

Abstract
The holE gene is an enterobacterial ORFan gene (open reading frame [ORF] with no detectable homology to other ORFs in a database). It encodes the θ subunit of the DNA polymerase III core complex. The precise function of the θ subunit within this complex is not well established, and loss of holE does not result in a noticeable phenotype. Paralogs of holE are also present on many conjugative plasmids and on phage P1 (hot gene). In this study, we provide evidence indicating that θ (HolE) exhibits structural and functional similarities to a family of nucleoid-associated regulatory proteins, the Hha/YdgT-like proteins that are also encoded by enterobacterial ORFan genes. Microarray studies comparing the transcriptional profiles of Escherichia coli holE, hha, and ydgT mutants revealed highly similar expression patterns for strains harboring holE and ydgT alleles. Among the genes differentially regulated in both mutants were genes of the tryptophanase (tna) operon. The tna operon consists of a transcribed leader region, tnaL, and two structural genes, tnaA and tnaB. Further experiments with transcriptional lacZ fusions (tnaL::lacZ and tnaA::lacZ) indicate that HolE and YdgT downregulate expression of the tna operon by possibly increasing the level of Rho-dependent transcription termination at the tna operon's leader region. Thus, for the first time, a regulatory function can be attributed to HolE, in addition to its role as structural component of the DNA polymerase III complex.
INTRODUCTION
The DNA polymerase III holoenzyme (Pol III) is the main replicative polymerase in Escherichia coli (1, 2). Pol III is composed of a three-subunit catalytic core plus seven additional subunits that act as accessory proteins for the core (3). Within the Pol III core, α (the dnaE gene product) is the DNA polymerase (4, 5), while ε (the dnaQ gene product) is the 3′-5′ proofreading exonuclease (6). The N-terminal domain of the ε subunit contains the binding site of the θ subunit (the holE gene product) (3). Genetic analysis of the Pol III core constituents has provided insight into the role of some of them. dnaE temperature-sensitive (TS) mutants display mutator or antimutator effects (7, 8), indicating the relevant fidelity role of this enzyme. The fact that several dnaQ mutants exhibit strong mutator phenotypes points out the importance of the 3′-exonuclease activity for replication fidelity (9).
In spite of both genetic and structural studies (3, 10), the function of the 9-kDa θ subunit has not unambiguously been designated. Loss of θ (ΔholE) results in apparently healthy cells with no morphology changes (10). The protein does, however, moderately stimulate the exonuclease activity of ε (11, 12), enhances the interaction of ε with α (13), and stabilizes ε under thermal inactivation (13, 14). The bacteriophage P1 hot gene encodes a homolog of the θ subunit. When expressed from the holE promoter, hot is able to complement a ΔholE defect (15). Interestingly, hot is expressed not only during the late stage of phage development but during the lysogenic state and early stages of a lytic induction (16). This complex expression pattern was interpreted as HOT affecting the replication machinery to benefit phage replication.
The Hha family includes a group of sequence-related low-molecular-mass proteins (molecular mass of about 8 kDa) involved in gene regulation. Remarkably, genes coding for such proteins can be present in one or more copies per chromosome or in transmissible elements such as plasmids in members of the Enterobacteriaceae family, but not in other genera such as Vibrio or Aeromonas. The first reported members of the family were the Hha protein of E. coli and the YmoA protein of Yersinia enterocolitica (17, 18). Studies on the mechanism of action of the Hha protein are based on the Hha-mediated environmental downregulation in E. coli of the hlyCABD operon, which encodes the α-hemolysin (18, 19). Hha does not bind directly to specific DNA sequences of the hly operon. Instead, Hha binds to the nucleoid-associated protein H-NS, which in turn binds to specific regions of regulatory sequences in this operon (20, 21). Coregulation of gene expression mediated by members of both families of proteins has been described for loci other than the hly operon (e.g., the inv gene of Y. enterocolitica [22] and the hilA gene of Salmonella enterica [23]). Hha-like proteins show structural mimicry to the H-NS oligomerization domain (the N-terminal domain) and can actually replace this domain, rendering a functional chimeric H-NS protein (24). Some enterobacterial chromosomes possess several hha-like genes. The YdgT protein, an Hha paralogue, was identified in E. coli, Salmonella, and Shigella bacteria. YdgT is overexpressed in hha mutants and partially compensates for Hha loss (25). It has also been shown that YdgT is a negative modulator of the S. enterica pathogenicity island 2 (SPI-2) (26). Outside of these examples, the regulatory role of YdgT is poorly characterized. Hha-like proteins can be also encoded by conjugative plasmids. These Hha orthologs modulate specific plasmid functions, as exemplified by the IncHI plasmid R27 hha-like gene that influences the temperature-dependent modulation of plasmid transfer (27). In this report, we provide evidence indicating that θ-like and Hha-like proteins exhibit a common phylogenetic distribution and share both structural and functional similarities. We show that θ (termed here HolE) modulates expression of a set of genes that are also modulated by the YdgT protein.
MATERIALS AND METHODS
Bacterial strains, plasmids, and culture conditions.
All bacterial strains and plasmids used in this study are listed in Table S1 in the supplemental material. Cultures were normally grown in Luria-Bertani (LB) medium (10 g NaCl, 10 g tryptone, and 5 g yeast extract per liter) at the indicated temperature with vigorous shaking at 200 rpm. (Innova 3100 water bath shaker; New Brunswick Scientific). Where indicated, LB medium with a higher NaCl concentration (60 g per liter), without NaCl (0 g per liter), or supplemented with l-tryptophan (1 mM final concentration) was used. Antibiotics were used at the following concentrations: 50 μg kanamycin ml−1, 100 μg ampicillin ml−1, 15 μg tetracycline ml−1, and 34 μg chloramphenicol ml−1.
3-[N-Morpholino]propanesulfonic acid (MOPS)-based minimal medium was prepared as described elsewhere (28). One percent acid-hydrolyzed casein (AHC) was added as carbon and energy source, and l-tryptophan was added at a final concentration of 0.5 mM, where indicated. Glucose used to elicit catabolite repression was filter sterilized and added, as indicated, at 20 mM.
To construct plasmids pBRHolE and pBRYdgT, the holE and ydgT genes of strain MG1655 were amplified using primers HOLE-Eco/HOLE-Bam and YDGT-Bam/YDGT-Sal, respectively (see Table S2 in the supplemental material), which add an EcoRI and a BamHI site or a BamHI and a SalI site, respectively, to the corresponding gene. Purified PCR products were then enzymatically digested with the corresponding restriction enzymes and cloned into pBR322 digested with the same enzymes, resulting in plasmids pBRHolE and pBRYdgT. pBRHolE and pBRYdgT were verified by DNA sequencing and then transformed into strains MG1655E, MG1655Y, and MG1655YE, respectively, for complementation studies.
To construct plasmids pET15bHolE and pET15bYdgT, the holE and ydgT genes of strain MG1655 were amplified using primers NDEHOLE_for/HOLEBAM_rev and NDEYDGT_for/YDGTBAM_rev, respectively (see Table S2 in the supplemental material), which add an NdeI and a BamHI site, respectively, to the corresponding gene. Purified PCR products were then enzymatically digested with NdeI and BamHI, cloned into the appropriate restriction sites in the polylinker of the pET15b expression vector, and transformed into E. coli DH5α cells. Recombinant plasmids were predicted to express an N-terminal His6-tagged fusion protein harboring a thrombin cleavage site between the His6 tag and the protein. Correct in-frame fusions of the genes in pET15b were confirmed by sequencing plasmid DNA with the T7 promoter and T7 terminator primers (see Table 2).
TABLE 2.
Expression of structural genes of the tna operon, tnaA and tnaB, in different mutant strains
| Strain | Mean ± SDa |
|||
|---|---|---|---|---|
| Exponential phaseb |
Stationary phaseb |
|||
| tnaA | tnaB | tnaA | tnaB | |
| MG1655E | +2.27 ± 0.15 | +2.93 ± 0.25 | −1.10 ± 0.10 | −1.37 ± 0.15 |
| MG1655Y | +2.37 ± 0.15 | +2.90 ± 0.20 | +1.23 ± 0.06 | −1.20 ± 0.10 |
| MG1655YE | +2.97 ± 0.31 | +3.53 ± 0.06 | +1.30 ± 0.10 | +1.47 ± 0.06 |
| MG1655E(pBRHolE) | −1.20 ± 0.10 | +1.53 ± 0.15 | +1.13 ± 0.12 | −1.27 ± 0.06 |
| MG1655YE(pBRHolE) | −1.23 ± 0.06 | +1.73 ± 0.15 | +1.27 ± 0.06 | +1.27 ± 0.06 |
| MG1655Y(pBRYdgT) | +1.30 ± 0.20 | +1.43 ± 0.06 | −1.02 ± 0.14 | −1.27 ± 0.21 |
| MG1655YE(pBRYdgT) | +1.63 ± 0.21 | +1.73 ± 0.21 | +1.30 ± 0.17 | +1.37 ± 0.15 |
The relative expression levels of tnaA and tnaB were determined by quantitative real-time PCR after growth in LB medium supplemented with l-tryptophan of the wt MG1655 strain and the indicated mutant strains. All mRNA levels were compared to the level in MG1655 wt. A value of 1 or −1 represents equal expression of the gene in the wt and the mutant strain. As an internal standard, 16S rRNA was used. Data are means and standard deviations of three independent experiments.
Samples were taken at the exponential phase of growth (OD600 of 0.6) and at the stationary phase of growth (OD600 of 2.0).
Genetic manipulations.
Standard molecular and genetic procedures were performed as described by Sambrook and Russell (29). Introduction of plasmids into bacteria was performed by electroporation of glycerol-washed cells using an Eppendorf gene pulser (Electroporator 2510). Table S1 in the supplemental material gives an overview of the methods used to obtain the different mutant strains constructed during this work.
Chromosomal deletions of genes holE and stpA were obtained by the λ Red recombinant method, as described by Datsenko and Wanner (30). The antibiotic resistance determinant of pKD3 (Cm) (for holE) or pKD4 (Kan) (for stpA) was amplified using primers HOLEH1/HOLEH2 and StpAP1/StpAP2, respectively (see Table S2 in the supplemental material). The PCR product was DpnI digested, purified, and used to electroporate strain MG1655 carrying plasmid pKD46, grown at 30°C in the presence of 10 mM arabinose. Mutants were selected on LB plates containing the appropriate selection marker, and successful deletion of the gene was confirmed by PCR. When necessary, the antibiotic resistance was then eliminated by transforming the mutant strain with plasmid pCP20 and incubation at 43°C for two passages. The other mutant strains were obtained by P1-vir-mediated transduction, which was performed as described elsewhere (31).
To construct the transcriptional lacZ fusion in tnaL, the gene was first disrupted in the E. coli strain AAG1 using the procedure of Datsenko and Wanner (30). The oligonucleotides used for this construction were tnaL_LacZ_for/tnaL_LacZ_rev (see Table S2 in the supplemental material), which amplified the antibiotic resistance of plasmid pKD3 with extensions corresponding to sequences of tnaL. After verification of the predicted deletion, the antibiotic resistance determinant was eliminated using an FLP/FLP recombination target (FRT)-mediated site-specific recombination method as described by Cherepanov and Wackernagel (32). The FRT-generated site was used to integrate plasmid pKG136 (33), thereby generating the transcriptional tnaL::lacZ fusion strain AAG1tnaL::lacZ. The AAG1tnaL::lacZ strain was then used as a donor strain to transfer the tnaL::lacZ fusion to strains AAG1E, AAG1Y, and AAG1YE using phage P1-vir (31).
RNA isolation.
Total RNA was extracted from bacteria using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. Potential traces of DNA were removed by digestion with DNase I (Turbo DNA-free; Ambion), according to the manufacturer's protocol. RNA concentration and RNA quality were measured using a Nano-Drop 1000 spectrophotometer (Thermo Fisher Scientific).
Real-time qRT-PCR.
Real-time quantitative reverse transcription-PCR (qRT-PCR), performed as previously described (34), was used to confirm microarray results and analyze expression of the tryptophanase operon, holE, hha, and ydgT. Briefly, 1 μg of total RNA was reverse transcribed to generate cDNA using the high-capacity cDNA reverse transcription kit (Applied Biosystems) as recommended by the manufacturer. As a control, parallel samples were run in which reverse transcriptase was omitted from the reaction mixture. Oligonucleotides complementary to the genes of interest were designed using primer3 software. Real-time PCR using SYBR green PCR master mix (Applied Biosystems) was carried out on the ABI Prism 7700 sequence detection system (Applied Biosystems). After analysis of amplification plots with the ABI Prism sds software package, relative quantification of gene transcription was performed using the comparative threshold cycle (CT) method. The relative amount of target cDNA was normalized using the 16S rRNA gene as an internal reference standard.
Semiquantitative RT-PCR.
Reverse transcription-PCR (RT-PCR) was used to verify restored transcription of holE in mutant strains carrying plasmid pBRHolE. Total RNA of strains MG1655, MG1655E, MG1655YE, MG1655E(pBRHolE), and MG1655YE(pBRHolE) was isolated. RT-PCR was performed with 10 ng RNA per reaction mixture using the Transcriptor One-Step RT-PCR kit (Roche) according to the manufacturer's instructions with sequence-specific primers. 16S rRNA was used as a control to confirm that equivalent quantities of templates were loaded. The PCR products were analyzed by 2% agarose gel electrophoresis.
Microarray.
cDNA library preparation and amplification were performed from 25 ng total RNA using WTA2 (Sigma-Aldrich) with 17 cycles of amplification. Eight micrograms of cDNA was subsequently fragmented by DNase I and biotinylated by terminal transferase obtained from the GeneChip Mapping 10Kv2 assay kit (Affymetrix). The hybridization mixture was prepared according to the Affymetrix protocol. Each sample was hybridized to an E. coli Genome 2.0 array (Affymetrix). The arrays were scanned in a GeneChip Scanner 3000 (Affymetrix). CEL files were generated from DAT files using GCOS software (Affymetrix). To generate the log2 expression estimates, overall array intensity was normalized between arrays and the probe intensity of all probes in a probe set summarized to a single value using the Robust Multichip Average (RMA) algorithm (35). All microarray experiments were performed in triplicate.
Overexpression and purification of HolE and YdgT protein.
For overexpression of HolE protein, E. coli strain BL21(DE3)/pLysS was used as a host. For overexpression of YdgT protein, E. coli strain BL21(DE3) Δhns/pLysS was used as a host. Plasmids (pET15b plasmids) containing the desired cloned genes (see Table 1) were introduced by transformation. Cultures (1 liter) were grown to an optical density at 600 nm (OD600) of 0.4 in LB medium containing ampicillin, then isopropyl-β-d-thiogalactopyranoside (IPTG) was added to an 0.5 mM final concentration, and incubation was carried out for additional 2 h. Cells were pelleted by centrifugation for 30 min at 7,500 rpm at 4°C (Beckman Coulter Avanti J-25) and resuspended in 20 ml of buffer A (20 mM HEPES, pH 7.9, 10% glycerol, 100 mM KCl, 5 mM MgCl2, 50 mM imidazole). The cells were lysed by 5 cycles of 1 min of sonication at 40% intensity (Branson Digital Sonifier), and the lysed extract was centrifuged at 12,000 × g for 30 min at 4°C (Beckman Coulter Avanti J-25). His-tagged proteins were purified by immobilized metal affinity chromatography by using Ni2+-nitrilotriacetic acid (NTA) technology (36), as described previously (21). Briefly, the protein-containing supernatant was mixed with 1 ml of Ni-NTA resin (Qiagen) previously equilibrated with buffer A. Mixing was carried out by agitating the samples for 2 h at 4°C, after which the resin was washed 10 times with 5 volumes of buffer A. Proteins were eluted 3 times with 1 ml of elution buffer (20 mM HEPES, pH 7.9, 10% glycerol, 100 mM KCl, 5 mM MgCl2, 200 mM imidazole). Then, the His6 tag was removed from the fusion protein by thrombin cleavage using the Thrombin CleanCleavage kit (Sigma) according to the manufacturer's protocol. Optimal cleavage conditions were determined before on pilot experiments. Cleavage of 1 mg of His-YdgT and His-HolE protein for 1 h at room temperature resulted in complete removal of the His tag without any nonspecific cleavage of the protein, as determined by visualizing the purified fusion protein both before and after thrombin cleavage by Coomassie blue staining of sodium dodecyl sulfate-polyacrylamide gels.
TABLE 1.
List of representative genes showing either up- or downregulation in the holE and ydgT mutants
| Transcript | Function | Functional group | Fold changea |
|
|---|---|---|---|---|
| holE mutant vs WT | ydgT mutant vs WT | |||
| flgA | Assembly of basal-body periplasmic P ring | Flagellar biosynthesis | −1.50 | −1.61 |
| flgD | Initiation of hook assembly | Flagellar biosynthesis | −1.92 | −2.33 |
| flgE | Hook protein | Flagellar biosynthesis | −1.95 | −2.45 |
| fliF | Basal-body MS ring and collar protein | Flagellar biosynthesis | −1.64 | −1.63 |
| fliI | Flagellum-specific ATP synthase | Flagellar biosynthesis | −1.96 | −2.01 |
| flgK | Hook-filament junction protein 1 | Flagellar biosynthesis | −2.21 | −2.36 |
| ivbL | ilvB operon leader; biosynthesis of branched-chain amino acids | Metabolism | −2.01 | −1.68 |
| tnaB | Tryptophan permease; degradation of tryptophan | Metabolism | +2.43 | +1.61 |
| hisD | Histidinol/histidinal dehydrogenase; biosynthesis of histidine | Metabolism | −1.51 | −1.56 |
| psuG | Pseudouridine 5′-phosphate glycosidase | Metabolism | +2.39 | +2.61 |
| trpE | Anthranilate synthase component; biosynthesis of tryptophan | Metabolism | −1.52 | −1.59 |
| hmpA | Nitric oxide dioxygenase, flavin reductase, and dihydropteridine reductase activity | Metabolism | +1.70 | +1.53 |
| dsrA | Antisense RNA; silencer of rcsA gene | Small RNA | −1.75 | −2.21 |
| oxyS | Global regulatory RNA | Small RNA | −2.83 | −2.32 |
| rttR | Small RNA; may regulate the stringent response | Small RNA | −2.32 | −2.88 |
| sokC | Antisense RNA blocking mokC and hokC translation | Small RNA | −2.01 | −3.05 |
| isrC | Small RNA | Small RNA | −1.76 | −2.13 |
| ryhA | Small regulatory RNA | Small RNA | −2.17 | −2.45 |
| araF | l-Arabinose-binding protein; arabinose ABC transporter component | Transport | +1.77 | +1.55 |
| mgtA | Mg2+ transport P-type ATPase | Transport | −1.96 | −1.79 |
| xylF | Xylose binding protein; component of xylose ABC transporter | Transport | +1.93 | +2.37 |
Data are from microarray experiments.
Electrophoretic mobility shift assay (EMSA).
A 411-bp fragment corresponding to the tna operon promoter region (nucleotides −165 to +246) was amplified using primers tnaEMSAfor and tnaEMSArev (see Table S2 in the supplemental material). For each reaction, 35 ng DNA was mixed with increasing concentrations of the purified protein in binding buffer (250 mM HEPES, pH 7.4, 350 mM KCl, 5 mM EDTA, 5 mM dithiothreitol [DTT], 500 μg bovine serum albumin [BSA] ml−1, 25% glycerol). After incubation for 30 min at different temperatures (4°C, 23°C, and 37°C), the samples (20 μl) were separated on 2% agarose gels in Tris-borate-EDTA buffer. The DNA was visualized by ethidium bromide staining.
β-Galactosidase assay.
β-Galactosidase assays were performed as described by Miller (31), using the CHCl3-sodium dodecyl sulfate permeabilization procedure. Data are mean values of duplicate determinations in three independent experiments.
Indole assay.
Overnight cultures of E. coli MG1655 wild-type (wt) and mutant strains were diluted 1/100 in LB medium containing 1 mM tryptophan. Cultures of 20 ml were grown at 37°C till reaching an OD600 of 0.6. One milliliter of the culture was then centrifuged at 6,000 rpm for 3 min in a tabletop centrifuge, and the bacterial pellet was removed. The supernatants of the samples were diluted 1:1 in fresh LB medium and mixed immediately with 100 μl of Kovac's reagent (5% [wt/vol] p-dimethylaminobenzaldehyde, 75% [wt/vol] methanol, 2.5 M HCl). The indole concentration was measured spectrophotometrically at 540 nm (UV-mini-1240 spectrophotometer; Shimadzu).
RESULTS
holE-like genes are, like hha-like genes, enterobacterial ORFan genes.
We showed previously that Hha-like proteins are an evolutive trait of the Enterobacteriaceae (37). This bacterial family includes several endosymbionts that have suffered genomic reductions. Some of them lack the hha gene, and others have retained this gene. When checking the presence of the hha gene in the enterobacterial endosymbionts, we noticed that those that still have a copy of the hha gene (e.g., Wigglesworthia glossinidia, Sodalis glossinidius, and Photorhabdus luminescens) also encode an additional enterobacterial ORFan gene (i.e., an annotated gene that exists exclusively within the Enterobacteriaceae) (ORFans are open reading frames [ORFs] with no detectable homology to other ORFs in a database), the holE gene. As is the case for hha, holE is solely present in the genomes of the Enterobacteriaceae (Fig. 1). We also searched for the presence of the holE gene in bacterial plasmids. We found that, similarly to hha (38, 39), holE orthologs are present in several plasmids. All of them have been exclusively isolated from enterobacterial hosts (Fig. 2).
FIG 1.

Distribution of H-NS, StpA, Hha, YdgT, and HolE proteins in the gammaproteobacteria. The dendrogram shows the phylogenetic relationship of the amino acid sequences of the N-terminal end of H-NS proteins from different bacterial species (see Table S3 in the supplemental material for definitions of the abbreviations). The presence of either hns-like or hha-like genes and the holE gene is indicated.
FIG 2.

Evolutionary relationship of plasmid-encoded HolE-like proteins. The phylogenetic relationship of the amino acid sequences of plasmid-encoded HolE-like proteins and the chromosome-encoded HolE of E. coli strain MG1655 was inferred using the neighbor-joining method. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Evolutionary analyses were conducted in the MEGA5 program.
Both Hha and HolE are low-molecular-mass proteins (molecular masses of 8 and 8.6 kDa, respectively). The three-dimensional (3-D) structure of HolE from E. coli has been solved by nuclear magnetic resonance (NMR) (3). As well, 3-D structures of Hha and its paralog YdgT also have been determined (40, 41). Interestingly, in spite of not exhibiting significant similarity in their amino acid sequences, the three proteins share a similar helical structure (Fig. 3). The core structure of Hha and YdgT is a three-helical bundle that, despite the differences in the length and orientation of the individual helical segments, closely resembles the three-dimensional structure of HolE. A short additional helix (helix 4) is present in the C-terminal end of Hha but not in the structures of YdgT or HolE (Fig. 3).
FIG 3.
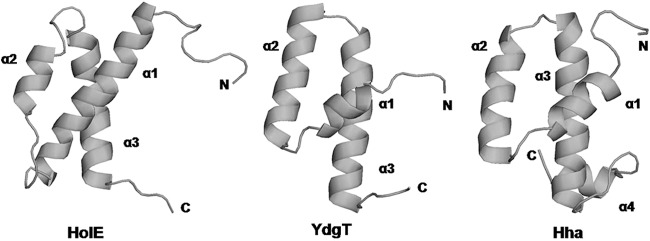
Comparison of the structures of HolE, YdgT, and Hha. The ribbons trace the backbones of ε186-bound HolE (PDB code 2AXD), YdgT (PDB code 2JQT), and Hha (PDB code 1JW2), respectively.
Given the noticeable similarities between Hha-like proteins and HolE, we decided to investigate whether there is any functional relationship between them.
Construction of single holE, double holE hha/holE ydgT, and triple holE hha ydgT mutants and analysis of growth kinetics.
In order to search for functional relationships between HolE and Hha/YdgT, we constructed an E. coli strain carrying a deletion of the holE gene. Using the method described by Datsenko and Wanner (30), the holE gene in E. coli strain MG1655 was knocked out, resulting in strain MG1655E. We also constructed double mutants carrying deletions of holE and hha (MG1655HE) or holE and ydgT (MG1655YE). An hha ydgT holE triple mutant (MG1655HYE) was also constructed.
To test whether the different mutant strains exhibit any alterations in the growth rate, we compared their growth kinetics with that of the MG1655 wild-type (wt) strain. Since temperature and osmolarity are environmental parameters that influence regulation of gene expression by the Hha protein, cultures of the wt and different mutant strains were grown in liquid LB medium at different temperatures (20°C and 37°C) and osmolarities. Regarding growth temperature, no differences between the growth rates of the mutant strains and the wt MG1655 strain could be observed (data not shown). To test bacterial growth at extreme osmolarities, all strains were grown at 37°C in LB medium containing either 60 g/liter NaCl or 0 g/liter NaCl. Even though at high NaCl concentrations all strains exhibited decelerated growth, the different mutants showed no alterations in the growth rate compared to the wt strain (data not shown).
Expression of holE, hha, and ydgT in E. coli MG1655 under different growth conditions.
We decided next to test whether expression of the holE gene in E. coli depends on the growth temperature and/or growth phase. For this purpose, strain MG1655 was grown in LB medium at 25°C and 37°C, respectively. At different stages during growth (OD600s of 0.5 and 3.0), samples were taken, total RNA was isolated, and the transcript level of holE was determined by quantitative real-time PCR. The relative expression of holE can be displayed as a function of temperature or as a function of growth phase (Fig. 4A and B).
FIG 4.

Expression of holE, ydgT, and hha in MG1655 wt strain obtained by quantitative real-time PCR depending on the growth temperature (37°C compared to 25°C) (A) and depending on the growth phase (exponential compared to stationary phase) (B). A value of 1 or −1 represents equal expression of the gene across a pair of cultures. As internal standard 16S rRNA was used. Data are means and standard deviations of three independent experiments.
As seen in Fig. 4A, the influence of growth temperature on expression of holE depends on the growth phase. During the exponential phase of growth, growth at low temperature (25°C) resulted in a slight increase of holE transcript levels compared with growth at high temperature (37°C). In contrast, at the stationary growth phase, holE is slightly more highly expressed at 37°C than at 25°C (Fig. 4A). Irrespective of growth temperature, holE transcription is significantly increased during the exponential phase of growth (OD600 of 0.5) compared to the stationary phase (OD600 of 3) (Fig. 4B).
In addition to holE, we also tested expression of ydgT and hha genes under the above-mentioned conditions (Fig. 4). As evidenced for holE, expression of ydgT and hha is higher during the exponential phase of growth than during the stationary phase (Fig. 4B). Nevertheless, the temperature influences hha and ydgT expression in a different way when cells grow exponentially. Whereas, as happens with holE, growth at low temperature slightly induces ydgT expression, it moderately represses hha expression (Fig. 4A).
Transcriptomic analysis of holE, hha, and ydgT mutants.
hha mutants exhibit significant alterations in the transcriptome (42). To try to determine if there exist any functional similarities between HolE and Hha/YdgT, we decided to obtain and compare the transcriptomic profiles of holE, hha, and ydgT mutants. Bacterial cells from the wt strain MG1655 and the three mutant strains were grown in LB medium at 37°C to the exponential growth phase (OD600 of 0.6). Samples were taken, and total RNA was isolated, reverse transcribed, labeled, and hybridized with the GeneChip E. coli Genome 2.0 Array commercial microchips (Affymetrix). When the transcriptional profiles of all three mutants were compared, it became apparent that, whereas loss of Hha protein mainly accounts for upregulation of gene expression, loss of YdgT mainly leads to downregulation of gene expression (Fig. 5A and B). Hitherto, the regulatory role of YdgT in E. coli had been poorly characterized. It was shown that, in hha mutants, overexpression of YdgT partially attenuates the hha phenotype (25). Strains MG1655H and MG1655Y share 61 commonly deregulated genes (6.4% of the genes altered in the hha mutant and 23.4% of those altered in the ydgT mutant). Remarkably, the comparison of the transcriptomic profiles of the E. coli hha and ydgT mutants shows that, apart from the reported role of YdgT partially compensating for Hha loss, YdgT influences gene expression of genes not modulated by Hha. In many of these genes, YdgT function upregulates their expression. Interestingly, a significant number of downregulated genes in strain MG1655Y overlapped the set of downregulated genes in strain MG1655E (Fig. 5B). These strains share 61 commonly regulated genes (23.4% for the ydgT mutant and 19.6% for the holE mutant). Those common genes upregulated in the ydgT mutant are upregulated in the holE mutant, and those downregulated in the ydgT mutant are downregulated in the holE mutant. Accordingly, the heat maps of genes exhibiting altered expression in the mutants holE (E), hha (H), and ydgT (Y), all with respect to the wt strain, clearly show that the expression patterns of the holE and ydgT mutants are more similar than the expression patterns of the holE and hha mutants (Fig. 5C). Surprisingly, the similarity between the gene expression patterns of holE and ydgT mutants is even higher than the similarity between the ydgT mutant and the hha mutant. Genes related to flagellar biosynthesis, metabolism, and small RNAs predominate among the genes that showed a significant alteration in their expression in both mutant strains, MG1655E and MG1655Y (see Tables S4 and S5 in the supplemental material). Some of those representative genes are shown in Table 1.
FIG 5.

Microarray analysis of strains MG1655 (wt), MG1655H (hha mutant), MG1655Y (ydgT mutant), and MG1655E (holE mutant). (A and B) Venn diagrams represent the number of genes upregulated (A) and downregulated (B) in the hha mutant (H; white circle), ydgT mutant (Y; light gray circle), and holE mutant (E; dark gray circle) compared to the wild-type strain (WT) (fold change [FC] of >1.5× and adjusted P value of <0.05). (C) Heat map representing the gene expression pattern of mutants hha (H), ydgT (Y), and holE (E) compared to the wild-type strain (WT) (FC of >1.5× and adjusted P value of <0.05). The genes that are downregulated are indicated in green, and those that are upregulated are indicated in red (respective to the wt strain). Genes marked in white do not show an altered expression.
YdgT and HolE proteins comodulate expression of the tryptophanase operon.
As shown in Table 1, the effect of the ydgT and holE alleles altering expression of their target genes in LB medium is modest in most of them. To further extend our studies on the modulatory role of HolE, we considered selecting a target gene whose expression could be increased under specific growth conditions. For this purpose, we selected the tryptophanase (tna) operon, which is upregulated in both mutant strains.
The enzyme tryptophanase catalyzes the degradation of l-tryptophan to indole, pyruvate, and ammonia (43). The tna operon of E. coli contains two structural genes: tnaA, encoding tryptophanase, and tnaB, encoding a tryptophan permease. Preceding tnaA in the tna operon is a transcribed regulatory region of 319 nucleotides, containing the coding region for a 24-residue leader peptide, TnaL (44). Since expression of the tna operon in E. coli is influenced by tryptophan, we decided to investigate the expression of the tna operon in LB medium supplemented with l-tryptophan. Using these culture conditions, we compared transcript levels of tnaA and tnaB in strains MG1655E and MG1655Y compared to those in wt MG1655 by quantitative real-time PCR. In addition, we included the double mutant strain MG1655YE in this study. As seen in Table 2, the expression of both genes is significantly increased in mutants lacking either HolE or YdgT. Notably, this effect is even more pronounced in the double mutant MG1655YE, supporting our hypothesis that HolE and YdgT may partially substitute for each other's function.
To correlate loss of either HolE or YdgT and alterations in tna operon expression, the mutations in MG1655E, MG1655Y, and MG1655YE were accordingly complemented with the wt holE and ydgT alleles in trans. The holE and ydgT genes of strain MG1655 were amplified and cloned into the vector pBR322, resulting in plasmids pBRHolE and pBRYdgT. Strains MG1655E and MG1655YE were transformed with pBRHolE, and strains MG1655Y and MG1655YE were transformed with pBRYdgT. Restored expression of holE or ydgT was confirmed in the resulting strains MG1655E(pBRHolE), MG1655Y(pBRYdgT), MG1655YE(pBRHolE), and MG1655YE(pBRYdgT) by RT-PCR (data not shown). As seen in Table 2, in strains MG1655E(pBRHolE), MG1655YE(pBRHolE), MG1655Y(pBRYdgT), and MG1655YE(pBRYdgT), tna operon expression levels were partially restored to levels observed in the wt strain. Transformation of MG1655E, MG1655Y, and MG1655YE with pBR322 alone did not alter tna transcript levels (data not shown).
The results of tna operon regulation presented so far were obtained from cells growing to exponential phase (OD600 of 0.6). We were curious whether we could observe the same regulation in cells at the stationary phase of growth. Bacteria were grown till an OD600 of 3.0 was reached, and transcript levels of tnaA and tnaB were determined. At the stationary phase of growth, no significant regulation of the genes of the tna operon by HolE or YdgT could be detected (Table 2). This result matches our data of holE and ydgT being predominantly expressed during the exponential phase of growth (Fig. 4B).
Degradation of l-tryptophan by the enzyme tryptophanase leads to indole production. We decided therefore to test whether the increased levels of tnaA and tnaB in the holE and ydgT mutants also result in higher production of indole in these strains. At the exponential phase of growth, samples were taken from cultures of MG1655 wt, MG1655E, MG1655Y, and MG1655YE, and the level of exogenous indole in the growth medium was determined. As Fig. 6 shows, in all three mutant strains the level of indole is increased over that in the wt strain. These results corroborate the results obtained when evaluating the role of holE and ydgT alleles in tna expression by real-time PCR.
FIG 6.
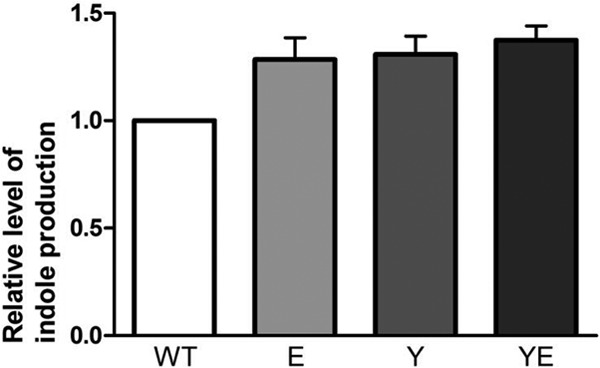
Level of exogenous indole, measured spectrophotometrically at 540 nm, in cultures of strains MG1655 wt, MG1655E, MG1655Y, and MG1655YE. All indole levels are relative to the level in MG1655 wt, which was set to 1. Data are means and standard deviations of three independent experiments.
Regulation of tryptophanase operon by HolE and YdgT is not mediated through interaction with H-NS or StpA.
It was shown that the E. coli YdgT protein can form heteromeric complexes with the nucleoid-associated protein H-NS and its paralogue StpA (25). Hence, a feasible hypothesis to consider is that at least some of the genes regulated by YdgT are also regulated by H-NS. In order to address the question whether regulation of the tna operon by HolE and YdgT is mediated through interaction with H-NS or StpA, we analyzed tryptophanase expression in hns and stpA mutant derivatives of strain MG1655, as well as in an hns stpA double mutant. Expression of the tryptophanase operon was not altered in any of the mutant strains (data not shown). This suggests that regulation of the tryptophanase operon by HolE and YdgT is not mediated through interaction of these proteins with either H-NS or StpA. We also constructed triple holE hns stpA and ydgT hns stpA mutants and analyzed tryptophanase expression in these mutant strains. In both triple mutants, we observed larger amounts of tnaA (1.77 ± 0.15 in the holE hns stpA mutant, 1.73 ± 0.20 in the ydgT hns stpA mutant) and tnaB (1.80 ± 0.20 in the holE hns stpA mutant, 1.83 ± 0.15 in the ydgT hns stpA mutant) transcripts than in the wild type. Compared to the corresponding single holE and ydgT mutants, the triple mutants did not show increased tna operon transcription. These data do not support the hypothesis that regulation of the tryptophanase operon by HolE and YdgT requires H-NS (StpA).
Another way how HolE and YdgT could exert their regulatory function on expression of target genes is by directly interacting with the regulatory region of the target gene. Even though the structure of neither protein shows typical DNA-binding domains, we decided to test this possibility by mobility shift assays. As target, we used a 411-bp DNA fragment corresponding to the promoter region of the tna operon, covering 165 bp upstream and 246 bp downstream of the transcriptional start site. Plasmids pET15bYdgT and pET15bHolE were used to overproduce and purify His-YdgT and His-HolE protein, respectively. Thrombin cleavage was used to remove the His6 tag, rendering native YdgT and HolE proteins (see Materials and Methods for details). The mobility of the 411-bp DNA fragment mixed with increasing concentrations of either YdgT or HolE proteins was analyzed on agarose gels. Even at the highest protein concentrations (47 μM HolE and 74 μM YdgT, respectively), no alterations in the mobility of the target DNA fragment were observed (data not shown). These results do not support the hypothesis that either YdgT or HolE directly binds to the tna promoter region.
YdgT and HolE influence termination at the tna operon.
Transcription of the tna structural genes tnaA and tnaB is regulated by catabolite repression at the promoter and by Rho-dependent transcription termination in the leader region. Antitermination is a main mechanism of regulation of the tna operon (28, 45, 46). Preceding the tnaA gene in the tna operon is a 319-nucleotide transcribed region that contains the coding sequence for a 24-amino-acid leader peptide, TnaL. The spacer sequence between tnaL and tnaA includes a Rho-dependent transcription terminator (Fig. 7A). Tryptophan-induced expression of the tna operon is based on relief from Rho-dependent transcription termination in the leader region. In the absence of tryptophan, Rho factor binds at the rut site in the leader RNA, leading to premature transcription termination. Induction of the operon requires synthesis of the leader peptide TnaL and high levels of free tryptophan (46). The mechanism by which the RNA polymerase can overcome the Rho-dependent transcription terminator and transcribe tnaA and tnaB requires TnaL-peptidyl-tRNA and a translating ribosome stalled at the tnaL stop codon. This blocks Rho factor's access to its binding site on the tna leader transcript (46).
FIG 7.
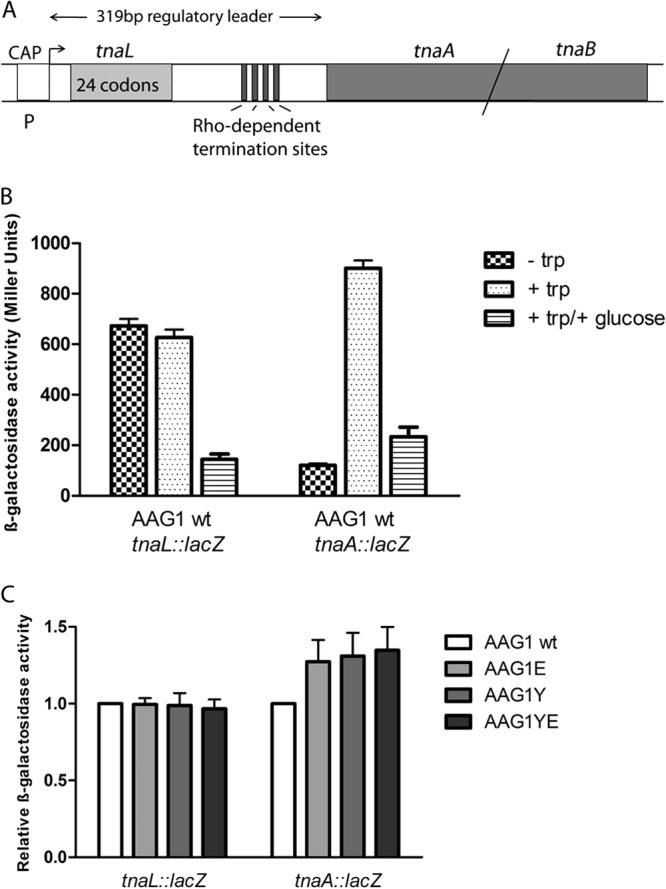
(A) Organization and features of the tna operon of E. coli. The tna operon consists of two structural genes, tnaA and tnaB, and a preceding transcribed 319-bp regulatory region. The leader transcript contains a coding sequence for a 24-residue leader peptide, TnaL; an RNA rut binding site for Rho; and several RNA pause sites required for Rho-dependent transcription termination. CAP indicates the presence of a catabolite gene activator protein (CAP) binding site at the tna promoter. (B) The tna operon transcriptional lacZ fusion constructs are sensitive to catabolite repression and tryptophan-dependent transcription antitermination. Strain AAG1 (MG1655ΔlacZ) containing either the tnaL::lacZ fusion or the tnaA::lacZ fusion was grown at 37°C in MOPS minimal medium alone (− trp), MOPS supplemented with tryptophan (+ trp), or MOPS supplemented with tryptophan and glucose (+ trp/+ glucose). At the exponential phase of growth (OD600 of 0.4), samples were taken and β-galactosidase activity was monitored. The data shown are the means and standard deviations of two independent experiments. (C) β-Galactosidase activity in strains AAG1 wt, AAG1E (holE mutant), AAG1Y (ydgT mutant), and AAG1YE (ydgT holE mutant) containing either the tnaL::lacZ fusion or the tnaA::lacZ fusion. The indicated strains were grown at 37°C in LB medium supplemented with tryptophan till an OD600 of 0.6 was reached. Samples were taken, and β-galactosidase activity was measured. The β-galactosidase activity levels for both lacZ fusion constructs are relative to the corresponding levels observed in AAG1 wt, which were set to 1. The data shown are the means and standard deviations of three independent experiments.
A recent report has provided evidence for a functional relationship between the YdgT protein and Rho-dependent transcription termination. Overexpression of YdgT restored the efficiency of termination in rho or nusG mutants (47). The mechanism by which YdgT influences transcription termination is not known, but the reported data show that this protein potentiates transcription termination at Rho sites. Indeed, depletion of YdgT exacerbated the polarity defects of rho mutants. With this in mind, we decided to investigate if alterations in transcription termination are underlying the increased expression of tnaA in ydgT and holE mutants. To do this, we constructed chromosomal transcriptional tna::lacZ gene fusions both at the tnaL leader region and within tnaA. To show that these constructs show the described antitermination effect due to the presence of tryptophan in the culture medium, tnaL::lacZ and tnaA::lacZ expression was monitored in the wt strain AAG1 (MG1655 ΔlacZ) grown in the presence and in the absence of tryptophan, as well as in the presence of glucose. As expected, tryptophan differentially influenced tnaL::lacZ and tnaA::lacZ expression. As well, the reported catabolite repression exerted by glucose could be evidenced for both transcriptional fusions (Fig. 7B). Upon having checked that both fusions report the antitermination regulation of the tna operon, we used them to monitor transcription at tnaL and at tnaA in the wt AAG1 strain and in ydgT, holE, and double ydgT holE mutant derivatives of AAG1 grown to the exponential growth phase (OD600 of 0.4). Compared to the wt strain, transcription in the tnaL leader region is not influenced by either the ydgT or the holE allele. In contrast, transcription in the tnaA gene is significantly influenced in the different mutant backgrounds (Fig. 7C). Hence, both YdgT and HolE influence tnaA expression by enhancing transcription termination at the leader DNA sequence.
DISCUSSION
Surprisingly, the holE gene, in spite of its gene product being a DNA Pol III core subunit, is encoded both in enterobacterial chromosomes and in plasmids and phage genomes that do not code for the rest of the Pol III core subunits (the dnaE and dnaQ gene products). This is the case for the genome of the phage P1. Strikingly, expression of the P1 hot gene is not strictly related to the replication cycle of the phage but is also related to the lysogenic growth stage (16). These data were interpreted as HOT benefiting phage replication, but a significant difference in phage yield upon the thermal induction of hot+ and hot-minus phages was not found (16). If the unique function of HolE is to act as a component of the Pol III core, the gene products of the corresponding plasmid-carried holE genes must alter the composition of Pol III core in those enterobacteria that randomly incorporate them by acquiring the corresponding plasmid. It remains elusive to understand how this would benefit the fitness of the recipient cell.
Moonlighting proteins are becoming recognized as a common phenomenon with relevant implications in systems biology (as reviewed in reference 48). Within bacteria, several examples of protein moonlighting have been reported (49–53), and several bacterial moonlighting proteins play a role as virulence determinants (as reviewed in reference 53). We provide in this work evidence for moonlighting activity of the holE gene product. Phenotypic analysis of mutant strains that combined the holE with hha/ydgT alleles did not provide evidence about any functional relationship between HolE and Hha/YdgT. Nevertheless, a comparative transcriptomic analysis of hha, ydgT, and holE mutants shed light on the moonlighting activity of HolE. When cells are grown in LB medium at 37°C, depletion of this protein significantly alters the expression of several E. coli genes. Both downregulation and upregulation occur, although downregulation predominates. This effect is inconsistent with a unique function of θ influencing chromosome replication. The fact that the effects of the holE mutation on the E. coli transcriptome significantly overlap the effects of the ydgT mutation further supports the assumption that HolE plays a regulatory role in E. coli. Remarkably, the influences of the growth conditions used on the expression patterns of YdgT and HolE are similar. The comparative transcriptomic analysis of holE, ydgT, and hha mutants shows a functional link between YdgT and HolE. In addition, it also shows that in E. coli, apart from providing a molecular backup for Hha (25), YdgT has specific modulatory functions. HolE playing regulatory functions is consistent with the presence of the holE gene in several conjugative plasmids and in the genome of the P1 bacteriophage.
In the transcriptomic analysis performed, the observed alterations in gene expression in both E. coli ydgT and holE mutants are generally modest. This could likely be due to the fact that the environmental signals that E. coli cells sense when they grow in LB medium at 37°C are not those required for HolE/YdgT exerting their main modulatory roles. This assumption is supported by the results obtained when analyzing the effect of both holE and ydgT alleles on expression of the tryptophanase operon. In LB medium containing tryptophan, the effect of either YdgT or HolE on tnaA or tnaB expression is more pronounced than the effect observed when cells grow in LB medium. The double ydgT holE mutant shows the highest derepression of the tna operon, thus confirming the cooperative role of the two proteins silencing this operon. The effect of these mutant alleles on indole production is in accordance with the observed increase in tna transcription. Outside of showing moonlighting activity for HolE, our data also show that in E. coli the tna operon is a target for the YdgT protein.
Whereas in Salmonella loss of YdgT has been shown to alter virulence gene expression (26), the regulatory role of YdgT in E. coli has remained hitherto uncharacterized. A recent report established a relationship between YdgT, H-NS, and Rho-dependent transcription termination (47). Overexpression of YdgT abolishes the polarity relief phenotype of rho or nusG mutants. The same effect was achieved by expressing a truncated H-NS variant. Recent reports strengthen the functional relationship between Rho and H-NS. Like H-NS, Rho has been shown to silence foreign DNA in E. coli (54). It has also been shown that H-NS aids in Rho suppression of pervasive antisense transcription in E. coli (55). Hence, a relevant physiological role for the H-NS family of proteins, to which Hha and YdgT can be considered to belong, is to influence transcription termination in Rho-dependent terminators. It is also important to point out here that some effects of YdgT on Rho-dependent transcription termination may be independent of H-NS (47).
By using the tryptophanase operon as a model, we provide further evidence about the effect of YdgT on transcription termination. As expected from the previous report (47), the presence of the protein would be required for a more efficient termination and its absence would result in transcriptional relief. We show this for the tna operon. Our results provide evidence that, in actively growing E. coli cells, YdgT influences either directly or indirectly transcription termination at tnaL and contributes to tna downregulation even if tryptophan is available in the medium. This effect does not take place in the stationary phase of growth and may be related to the need for a differential indole production in exponentially growing and in resting E. coli cells. Indole production is critical to regulate biofilm formation (56). The holE gene product is a molecular backup for the YdgT protein and contributes to silencing the tna operon in exponentially growing cells. Remarkably, both YdgT and HolE show the highest expression in exponentially growing cells.
A yet-unanswered question is the mechanism by which YdgT or HolE influences transcription termination. Whereas, as shown in this work, these proteins do not directly bind the tna regulatory region or interact with H-NS to modulate tna, they might form complexes either with Rho or with RNA. These aspects deserve future research.
Supplementary Material
ACKNOWLEDGMENTS
We thank C. Balsalobre and M. Gibert for strains AAG1tnaA::lacZ and AAG1hns. We also thank Sergi Beltran (Bioinformatics Unit at CCiTUB) for the bioinformatic analysis of the microarrays.
This work was supported by funds from the Spanish MICINN-FEDER (BFU2010-21836-C02-01, CSD2008-00013, and BIO2010-15683) and the Generalitat de Catalunya (2009SGR66).
Footnotes
Published ahead of print 27 December 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01448-13.
REFERENCES
- 1.Kelman Z, O'Donnell M. 1995. DNA polymerase III holoenzyme: structure and function of a chromosomal replicating machine. Annu. Rev. Biochem. 64:171–200. 10.1146/annurev.bi.64.070195.001131 [DOI] [PubMed] [Google Scholar]
- 2.McHenry CS. 2003. Chromosomal replicases as asymmetric dimers: studies of subunit arrangement and functional consequences. Mol. Microbiol. 49:1157–1165. 10.1046/j.1365-2958.2003.03645.x [DOI] [PubMed] [Google Scholar]
- 3.Keniry MA, Park AY, Owen EA, Hamdan SM, Pintacuda G, Otting G, Dixon NE. 2006. Structure of the theta subunit of Escherichia coli DNA polymerase III in complex with the epsilon subunit. J. Bacteriol. 188:4464–4473. 10.1128/JB.01992-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maki H, Kornberg A. 1985. The polymerase subunit of DNA polymerase III of Escherichia coli. II. Purification of the alpha subunit, devoid of nuclease activities. J. Biol. Chem. 260:12987–12992 [PubMed] [Google Scholar]
- 5.Maki H, Kornberg A. 1987. Proofreading by DNA polymerase III of Escherichia coli depends on cooperative interaction of the polymerase and exonuclease subunits. Proc. Natl. Acad. Sci. U. S. A. 84:4389–4392. 10.1073/pnas.84.13.4389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheuermann R, Tam S, Burgers PM, Lu C, Echols H. 1983. Identification of the epsilon-subunit of Escherichia coli DNA polymerase III holoenzyme as the dnaQ gene product: a fidelity subunit for DNA replication. Proc. Natl. Acad. Sci. U. S. A. 80:7085–7089. 10.1073/pnas.80.23.7085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fijalkowska IJ, Dunn RL, Schaaper RM. 1993. Mutants of Escherichia coli with increased fidelity of DNA replication. Genetics 134:1023–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maki H, Mo JY, Sekiguchi M. 1991. A strong mutator effect caused by an amino acid change in the alpha subunit of DNA polymerase III of Escherichia coli. J. Biol. Chem. 266:5055–5061 [PubMed] [Google Scholar]
- 9.Taft-Benz SA, Schaaper RM. 1998. Mutational analysis of the 3′→5′ proofreading exonuclease of Escherichia coli DNA polymerase III. Nucleic Acids Res. 26:4005–4011. 10.1093/nar/26.17.4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Slater SC, Lifsics MR, O'Donnell M, Maurer R. 1994. holE, the gene coding for the theta subunit of DNA polymerase III of Escherichia coli: characterization of a holE mutant and comparison with a dnaQ (epsilon-subunit) mutant. J. Bacteriol. 176:815–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perrino FW, Harvey S, McNeill SM. 1999. Two functional domains of the epsilon subunit of DNA polymerase III. Biochemistry 38:16001–16009. 10.1021/bi991429+ [DOI] [PubMed] [Google Scholar]
- 12.Studwell-Vaughan PS, O'Donnell M. 1993. DNA polymerase III accessory proteins. V. Theta encoded by holE. J. Biol. Chem. 268:11785–11791 [PubMed] [Google Scholar]
- 13.Taft-Benz SA, Schaaper RM. 2004. The theta subunit of Escherichia coli DNA polymerase III: a role in stabilizing the epsilon proofreading subunit. J. Bacteriol. 186:2774–2780. 10.1128/JB.186.9.2774-2780.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamdan S, Bulloch EM, Thompson PR, Beck JL, Yang JY, Crowther JA, Lilley PE, Carr PD, Ollis DL, Brown SE, Dixon NE. 2002. Hydrolysis of the 5′-p-nitrophenyl ester of TMP by the proofreading exonuclease (epsilon) subunit of Escherichia coli DNA polymerase III. Biochemistry 41:5266–5275. 10.1021/bi0159480 [DOI] [PubMed] [Google Scholar]
- 15.Chikova AK, Schaaper RM. 2005. The bacteriophage P1 hot gene product can substitute for the Escherichia coli DNA polymerase III θ subunit. J. Bacteriol. 187:5528–5536. 10.1128/JB.187.16.5528-5536.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chikova AK, Schaaper RM. 2007. The bacteriophage P1 hot gene, encoding a homolog of the E. coli DNA polymerase III theta subunit, is expressed during both lysogenic and lytic growth stages. Mutat. Res. 624:1–8. 10.1016/j.mrfmmm.2007.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornelis GR, Sluiters C, Delor I, Geib D, Kaniga K, Lambert de Rouvroit C, Sory MP, Vanooteghem JC, Michiels T. 1991. ymoA, a Yersinia enterocolitica chromosomal gene modulating the expression of virulence functions. Mol. Microbiol. 5:1023–1034. 10.1111/j.1365-2958.1991.tb01875.x [DOI] [PubMed] [Google Scholar]
- 18.Nieto JM, Carmona M, Bolland S, Jubete Y, de la Cruz F, Juarez A. 1991. The hha gene modulates haemolysin expression in Escherichia coli. Mol. Microbiol. 5:1285–1293. 10.1111/j.1365-2958.1991.tb01902.x [DOI] [PubMed] [Google Scholar]
- 19.Carmona M, Balsalobre C, Munoa F, Mourino M, Jubete Y, De la Cruz F, Juarez A. 1993. Escherichia coli hha mutants, DNA supercoiling and expression of the haemolysin genes from the recombinant plasmid pANN202-312. Mol. Microbiol. 9:1011–1018. 10.1111/j.1365-2958.1993.tb01230.x [DOI] [PubMed] [Google Scholar]
- 20.Madrid C, Nieto JM, Paytubi S, Falconi M, Gualerzi CO, Juarez A. 2002. Temperature- and H-NS-dependent regulation of a plasmid-encoded virulence operon expressing Escherichia coli hemolysin. J. Bacteriol. 184:5058–5066. 10.1128/JB.184.18.5058-5066.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nieto JM, Madrid C, Prenafeta A, Miquelay E, Balsalobre C, Carrascal M, Juarez A. 2000. Expression of the hemolysin operon in Escherichia coli is modulated by a nucleoid-protein complex that includes the proteins Hha and H-NS. Mol. Gen. Genet. 263:349–358. 10.1007/s004380051178 [DOI] [PubMed] [Google Scholar]
- 22.Ellison DW, Miller VL. 2006. H-NS represses inv transcription in Yersinia enterocolitica through competition with RovA and interaction with YmoA. J. Bacteriol. 188:5101–5112. 10.1128/JB.00862-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olekhnovich IN, Kadner RJ. 2006. Crucial roles of both flanking sequences in silencing of the hilA promoter in Salmonella enterica. J. Mol. Biol. 357:373–386. 10.1016/j.jmb.2006.01.007 [DOI] [PubMed] [Google Scholar]
- 24.Rodriguez S, Nieto JM, Madrid C, Juarez A. 2005. Functional replacement of the oligomerization domain of H-NS by the Hha protein of Escherichia coli. J. Bacteriol. 187:5452–5459. 10.1128/JB.187.15.5452-5459.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paytubi S, Madrid C, Forns N, Nieto JM, Balsalobre C, Uhlin BE, Juarez A. 2004. YdgT, the Hha paralogue in Escherichia coli, forms heteromeric complexes with H-NS and StpA. Mol. Microbiol. 54:251–263. 10.1111/j.1365-2958.2004.04268.x [DOI] [PubMed] [Google Scholar]
- 26.Coombes BK, Wickham ME, Lowden MJ, Brown NF, Finlay BB. 2005. Negative regulation of Salmonella pathogenicity island 2 is required for contextual control of virulence during typhoid. Proc. Natl. Acad. Sci. U. S. A. 102:17460–17465. 10.1073/pnas.0505401102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forns N, Banos RC, Balsalobre C, Juarez A, Madrid C. 2005. Temperature-dependent conjugative transfer of R27: role of chromosome- and plasmid-encoded Hha and H-NS proteins. J. Bacteriol. 187:3950–3959. 10.1128/JB.187.12.3950-3959.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stewart V, Yanofsky C. 1985. Evidence for transcription antitermination control of tryptophanase operon expression in Escherichia coli K-12. J. Bacteriol. 164:731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sambrook J, Russell DW. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 30.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97:6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Plainview, NY [Google Scholar]
- 32.Cherepanov PP, Wackernagel W. 1995. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158:9–14. 10.1016/0378-1119(95)00193-A [DOI] [PubMed] [Google Scholar]
- 33.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. 10.1016/S0378-1119(02)00551-6 [DOI] [PubMed] [Google Scholar]
- 34.Dietrich M, Mollenkopf H, So M, Friedrich A. 2009. Pilin regulation in the pilT mutant of Neisseria gonorrhoeae strain MS11. Lett. 296:248–256. 10.1111/j.1574-6968.2009.01647.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. 2003. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 31:e15. 10.1093/nar/gng015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoffmann A, Roeder RG. 1991. Purification of his-tagged proteins in non-denaturing conditions suggests a convenient method for protein interaction studies. Nucleic Acids Res. 19:6337–6338. 10.1093/nar/19.22.6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madrid C, Garcia J, Pons M, Juarez A. 2007. Molecular evolution of the H-NS protein: interaction with Hha-like proteins is restricted to Enterobacteriaceae. J. Bacteriol. 189:265–268. 10.1128/JB.01124-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madrid C, Nieto JM, Juarez A. 2002. Role of the Hha/YmoA family of proteins in the thermoregulation of the expression of virulence factors. Int. J. Med. Microbiol. 291:425–432. 10.1078/1438-4221-00149 [DOI] [PubMed] [Google Scholar]
- 39.Takeda T, Yun CS, Shintani M, Yamane H, Nojiri H. 2011. Distribution of genes encoding nucleoid-associated protein homologs in plasmids. Int. J. Evol. Biol. 2011:685015. 10.4061/2011/685015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bae SH, Liu D, Lim HM, Lee Y, Choi BS. 2008. Structure of the nucleoid-associated protein Cnu reveals common binding sites for H-NS in Cnu and Hha. Biochemistry 47:1993–2001. 10.1021/bi701914t [DOI] [PubMed] [Google Scholar]
- 41.Yee A, Chang X, Pineda-Lucena A, Wu B, Semesi A, Le B, Ramelot T, Lee GM, Bhattacharyya S, Gutierrez P, Denisov A, Lee CH, Cort JR, Kozlov G, Liao J, Finak G, Chen L, Wishart D, Lee W, McIntosh LP, Gehring K, Kennedy MA, Edwards AM, Arrowsmith CH. 2002. An NMR approach to structural proteomics. Proc. Natl. Acad. Sci. U. S. A. 99:1825–1830. 10.1073/pnas.042684599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vivero A, Banos RC, Mariscotti JF, Oliveros JC, Garcia-del Portillo F, Juarez A, Madrid C. 2008. Modulation of horizontally acquired genes by the Hha-YdgT proteins in Salmonella enterica serovar Typhimurium. J. Bacteriol. 190:1152–1156. 10.1128/JB.01206-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Snell EE. 1975. Tryptophanase: structure, catalytic activities, and mechanism of action. Adv. Enzymol. Relat. Areas Mol. Biol. 42:287–333 [DOI] [PubMed] [Google Scholar]
- 44.Yanofsky C. 2007. RNA-based regulation of genes of tryptophan synthesis and degradation, in bacteria. RNA 13:1141–1154. 10.1261/rna.620507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong F, Yanofsky C. 2001. Reproducing tna operon regulation in vitro in an S-30 system. Tryptophan induction inhibits cleavage of TnaC peptidyl-tRNA. J. Biol. Chem. 276:1974–1983. 10.1074/jbc.M008892200 [DOI] [PubMed] [Google Scholar]
- 46.Gong F, Yanofsky C. 2003. A transcriptional pause synchronizes translation with transcription in the tryptophanase operon leader region. J. Bacteriol. 185:6472–6476. 10.1128/JB.185.21.6472-6476.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saxena S, Gowrishankar J. 2011. Modulation of Rho-dependent transcription termination in Escherichia coli by the H-NS family of proteins. J. Bacteriol. 193:3832–3841. 10.1128/JB.00220-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Copley SD. 2012. Moonlighting is mainstream: paradigm adjustment required. Bioessays 34:578–588. 10.1002/bies.201100191 [DOI] [PubMed] [Google Scholar]
- 49.Das P, Lahiri A, Chakravortty D. 2010. Modulation of the arginase pathway in the context of microbial pathogenesis: a metabolic enzyme moonlighting as an immune modulator. PLoS Pathog. 6:e1000899. 10.1371/journal.ppat.1000899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Backert S, Feller SM, Wessler S. 2008. Emerging roles of Abl family tyrosine kinases in microbial pathogenesis. Trends Biochem. Sci. 33:80–90. 10.1016/j.tibs.2007.10.006 [DOI] [PubMed] [Google Scholar]
- 51.Madureira P, Baptista M, Vieira M, Magalhaes V, Camelo A, Oliveira L, Ribeiro A, Tavares D, Trieu-Cuot P, Vilanova M, Ferreira P. 2007. Streptococcus agalactiae GAPDH is a virulence-associated immunomodulatory protein. J. Immunol. 178:1379–1387 [DOI] [PubMed] [Google Scholar]
- 52.Kunert A, Losse J, Gruszin C, Huhn M, Kaendler K, Mikkat S, Volke D, Hoffmann R, Jokiranta TS, Seeberger H, Moellmann U, Hellwage J, Zipfel PF. 2007. Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J. Immunol. 179:2979–2988 [DOI] [PubMed] [Google Scholar]
- 53.Henderson B, Martin A. 2011. Bacterial virulence in the moonlight: multitasking bacterial moonlighting proteins are virulence determinants in infectious disease. Infect. Immun. 79:3476–3491. 10.1128/IAI.00179-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cardinale CJ, Washburn RS, Tadigotla VR, Brown LM, Gottesman ME, Nudler E. 2008. Termination factor Rho and its cofactors NusA and NusG silence foreign DNA in E. coli. Science 320:935–938. 10.1126/science.1152763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peters JM, Mooney RA, Grass JA, Jessen ED, Tran F, Landick R. 2012. Rho and NusG suppress pervasive antisense transcription in Escherichia coli. Genes Dev. 26:2621–2633. 10.1101/gad.196741.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Martino PD, Fursy R, Bret L, Sundararaju B, Phillips RS. 2003. Indole can act as an extracellular signal to regulate biofilm formation of Escherichia coli and other indole-producing bacteria. Can. J. Microbiol. 49:443–449. 10.1139/w03-056 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.