

Abstract
The organosulfur compound dimethylsulfoniopropionate (DMSP) is produced by phytoplankton and is ubiquitous in the surface ocean. Once released from phytoplankton, marine bacteria degrade DMSP by either the cleavage pathway to form the volatile gas dimethylsulfide (DMS) or the demethylation pathway, yielding methanethiol (MeSH), which is readily assimilated or oxidized. The enzyme DmdB, a methylmercaptopropionate (MMPA)-coenzyme A (CoA) ligase, catalyzes the second step in the demethylation pathway and is a major regulatory point. The two forms of DmdB present in the marine roseobacter Ruegeria pomeroyi DSS-3, RPO_DmdB1 and RPO_DmdB2, and the single form in the SAR11 clade bacterium “Candidatus Pelagibacter ubique” HTCC1062, PU_DmdB1, were characterized in detail. DmdB enzymes were also examined from Ruegeria lacuscaerulensis ITI-1157, Pseudomonas aeruginosa PAO1, and Burkholderia thailandensis E264. The DmdB enzymes separated into two phylogenetic clades. All enzymes had activity with MMPA and were sensitive to inhibition by salts, but there was no correlation between the clades and substrate specificity or salt sensitivity. All Ruegeria species enzymes were inhibited by physiological concentrations (70 mM) of DMSP. However, ADP reversed the inhibition of RPO_DmdB1, suggesting that this enzyme was responsive to cellular energy charge. MMPA reversed the inhibition of RPO_DmdB2 as well as both R. lacuscaerulensis ITI-1157 DmdB enzymes, suggesting that a complex regulatory system exists in marine bacteria. In contrast, the DmdBs of the non-DMSP-metabolizing P. aeruginosa PAO1 and B. thailandensis E264 were not inhibited by DMSP, suggesting that DMSP inhibition is a specific adaptation of DmdBs from marine bacteria.
INTRODUCTION
Dimethylsulfoniopropionate (DMSP) is ubiquitous in the ocean, where its degradation impacts global carbon and sulfur cycles. Marine phytoplankton produce DMSP as an osmolyte, predator deterrent, and antioxidant (1–3). It accounts for 10% of carbon fixed by marine phytoplankton in some parts of the ocean and 12 to 103 Tg of reduced sulfur each year (4–6). The significance of DMSP is partially due to its role as a precursor to the climatically active gas dimethylsulfide (DMS), but it is also a major carbon and energy source for microorganisms (6, 7). DMS is the main biogenic source of sulfur to the atmosphere, with emissions ranging from 0.6 to 1.6 Tg of sulfur per year (8, 9). Once in the atmosphere, DMS is oxidized and enhances the formation of cloud condensation nuclei (CCN) (1, 6, 10).
DMSP is metabolized by marine bacteria via one of two competing pathways, the cleavage or the demethylation pathway (11, 12). Certain microorganisms, such as Ruegeria pomeroyi DSS-3, possess both pathways (11, 13). Approximately 10% of DMSP is degraded via the cleavage pathway, resulting in the formation of DMS and acrylate (14–16). However, the majority of DMSP is degraded by the demethylation pathway, which forms methyl-tetrahydrofolate (methyl-THF), methanethiol (MeSH), CO2, and acetaldehyde (Fig. 1) (11). In this pathway, DMSP is demethylated to form methylmercaptopropionate (MMPA), precluding the formation of DMS and allowing DMSP to be used as both a carbon and reduced sulfur source by bacterioplankton (7, 11, 14). Four previously uncharacterized enzymes catalyze this pathway, DmdA, DmdB, DmdC, and DmdD (11). In R. pomeroyi DSS-3, a model organism for DMSP metabolism, there are two forms of the demethylation pathway MMPA-coenzyme A (CoA) ligase enzyme DmdB, RPO_DmdB1 and RPO_DmdB2 (Fig. 1). These isozymes are representative of each of the two phylogenetic clades of DmdB, B1 and B2. The ubiquitous SAR11 clade bacterium “Candidatus Pelagibacter ubique” HTCC1062 possesses a single form of this enzyme, a member of the B1 clade designated PU_DmdB1 (Fig. 2) (11).
FIG 1.
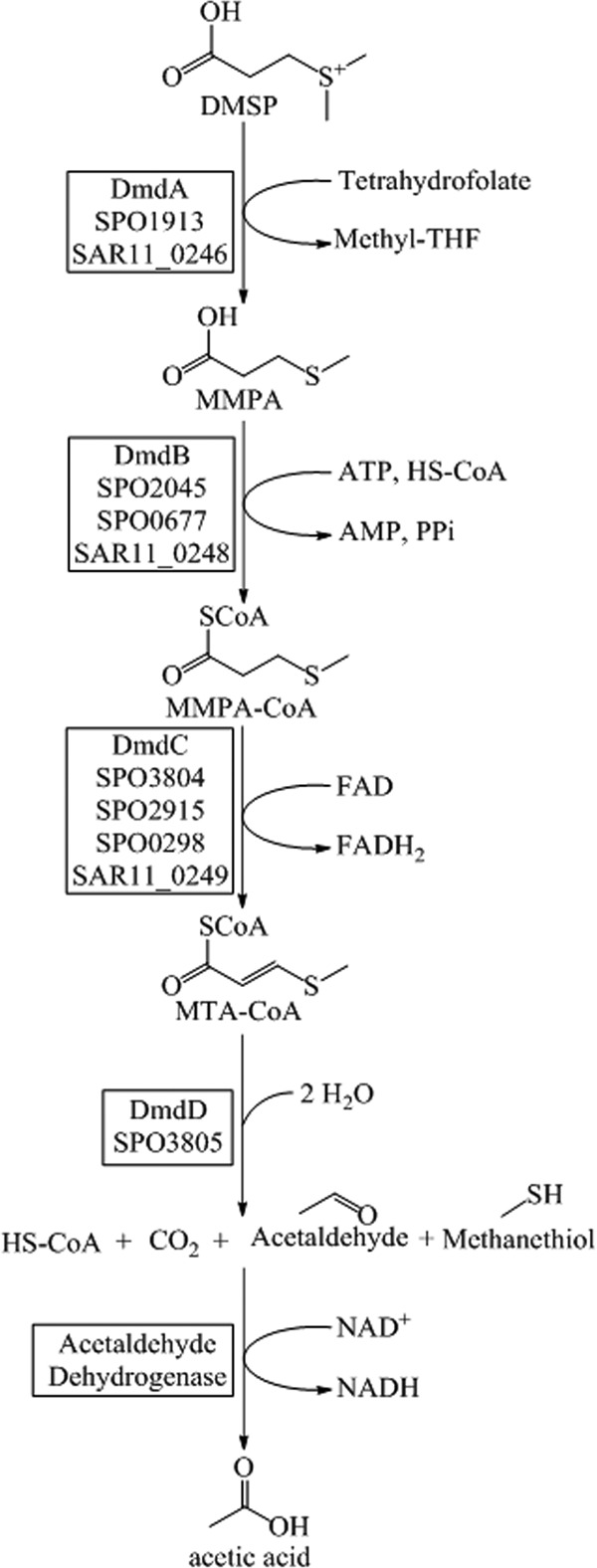
DMSP demethylation pathway from R. pomeroyi DSS-3 and “Ca. Pelagibacter ubique” HTCC1062 (11).
FIG 2.

Phylogenetic tree displaying the two DmdB clades. The phylogenetic tree was constructed using the maximum likelihood method in MEGA 5.2. DmdBs investigated for this study are indicated (●). Nomenclature for DmdBs used in this study is as follows: BTHI2141, BTH_DmdB2; PA4198, PA_DmdB1; SAR11_0248, PU DmdB1; SL1157 1815, RL_DmdB1; SL1157 2728, RL_DmdB2; SPO0677, RPO_DmdB1; SPO2045, RPO_DmdB2.
The goal of the current work is to gain better understanding of the DmdB isozymes involved in the demethylation pathway. Marine bacteria use DMSP as a carbon and sulfur source, incorporating 15 to 40% of DMSP-S for amino acid and protein synthesis (7, 15, 16). DMSP may also be an osmolyte and antioxidant for bacteria as well as phytoplankton (17). DMSP being used in any of these capacities may alter the regulation of the pathways or the isozymes (7, 14, 17). To gain further insight into the roles and regulation of the DmdB isozymes, the purified recombinant PU_DmdB1, RPO_DmdB1, and RPO_DmdB2 enzymes were further characterized. To determine if functional characteristics of DmdB enzymes were predictable based upon their clade designation, four additional members of the two DmdB clades were also purified and characterized. Two enzymes from Ruegeria lacuscaerulensis ITI-1157 (RL_DmdB1 and RL_DmdB2) were chosen to represent a sister organism to R. pomeroyi DSS-3. The enzymes from Pseudomonas aeruginosa PAO1 (PA_DmdB1) and Burkholderia thailandensis E264 (BTH_DmdB2) were chosen to represent nonmarine organisms capable of metabolizing MMPA to MeSH (11). As a group, the enzymes represent a range of amino acid identities of 86 to 51% within and 35 to 33% between the clades (see Table S1 in the supplemental material).
MATERIALS AND METHODS
Materials/substrate synthesis.
MMPA, marketed as 3-(methylthio)propionic acid (product no. L12861), was purchased from A. Aesar (Ward Hill, MA). DMSP was synthesized as described previously by Chambers et al. from dimethyl sulfide and acrylic acid (Sigma-Aldrich) (18).
Expression of recombinant proteins.
The genes from R. pomeroyi DSS-3 (RPO_DmdB1, SPO0677 [YP_165932.1]; RPO_DmdB2, SPO2045 [YP_167275.1]) (19, 20), P. aeruginosa PAO1 (PA_DmdB1, PA4198 [NP_252887.1]) (21), R. lacuscaerulensis ITI-1157 (RL_DmdB1, SL1157_1815 [WP_005981195.1]; RL_DmdB2, SL1157_2728 [WP_005982887.1]) (20, 22), and B. thailandensis E264 (BTH_DmdB2, BTH_I2141 [YP_442662.1]) (23) were PCR amplified from genomic DNA. The “Ca. Pelagibacter ubique” HTCC1062 gene PU_DmdB1 (SAR11_0248 [YP_265673.1]) (24) was previously synthesized by using Escherichia coli codon usage by the GenScript Corporation (11). Amplified genes were cloned into the pET101 expression vector per Invitrogen-recommended methods. Constructed plasmids and relevant primers are summarized in Tables S2 and S3 in the supplemental material. Clones were then transformed into BL21(DE3) or Rosetta (DE3) E. coli cells for protein expression. P. aeruginosa PAO1, R. lacuscaerulensis ITI-1157, and B. thailandensis E264 dmdB genes were cloned to include the 6× histidine tag from the pET101 vector. The recombinant proteins were given the RPO, PU, RL, PA, or BTH designations before the gene product name to allow for a clear indication of the genus and species of the source bacterium.
Cells for protein expression were grown in Luria-Bertani (LB) broth at 30°C until an optical density at 600 nm of 0.4 to 0.5 was reached. Cultures were then induced with 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated for 4 h at 30°C. Cultures were harvested by centrifugation at 8,000 × g for 10 min, and pellets were washed with 50 mM Tris (pH 7.5). Cells resuspended in 50 mM Tris were lysed by French press or sonication. Lysed cells were centrifuged at 17,000 × g for 5 min to remove cell debris.
Chromatography techniques.
Six forms of column chromatography were used to purify the various DmdB enzymes. Fractions including the DmdB proteins were assayed for the formation of MMPA-CoA by the high-performance liquid chromatography (HPLC)-based assay described below. A Q-Sepharose (GE Healthcare) column (1.6 by 7 cm) was used for anion exchange chromatography. The column was equilibrated with 50 mM Tris (pH 7.5), and proteins were eluted using a linear gradient from 0 M NaCl to1 M NaCl at a flow rate of 2 ml min−1. A phenyl Superose (GE Healthcare) column (1.0 by 10 cm) was equilibrated with 50 mM Tris (pH 7.5) plus 1.7 M (NH4)2SO4, and proteins were eluted with a linear gradient from 1.7 M to 0 M (NH4)2SO4 at a flow rate of 1 ml min−1. A HiTrap Blue HP (GE Healthcare) column (1.6 by 2.5 cm) was used as an affinity chromatography step. The column was equilibrated with 50 mM KHPO4 (pH 7.5), and proteins were eluted with a linear gradient of KCl increasing from 0 to 2 M KCl at a flow rate of 1 ml min−1. A CHT ceramic hydroxyapatite type 1 (Bio-Rad) column (1.0 by 7.0 cm) was equilibrated with 5 mM NaHPO4 (pH 7.5) and 150 mM NaCl. Proteins were eluted with a linear gradient from 5 mM to 500 mM NaHPO4 plus 150 mM NaCl at a flow rate of 0.5 ml min−1. Gel filtration was performed using a Sephacryl (25)S200 HR (GE Healthcare) column (1.6 by 34 cm) equilibrated with 50 mM Tris (pH 7.5) and 250 mM NaCl. Proteins were eluted in the same buffer at a flow rate of 0.75 ml min−1.
Purification of PU_DmdB1.
Extracts of 2.3 g protein containing recombinant PU_DmdB1 were applied to a Q-Sepharose anion exchange column. Active fractions eluted from 0.21 to 0.26 M NaCl. Active fractions were pooled, adjusted to 1.7 M (NH4)2SO4 using solid (NH4)2SO4, and applied to a phenyl Superose hydrophobic interaction column. Active fractions eluted from 0.35 to 0.28 M (NH4)2SO4. These fractions were pooled and concentrated at 6,000 × g for 15 min using Amicon Ultra-4 10K centrifugal filter units (Millipore) to remove excess (NH4)2SO4. Protein samples were diluted two times with 50 mM KHPO4 (pH 7.5) and then applied to the HiTrap Blue column (GE Healthcare). Active fractions eluted from 0.75 to 1.0 M KCl. These fractions were concentrated using the Amicon Ultra-4 10K centrifugal filter units and brought up to 2 ml using 100 mM HEPES (pH 7.5). The enzyme was then stored at −20°C.
Purification of RPO_DmdB1.
Extracts of 2.5 g protein containing recombinant RPO_DmdB1 were applied to a Q-Sepharose anion exchange column, and active fractions were pooled and concentrated as described above. Active fractions from the Q-Sepharose column eluted from 0.42 to 0.46 M NaCl. Protein samples were diluted two times using 50 mM KHPO4 (pH 7.5) and then applied to the HiTrap Blue column. Fractions containing RPO_DmdB1 eluted from 1.0 to 1.2 M KCl. Active fractions were pooled, concentrated as described before, and applied to the gel filtration Sephacryl S200 HR column. Final protein samples were concentrated as described above, brought to 2 ml using 100 mM KHPO4 (pH 7.5), and stored at −20°C.
Purification of RPO_DmdB2.
Extracts of 2.7 g protein containing recombinant RPO_DmdB2 were applied to a Q-Sepharose anion exchange column, and active fractions were pooled and concentrated as described previously. Active fractions eluted from 0.46 to 0.50 M NaCl. Protein samples were diluted two times using 50 mM NaHPO4 (pH 7.5) and then applied to the CHT ceramic hydroxyapatite type 1 column. Active fractions eluted from 0.06 to 0.08 M NaHPO4. These fractions were pooled, concentrated as described before, and applied to the HiTrap Blue column. Active fractions from the HiTrap Blue column eluted from 1.3 to 1.5 M KCl. Final protein samples were concentrated, brought to 2 ml using 100 mM KHPO4 (pH 7.5), and stored at −20°C.
Purification of His-tagged DmdBs.
His-tagged DmdBs from R. lacuscaerulensis ITI-1157, P. aeruginosa PAO1, and B. thailandensis E264 were purified using a HisTrap HP (GE Healthcare) column (7 by 25 mm). The column was equilibrated with 20 mM NaHPO4 (pH 7.5) and 5 mM imidazole, and proteins were eluted using a linear imidazole gradient from 5 to 500 mM imidazole at a flow rate of 0.5 ml min−1. In all cases, the desired protein eluted from 0.11 to 0.16 M imidazole. The NaHPO4 buffer was exchanged with 100 mM KHPO4 (pH 7.5) using Amicon Ultra-4 10K centrifugal filter units. Final protein samples were concentrated as described above, brought to 2 ml using 100 mM KHPO4 (pH 7.5), and stored at −20°C.
Protein concentration and purity.
Protein concentrations were determined using either the Bio-Rad Bradford reagent or the BCA protein assay kit (Thermo Scientific) with bovine serum albumin standard. These methods yielded similar results for these proteins. Protein purity was assessed using SDS-PAGE gels and the Gel Pro Analyzer program version 4.0 from Media Cybernetics, L.P.
SDS-PAGE.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was accomplished using Mini-PROTEAN TGX precast gels (Bio-Rad; 4 to 15% polyacrylamide gradient minigels). Gels were stained using SimplyBlue safe stain (Invitrogen). SDS-PAGE images of purified recombinant DmdB enzymes are shown in Fig. S1 and S2 in the supplemental material.
Enzyme assays.
Two types of enzyme assays were performed. In both assays, one unit was defined as 1 μmol product formed min−1, and specific activity was defined as 1 unit per mg of protein. All enzyme assays were performed at room temperature, because preliminary studies of PU_DmdB1 and RPO_DmdB2 possessed good activity at this temperature. Their activities at 10°C to 30°C were nearly the same as those at room temperature.
In the first assay, product formation was analyzed using a Waters Alliance 2695 HPLC by using a Hypersil Gold C18 reverse-phase column with a 3-μm particle size (4.6 by 150 mm; Thermo Scientific). The column was developed with a linear gradient from 3% to 25% acetonitrile with 50 mM ammonium acetate at a flow rate of 1.0 ml min−1 over 10 min. This HPLC assay was less sensitive to interference by salts and contained fewer potential effectors, such as ADP, than the coupled assay. Thus, it was used to perform kinetic experiments, salt sensitivity analysis with 0.4 M salts, DMSP sensitivity, and specific activity analyses. Unless stated otherwise, the HPLC assay contained 2 mM ATP, 2 mM MgCl2, 2 mM MMPA, 0.1 mM CoA, and 100 mM HEPES buffer (pH 7.5) in a total volume of 100 μl. Assays were run for 2 min and quenched by the addition of 4 μl of 85% phosphoric acid. The HPLC assay was linear over the 2-min time course.
A spectrophotometric-coupled assay was used for all other experiments (25). ATP, MgCl2, and MMPA were all supplied at 2 mM, CoA at 0.3 mM, phosphoenolpyruvate at 3 mM, and NADH at 0.1 mM in 100 mM HEPES buffer (pH 7.5). One unit of rabbit myokinase (Sigma) and two units of pyruvate kinase-lactate dehydrogenase (Sigma) were used. At these levels, the coupling enzymes were not rate limiting. The reaction was initiated by the addition of either MMPA or ATP, and reaction progress was monitored at 340 nm. Rates were calculated using the NADH extinction coefficient of 6,220 M−1 cm−1 (26). This assay was used for the determination of substrate specificity and pH optima. Substrate specificity was examined by replacing MMPA with 2 mM each substrate tested. Initial screening of substrates using the HPLC assay showed that no other products were formed than the expected CoA-thioesters, which confirmed the identity and purity of the substrates.
Apparent Michaelis-Menten kinetic constants were calculated by varying one of three substrates (MMPA or the fatty acid, CoA, or ATP) while keeping the other two at the concentrations of the standard assay. MMPA, butyrate, propionate, and acrylate were varied from 0.01 mM to 20 mM. ATP was varied from 0.01 to 2 mM, and CoA was varied from 0.005 to 0.2 mM. Kinetic data analyses, other calculations, and statistical analyses were performed using SigmaPlot 11.0 (Systat Software Inc.).
The pH optimum was determined using the following buffers and pH values: sodium HEPES (6.5, 7.0, 7.5, and 8.0,), Tris base (8.0, 8.5, and 9.0), sodium 2-(N-morpholino)ethanesulfonic acid (MES) (5.5, 6.0, and 6.5), 1,3-bis(Tris)propane (6.5, 7.0, 7.5, 8.0, 8.5, and 9.0), morpholinepropanesulfonic acid (MOPS) (6.5, 7.0, and 7.5), and sodium 2-(cyclohexylamino)ethanesulfonic acid (CHES) (8.5, 9.0, 9.5, 10.0). Controls established that the coupling enzymes were not rate limiting in each of the buffers. All proteins had the highest activity in 100 mM HEPES, compared with that in 100 mM Tris, MES, CHES, MOPS, and Bis-Tris-propane (data not shown). For this reason, subsequent assays were performed using 100 mM HEPES at pH 7.5 for PU_DmdB1 and RPO_DmdB2 and at pH 7.0 for RPO_DmdB1.
Native molecular weight.
The native molecular weights of the enzymes were determined using a Sephacryl S200 HR gel filtration column, as described above. β-Amylase (molecular weight of 200), alcohol dehydrogenase (150), bovine albumin (66), carbonic anhydrase (29), and cytochrome c (12.4) served as molecular weight standards.
Phylogenetic analysis.
The DmdB from “Ca. Pelagibacter ubique” HTCC1062 (SAR11_0248:PU_DmdB1) was used as a query sequence for the BLASTp search. The PU_DmdB1 sequence was queried against the genomes of Escherichia coli BL21(DE3), Burkholderia thailandensis, Deinococcus radiodurans, Dinoroseobacter shibae, marine gammaproteobacterium HTCC2143, SAR11 HIMB59, alphaproteobacterium HIMB114, Myxococcus xanthus, Pseudomonas aeruginosa, Pseudoalteromonas atlantica, “Ca. Pelagibacter ubique” HTCC7211, “Ca. Pelagibacter ubique” HTCC1062, Puniceispirillum marinum IMCC1322, SAR11 HIMB5, Ruegeria lacuscaerulensis 1157, and Ruegeria pomeroyi DSS-3. Sequences with an expected value of <e−10 were aligned using the MUSCLE algorithm, and the phylogenetic tree was built using the maximum likelihood method in MEGA 5.2. Bootstrap values of 100 were used for the analysis.
Chemostat cultures.
R. pomeroyi DSS-3 was grown in carbon-limited chemostats containing marine basal medium (MBM) (12, 27) with 6 mM acetate, 2 mM DMSP, 4 mM MMPA, or 3 mM methionine as the sole carbon source. The concentration of each carbon source was chosen to ensure that all four chemostats obtained similar cell densities at the chosen growth rate. Although tested empirically, the concentrations chosen reflect the number of electrons available for growth on each substrate. Chemostats were maintained at 30°C with a dilution rate of 0.0416 h−1 and a 14-h doubling time. Five exchanges of the 144-ml chemostat volume were completed prior to collection of the first sample. Subsequently, 100-ml samples were taken daily from each chemostat, centrifuged immediately at 8,000 × g for 10 min, and stored at −80°C for RNA extraction.
RNA extraction and mRNA enrichment.
RNA extractions from chemostat samples were performed using the Illustra RNAspin minikit (GE Life Sciences). Two samples from each chemostat were further treated with the MICROBExpress bacterial mRNA enrichment kit (Ambion) and Terminator 5′-phosphate-dependent exonuclease (Epicenter) to enrich the mRNA and deplete rRNA.
Sequencing and read mapping to the R. pomeroyi DSS-3 genome.
The TruSeq RNA sample preparation kit v2 (Illumina) was used to form cDNA from the enriched mRNA samples, add barcodes to each cDNA sample, and prepare the Illumina library for sequencing. The enriched mRNA and Illumina libraries were quantified using a 2100 Bioanalyzer (Agilent). RNA samples included two biological replicates, with the exception of the acetate RNA sample, which had only one biological sample, and a technical replicate. Sequencing was performed by the Genome Services Laboratory at the Hudson Alpha Institute for Biotechnology, Huntsville, AL, on a single lane of the SE50 HiSeq 2000 sequencer. High coverage was attained from the resultant nearly 2 million 50-bp reads. Reads were mapped to the R. pomeroyi DSS-3 genome using Bowtie2 (28). Differential expression was determined using the CuffDiff program from the Cufflinks package. P values for differential expression were calculated using a negative binomial distribution using CuffDiff (28, 29). Between 2.4 and 5.4% of the reads from each sample mapped exactly once to the R. pomeroyi DSS-3 genome and were used for further analyses. The other 95% of reads mapped identically to multiple places in the genome, in particular rRNA islands.
RESULTS
DmdBs are dimers with MMPA-CoA ligase activity.
The genes encoding the DmdB isozymes were recombinantly expressed in E. coli BL21(DE3) and purified to over 90% purity (see Fig. S1 and S2 in the supplemental material). The PU_DmdB1 enzyme was stable at −20°C for up to 6 months when stored in 100 mM HEPES. The RPO_DmdB1 and RPO_DmdB2 enzymes were stable in a solution of 100 mM KHPO4 at −20°C for up to 2 months. The Mrs of the isozyme subunits, as determined by SDS-PAGE, were consistent with the predicted molecular weights based on their amino acid sequences (19, 24). The native Mrs as determined by gel filtration chromatography were consistent with these proteins being dimers (Table 1).
TABLE 1.
General characteristics of the recombinant DmdB isozymes from “Ca. Pelagibacter ubique” HTCC1062 and R. pomeroyi DSS-3
| Characteristic | Value |
||
|---|---|---|---|
| PU_DmdB1 | RPO_DmdB1 | RPO_DmdB2 | |
| Mol wt | |||
| Predicted | 61,000 | 59,000 | 59,000 |
| Observed denatured | 59,000 | 62,000 | 58,000 |
| Observed native | 108,000 | 112,000 | 122,000 |
| % purity | 93 | 90 | 92 |
| Sp act (μmol min−1 mg−1) | 28 | 15 | 16 |
| pH | |||
| Optimum | 7.5 | 7.0 | 7.5 |
| Range | 6.0–8.5 | 6.0–8.0 | 6.5–9.0 |
The specific activity of PU_DmdB1 with MMPA was nearly 2-fold higher than those of RPO_DmdB1 and RPO_DmdB2 (Table 1). “Ca. Pelagibacter ubique” HTCC1062 is an oligotroph with a minimal genome that encodes only four other CoA ligase-type enzymes, while R. pomeroyi DSS-3 is an opportunotroph that maintains 26 CoA ligase homologs (19, 24). Selection for greater efficiency in the oligotrophs potentially results in higher specific activity enzymes. The isozymes all exhibited a similar pH range for optimal activity (Table 1).
DmdBs show various activities in the presence of salts.
The DmdB isozymes responded differently to the presence of some salts (Table 2). To determine if the responses were characteristic of the clades, they were examined in more detail. Because Mg2+ was required for activity, divalent cations were not tested. In most cases, the enzymes were either inhibited by salts or showed no change in activity. However, LiCl and NH4Cl stimulated RPO_DmdB2, RL_DmdB1, and RL_DmdB2. This stimulation was sufficient to yield specific activities comparable to that of PU_DmdB1 in the absence of salt. The other effects of salts were complex. For instance, Cl− stimulated both RL_DmdB1 and PA_DmdB1, and Na+ inhibited both the PA_DmdB1 and BTH_Dmd2, but even in these cases the counterions played significant roles. Moreover, the response to salts for members of the same DmdB clade possessed no more similarity than responses of members of different clades. Even though the R. pomeroyi DSS-3 and R. lacuscaerulensis ITI-1157 DmdB2s possessed 86% amino acid sequence identity, their responses to salts were very different. Thus, the salt response was highly individual among these enzymes.
TABLE 2.
Effect of salts on the activity of the DmdB isozymesa
| Salt | Relative activity (%) |
||||||
|---|---|---|---|---|---|---|---|
| PU_DmdB1 | RPO_DmdB1 | RPO_DmdB2 | RL_DmdB1 | RL_DmdB2 | PA_DmdB1 | BTH_DmdB2 | |
| No Salt | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| LiCl | 9 | 41 | 301 | 135 | 138 | 97 | 13 |
| NH4Cl | 68 | 6 | 210 | 164 | 50 | 95 | 45 |
| KCl | 92 | 30 | 45 | 104 | 141 | 103 | 27 |
| NaCl | 43 | 40 | 50 | 86 | 90 | 49 | 8 |
| Na acetate | 39 | 14 | 34 | 32 | 12 | 17 | 55 |
| K acetate | 84 | 27 | 49 | 44 | 103 | 15 | 93 |
| Li2SO4 | 8 | 18 | 18 | 43 | 59 | 19 | 8 |
| (NH4)2SO4 | 86 | 25 | 154 | 57 | 71 | 50 | 7 |
| K2SO4 | 84 | 32 | 157 | 95 | 10 | 104 | 133 |
| Na2SO4 | 40 | 32 | 117 | 42 | 34 | 36 | 16 |
All salts were used at a concentration of 0.4 M. The HPLC-based assay was used to avoid salt effects on coupling enzymes. Relative activity (μmol min−1 mg−1) values expressed as a percentage of the total specific activity as measured without salt additions (100%). The standard errors are from three independent experiments and were within 3%. Specific activities in units mg−1 of protein (±SE) defined as 100% were as follows: PU_DmdB1, 26 ± 3; RPO_DmdB1, 17 ± 2; RPO_DmdB2, 16 ± 3; RL_DmdB1, 21 ± 3; RL_DmdB2, 18 ± 2; PA_DmdB1, 31 ± 2; and BTH_DmdB2, 25 ± 4.
DmdBs have activity with MMPA and short-chain fatty acids.
All of the recombinant DmdBs possessed high MMPA-CoA ligase activity (Table 3). In addition, all of the enzymes were active with carboxylic acids up to six carbons in length, indicating they were short-chain-fatty-acid–CoA ligases. The highest activities were observed with MMPA or substrates between three and five carbons in length. DMSP was included in these tests, but no activity for any of the enzymes was detected at a sensitivity of 0.1% of the MMPA-CoA ligase activity (data not shown). Two of the three DmdB clade 2 enzymes tested, RPO_DmdB2 and BTH_DmdB2, had their highest activity under these conditions with MMPA and little to no activity with acetate. None of the DmdB clade 1 enzymes had their highest activity with MMPA. RPO_DmdB1 had similar levels of activity with MMPA and crotonate. PU_DmdB1 had the highest activity with butyrate, almost twice of that with MMPA. However, as shown below, the levels of CoA used in these tests were below the Km for MMPA-dependent but not the butyrate-dependent ligase activity of this enzyme. Thus, the values reported here do not necessarily reflect the maximum activities for these substrates. RL_DmdB1 and PA_DmdB1 had high levels of activity with acetate and propionate. However, there were no consistent differences between the clade 1 and 2 enzymes.
TABLE 3.
Substrate specificities of the DmdB isozymesa
| Substrate | Relative activity (%) |
||||||
|---|---|---|---|---|---|---|---|
| PU_DmdB1 | RPO_DmdB1 | RPO_DmdB2 | RL_DmdB1 | RL_DmdB2 | PA_DmdB1 | BTH_DmdB2 | |
| MMPA | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Acetate | 10 | 0.0b | 10 | 151 | 0.0b | 110 | 0.0b |
| Propionate | 13 | 77 | 16 | 126 | 72 | 124 | 45 |
| Acrylate | 50 | 79 | 56 | 6 | 29 | 65 | 15 |
| Butyrate | 160 | 36 | 73 | 81 | 113 | 121 | 93 |
| Isobutyrate | 13 | 30 | 12 | 10 | 7 | 48 | 5 |
| Crotonate | 2 | 109 | 63 | 32 | 41 | 63 | 87 |
| Methylbutyrate | 8 | 10 | 39 | 20 | 18 | 24 | 23 |
| Valerate | 18 | 31 | 51 | 49 | 179 | 91 | 68 |
| Isovalerate | 8 | 19 | 14 | 15 | 5 | 18 | 4 |
| Hexanoate | 5 | 17 | 27 | 17 | 62 | 12 | 94 |
| Caprylate | 0.0b | 0.0b | 0.0b | 0.0b | 0.0b | 0.0b | 1 |
| Caprate | 0.0b | 0.0b | 0.0b | 0.0b | 0.0b | 0.0b | 0.0b |
Relative activity values expressed as a percentage of the total specific activity as measured with MMPA (100%). All standard errors are from three independent experiments and are within 3%. Specific activities in units mg−1 of protein (±SE) defined as 100% were as follows: PU_DmdB1, 28 ± 1; RPO_DmdB1, 15 ± 3; RPO_DmdB2, 17 ± 3; RL_DmdB1, 24 ± 2; RL_DmdB2, 16 ± 2; PA_DmdB1, 32 ± 2; and BTH_DmdB2, 25 ± 4.
<0.1%
Kinetic analysis of DmdB reveals substrate preferences.
The DmdB isozymes PU_DmdB1, RPO_DmdB1, and RPO_DmdB2 all possessed low apparent Michaelis-Menten constants and high catalytic efficiencies (kcat/Km) for MMPA, consistent with their roles in DMSP metabolism (Table 4). Of the three enzymes, PU_DmdB1 had the lowest Km and the highest catalytic efficiency for MMPA and all three fatty acid substrates tested. The catalytic efficiencies of this enzyme for MMPA, butyrate, and propionate were within the range expected for physiological activities. In contrast, the catalytic efficiencies of RPO_DmdB1 and RPO_DmdB2 were similar for MMPA, 233 mM−1 s−1 and 213 mM−1 s−1, respectively. Moreover, the higher catalytic efficiencies for butyrate (1,031 mM−1 s−1) and propionate (271 mM−1 s−1) of RPO_DmdB1 were consistent with these activities being physiologically relevant (30, 31). RPO_DmdB2 possessed much lower values for these fatty acids, consistent with the conclusion that this DmdB was a specialized MMPA-CoA ligase.
TABLE 4.
Apparent kinetic constants for “Ca. Pelagibacter ubique” HTCC1062 and R. pomeroyi DSS-3 DmdBsa
| Substrate | Kinetic constant | Value |
||
|---|---|---|---|---|
| PU_DmdB1 | RPO_DmdB1 | RPO_DmdB2 | ||
| MMPA | Km | 0.04 ± 0.01 | 0.08 ± 0.02 | 0.07 ± 0.02 |
| Vmax | 31.4 ± 5.3 | 19.3 ± 3.3 | 15.4 ± 2.5 | |
| kcat | 29.4 | 18.7 | 14.9 | |
| kcat/Km | 735 | 233 | 213 | |
| Butyrate | Km | 0.01 ± 0.01 | 0.02 ± 0.01 | 0.12 ± 0.03 |
| Vmax | 46.8 ± 7.7 | 14.9 ± 3.6 | 7.4 ± 2.1 | |
| kcat | 43.8 | 14.4 | 7.2 | |
| kcat/Km | 3,370 | 1,031 | 71 | |
| Propionate | Km | 0.04 ± 0.02 | 0.04 ± 0.01 | 3.11 ± 1.13 |
| Vmax | 18.9 ± 3.0 | 11.2 ± 2.5 | 3.8 ± 1.4 | |
| kcat | 17.7 | 10.8 | 3.7 | |
| kcat/Km | 505 | 271 | 1.2 | |
| Acrylate | Km | 0.44 ± 0.04 | 0.9 ± 0.2 | 5.25 ± 2.1 |
| Vmax | 23.8 ± 5.3 | 10.5 ± 2.0 | 1.0 ± 0.2 | |
| kcat | 22.3 | 14.3 | 1.0 | |
| kcat/Km | 50 | 16 | 0.2 | |
Km (mM) and Vmax (μmol min−1 mg−1) are shown (±SE) from three independent experiments. kcat is expressed in units of s−1 and kcat/Km in units of mM−1 s−1.
The apparent Kms of the DmdB isozymes for ATP and CoA also depended to some extent on whether the substrate was MMPA or butyrate (Table 5). PU_DmdB1 had a higher Km for CoA with MMPA (0.58 mM) than with butyrate (0.11 mM). As cellular levels of CoA are typically between 0.01 to 0.6 mM, PU_DmdB1 may have a lower activity with MMPA when other substrates, like butyrate, are available and the CoA pool is limited (32, 33).
TABLE 5.
Apparent kinetic constants for “Ca. Pelagibacter ubique” HTCC1062 and R. pomeroyi DSS-3 DmdBs for ATP and CoA in the presence of MMPA and butyrate
| Substrate | Kinetic constanta | Value |
||
|---|---|---|---|---|
| PU_DmdB1 | RPO_DmdB1 | RPO_DmdB2 | ||
| MMPA-ATP | Km | 0.03 ± 0.01 | 0.01 ± 0.005 | 0.03 ± 0.01 |
| Vmax | 24.4 ± 4.3 | 18.9 ± 4.2 | 7.6 ± 2.3 | |
| MMPA-CoA | Km | 0.58 ± 0.02 | 0.01 ± 0.007 | 0.02 ± 0.01 |
| Vmax | 46.1 ± 5.8 | 18.4 ± 3.4 | 15.4 ± 2.5 | |
| Butyrate-ATP | Km | 0.06 ± 0.02 | 0.01 ± 0.007 | 0.08 ± 0.03 |
| Vmax | 24.0 ± 4.4 | 12.4 ± 2.6 | 3.8 ± 1.4 | |
| Butyrate-CoA | Km | 0.11 ± 0.03 | 0.14 ± 0.05 | 0.02 ± 0.01 |
| Vmax | 52.2 ± 3.5 | 8.5 ± 2.7 | 3.6 ± 0.7 | |
Km (mM) and Vmax (μmol min−1 mg−1) are shown (±SE) from three independent experiments.
DMSP inhibition of marine DmdBs.
During growth on DMSP, R. pomeroyi DSS-3 maintains an intracellular concentration of 70 mM DMSP (12). However, DMSP strongly inhibited both RPO_DmdB1 and RPO_DmdB2 (Fig. 3). While internal concentrations of DMSP in “Ca. Pelagibacter ubique” HTCC1062 are not known, PU_DmdB1 was also inhibited. The inhibition of RPO_DmdB1 and RPO_DmdB2 was so severe that no activity would be expected under the cellular concentrations of DMSP. Similar to the R. pomeroyi DSS-3 enzymes, RL_DmdB1 and RL_DmdB2 were also sensitive to DMSP. In contrast, the DmdB enzymes from nonmarine organisms, PA_DmdB1 and BTH_DmdB2, were not inhibited by DMSP (Fig. 4). This result suggests that DMSP sensitivity is a specific adaptation of the DmdBs from marine bacteria.
FIG 3.
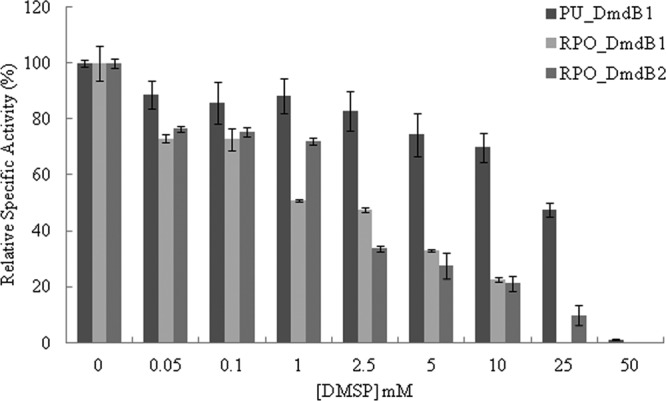
Inhibition of the MMPA-CoA ligases in the presence of DMSP for the DmdB isozymes from “Ca. Pelagibacter ubique” HTCC1062 and R. pomeroyi DSS-3. Relative activity values expressed as a percentage of the total measured activity in the absence of DMSP (100%). Specific activities in units mg−1 of protein defined as 100% were as follows: PU_DmdB1, 31 ± 1; RPO_DmdB1, 18 ± 4; RPO_DmdB2, 15 ± 2. Errors are ±standard errors (SE) from three replicates.
FIG 4.
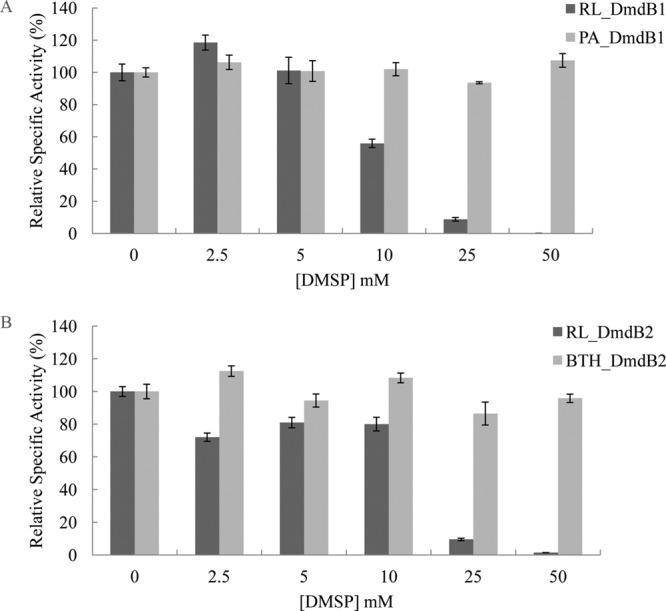
DMSP inhibition of members of each DmdB clade. (A) Inhibition of DmdB activity of DmdB clade 1 members RL_DmdB1 and PA_DmdB1. (B) Inhibition of DmdB clade 2 members RL_DmdB2 and BTH_DmdB2. Relative activity values expressed as a percentage of the total measured in the absence of DMSP (100%). Specific activities in units mg−1 of protein defined as 100% were as follows: RL_DmdB1, 25 ± 5; PA_DmdB1, 35 ± 2; RL_DmdB2, 16 ± 3; and BTH_DmdB2, 24 ± 4. Errors are ±SE from three replicates.
ADP and MMPA relieved inhibition by DMSP in R. pomeroyi DSS-3.
ADP relieved the DMSP inhibition of RPO_DmdB1 (Fig. 5). ATP, which is a substrate of the MMPA-CoA ligase reaction, had no effect on DMSP inhibition (data not shown). AMP inhibited RPO_DmdB1, yielding 50% inhibition at concentrations greater than 0.1 mM, presumably due to product inhibition (data not shown). In the presence of 50 mM DMSP, up to 65% of RPO_DmdB1 activity was regained at 4 mM ADP, which was comparable to the cellular concentration in E. coli during energy limitation (Fig. 5) (34). In contrast, ADP had no effect on the DMSP inhibition of RPO_DmdB2, indicating that it was regulated differently. However, MMPA concentrations above 2 mM relieved the DMSP inhibition of RPO_DmdB2, and 80% of activity was regained at an MMPA concentration of 8 mM (Fig. 5).
FIG 5.

Protection of DmdB from DMSP inhibition by ADP and MMPA. (A) Effect of ADP on RPO_DmdB1. Effect of MMPA on RPO_DmdB2 (B), RL_DmdB1 (C), and RL_DmdB2 (D). Relative activity expressed as a percentage of the total activity in the absence of DMSP. Specific activities in units mg−1 of protein defined as 100% were as follows: RPO_DmdB1, 14 ± 2; RPO_DmdB2, 13 ± 2; RL_DmdB1, 18 ± 3; and RL_DmdB2, 10 ± 3. Errors are ±SE from three replicates.
The reversals of DMSP inhibition by ADP or MMPA were not properties of the R. lacuscaerulensis and “Ca. Pelagibacter ubique” DmdB enzymes. Additions of ADP concentration above 0.5 mM inhibited PU_DmdB1's activity by 50% even in the absence of DMSP. This inhibition was enhanced in the presence of DMSP. Additions of increasing concentrations of MMPA to the PU_DmdB1 assays had no effect on the level of DMSP inhibition (data not shown). Similarly, the addition of MMPA above 8 mM restored only 25% of RL_DmdB1 activity and 35% of RL_DmdB2 activity (Fig. 5). ADP had no effect on DMSP inhibition of these enzymes. However, it is not known if R. lacuscaerulensis or “Ca. Pelagibacter ubique” accumulate DMSP to high levels, and the response of R. pomeroyi DSS-3 to DMSP may be specific to its DmdBs.
RPO_dmdB1 and RPO_dmdB2 are differentially expressed depending on carbon source.
RNA extracted from chemostat-grown cells given four different sole carbon sources was analyzed for variations in RPO_dmdB1 and RPO_dmdB2 expression levels. Cells were grown in chemostats containing 2 mM DMSP, 4 mM MMPA, 3 mM methionine, or 6 mM acetate as the sole source of carbon. DmdB activity was expected to be required for metabolism of all of these substrates except acetate, while DmdA was required only for the catabolism of DMSP. As expected, the steady-state levels of RPO_dmdA were elevated only during growth on DMSP (Table 6). RPO_dmdB1 exhibited overall lower steady-state levels of mRNA than RPO_dmdB2 for all substrates except acetate (Table 6). Although the levels of the RPO_dmdB1 transcripts increased during growth on DMSP and methionine, it was always lower than those of RPO_dmdB2. For that reason, RPO_dmdB2 appeared to be the major DmdB during growth on compounds leading to MMPA formation (Table 6).
TABLE 6.
Expression of RPO_dmdA, RPO_dmdB1, and RPO_dmdB2 during steady-state growth on DMSP and other carbon sources
| Carbon source | Valuea |
||
|---|---|---|---|
| RPO_dmdA | RPO_dmdb1 | RPO_dmdB2 | |
| DMSP | 33.5 A | 6.3 A | 19.5 A |
| MMPA | 3.6 B | 2.2 C | 17.8 A |
| Methionine | 2.6 B | 4.5 AB | 16.6 A |
| Acetate | 4.1 B | 2.7 BC | 3.7 B |
Values within a column with the same letter were not significantly different with a P value of <0.05. P values were calculated using a negative binominal distribution using the CuffDiff program (29). Expression based on Illumina RNAseq reads mapped to RPO_dmdA, RPO_dmdB1, and RPO_dmdB2. Given values are fragments per kilobase per million (FPKM), indicating reads/length of transcript in kb/total number of reads.
DISCUSSION
DmdB is an acyl-CoA ligase that catalyzes the second reaction of the DMSP demethylation pathway. In this study, DmdB enzymes from R. pomeroyi DSS-3 and “Ca. Pelagibacter ubique” HTCC1062 were characterized in vitro to gain insight into their functional roles and regulation. Four additional DmdB orthologs from R. lacuscaerulensis ITI-1157, P. aeruginosa PAO1, and B. thailandensis E264 were also evaluated for functional similarity to the “Ca. Pelagibacter ubique” HTCC1062 DmdB and R. pomeroyi DSS-3 isozymes. Phylogenetic analysis showed that these enzymes are part of a family which forms two distinct clades, with amino acid sequence similarity varying from 33% between members of different clades and up to 86% among members of the same clade (11). All enzymes catalyzed the MMPA-CoA ligase reaction with a high activity, but members of each clade varied greatly in other characteristics.
All seven enzymes possessed broad substrate specificities encompassing a range of short-chain fatty acids and MMPA. This verified that the members of both DmdB clades can catalyze the MMPA-CoA ligase reaction as predicted. However, the activity levels of the DmdB enzymes with fatty acids varied greatly, revealing no pattern between enzymes from the same clade. The same was true for the effect of salt ions on enzyme activity. This study emphasizes the broad-ranging activities and functional characteristics of the CoA ligase enzymes. Thus, the level of amino acid sequence similarity between two enzymes does not accurately predict many of the functional characteristics beyond the general range of substrates. For that reason, more isozymes from other bacteria were not characterized. This result is consistent with studies of CoA ligases from Burkholderia xenovorans and Geobacillus thermodenitrificans, which found that sequence and even structural similarity did not necessarily infer functional similarity (35, 36). In the case of the two benzoate-CoA ligases from B. xenovorans, the activities and affinities of the two enzymes varied even though the active sites in both enzymes were highly conserved and they shared 83% amino acid sequence identity overall (36). This was further demonstrated with the long-chain acyl-CoA ligase derived from rat liver, yeast, and E. coli. These CoA ligases have 48 to 51% sequence identity but vary in substrate specificities and sensitivity to inhibitors (37–39).
The DmdB enzymes investigated here appear to be adapted to the individual lifestyle of the bacterium regardless of the phylogenetic clade. For instance, PU_DmdB1 from the obligate oligotroph “Ca. Pelagibacter ubique” HTCC1062 possesses many features absent from other members of the DmdB1 clade. At 1.3 Mbp, this microorganism has one of the smallest genomes of any free-living bacterium and may be under extreme selective pressure to maximize its growth efficiency (24). This hypothesis is consistent with the properties of its DmdB. The high specific activity of the enzyme may minimize the amount of enzyme needed to maintain physiological levels of activity, while the broad substrate specificity may allow a single enzyme to perform multiple functions. This would also reduce the number of genes encoding this family of enzymes, consistent with the “Ca. Pelagibacter ubique” HTCC1062 genome encoding only five CoA ligase-like enzymes (24). In comparison, the opportunitroph R. pomeroyi DSS-3 has a genome of 4.1 Mb, plus a 0.4-Mb megaplasmid, and encodes 26 CoA ligases (19). Likewise, the high affinity of the PU_DmdB1 for substrates may allow the cell to lower the pool sizes of intermediates, lowering the difference between intracellular and extracellular concentrations to reduce the energy needed for transport and minimize leakage across the cytoplasmic membrane.
DMSP may be an important regulatory effector of the DmdB enzymes from marine bacteria. All of the enzymes from marine bacteria were sensitive to concentrations of DMSP well below the 70 mM DMSP found to accumulate in R. pomeroyi DSS-3 (12). In contrast, the two enzymes from terrestrial bacteria were not affected by DMSP. Neither of these bacteria possesses dmdA homologs, and they are unlikely to metabolize or encounter DMSP. For that reason, DMSP sensitivity appears to be a specific adaptation in marine bacteria. However, only R. pomeroyi DSS-3 is known to accumulate high intracellular concentrations of DMSP, and mechanisms to reverse DMSP inhibition were clearly identified only in the DmdBs from this bacterium. Thus, the physiological significance of DMSP inhibition in other marine bacteria is not certain at this time.
The presence of two DmdB isozymes in R. pomeroyi DSS-3 raises additional questions concerning their regulation. Indeed, the levels of both RPO_dmdB1 and RPO_dmdB2 transcripts increased during growth on DMSP even though the intracellular levels of DMSP would be expected to be strongly inhibitory for both enzymes. This suggests that there is a complex regulatory network for their activity, and a working model of DmdB regulation has been developed to illustrate how the properties of the DmdB isozymes might affect DMSP metabolism (Fig. 6).
FIG 6.
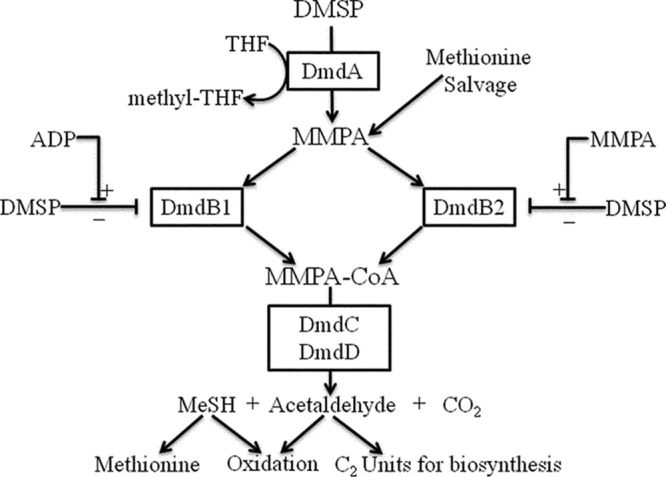
Model for the regulation of DmdB activity in Ruegeria pomeroyi DSS-3. During carbon and energy limitation, DmdB1 overcomes DMSP inhibition as ADP concentrations increase as a result of decreasing cellular energy charge. The resulting stimulation of the demethylation pathway increases the energy charge and lowers ADP concentrations until DMSP inhibition is restored. A different chain of events may explain the role of DmdB2. The levels of DmdA activity and MMPA are likely set by the availability of free THF. During rapid growth, methyl-THF is oxidized, yielding high intracellular levels of free THF and MMPA. The increased levels of MMPA reverse the DMSP inhibition of DmdB2 and lead to rapid production of acetaldehyde and MeSH. These additional energy sources spare the pools of methyl-THF, leading to a depletion of free THF and a decline in MMPA production. In addition, the oxidation of MeSH leads to H2O2 production, resulting in oxidative stress and slowing DMSP-dependent growth. The slowdown in growth reduces the demand for methyl-THF, causing a depletion of free THF. Alternatively, both enzymes may be active during growth on methionine when MMPA is produced in the absence of DMSP.
The central question is how cells can possess high levels of the enzymes in a catabolic pathway, such as DmdA and DmdB, when they also maintain very high levels of the substrate DMSP. RPO_DmdB1 increased activity in response to increasing ADP levels, indicating that the enzyme is sensitive to the energy charge of the cell. Energy charge is a measurement of the amount of metabolic energy contained in the adenylate pool, described by the ratio (ATP + 1/2 ADP)/(ATP + ADP + AMP) (40). During normal cellular growth, the majority of the adenylate pool is ATP, and an energy charge of about 0.9 is common (41). When the cell is energy and carbon limited, ADP and AMP accumulate, causing a decrease in energy charge (40). Documented cellular concentrations of ADP are between 0.8 and 3 mM in E. coli, within the range of ADP concentrations sufficient to reverse DMSP inhibition of RPO_DmdB1 activity (42, 43). In this model, as DMSP accumulates to high levels presumably for use as an osmolyte, it can also serve as a pool of readily available carbon and energy in the cell (1, 7, 12). Should other carbon and energy sources become depleted, the levels of ADP would increase, RPO_DmdB1 activity would increase, and MMPA (and DMSP) would be degraded as an energy source. This conclusion is supported by the increased level of RPO_dmdB1 transcripts during growth on DMSP but not on MMPA.
Based upon the high levels of transcripts during growth on DMSP, RPO_DmdB2 appears to be the major form of DmdB utilized during growth on DMSP. This conclusion is supported by kinetic analysis which confirms that RPO_DmdB2 has a higher specificity for MMPA than RPO_DmdB1. However, RPO_DmdB2 is expected to become active only when levels of MMPA are sufficient to relieve the inhibition caused by high intracellular concentrations of DMSP. Thus, MMPA appears to be a second important effector for the regulation of DMSP demethylation. Because DmdA activity is not directly affected by MMPA, presumably the DmdA reaction and the accumulation of MMPA are controlled by the availability of free THF and the turnover of methyl-THF (12). Methyl-THF is probably rapidly oxidized to provide electrons for respiration. This hypothesis is consistent with the increased levels of transcripts for the enzymes involved in methyl-THF oxidation observed during growth with DMSP and MMPA (unpublished data). Methyl-THF also provides C1 intermediates for biosynthesis of purine nucleotides and methionine (44, 45). In the case of DMSP demethylation, when methyl-THF is rapidly metabolized during growth on DMSP, free THF is available for DmdA, leading to increased levels of MMPA. As MMPA accumulates, RPO_DmdB2 becomes active, and MMPA metabolism could provide an additional source of electrons for respiration and carbon for biosynthesis. This could lower the demand for methyl-THF, decrease levels of free THF, and decrease the rate of MMPA formation. Thus, the rate of MMPA metabolism may feedback on the rate of DMSP consumption via the levels of free THF. In addition, MMPA metabolism leads to the formation of MeSH and acetaldehyde, which is toxic. MeSH oxidation also leads to the formation of hydrogen peroxide, which may cause oxidative stress and inhibit growth (46, 47). In this variation of the model, growth inhibition by oxidative stress and acetaldehyde toxicity limits methyl-THF metabolism, leading to a decrease in the levels of MMPA and lower RPO_DmdB2 activity, preventing the further metabolism of DMSP. Because DMSP is also a potent antioxidant, its accumulation would also be protective (2, 17).
The potential for DMSP metabolism to lead to oxidative stress may also explain the response of RPO_DmdB1 to ADP. In this model, DMSP is needed as a carbon and energy source when other sources are depleted but also to protect against oxidative and osmotic stress. Although speculative, this model illustrates the need for cells to balance the multiple demands for DMSP as an osmolyte and antioxidant during its metabolism with its value as a carbon and sulfur source. Because DMSP inhibition of the DmdBs from R. lacuscaerulensis ITI-1157 is also affected by the levels of MMPA, this may be a more general model for DMSP regulation in marine bacteria. However, it is not yet known if this bacterium accumulates high levels of DMSP, so this conclusion is less certain at this time.
While DMSP is likely the main source of MMPA in marine bacteria, the methionine salvage pathway may provide another source of MMPA (48, 49). During the transformation of 1,2-dihydroxy 3-oxomethylthiopentene, if the acireductone dioxygenase is bound to Ni2+ instead of Fe2+, MMPA is formed rather than 4-methylthio-2-oxobutyrate (48, 50). This pathway may also be the source of MMPA in nonmarine microorganisms, such as P. aeruginosa PAO1 and B. thailandensis E264. The physiological relevance of this off-pathway has not yet been demonstrated; however, Klebsiella pneumonia-derived acireductone dioxygenases bound to Fe2+ and Ni2+ have been purified and studied (51, 52). The role of MMPA in methionine salvage is supported by the expression data indicating that RPO_dmdB1 and RPO_dmdB2 are highly expressed during growth on methionine. Furthermore, during growth on methionine, R. pomeroyi DSS-3 produced MeSH (unpublished observation), suggesting a link between the methionine salvage and demethylation pathways that still needs to be investigated.
In conclusion, DmdB appears to be a major regulatory point in DMSP metabolism by R. pomeroyi DSS-3. However, regulation of DmdB activity by itself is not sufficient to explain what is likely to be a complex system balancing the cellular requirements for carbon and sulfur for growth, maintaining the cellular osmotic balance, and protection from oxidative stress. Further study will be necessary to fully understand all of the regulatory aspects of DMSP demethylation and the enzymes involved. Nevertheless, the work here provides valuable insight into the regulation of the DmdB isozymes and expands our knowledge of this multifaceted regulatory system.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the National Science Foundation (grant MCB-1158037) for providing the funding for this project.
Footnotes
Published ahead of print 17 January 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.00026-14.
REFERENCES
- 1.Stefels J. 2000. Physiological aspects of the production and conversion of DMSP in marine algae and higher plants. J. Sea Res. 43:183–197. 10.1016/S1385-1101(00)00030-7 [DOI] [Google Scholar]
- 2.Sunda W, Kieber DJ, Kiene RP, Huntsman S. 2002. An antioxidant function for DMSP and DMS in marine algae. Nature 418:317–320. 10.1038/nature00851 [DOI] [PubMed] [Google Scholar]
- 3.Steinke M, Wolfe GV, Kirst GO. 1998. Partial characterization of dimethylsulfoniopropionate (DMSP) lyase isozymes in 6 strains of Emiliania huxleyi. Mar. Ecol. Prog. Ser. 175:215–225. 10.3354/meps175215 [DOI] [Google Scholar]
- 4.Reisch CR, Moran MA, Whitman WB. 2011. Bacterial catabolism of dimethylsulfoniopropionate (DMSP). Front. Microbiol. 2:172. 10.3389/fmicb.2011.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simo R, Pedros-Alio C, Malin G, Grimalt JO. 2000. Biological turnover of DMS, DMSP and DMSO in contrasting open-sea waters. Mar. Ecol. Prog. Ser. 203:1–11. 10.3354/meps203001 [DOI] [Google Scholar]
- 6.Simo R, Archer SD, Pedros-Alio C, Gilpin L, Stelfox-Widdicombe CE. 2002. Coupled dynamics of dimethylsulfoniopropionate and dimethylsulfide cycling and the microbial food web in surface waters of the North Atlantic. Limnol. Oceanogr. 47:53–61. 10.4319/lo.2002.47.1.0053 [DOI] [Google Scholar]
- 7.Kiene RP, Linn LJ, Bruton JA. 2000. New and important roles for DMSP in marine microbial communities. J. Sea Res. 43:209–224. 10.1016/S1385-1101(00)00023-X [DOI] [Google Scholar]
- 8.Andreae MO. 1990. Ocean-atmosphere interactions in the global biogeochemical sulfur cycle. Mar. Chem. 30:1–29. 10.1016/0304-4203(90)90059-L [DOI] [Google Scholar]
- 9.Bruhl C, Lelieveld J, Crutzen PJ, Tost H. 2012. The role of carbonyl sulphide as a source of stratospheric sulphate aerosol and its impact on climate. Atmos. Chem. Phys. 12:1239–1253. 10.5194/acp-12-1239-2012 [DOI] [Google Scholar]
- 10.Charlson RJ, Lovelock JE, Andreae MO, Warren SG. 1987. Oceanic phytoplankton, atmospheric sulfur, cloud albedo and climate. Nature 326:655–661. 10.1038/326655a0 [DOI] [Google Scholar]
- 11.Reisch CR, Stoudemayer MJ, Varaljay VA, Amster IJ, Moran MA, Whitman WB. 2011. Novel pathway for assimilation of dimethylsulphoniopropionate widespread in marine bacteria. Nature 473:208–211. 10.1038/nature10078 [DOI] [PubMed] [Google Scholar]
- 12.Reisch CR, Moran MA, Whitman WB. 2008. Dimethylsulfoniopropionate-dependent demethylase (DmdA) from Pelagibacter ubique and Silicibacter pomeroyi. J. Bacteriol. 190:8018–8024. 10.1128/JB.00770-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gonzalez JM, Kiene RP, Moran MA. 1999. Transformation of sulfur compounds by an abundant lineage of marine bacteria in the alpha-subclass of the class Proteobacteria. Appl. Environ. Microbiol. 65:3810–3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howard EC, Henriksen JR, Buchan A, Reisch CR, Burgmann H, Welsh R, Ye W, Gonzalez JM, Mace K, Joye SB, Kiene RP, Whitman WB, Moran MA. 2006. Bacterial taxa that limit sulfur flux from the ocean. Science 314:649–652. 10.1126/science.1130657 [DOI] [PubMed] [Google Scholar]
- 15.Kiene RP, Linn LJ, Gonzalez J, Moran MA, Bruton JA. 1999. Dimethylsulfoniopropionate and methanethiol are important precursors of methionine and protein-sulfur in marine bacterioplankton. Appl. Environ. Microbiol. 65:4549–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kiene RP, Linn LJ. 2000. The fate of dissolved dimethylsulfoniopropionate (DMSP) in seawater: tracer studies using 35S-DMSP. Geochim. Cosmochim. Acta 64:2797–2810. 10.1016/S0016-7037(00)00399-9 [DOI] [Google Scholar]
- 17.Lesser MP. 2006. Oxidative stress in marine environments: biochemistry and physiological ecology. Annu. Rev. Physiol. 68:253–278. 10.1146/annurev.physiol.68.040104.110001 [DOI] [PubMed] [Google Scholar]
- 18.Chambers ST, Kunin CM, Miller D, Hamada A. 1987. Dimethylthetin can substitute for glycine betaine as an osmoprotectant molecule for Escherichia coli. J. Bacteriol. 169:4845–4847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moran MA, Buchan A, Gonzalez JM, Heidelberg JF, Whitman WB, Kiene RP, Henriksen JR, King GM, Belas R, Fuqua C, Brinkac L, Lewis M, Johri S, Weaver B, Pai G, Eisen JA, Rahe E, Sheldon WM, Ye WY, Miller TR, Carlton J, Rasko DA, Paulsen IT, Ren QH, Daugherty SC, Deboy RT, Dodson RJ, Durkin AS, Madupu R, Nelson WC, Sullivan SA, Rosovitz MJ, Haft DH, Selengut J, Ward N. 2004. Genome sequence of Silicibacter pomeroyi reveals adaptations to the marine environment. Nature 432:910–913. 10.1038/nature03170 [DOI] [PubMed] [Google Scholar]
- 20.Yi H, Lim YW, Chun J. 2007. Taxonomic evaluation of the genera Ruegeria and Silicibacter: a proposal to transfer the genus Silicibacter Petursdottir and Kristjansson 1999 to the genus Ruegeria Uchino et al. Int. J. Syst. Evol. Microbiol. 57:815–819 [DOI] [PubMed] [Google Scholar]
- 21.Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FSL, Hufnagle WO, Kowalik DJ, Lagrou M, Garber RL, Goltry L, Tolentino E, Westbrock-Wadman S, Yuan Y, Brody LL, Coulter SN, Folger KR, Kas A, Larbig K, Lim R, Smith K, Spencer D, Wong GKS, Wu Z, Paulsen IT, Reizer J, Saier MH, Hancock REW, Lory S, Olson MV. 2000. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406:959–964. 10.1038/35023079 [DOI] [PubMed] [Google Scholar]
- 22.Zinser BA, Ferriera SF, Johnson JJ, Kravitz SK, Beeson KB, Sutton GS, Roger YHR, Friedman RF, Frazier MF, Venter JC. 2008. Silicibacter lacuscaerulensis ITI-1157 genome sequence. EMBL/GenBank/DDBJ databases. The J. Craig Venter Institute, Rockville, MD [Google Scholar]
- 23.Kim HS, Schell MA, Yu Y, Ulrich RL, Sarria SH, Nierman WC, DeShazer D. 2005. Bacterial genome adaptation to niches: divergence of the potential virulence genes in three Burkholderia species of different survival strategies. BMC Genomics 6:174. 10.1186/1471-2164-6-174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giovannoni SJ, Tripp HJ, Givan S, Podar M, Vergin KL, Baptista D, Bibbs L, Eads J, Richardson TH, Noordewier M, Rappe MS, Short JM, Carrington JC, Mathur EJ. 2005. Genome streamlining in a cosmopolitan oceanic bacterium. Science 309:1242–1245. 10.1126/science.1114057 [DOI] [PubMed] [Google Scholar]
- 25.Shimizu S, Inoue K, Tani Y, Yamada H. 1979. Enzymatic microdetermination of serum free fatty acids. Anal. Biochem. 98:341–345. 10.1016/0003-2697(79)90151-9 [DOI] [PubMed] [Google Scholar]
- 26.Bergmeyer H, Hagen A. 1972. New principle of enzymatic analysis. Fresenius J. Anal. Chem. 261:333–336. 10.1007/BF00786990 [DOI] [Google Scholar]
- 27.Gonzalez JM, Mayer F, Moran MA, Hodson RE, Whitman WB. 1997. Microbulbifer hydrolyticus gen nov, sp nov, and Marinobacterium georgiense gen nov, sp nov, two marine bacteria from a lignin-rich pulp mill waste enrichment community. Int. J. Syst. Bacteriol. 47:369–376. 10.1099/00207713-47-2-369 [DOI] [PubMed] [Google Scholar]
- 28.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31:46–53. 10.1038/nbt.2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumari S, Tishel R, Eisenbach M, Wolfe AJ. 1995. Cloning, characterization, and functional expression of acs, the gene which encodes acetyl-coenzyme A synthetase in Escherichia coli. J. Bacteriol. 177:2878–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watkins PA, Lu JF, Steinberg SJ, Gould SJ, Smith KD, Braiterman LT. 1998. Disruption of the Saccharomyces cerevisiae FAT1 gene decreases very long-chain fatty acyl-CoA synthetase activity and elevates intracellular very long-chain fatty acid concentrations. J. Biol. Chem. 273:18210–18219. 10.1074/jbc.273.29.18210 [DOI] [PubMed] [Google Scholar]
- 32.Chohnan S, Furukawa H, Fujio T, Nishihara H, Takamura Y. 1997. Changes in the size and composition of intracellular pools of nonesterified coenzyme A and coenzyme A thioesters in aerobic and facultatively anaerobic bacteria. Appl. Environ. Microbiol. 63:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackowski S, Rock C. 1986. Consequences of reduced intracellular coenzyme A content in Escherichia coli. J. Bacteriol. 166:866–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckstein MH, He J, Rubin H. 2008. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J. Bacteriol. 190:718–726. 10.1128/JB.01020-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dong YP, Du HQ, Gao CX, Ma T, Feng L. 2012. Characterization of two long-chain fatty acid CoA ligases in the Gram-positive bacterium Geobacillus thermodenitrificans NG80-2. Microbiol. Res. 167:602–607. 10.1016/j.micres.2012.05.001 [DOI] [PubMed] [Google Scholar]
- 36.Bains J, Boulanger MJ. 2007. Biochemical and structural characterization of the paralogous benzoate CoA ligases from Burkholderia xenovorans LB400: defining the entry point into the novel benzoate oxidation (box) pathway. J. Mol. Biol. 373:965–977. 10.1016/j.jmb.2007.08.008 [DOI] [PubMed] [Google Scholar]
- 37.Black PN, Zhang Q, Weimar JD, DiRusso CC. 1997. Mutational analysis of a fatty acyl-coenzyme A synthetase signature motif identifies seven amino acid residues that modulate fatty acid substrate specificity. J. Biol. Chem. 272:4896–4903. 10.1074/jbc.272.8.4896 [DOI] [PubMed] [Google Scholar]
- 38.Black PN, Dirusso CC, Metzger AK, Heimert TL. 1992. Cloning, sequencing, and expression of the fadD gene of Escherichia coli encoding acyl coenzyme A synthetase. J. Biol. Chem. 267:25513–25520 [PubMed] [Google Scholar]
- 39.Caviglia JM, Li LO, Wang SL, DiRusso CC, Coleman RA, Lewin TM. 2004. Rat long chain acyl-CoA synthetase 5, but not 1, 2, 3, or 4, complements Escherichia coli fadD. J. Biol. Chem. 279:11163–11169. 10.1074/jbc.M311392200 [DOI] [PubMed] [Google Scholar]
- 40.Atkinson DE. 1968. Energy charge of adenylate pool as a regulatory parameter. interaction with feedback modifiers. Biochemistry 7:4030–4034. 10.1021/bi00851a033 [DOI] [PubMed] [Google Scholar]
- 41.Bennett BD, Kimball EH, Gao M, Osterhout R, Van Dien SJ, Rabinowitz JD. 2009. Absolute metabolite concentrations and implied enzyme active site occupancy in Escherichia coli. Nat. Chem. Biol. 5:593–599. 10.1038/nchembio.186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albe KR, Butler MH, Wright BE. 1990. Cellular concentrations of enzymes and their substrates. J. Theor. Biol. 143:163–195. 10.1016/S0022-5193(05)80266-8 [DOI] [PubMed] [Google Scholar]
- 43.Henry CS, Broadbelt LJ, Hatzimanikatis V. 2007. Thermodynamics-based metabolic flux analysis. Biophys. J. 92:1792–1805. 10.1529/biophysj.106.093138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagy PL, Marolewski A, Benkovic SJ, Zalkin H. 1995. Formyltetrahydrofolate hydrolase, a regulatory enzyme that functions to balance pools of tetrahydrofolate and one-carbon tetrahydrofolate adducts in Escherichia coli. J. Bacteriol. 177:1292–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang EY, Mohler AM, Rohlman CE. 1997. Protein expression in response to folate stress in Escherichia coli. J. Bacteriol. 179:5648–5653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim JS, Kim YC, Lee DS, Yang JW. 2000. Isolation and purification of methyl mercaptan oxidase from Rhodococcus rhodochrous for mercaptan detection. Biotechnol. Bioprocess Eng. 5:465–468. 10.1007/BF02931949 [DOI] [Google Scholar]
- 47.Gould WD, Kanagawa T. 1992. Purification and properties of methyl mercaptan oxidase from Thiobacillus thioparus TK-M. J. Gen. Microbiol. 138:217–221. 10.1099/00221287-138-1-217 [DOI] [Google Scholar]
- 48.Albers E. 2009. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB Life 61:1132–1142. 10.1002/iub.278 [DOI] [PubMed] [Google Scholar]
- 49.Sekowska A, Ashida V DH, Michoud K, Haas D, Yokota A, Danchin A. 2004. Bacterial variations on the methionine salvage pathway. BMC Microbiol. 4:9. 10.1186/1471-2180-4-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dai Y, Wensink PC, Abeles RH. 1999. One protein, two enzymes. J. Biol. Chem. 274:1193–1195. 10.1074/jbc.274.3.1193 [DOI] [PubMed] [Google Scholar]
- 51.Dai Y, Pochapsky TC, Abeles RH. 2001. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry 40:6379–6387. 10.1021/bi010110y [DOI] [PubMed] [Google Scholar]
- 52.Al-Mjeni F, Ju T, Pochapsky TC, Maroney MJ. 2002. XAS investigation of the structure and function of Ni in acireductone dioxygenase. Biochemistry 41:6761–6769. 10.1021/bi012209a [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.