self-activating expression in addition to activating expression of one another and of hilA. Of these three regulators, HilD is considered to be dominant, as deletion of hilD nearly abolishes hilA expression (9–11), whereas deletion of either hilC or rtsA does not. Additionally, HilD serves as the integration point of various activating environmental signals, including low oxygen, iron limitation, and osmolarity, which together indicate the intestinal microenvironment of the host appropriate for invasion gene expression (12, 13).
HilD has mainly been studied in the context of hilA regulation (7) and regulation of other known virulence genes (14). Previously described HilD regulatory targets are hilD itself, hilA, hilC, rtsA, invF, and downstream genes (the inv-spa and sic-sip operons), invR, dsbA, slrP, ssrAB (6), and genes in SPI-4 (15). Most of these genes were identified as being regulated by HilD by using targeted experiments. Next-generation sequencing methods, such as chromatin immunoprecipitation-sequencing (ChIP-seq), can be used to probe regulatory effects in a much more comprehensive and unbiased manner. ChIP with microarray technology (ChIP-chip) and ChIP-seq enable quantitative, genome-wide mapping of both the position and magnitude of transcription factor binding sites (16). Even for well-studied transcription factors, ChIP-chip and ChIP-seq experiments have repeatedly revealed novel binding sites, including those previously considered to be noncanonical in nature (e.g., within genes and upstream of seemingly unrelated genes) (17–19). To date, ChIP-seq and other genome-scale approaches have been sparingly applied to study the layered regulation of S. Typhimurium virulence (19–25).
In this work, we combine ChIP-seq and targeted reporter gene assays to map the regulon of HilD in S. Typhimurium and identify regulation upon HilD binding. We identify 11 novel HilD-bound regions and four novel HilD-regulated genes. We also show regulation of many HilD target genes by HilC and RtsA. To our knowledge, this is the first study to globally map the regulon of this transcription factor essential to S. Typhimurium virulence.
MATERIALS AND METHODS
Strains and plasmids.
All strains and plasmids used in this work are listed in Table 1. All oligonucleotides used in this work are listed in Table S1 in the supplemental material.
TABLE 1.
List of strains and plasmids used in this study
| Strain or plasmid | Description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli AMD054 | MG1655 ΔlacZ | 27 |
| S. Typhimurium | ||
| 14028s | Wild type | 59 |
| BLP006 | 14028s hilD-FLAG3 | 31 |
| NR-40655 | 14028s ΔhilD | BEI Resources |
| Plasmids | ||
| pAMD-BA-lacZ | Single-copy lacZ expression vector, encodes chloramphenicol resistance | 27 |
| pBAD24 | Expression plasmid with PBAD promoter | 26 |
| pBLP013 | pBAD24-hilD | This work |
| pBLP011 | pBAD24-hilC | This work |
| pBLP010 | pBAD24-rtsA | This work |
| pBLP002 | pAMD-BA-lacZ with STM14_1282 upstream region | This work |
| pBLP003 | pAMD-BA-lacZ with STM14_2342 upstream region | This work |
| pBLP005 | pAMD-BA-lacZ with hilD upstream region | This work |
| pBLP006 | pAMD-BA-lacZ with hilA upstream region | This work |
| pBLP008 | pAMD-BA-lacZ with siiA upstream region | This work |
| pBLP009 | pAMD-BA-lacZ with rtsA upstream region | This work |
| pAMD144 | pAMD-BA-lacZ with lpxR upstream region | This work |
| pAMD151 | pAMD-BA-lacZ with ytfK upstream region | This work |
| pAMD145 | pAMD-BA-lacZ with STM14_1613 upstream region | This work |
| pAMD147 | pAMD-BA-lacZ with mcpC upstream region | This work |
| pAMD149 | pAMD-BA-lacZ with ytfJ upstream region | This work |
| pAMD150 | pAMD-BA-lacZ with STM14_5291 upstream region | This work |
| pAMD152 | pAMD-BA-lacZ with flhD upstream region | This work |
| pBLP001 | pAMD-BA-lacZ with sinR upstream region | This work |
| pBLP004 | pAMD-BA-lacZ with STM14_2343 upstream region | This work |
| pBLP007 | pAMD-BA-lacZ with STM14_5116 upstream region | This work |
| pAMD148 | pAMD-BA-lacZ with STM14_5184 upstream region | This work |
hilD, hilC, and rtsA were cloned into pBAD24 (26) for arabinose-inducible expression. PCR products for each gene were generated and cloned into NheI and SphI restriction sites using standard ligation. PCR products were generated with the following oligonucleotides: plasmid pBLP013 (hilD) with oligonucleotides JW2401 and JW2402, plasmid pBLP011 (hilC) with oligonucleotides JW2399 and JW2400, and plasmid pBLP010 (rtsA) with oligonucleotides JW2403 and JW2404.
Regions upstream of candidate HilD-regulated genes were cloned upstream of lacZ in the single-copy plasmid pAMD-BA-lacZ (27). All upstream regions are described in Fig. S2 and S3 in the supplemental material. PCR products for each upstream region were generated and cloned into SphI and HindIII restriction sites. Cloning was performed using either standard ligation or using an In-Fusion kit (Clontech). PCR products were generated using the primers listed in Table S2 in the supplemental material.
ChIP-qPCR and ChIP-seq.
The ChIP methods are based on those described previously (28). Wild-type 14028s or BLP006 (14028s hilD-FLAG3) S. Typhimurium cells were grown overnight in LB (1% NaCl, 1% tryptone, 0.5% yeast extract), subcultured 1/100 in 20 ml, and grown in screw-cap 50-ml tubes with shaking (250 rpm) to an optical density at 600 nm (OD600) of ∼1.0. Expression of SPI-1 genes has been observed previously under similar growth conditions (14). Cells were cross-linked for 20 min with formaldehyde (1% final concentration), pelleted by centrifugation, and washed once with Tris-buffered saline (TBS). Cell pellets were resuspended in 1 ml FA lysis buffer (50 mM HEPES-KOH [pH 7], 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS) with 2 mg/ml lysozyme and incubated at 37°C for 30 min. Samples were then chilled and sonicated for 30 min in a Bioruptor sonicator (Diagenode) with 30-s-on/30-s-off pulsing at maximum amplitude. Samples were pelleted in a microcentrifuge to remove debris, and supernatants (chromatin) were saved, 1 ml FA lysis buffer was added, and samples were stored indefinitely at −20°C.
For each immunoprecipitation (IP), 500 μl chromatin was incubated with 300 μl FA lysis buffer, 20 μl protein A-Sepharose slurry (50%) in TBS, and 2 μl M2 anti-FLAG antibody (Sigma) for 90 min at room temperature with gentle mixing on a LabQuake rotisserie rotator (Thermo Scientific). Beads were then pelleted at 1,500 × g in a microcentrifuge for 1 min. The supernatant was removed, and the beads were resuspended in 750 μl FA lysis buffer and transferred to a Spin-X column (Corning). Beads were then incubated for 3 min with gentle mixing on a rotisserie rotator before being pelleted at 1,500 × g in a microcentrifuge for 1 min. Equivalent washes were performed with FA lysis buffer, high-salt FA lysis buffer (50 mM HEPES-KOH [pH 7], 500 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS), ChIP wash buffer (10 mM Tris-HCl [pH 8.0], 250 mM LiCl, 1 mM EDTA, 0.5% Nonidet P-40, 0.5% sodium deoxycholate), and TE buffer (10 mM Tris-HCl [pH 7.5], 1 mM EDTA). After the TE wash, beads were transferred to a fresh Spin-X column and eluted with 100 μl ChIP elution buffer (50 mM Tris-HCl [pH 7.5], 10 mM EDTA, 1% SDS) for 10 min at 65°C with occasional agitation. Eluted samples were centrifuged at 1,500 × g in a microcentrifuge for 1 min. Supernatants were de-cross-linked by boiling for 10 min and cleaned up using a PCR purification kit (Qiagen). For all ChIP-quantitative PCR (qPCR) experiments, 20 μl untreated chromatin was de-cross-linked by boiling for 10 min and cleaned up using a PCR purification kit (Qiagen). This sample served as the “input” control.
For qPCR, ChIP and input samples were analyzed using an ABI 7500 fast real-time PCR machine, as described previously (17). Enrichment of ChIP samples was calculated relative to the control region within sbcC and normalized to input DNA. Occupancy units represent background-subtracted fold enrichment. The oligonucleotides used for real-time PCR were JW1495 and JW1496 (sbcC), JW2412 and JW2413 (sinR), JW2414 and JW2415 (STM14_1282), JW2418 and JW2419 (lpxR/STM14_1613), JW2424 and JW2425 (STM14_2342/3), JW2430 and JW2431 (hilC), JW2434 and JW2435 (prgH/hilD), JW2436 and JW2437 (hilA), JW2442 and JW2443 (invF/H), JW3454 and JW3455 (mcpC), JW3458 and JW3459 (siiA), JW2458 and JW2459 (rtsA/STM14_5189), JW3460 and JW3461 (ytfJ/K), JW2410 and JW2411 (ribF), JW2420 and JW2421 (STM14_1833), JW2428 and JW2429 (sprB), JW2438 and JW2439 (sicP), JW2440 and JW2441 (invA), JW2448 and JW2449 (ygiK), JW2452 and JW2453 (STM14_4640), JW2456 and JW2457 (STM14_5184), and JW2462 and JW2463 (STM14_5405).
For ChIP-seq, 32 ChIP samples for HilD-FLAG3 cells were pooled (32 were needed to generate enough DNA for a Helicos sequencing run), ethanol precipitated, and resuspended in water to a final volume of 11 μl. For the control sample, 2 μl from each of the 32 chromatin samples was de-cross-linked by boiling for 10 min and cleaned up using a PCR purification kit (Qiagen). DNA from ChIP or control samples was then treated using the NEB quick blunting kit using 1 μl blunting enzyme mix (NEB) according to the manufacturer's instructions. Samples were incubated for 30 min at room temperature, followed by inactivation of the enzyme by heating samples to 70°C for 10 min. Once cooled, samples were treated with RNase A, incubated for 15 min at 37°C, and purified using the Qiagen PCR purification kit, eluting in 11 μl. DNA (10.8 μl) was then poly(A) tailed by adding 2 μl 2.5 mM CoCl2, 2 μl 10× terminal transferase buffer, heating at 95°C for 5 min, cooling at 4°C, adding 4 μl 50 μM dATP, 0.2 μl bovine serum albumin (BSA), 0.75 μl water, and 0.25 μl terminal transferase and incubating the mixture at 37°C for 1 h and then 70°C for 10 min, followed by cooling to 4°C. Samples were blocked using biotin-ddATP by heating 20 μl poly(A)-tailing reaction mixture at 95°C for 5 min, cooling to 4°C, adding 1 μl 2.5 mM CoCl2, 1 μl 10× terminal transferase buffer, 0.5 μl 200 μM biotin-ddATP, 7.25 μl water, and 0.25 μl terminal transferase, incubating the mixture at 37°C for 1 h and then 70°C for 20 min, followed by cooling to 4°C.
Poly(A)-tailed, ddATP-blocked samples were sequenced using a HeliScope single-molecule sequencer (Helicos Biosciences). Sequence reads were mapped to the S. Typhimurium 14028s genome sequence using HeliSphere software (Helicos Biosciences). Putative HilD-bound regions were identified using model-based analysis of ChIP-seq (MACS) (default settings), with both ChIP and input samples (29). The coordinate of the peak center was defined as the position midway between the highest-scoring position on the top strand and the highest-scoring position on the bottom strand. Secondary peaks within two regions were identified by virtue of having >100 mapped sequence reads and being separated from the major peak and other secondary peaks by genome positions with <10% of the number of reads of the major peak.
β-Galactosidase assays.
For Escherichia coli, 2 to 3 ml of cells were grown in LB ± 0.2% arabinose at 37°C to an OD600 of 0.7 to 1.0, and the OD600 was recorded. For S. Typhimurium, 2 to 3 ml of cells were grown in LB to an OD600 of ~1.0. Cells (800 μl) were pelleted at full speed in a microcentrifuge for 1 min. (Eighty microliters was used for strongly active fusions, and this was corrected for at the final calculation step.) Cell pellets were resuspended in 800 μl Z buffer (0.06 M Na2HPO4, 0.04 M NaH2PO4, 0.01 M KCl, 0.001 M MgSO4) plus 50 mM β-mercaptoethanol (added fresh). Twenty microliters chloroform and 10 μl 0.1% SDS were added to the cells, followed by vortexing for 5 s. Assays were started by addition of 160 μl o-nitrophenyl-β-d-galactopyranoside (ONPG) (4 mg/ml in distilled water [dH2O]) and stopped by addition of 400 μl 1 M Na2CO3, upon development of an appropriate yellow color. The reaction time was noted. Samples were centrifuged at full speed in a microcentrifuge to separate the chloroform phase. The OD420 of the supernatant was recorded. Assay units were calculated as 1,000 × A420/(A600)(total time).
Multiple sequence alignment.
Peptide sequences for the HilD, HilC, and RtsA DNA-binding domains were aligned using ClustalW (30).
Microarray data accession number.
Raw ChIP-seq data are available through the EMBL-EBI ArrayExpress site under accession no. E-MTAB-1848.
RESULTS
To identify HilD-regulated genes, we mapped the genome-wide association of HilD using ChIP-seq. HilD was C-terminally epitope tagged with three FLAG tags, as described previously (31). We confirmed that FLAG-tagged HilD is fully active by measuring expression from the hilA promoter (see Fig. S1 in the supplemental material). We used model-based analysis of ChIP-seq (MACS) (29) to analyze the ChIP-seq data in combination with a control “input” data set from DNA taken prior to immunoprecipitation. Thus, we identified 51 candidate HilD-bound regions (see Table S3 in the supplemental material). We manually subdivided two of these regions (see Materials and Methods), since by visual inspection of aligned sequence reads, it was clear that they included multiple, independent ChIP-seq peaks. To validate the ChIP-seq data, we used ChIP coupled with quantitative real-time PCR (ChIP-qPCR) to measure association of HilD with 21 of the top-scoring ChIP-seq peaks identified by MACS. As a control, we performed ChIP-qPCR with an untagged strain. We confirmed association of HilD with 17 of the 21 regions, with significantly greater enrichment observed for the ChIP sample than for the sample from the untagged strain (Fig. 1A) (t test, P < 0.02 in all cases). Our data suggest that regions identified by MACS with a score of <600 (there are 32 such regions) are likely to be false positives. One of the highest-scoring regions from the MACS analysis showed no significant enrichment in the targeted ChIP-qPCR region. Closer inspection of this region revealed a long, inverted repeat, which is likely to result in anomalous sequence read mapping. Consistent with this, the vast majority of sequence reads mapped to only one of the two DNA strands in this region.
FIG 1.
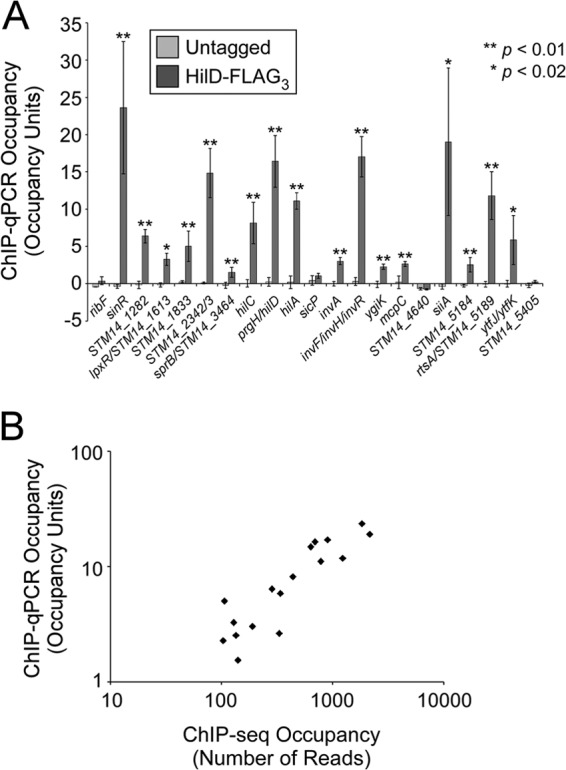
Sites of HilD association identified by ChIP-seq. (A) Validation of putative S. Typhimurium HilD target regions identified by ChIP-seq. Data are from ChIP of untagged 14028s or FLAG-tagged HilD, followed by quantitative real-time PCR. Occupancy units represent background-subtracted fold enrichment relative to a control genomic region within the sbcC gene. Error bars represent 1 standard deviation from the mean, based on three independent biological replicates, with the exception of lpxR/STM14_1613, for which only two replicates were performed. Asterisks indicate a significant difference between the occupancy unit values for tagged and untagged strains (**, P < 0.01; *, P < 0.02). (B) Comparison of ChIP-seq and ChIP-qPCR data for HilD-FLAG3. Each data point represents a HilD target region identified by ChIP-seq and confirmed by ChIP-qPCR. ChIP-seq scores are the sum of the read counts on both strands at the highest-scoring position. ChIP-qPCR scores are in occupancy units.
The 17 experimentally confirmed targets are listed in Table 2, and ChIP-seq data are shown for each corresponding MACS region in Fig. S2 in the supplemental material. Six of the confirmed HilD-bound regions are associated with genes that have been previously described as HilD targets: hilC (STM14_3465), hilD (STM14_3474), hilA (STM14_3475), invF (STM14_3498)/invR (ChIP-seq peak too broad to separate two distinct bound regions), siiA (STM14_5117; regulation was shown only for the downstream gene siiE), and rtsA (STM14_5188) (6, 15, 32, 33). We did not identify HilD binding in regions corresponding to the described HilD targets dsbA (STM14_4806), slrP (STM14_0928), and ssrAB (STM14_1687-STM14_1686) (14, 33, 34). The remaining 11 regions have not previously been linked to HilD, and 9 are located outside SPI-1. Most ChIP-seq peaks are located close to the start of an annotated gene, with the exception of peaks that fall well within the STM14_1833, invA (STM14_3495), and ygiK (STM14_3843) genes. The levels of association determined by ChIP-qPCR correspond well with those determined by ChIP-seq (Fig. 1B and Table 2). We attempted to identify enriched DNA sequence motifs at the confirmed sites of HilD association using MEME (default settings) (35), but were unable to detect any significant hits, suggesting that HilD binds to a degenerate sequence motif.
TABLE 2.
List of ChIP-seq peaks for HilDa
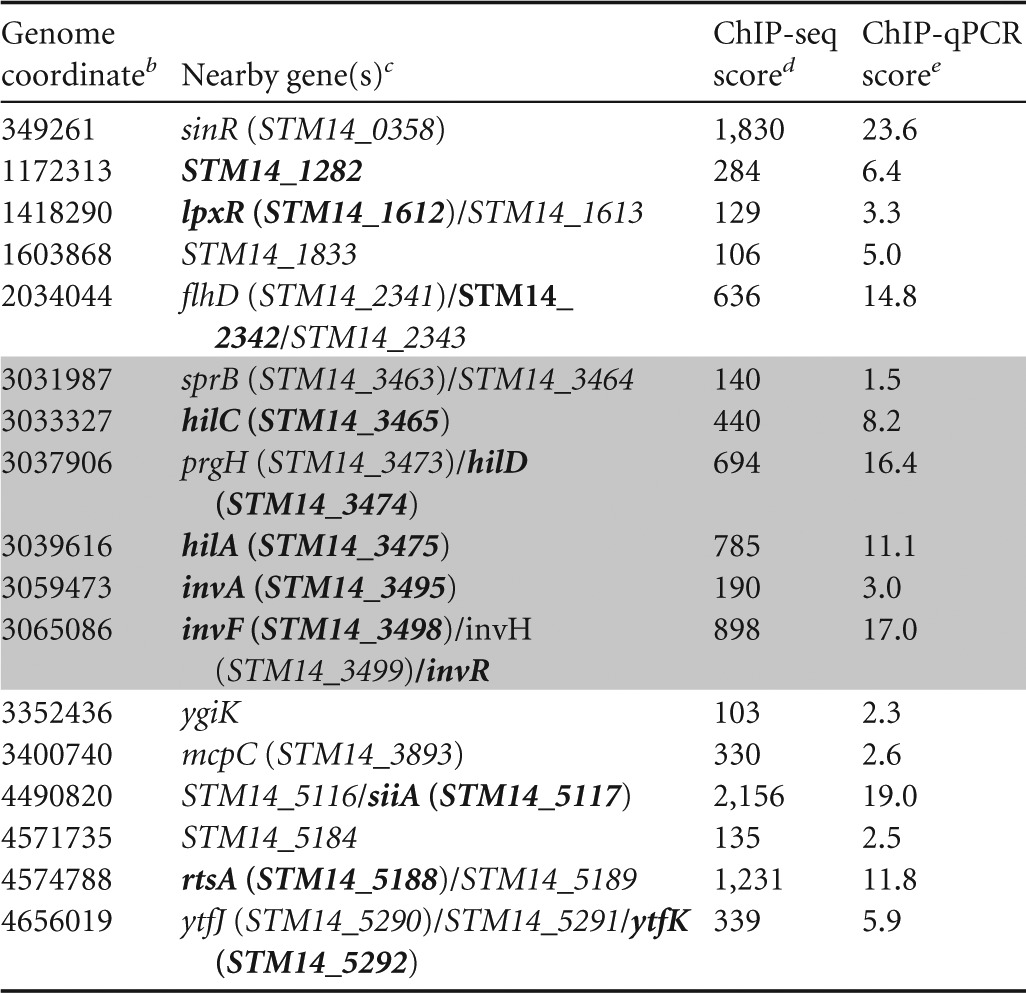
SPI-1 regions are shaded in gray.
Position (bp) in the genome of the ChIP-seq peak.
Genes confirmed to be regulated by HilD in this or previous studies are indicated in bold text.
Number of sequence reads at the genome coordinate with the most signal in the region.
Occupancy units.
ChIP-seq identifies sites of association with DNA but provides no direct information on regulation; for most HilD-bound regions, the likely target gene was unclear from ChIP-seq data alone. Hence, we selected one or more candidate genes that are located close to each confirmed ChIP-seq peak. In total, we identified 17 candidate genes located within or near the confirmed HilD-bound regions and constructed translational fusions of their upstream regions to a lacZ reporter gene (see Fig. S2 and S3 in the supplemental material). We then determined the expression of lacZ from the 17 constructs in Escherichia coli for cells containing either empty vector or plasmid expressing hilD from an arabinose-inducible promoter under inducing conditions. We reasoned that measuring expression in E. coli with heterologous expression of HilD would avoid the potential cross talk and redundancy that occur in S. Typhimurium between HilD and other regulators, such as HilC, RtsA, and HilA. For 9 of the 17 constructs, we detected no effect of HilD on their expression (see Table S4 in the supplemental material). For the remaining 8 constructs, we detected HilD-dependent transcription activation (Fig. 2; see Table S4). In all 8 cases, transcription activation required both the inducible HilD plasmid and the inducer, arabinose. The level of induction by HilD varied from <2-fold for hilD itself to 146-fold for hilA. We conclude that these 8 genes are direct regulatory targets of HilD. The 8 genes include four described HilD-activated genes: hilD, hilA, siiA, and rtsA. We did not test the other described HilD-activated genes within SPI-1, hilC and invF. Four of the direct regulatory targets of HilD have not been described previously: STM14_1282, STM14_2342, lpxR (STM14_1612), and ytfK (STM14_5292).
FIG 2.
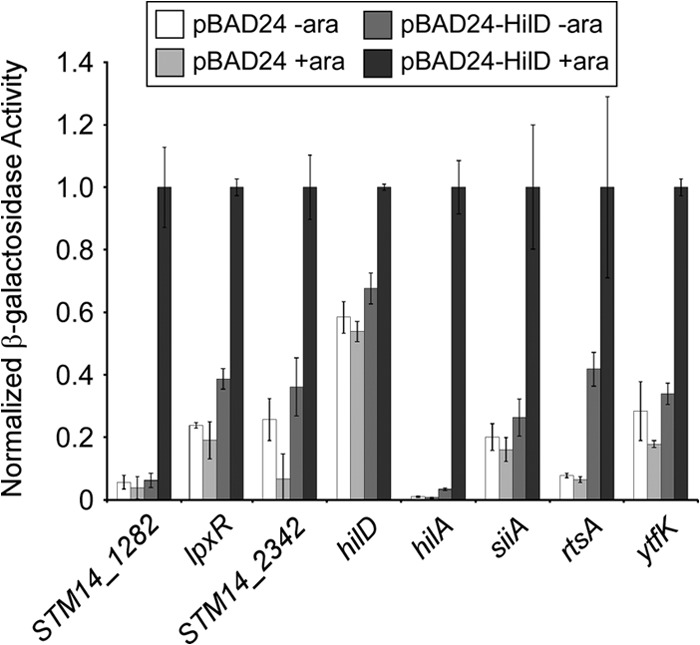
β-Galactosidase assay showing HilD activation of target genes. β-Galactosidase assay of fusions of the indicated upstream regions fused translationally to a lacZ reporter gene on a single-copy plasmid. Assays were performed with E. coli strain AMD054 containing either empty pBAD24 plasmid or pBLP013 (hilD), under noninducing (no arabinose) or inducing (with arabinose) conditions, as indicated. Data are shown normalized to the values in the presence of pBLP013 (hilD) and arabinose. Error bars represent 1 standard deviation from the mean, based on at least three independent biological replicates.
HilC and RtsA are homologues of HilD with 62% and 61% identity with HilD, respectively, in their DNA-binding domains (Fig. 3A). HilD, HilC, and RtsA are members of the AraC family of transcription factors. The crystal structure of MarA, another AraC family member, has been solved in its DNA-bound state. By aligning the sequences of HilD, HilC, and RtsA DNA-binding domains with that of MarA using Pfam (36), we predicted the amino acids that make base-specific contacts with DNA. Strikingly, all but one of these positions are identical in all three proteins (Fig. 3A), suggesting similar cognate DNA-binding sites. Consistent with this, HilD, HilC, and RtsA have been shown to directly regulate several of the same target genes, including hilA (33, 34, 37, 38), invF (33, 39), dsbA (34), and slrP (33). Hence, we predicted that the HilD-regulated genes are also regulated by HilC and RtsA. To test this hypothesis, we measured expression of the lacZ fusion constructs in E. coli containing either plasmid expressing hilC or rtsA from an arabinose-inducible promoter under inducing and noninducing conditions. We detected positive regulation of 7/8 genes by both HilC and RtsA (Fig. 3B and C; raw data, including for empty vector controls, shown in Table S4 in the supplemental material). The levels of induction by HilD, HilC, and RtsA were similar for all genes tested (c.f. Fig. 2 and Fig. 3B and C). Transcription of one gene, ytfK, was activated by HilC but not RtsA (Fig. 3B and C).
FIG 3.

β-Galactosidase assay showing activation of HilD target genes by HilC and RtsA. (A) Sequence alignment of predicted DNA-binding domains of HilD, HilC, and RtsA, shown in the CLUSTAL format (30). Shaded residues indicate those predicted to make base-specific contacts with DNA. (B) β-Galactosidase assay of fusions of the indicated upstream regions fused translationally to a lacZ reporter gene on a single-copy plasmid. Assays were performed with E. coli strain AMD054 containing pBLP011 (hilC), under noninducing (no arabinose) or inducing (with arabinose) conditions, as indicated. Data are shown normalized to the values for cells grown in the presence of arabinose. Error bars represent 1 standard deviation from the mean, based on at least three independent biological replicates. (C) As described above, but with pBLP010 (rtsA) used in place of pBLP011 (hilC).
DISCUSSION
Much of the HilD regulon is outside SPI-1.
The known HilD regulon, as determined in this and previous studies, is summarized in Fig. 4. Using a combination of ChIP-seq and targeted expression analysis, we have identified four novel HilD-activated genes: STM14_1282, STM14_2342, lpxR, and ytfK. In addition, we confirmed association of HilD upstream of previously described targets hilC, hilD, hilA, invF, siiA, and rtsA (Fig. 1), and we confirmed regulation of previously described targets hilD, hilA, siiA, and rtsA by HilD (Fig. 2). We did not detect HilD upstream of the previously described target ssrAB, the two-component regulatory system of SPI-2. This could be due to condition-specific binding of HilD upstream of ssrA, since ssrAB expression is induced by HilD in the late stationary phase, well beyond the time point of our ChIP-seq experiment (14). We also did not detect HilD binding upstream of previously described regulatory targets dsbA and slrP. Although direct association of HilD with these regions has not been demonstrated, transcription activation of both dsbA and slrP by HilD has been observed in strains containing deletions of both hilC and rtsA (33, 34), ruling out the possibility of indirect regulation through cross talk between HilD and HilC/RtsA. We propose that binding of HilD upstream of these genes and regulation by HilD occur in a condition-specific manner.
FIG 4.
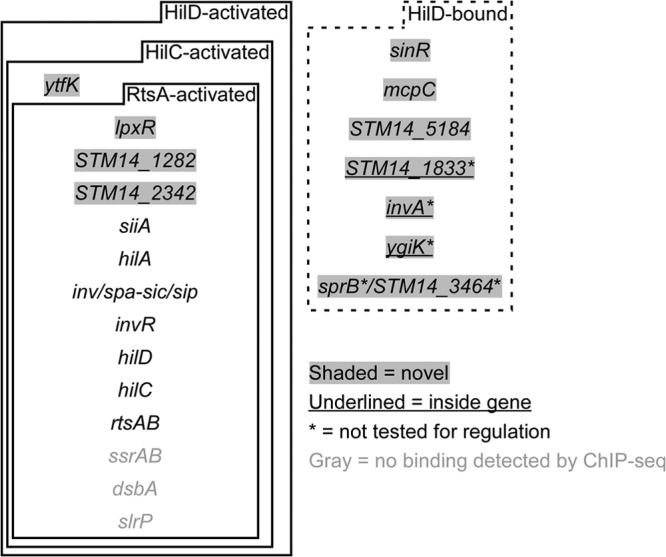
Summary of HilD-bound regions and HilD-regulated genes. Shown are genes identified as being directly regulated by HilD and regions identified as being bound by HilD, based on this and previous studies. Genes given in gray text were not identified as being HilD bound in this study. Genes with gray shading have not previously been described as being HilD regulated or bound. The dashed box indicates genes (i) that are not regulated by HilD or for which regulation has not been tested and (ii) whose upstream region is bound by HilD or for which HilD binds within the gene. (Underlined text indicates intragenic binding.) Asterisks indicate HilD-associated regions for which regulation by HilD was not tested in this or previous studies.
The function of novel HilD-regulated genes.
Most previously described HilD targets are located within SPI-1 (6). In contrast, all but two of the novel HilD targets are located outside SPI-1. In fact, the majority of HilD-bound regions are outside SPI-1, and the same is true for HilD-activated genes. Thus, HilD coordinates expression of SPI-1 and non-SPI-1 genes. lpxR, a novel HilD-regulated gene, encodes an outer membrane protein that removes the 3′-acyloxyacyl group of lipid A, the hydrophobic anchor of lipopolysaccharide (LPS) (40). Modification of lipid A by LpxR increases the ability of S. Typhimurium to evade the innate immune response (41) and promotes survival within macrophages (42). LpxR is conserved in other pathogenic bacterial species, and lpxR mutants of Yersinia enterocolitica are attenuated for virulence in a mouse model (43). (Note that lpxR in Y. enterocolitica is known as sfpA.) Regulation of lpxR by HilD, HilC, and RtsA likely coordinates the timing of lipid A deacylation with epithelial cell invasion, suggesting a selective advantage for acylated lipid A prior to epithelial cell invasion. Three of the novel HilD-activated genes identified in this study, STM14_1282, STM14_2342, and ytfK, have no known or predicted function. Given that all other described HilD-activated genes have an established connection to virulence, regulation of STM14_1282, STM14_2342, and ytfK by HilD implicates these genes in virulence-associated processes.
HilD binding that is not associated with detectable regulation.
Three of the HilD-bound regions we identified by ChIP-seq can be unambiguously associated with nearby genes that are not detectably regulated by HilD: mcpC (STM14_3893), sinR (STM14_0358), and STM14_5184. mcpC is a member of the flagellar regulon (44) and encodes a protein involved in chemotaxis (44, 45). sinR encodes a LysR family transcription factor and is conserved only in serovars of Salmonella that infect warm-blooded hosts (46). The function of STM14_5184 is unknown. None of these genes has been previously linked to SPI-1 gene regulation. However, SinR has a virulence-associated function since sinR mutants are defective in survival within macrophages (47). Furthermore, sinR is located within a horizontally acquired segment of DNA that encodes a fimbrial apparatus (46). The level of association of HilD upstream of sinR is greater than with any other genomic location. We speculate that mcpC, sinR, and STM14_5184 are regulatory targets of HilD and possibly also of HilC and RtsA, but require additional transcription factors that are not present or not expressed in E. coli under the conditions used in our expression assays. This model suggests that HilD functions cooperatively with one or more additional transcription factors that may be condition or species specific.
HilD binding within genes.
Three of the HilD-bound regions we identified by ChIP-seq fall well within annotated genes, far from the start of the overlapping gene or any other nearby gene (Fig. 4; see Fig. S2 in the supplemental material). These include a peak within a SPI-1 gene, invA. We did not test regulation of these overlapping genes. Little is known about the function of transcription factor binding sites within genes, although there are a few examples of such binding sites that have been shown to serve as roadblocks to elongating RNA polymerase (48–50). Recent ChIP-chip and ChIP-seq studies have identified numerous intragenic binding sites for some transcription factors (17, 51, 52), including in S. Typhimurium (19), suggesting an important regulatory function for these sites.
Cross talk between HilD, HilC, and RtsA.
HilD, HilC, and RtsA are homologous AraC family transcription factors. Their DNA-binding domains are each ∼60% identical, with the predicted base-specific contacts being largely conserved (Fig. 3A). Previous studies have identified shared regulatory targets of HilD, HilC, and RtsA, and it has been suggested that these transcription factors bind their targets in vivo as heterodimers (7). HilD, HilC, and RtsA all activate transcription of hilA, hilC, hilD, dsbA, invF, slrP, rtsA, and genes in SPI-4 (6, 15, 32, 33). Furthermore, HilD and HilC bind the same DNA sites upstream of hilD and hilC (38). Our data confirm a large degree of overlap between the HilD, HilC, and RtsA regulons and extend this overlap to include the novel HilD targets; we observed transcription activation of all 8 of the HilD-activated genes by HilC and all but one by RtsA (Fig. 3). It is important to note that these assays were performed in E. coli, in which only one of the three regulators is expressed in any given experiment; there are no close homologues of HilD, HilC, or RtsA in E. coli. Hence, regulation by HilD, HilC, and RtsA must be direct. We conclude that HilD, HilC, and RtsA bind very similar DNA sites, although the failure of RtsA to regulate ytfK (Fig. 3C) indicates that there are subtle differences in the sequence specificities of HilD, HilC, and RtsA. Consistent with this, RtsA has been shown to be a substantially stronger activator of slrP than HilD or HilC (33). Furthermore, a previous study suggested that although HilD and HilC have similar consensus DNA-binding sites, HilD is able to bind only a subset of HilC sites (38).
Overlap of the HilD, HilA, and SprB regulons.
In addition to HilD and HilC, three other transcription factors are encoded within SPI-1: HilA, SprB, and InvF. Unlike HilC and RtsA, these three regulators are not homologues of HilD; hence, they are expected to bind DNA with different sequence specificities. The HilD targets we identified have no overlap with the known targets of InvF (53). However, there is overlap of the HilD targets with those of HilA and SprB. HilA has been shown to directly regulate the previously described HilD target invF (21, 54). A HilA ChIP-chip study also identified a HilA binding site in the vicinity of the SL1344 homologue of STM14_2342. However, microarray analysis suggested that binding of HilA at this site regulates expression of the downstream gene flhD (21), for which we did not detect regulation by HilD (see Table S4 in the supplemental material).
We observed direction regulation of siiA by HilD. siiA is the first gene in Salmonella pathogenicity island 4 (SPI-4), and it is likely to be the first gene in an operon that includes all SPI-4 genes. SPI-4 genes encode a nonfimbrial adhesin and an associated type I secretion system (55) that are required for maximal virulence of S. Typhimurium in some, but not all, hosts (56, 57). Regulation of SPI-4 genes by HilD, HilC, and RtsA has been described previously (15, 32, 33) and was suggested to be dependent upon HilA (15, 33). However, modest HilA-independent regulation by HilD and HilC was observed (15), consistent with our data. HilA has been shown by ChIP-chip to bind upstream of siiA (21), suggesting that HilD/HilC/RtsA and HilA bind to neighboring sites. Thus, regulation of SPI-4 by HilD, HilC, and RtsA likely coordinates expression of SPI-4 and SPI-1 genes, which are required for attachment to and invasion of epithelial cells, respectively. SprB has been shown to activate transcription of siiA (58), although the same study showed no regulation of siiA by HilA or HilD, in contrast to our data. We propose that siiA is regulated by HilD, HilC, RtsA, HilA, and SprB, which would indicate that SPI-1 and SPI-4 gene expression is tightly linked. Intriguingly, our ChIP-seq analysis identified a weak HilD binding site upstream of sprB (Fig. 4; see Fig. S2 in the supplemental material), suggesting an added layer of feedback regulation.
Conclusions.
Our data demonstrate that the HilD regulon extends well beyond SPI-1, strengthening links to SPI-4 regulation and adding a novel connection between invasion and lipid A modification. Our findings also broaden the regulatory overlap between HilD and its homologues, HilC and RtsA. The impact of this work on our understanding of invasion gene regulation in S. Typhimurium highlights the utility of genome-scale investigation of transcription factor binding, particularly in the context of layered regulatory networks.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Wadsworth Center Applied Genomic Technologies Core Facility for technical assistance. We thank Zach Herbert and the Dana-Farber Cancer Institute Molecular Biology Core Facilities for Helicos sequencing and read mapping. We thank Todd Gray, Keith Derbyshire, David Grainger, James Galagan, and members of the Wade group for helpful discussions.
This work was funded by the National Institutes of Health through the NIH Director's New Innovator Award Program (1DP2OD007188) and by an appointment (B.P.) to the Emerging Infectious Diseases (EID) Fellowship Program administered by the Association of Public Health Laboratories (APHL) and funded by the Centers for Disease Control and Prevention (CDC).
Footnotes
Published ahead of print 27 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01449-13.
REFERENCES
- 1.Hohmann EL. 2001. Nontyphoidal salmonellosis. Clin. Infect. Dis. 32:263–269. 10.1086/318457 [DOI] [PubMed] [Google Scholar]
- 2.Ohl ME, Miller SI. 2001. Salmonella: a model for bacterial pathogenesis. Annu. Rev. Med. 52:259–274. 10.1146/annurev.med.52.1.259 [DOI] [PubMed] [Google Scholar]
- 3.Marcus SL, Brumell JH, Pfeifer CG, Finlay BB. 2000. Salmonella pathogenicity islands: big virulence in small packages. Microbes Infect. 2:145–156. 10.1016/S1286-4579(00)00273-2 [DOI] [PubMed] [Google Scholar]
- 4.Lostroh CP, Lee CA. 2001. The Salmonella pathogenicity island-1 type III secretion system. Microbes Infect. 3:1281–1291. 10.1016/S1286-4579(01)01488-5 [DOI] [PubMed] [Google Scholar]
- 5.Zhou D, Galán J. 2001. Salmonella entry into host cells: the work in concert of type III secreted effector proteins. Microbes Infect. 3:1293–1298. 10.1016/S1286-4579(01)01489-7 [DOI] [PubMed] [Google Scholar]
- 6.Ellermeier JR, Slauch JM. 2007. Adaptation to the host environment: regulation of the SPI1 type III secretion system in Salmonella enterica serovar Typhimurium. Curr. Opin. Microbiol. 10:24–29. 10.1016/j.mib.2006.12.002 [DOI] [PubMed] [Google Scholar]
- 7.Ellermeier CD, Ellermeier JR, Slauch JM. 2005. HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 57:691–705. 10.1111/j.1365-2958.2005.04737.x [DOI] [PubMed] [Google Scholar]
- 8.Saini S, Ellermeier JR, Slauch JM, Rao CV. 2010. The role of coupled positive feedback in the expression of the SPI1 type three secretion system in Salmonella. PLoS Pathog. 6:e1001025. 10.1371/journal.ppat.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones BD. 2005. Salmonella invasion gene regulation: a story of environmental awareness. J. Microbiol. 43:110–117 http://www.msk.or.kr/jsp/downloadPDF1.jsp?fileName=p%5B1%5D.110-117.pdf [PubMed] [Google Scholar]
- 10.Schechter LM, Damrauer SM, Lee CA. 1999. Two AraC/XylS family members can independently counteract the effect of repressing sequences upstream of the hilA promoter. Mol. Microbiol. 32:629–642. 10.1046/j.1365-2958.1999.01381.x [DOI] [PubMed] [Google Scholar]
- 11.Boddicker JD, Knosp BM, Jones BD. 2003. Transcription of the Salmonella invasion gene activator, hilA, requires HilD activation in the absence of negative regulators. J. Bacteriol. 185:525–533. 10.1128/JB.185.2.525-533.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellermeier JR, Slauch JM. 2008. Fur regulates expression of the Salmonella pathogenicity island 1 type III secretion system through HilD. J. Bacteriol. 190:476–486. 10.1128/JB.00926-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Golubeva YA, Sadik AY, Ellermeier JR, Slauch JM. 2012. Integrating global regulatory input into the Salmonella pathogenicity island 1 type III secretion system. Genetics 190:79–90. 10.1534/genetics.111.132779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bustamante VH, Martínez LC, Santana FJ, Knodler LA, Steele-Mortimer O, Puente JL. 2008. HilD-mediated transcriptional cross-talk between SPI-1 and SPI-2. Proc. Natl. Acad. Sci. U. S. A. 105:14591–14596. 10.1073/pnas.0801205105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Main-Hester KL, Colpitts KM, Thomas GA, Fang FC, Libby SJ. 2008. Coordinate regulation of Salmonella pathogenicity island 1 (SPI1) and SPI4 in Salmonella enterica serovar Typhimurium. Infect. Immun. 76:1024–1035. 10.1128/IAI.01224-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wade JT, Struhl K, Busby SJ, Grainger DC. 2007. Genomic analysis of protein-DNA interactions in bacteria: insights into transcription and chromosome organization. Mol. Microbiol. 65:21–26. 10.1111/j.1365-2958.2007.05781.x [DOI] [PubMed] [Google Scholar]
- 17.Wade JT, Reppas NB, Church GM, Struhl K. 2005. Genomic analysis of LexA binding reveals the permissive nature of the Escherichia coli genome and identifies unconventional target sites. Genes Dev. 19:2619–2630. 10.1101/gad.1355605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Laub MT, Chen SL, Shapiro L, McAdams HH. 2002. Genes directly controlled by CtrA, a master regulator of the Caulobacter cell cycle. Proc. Natl. Acad. Sci. U. S. A. 99:4632–4637. 10.1073/pnas.062065699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tomljenovic-Berube AM, Mulder DT, Whiteside MD, Brinkman FS, Coombes BK. 2010. Identification of the regulatory logic controlling Salmonella pathoadaptation by the SsrA-SsrB two-component system. PLoS Genet. 6:e1000875. 10.1371/journal.pgen.1000875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucchini S, Rowley G, Goldberg MD, Hurd D, Harrison M, Hinton JC. 2006. H-NS mediates the silencing of laterally acquired genes in bacteria. PLoS Pathog. 2:e81. 10.1371/journal.ppat.002008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thijs IM, De Keersmaecker SC, Fadda A, Engelen K, Zhao H, McClelland M, Marchal K, Vanderleyden J. 2007. Delineation of the Salmonella enterica serovar Typhimurium HilA regulon through genome-wide location and transcript analysis. J. Bacteriol. 189:4587–4596. 10.1128/JB.00178-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kröger C, Dillon SC, Cameron AD, Papenfort K, Sivasankaran SK, Hokamp K, Chao Y, Sittka A, Hébrard M, Händler K, Colgan A, Leekitcharoenphon P, Langridge GC, Lohan AJ, Loftus B, Lucchini S, Ussery DW, Dorman CJ, Thomson NR, Vogel J, Hinton JC. 2012. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U. S. A. 109:E1277–E1286. 10.1073/pnas.1201061109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Navarre WW, Porwollik S, Wang Y, McClelland M, Rosen H, Libby SJ, Fang FC. 2006. Selective silencing of foreign DNA with low GC content by the H-NS protein in Salmonella. Science 313:236–238. 10.1126/science.1128794 [DOI] [PubMed] [Google Scholar]
- 24.Lucchini S, McDermott P, Thompson A, Hinton JC. 2009. The H-NS-like protein StpA represses the RpoS (s38) regulon during exponential growth of Salmonella Typhimurium. Mol. Microbiol. 74:1169–1186. 10.1111/j.1365-2958.2009.06929.x [DOI] [PubMed] [Google Scholar]
- 25.Dillon SC, Espinosa E, Hokamp K, Ussery DW, Casadesús J, Dorman CJ. 2012. LeuO is a global regulator of gene expression in Salmonella enterica serovar Typhimurium. Mol. Microbiol. 85:1072–1089. 10.1111/j.1365-2958.2012.08162.x [DOI] [PubMed] [Google Scholar]
- 26.Guzman L-M, Belin D, Carson MJ, Beckwith JR. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177:4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dornenburg JE, DeVita AM, Palumbo MJ, Wade JT. 2010. Widespread antisense transcription in Escherichia coli. mBio 1(1):e00024–10. 10.1128/mBio.00024-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rhodius VA, Wade JT. 2009. Technical considerations in using DNA microarrays to define regulons. Methods 47:63–72. 10.1016/j.ymeth.2008.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stringer AM, Singh N, Yermakova A, Petrone BL, Amarasinghe JJ, Reyes-Diaz L, Mantis NJ, Wade JT. 2012. FRUIT, a scar-free system for targeted chromosomal mutagenesis, epitope tagging, and promoter replacement in Escherichia coli and Salmonella enterica. PLoS One 7:e44841. 10.1371/journal.pone.0044841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Keersmaecker SC, Marchal K, Verhoeven TL, Engelen K, Vanderleyden J, Detweiler CS. 2005. Microarray analysis and motif detection reveal new targets of the Salmonella enterica serovar Typhimurium HilA regulatory protein, including hilA itself. J. Bacteriol. 187:4381–4391. 10.1128/JB.187.13.4381-4391.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellermeier CD, Slauch JM. 2003. RtsA and RtsB coordinately regulate expression of the invasion and flagellar genes in Salmonella enterica serovar Typhimurium. J. Bacteriol. 185:5096–5108. 10.1128/JB.185.17.5096-5108.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellermeier CD, Slauch JM. 2004. RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J. Bacteriol. 186:68–79. 10.1128/JB.186.1.68-79.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 2:28–36 [PubMed] [Google Scholar]
- 36.Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. 2012. The Pfam protein families database. Nucleic Acids Res. 40:D290–D301. 10.1093/nar/gkr1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schechter LM, Lee CA. 2001. AraC/XylS family members, HilC and HilD, directly bind and derepress the Salmonella Typhimurium hilA promoter. Mol. Microbiol. 40:1289–1299. 10.1046/j.1365-2958.2001.02462.x [DOI] [PubMed] [Google Scholar]
- 38.Olekhnovich IN, Kadner RJ. 2002. DNA-binding activities of the HilC and HilD virulence regulatory proteins of Salmonella enterica serovar Typhimurium. J. Bacteriol. 184:4148–4160. 10.1128/JB.184.15.4148-4160.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akbar S, Schechter LM, Lostroh CP, Lee CA. 2003. AraC/XylS family members, HilD and HilC, directly activate virulence gene expression independently of HilA in Salmonella Typhimurium. Mol. Microbiol. 47:715–728. 10.1046/j.1365-2958.2003.03322.x [DOI] [PubMed] [Google Scholar]
- 40.Reynolds CM, Ribeiro AA, McGrath SC, Cotter RJ, Raetz CR, Trent MS. 2006. An outer membrane enzyme encoded by Salmonella Typhimurium lpxR that removes the 3′-acyloxyacyl moiety of lipid A. J. Biol. Chem. 281:21974–21987. 10.1074/jbc.M603527200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kawasaki K, Teramoto M, Tatsui R, Amamoto S. 2012. Lipid A 3′-O-deacylation by Salmonella outer membrane enzyme LpxR modulates the ability of lipid A to stimulate Toll-like receptor 4. Biochem. Biophys. Res. Commun. 428:343–347. 10.1016/j.bbrc.2012.10.054 [DOI] [PubMed] [Google Scholar]
- 42.Kawano M, Manabe T, Kawasaki K. 2010. Salmonella enterica serovar Typhimurium lipopolysaccharide deacylation enhances its intracellular growth within macrophages. FEBS Lett. 584:207–212. 10.1016/j.febslet.2009.11.062 [DOI] [PubMed] [Google Scholar]
- 43.Mildiner-Earley S, Miller VL. 2006. Characterization of a novel porin involved in systemic Yersinia enterocolitica infection. Infect. Immun. 74:4361–4365. 10.1128/IAI.00154-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frye J, Karlinsey JE, Felise HR, Marzolf B, Dowidar N, McClelland M, Hughes KT. 2006. Identification of new flagellar genes of Salmonella enterica serovar Typhimurium. J. Bacteriol. 188:2233–2243. 10.1128/JB.188.6.2233-2243.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazova MD, Butler MT, Shimizu TS, Harshey RM. 2012. Salmonella chemoreceptors McpB and McpC mediate a repellent response to L-cystine: a potential mechanism to avoid oxidative conditions. Mol. Microbiol. 84:697–711. 10.1111/j.1365-2958.2012.08051.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Folkesson A, Advani A, Sukupolvi S, Pfeifer JD, Normark S, Löfdahl S. 1999. Multiple insertions of fimbrial operons correlate with the evolution of Salmonella serovars responsible for human disease. Mol. Microbiol. 33:612–622. 10.1046/j.1365-2958.1999.01508.x [DOI] [PubMed] [Google Scholar]
- 47.Klumpp J, Fuchs TM. 2007. Identification of novel genes in genomic islands that contribute to Salmonella Typhimurium replication in macrophages. Microbiology 153:1207–1220. 10.1099/mic.0.2006/004747-0 [DOI] [PubMed] [Google Scholar]
- 48.Belitsky BR, Sonenshein AL. 2011. Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 411:729–743. 10.1016/j.jmb.2011.06.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.He B, Zalkin H. 1992. Repression of Escherichia coli purB is by a transcriptional roadblock mechanism. J. Bacteriol. 174:7121–7127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Deuschle U, Gentz R, Bujard H. 1986. lac repressor blocks transcribing RNA polymerase and terminates transcription. Proc. Natl. Acad. Sci. U. S. A. 83:4134–4137. 10.1073/pnas.83.12.4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shimada T, Ishihama A, Busby SJ, Grainger DC. 2008. The Escherichia coli RutR transcription factor binds at targets within genes as well as intergenic regions. Nucleic Acids Res. 36:3950–3955. 10.1093/nar/gkn339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Belitsky BR, Sonenshein AL. 2013. Genome-wide identification of Bacillus subtilis CodY-binding sites at single-nucleotide resolution. Proc. Natl. Acad. Sci. U. S. A. 110:7026–7031. 10.1073/pnas.1300428110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Darwin KH, Miller VL. 1999. InvF is required for expression of genes encoding proteins secreted by the SPI1 type III secretion apparatus in Salmonella Typhimurium. J. Bacteriol. 181:4949–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bajaj V, Lucas RL, Hwang C, Lee CA. 1996. Co-ordinate regulation of Salmonella Typhimurium invasion genes by environmental and regulatory factors is mediated by control of hilA expression. Mol. Microbiol. 22:703–714. 10.1046/j.1365-2958.1996.d01-1718.x [DOI] [PubMed] [Google Scholar]
- 55.Gerlach RG, Jäckel Stecher DB, Wagner C, Lupas A, Hardt WD, Hensel M. 2007. Salmonella pathogenicity island 4 encodes a giant non-fimbrial adhesin and the cognate type 1 secretion system. Cell. Microbiol. 9:1834–1850. 10.1111/j.1462-5822.2007.00919.x [DOI] [PubMed] [Google Scholar]
- 56.Morgan E, Campbell JD, Rowe SC, Bispham J, Stevens MP, Bowen AJ, Barrow PA, Maskell DJ, Wallis TS. 2004. Identification of host-specific colonization factors of Salmonella enterica serovar Typhimurium. Mol. Microbiol. 54:994–1010. 10.1111/j.1365-2958.2004.04323.x [DOI] [PubMed] [Google Scholar]
- 57.Kiss T, Morgan E, Nagy G. 2007. Contribution of SPI-4 genes to the virulence of Salmonella enterica. FEMS Microbiol. Lett. 275:153–159. 10.1111/j.1574-6968.2007.00871.x [DOI] [PubMed] [Google Scholar]
- 58.Saini S, Rao CV. 2010. SprB is the molecular link between Salmonella pathogenicity island 1 (SPI1) and SPI4. J. Bacteriol. 192:2459–2462. 10.1128/JB.00047-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarvik T, Smillie C, Groisman EA, Ochman H. 2010. Short-term signatures of evolutionary change in the Salmonella enterica serovar Typhimurium 14028 genome. J. Bacteriol. 192:560–567. 10.1128/JB.01233-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.