

Short abstract
Many non-coding sequences transcribed from the mammalian genome are proving to have important regulatory roles, but the functions of the majority remain mysterious.
Abstract
For decades, researchers have focused most of their attention on protein-coding genes and proteins. With the completion of the human and mouse genomes and the accumulation of data on the mammalian transcriptome, the focus now shifts to non-coding DNA sequences, RNA-coding genes and their transcripts. Many non-coding transcribed sequences are proving to have important regulatory roles, but the functions of the majority remain mysterious.
Initial understanding of the profound differences between the mammalian proteome and the underlying transcriptome emerged in the 1970s, with the discovery of RNA splicing and of the complex intron-exon structure of primary RNA transcripts. The importance of non-coding DNA was not readily accepted by the scientific community at the time of the discovery of the seemingly wasteful mechanisms of RNA processing, whereby most of the primary transcript is edited out of the mature messenger RNA [1]. As the biological function of non-protein-coding DNA sequences was not understood, the term 'junk DNA', was coined and applied to most of the mammalian genome. The historic achievement of sequencing and annotating the complete human genome has revealed the complex landscape of mammalian non-coding DNA [2]. The subsequent sequencing of the complete genome of the mouse has not only provided a genetic platform for biomedical studies on this model mammal, but also promoted better understanding of the human genome through detailed comparative analysis [3].
Large-scale sequencing and initial analysis of mouse and human cDNA libraries has provided the first in-depth look into the mammalian transcriptome [4-6]. An assembly of the rat genome is also now available online [7]. These accomplishments allow researchers to address some unanswered questions using genome-wide comparisons. How many genes - separately regulated transcriptional units, encoding distinct transcripts - are there in the mammalian genome, and what is the proportion of the protein-coding and non-coding genes? What part of the mammalian genome is transcribed? What is the function of non-coding RNA transcripts and non-coding DNA regions? And what structural elements in the genomes of mammals are responsible for the increased complexity of mammals relative to other organisms?
The transcribed part of the mammalian genome
Early estimations of the level of transcription in mammals were based on the hybridization of primary nuclear transcripts to genomic DNA. The major part of the mammalian genome was found to be expressed as nuclear transcripts, from one strand or the other. Hybridization experiments demonstrated that, in rat embryos, primary nuclear transcripts contained both unique and moderately repetitive sequences transcribed from 32.8% and 32.9% of genomic DNA, respectively [8]. The most transcriptionally active rat tissue is the adult brain, where transcribed unique and moderately repetitive DNA represent 46.6% and 13.7% of the whole genome, respectively [8]. Similar results were obtained for mouse brain tissues, where 42% of the genome represented by unique sequences was found to be transcribed [9].
Maximal transcription levels are difficult to measure with hybridization experiments because not all genes may be expressed under particular physiological conditions, and also because of difficulties in the isolation of rare transcripts. Experimental determination of the transcribed part of the well-annotated genomes of Escherichia coli (73% [10]) and Saccharomyces cerevisiae (40% [11]) yielded smaller numbers than calculations based on genomic annotation for the same species (88.6% [12] and 78% [13], respectively; see Figure 1). According to a recent detailed analysis of the length of sequence occupied by the annotated genes on several chromosomes in the human genome, primary transcripts cover 42.2%, 46.5%, 43.6%, 42.4% and 51% of chromosomes 6, 7,14, 20 and 22, respectively (reviewed in [14]). But these numbers do not represent the full transcriptional potential of the human genome.
Figure 1.
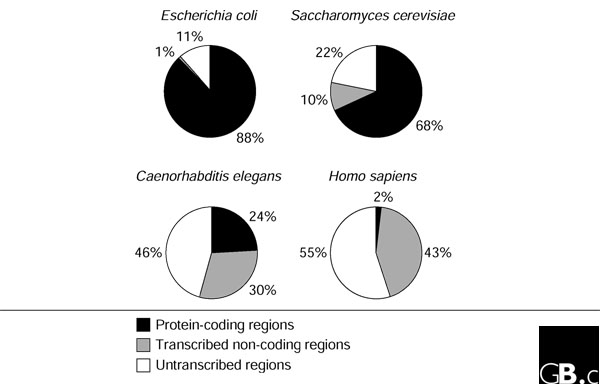
Ratios of the protein-coding, non-coding, and untranscribed sequences in bacterial, yeast, nematode and mammalian genomes. Estimations of the transcribed and protein-coding parts of genomes are based on the sequence length of annotated genes [3,12,13,73]. Estimation of the transcribed portion of the human genome is based on the sequence length occupied by the annotated genes on chromosomes 6, 7, 14, 20, and 22 [5].
The annotation of the human genome mostly comprises data on identified protein-coding genes, while a substantial part of the transcriptome has not yet been identified and annotated. Whole-chromosome analysis with oligonucleotide arrays has demonstrated that the level of transcription from human chromosomes 21 and 22 is significantly higher than can be accounted for by known or predicted sequence annotations [15]. The unmapped part of the mammalian transcriptome may contain numerous non-protein-coding genes, as evidenced by the high proportion of non-protein-coding transcripts in human and mouse cDNA libraries [4,6]. Estimations of the relative complexity of heterogeneous nuclear (hn) RNA versus mature mRNA, based on analysis of the kinetics of hybridization, suggest that non-protein-coding transcripts could represent half, or more, of all transcriptional output from the genomes of eukaryotic organisms [16,17]. We might expect that in mammals about half of the genome is transcribed.
The question of how many genes there are in the mammalian genome remains open. Pregenomic estimates of the number of human genes ranged between 30,000 and 120,000 [18-20]. Recent analysis of the mouse transcriptome on the basis of annotation of full-length cDNA collections enabled identification of 33,409 unique full-length transcripts, with an estimated total number of independent transcriptional units in the mouse genome of around 70,000 [4]. Large-scale annotation of the human genome with the UniGene assembly of individual expressed sequence tags (ESTs) and cDNAs revealed 59,500 nonredundant clusters representing putative transcriptional units [21,22]. Thus, the total number of genes in the mammalian transcriptome could be as high as 60,000-70,000.
Protein-coding genes and their untranslated regions
A detailed inventory of the protein-coding genes was made upon the completion of the human and mouse genome projects [3]. Overall, the mouse proteome is similar to that of the human, and about 99% of the mouse protein-coding genes have a homolog in the human genome [3]. The number of protein-coding genes in the mammalian genome was calculated on the basis of known cDNAs and genes predicted by similarity to protein-coding genes in other organisms, and was extended by computer predictions that are supported by experimental evidence such as ESTs. Catalogs of human and mouse protein-coding genes contain slightly more than 22,000 genes for each species [3]. Recent large-scale sequencing and analysis of the large Japanese collection of human cDNA clones added around 2,000 more new sequences to the human protein catalog [6]. Current approaches to gene identification are likely to miss a substantial number of small genes, such as those encoding neuropeptides, antimicrobial peptides, and small adaptor and regulatory proteins. Taking into account the small genes that have yet to be discovered, the total number of protein-coding genes in the mammalian genome is estimated to be around 30,000 [3]. This upper estimate is still surprisingly close to the number of protein-coding genes in the nematode genome (Table 1). The average size of mammalian protein-coding genes far exceeds the average size of the nematode and yeast protein-coding genes, however, mostly on account of the increased length of introns.
Table 1.
General features of bacterial, yeast, nematode and mammalian genomes
| Species | Genome size (Mbp) | Repetitive sequences (%) | Transcriptional units | Protein-coding genes | Introns | References |
| Escherichia coli | 4.6 | 0.7 | 5,471 | 4,288 | [12,74] | |
| Saccharomyces cerevisiae | 12 | 3.2 | 6,682 | 6,183 | 233 | [13] |
| Caenorhabditis elegans | 100.3 | 16.5 | 19,646 | 18,808 | 99,237 | [73] |
| Caenorhabditis briggsae | 104 | 22.4 | 20,469 | 19,507 | 94,832 | [73] |
| Mus musculus | 2,500 | 40 | 33,409 | 22,011 | 191,500 | [3,4] |
| Homo sapiens | 2,900 | 44 | 25,003 | 22,808 | 177,000 | [3,5,14] |
The 5' and 3' untranslated regions
Gene expression in eukaryotic organisms is tightly controlled at various levels, and critical cis-regulatory elements for posttranscriptional control are encoded in the 5' and 3' untranslated regions (UTRs). On average, 5' and 3' UTRs are less conserved than protein-coding sequences across species, but more conserved than untranscribed sequences [23,24]. Highly conserved nucleotide blocks have been detected in 5' UTRs and, especially, in the 3' UTRs of orthologous genes from different mammalian orders, and even between mammals and birds or fish [25,26]. For some genes, the conservation of UTRs exceeds the conservation of the corresponding coding regions [27]. Many conserved sequence elements in UTRs have been identified as binding sites for proteins or antisense RNAs, which contribute to the regulation of nucleocytoplasmic transport, subcellular localization, translation and the stability of mRNAs [28-31]. The nucleotide context around the principal functional signals, such as start and stop codons, is also an important determinant of expression level [32,33].
According to the current, scanning model of translation initiation, the eukaryotic ribosome binds to the 5'-terminal cap of an mRNA and starts scanning the mRNA until it detects the first AUG start codon, where it initiates translation [34,35]. The 5' UTRs contain binding sites for components of multiprotein transcription complexes and also participate in the recruitment of the 40S ribosomal subunit and translation initiation. The length of 5' UTRs, and the presence of additional upstream transcription start codons, may be important for regulating the basal translation level of an mRNA, It has been shown that transcripts with an optimal start codon context tend to have shorter 5' UTRs, whereas an increased length of 5' UTR correlates with a 'weak' start codon context and with the presence of additional upstream start codons [36]. A reduced level of basal translation also correlates with the presence of minor open reading frames located within 5' UTRs and upstream of the main start codon in some genes. Other sequence elements within 5' UTRs act as internal ribosome entry sites (IRESs); these elements have been found in many cellular mRNAs encoding regulatory proteins [28].
It is widely accepted that 3' UTRs play crucial roles in transcript cleavage, polyadenylation and nuclear export, and in regulating the level of transcription and the stability of transcripts. The 3' UTRs may contain sequence elements that mediate negative posttranscriptional regulation. Increasing numbers of publications describe suppression of mRNA translation by small RNA molecules through base-pairing interactions with complementary sequence motifs within 3' UTRs [37]. It has also been shown that the turnover of mRNA is regulated by cis-acting AU-rich elements that promote mRNA degradation, and such motifs are found in the conserved 3' UTRs of many mRNAs encoding regulatory proteins [38].
In addition to motifs that have a negative effect on translation, 3' UTRs carry binding sites for factors involved in translation termination and the release of the synthesized polypeptide, processes that are understood much less thoroughly than the initiation of translation [39]. Binding of regulatory proteins to cis-acting elements within a 3' UTR can be either sequence-specific or facilitated by stem-loop structural elements formed within the mRNA. The importance of the secondary structure of the 3' UTR is exemplified by the family of selenoprotein mRNAs. All mammalian selenoproteins identified so far contain a selenocysteine residue encoded by the stop codon UGA. Incorporation of selenocysteine into the growing polypeptide depends on a conserved stem-loop structure within the mRNA formed by the selenocysteine insertion sequence (SECIS), which is necessary for decoding UGA as selenocysteine rather than as a stop signal [40].
Genomic regions corresponding to the UTRs of mRNAs may contain introns, which leads to the formation of alternative UTRs. Introns are more frequently found in 5' UTRs, although 3' UTRs are generally much longer than 5' UTRs. Alternative UTRs can be formed by the use of different transcription start sites, different donor/acceptor splice sites, and different polyadenylation sites. These have been shown to vary with the tissue and the stage of development, and can significantly affect patterns of gene expression [28,41].
Introns
The origin of eukaryotic introns is the subject of much debate. One hypothesis argues that modern nuclear introns are evolutionary descendants of bacterial self-catalytic introns that penetrated into the eukaryotic lineage and gained biological function in eukaryotes in the process of co-evolution with their hosts through their involvement in the splicing of primary RNA transcripts. An alternative notion is that the vast majority of introns arose within multicellular eukaryotes and were randomly inserted into eukaryotic genes (reviewed in [42,43]). Introns, which are few in unicellular eukaryotes, are greatly increased in numbers and size within the genomes of higher eukaryotes (Table 1). Nematodes contain more DNA in introns than in exons, while in mammalian genomes introns comprise about 95% of the sequence within protein-coding genes [2,3]. Interspecies sequence conservation studies have demonstrated that introns are generally high in sequence complexity, although they are less conserved than protein-coding sequences; introns contain blocks of conserved sequences and a significant number of selectively constrained nucleotides that remain invariant as a result of stabilizing selection. Genomic sequencing of different taxa has allowed large-scale analysis of homologous intron sequences between related species, such as between Caenorhabditis species or Drosophila species, or between human and mouse or rat, and human and whale or seal [44-51]. Using different alignment methods, these studies estimate that the level of selective constraint in introns is between 5% and 28%, as compared to around 60-70% in exons.
One established biological role for introns is their involvement in nucleosome formation and chromatin organization. Introns have higher potential for nucleosome formation than exons or Alu repeats [52]. Other functional elements identified in mammalian introns are scaffold/matrix-attachment regions (S/MARs), which are thought to anchor chromatin loops to the nuclear matrix and to chromosome scaffolds [53,54]. These elements account for only a small proportion of constrained nucleotides in introns, however.
Alternative splicing is an important source of proteome complexity in higher eukaryotes; it amplifies the number of proteins encoded by a single gene by generating isoforms differing in amino-acid sequence. Nevertheless, the dominance of intronic sequences in the protein-coding genes of higher organisms cannot be fully explained by their role in alternative splicing. Although the vast majority of human and mouse protein-coding genes have introns, only about 40% of them show evidence of alternative splicing [4,55]. As a rule, internal introns within protein-coding regions are not involved in alternative splicing, unlike those in UTRs, and splicing signals located at intron-exon boundaries are relatively short. The significant levels of nucleotide conservation within introns suggest that introns may have other important functional roles, probably at the RNA level. It has been suggested that the products of intron degradation generated during splicing of pre-mRNA transcripts serve as endogenous control molecules of an RNA-based gene-regulatory network [16,17]; but to date, no experimental data confirm or disprove this idea.
In modern eukaryotes, the transcription and processing of mRNA are highly coupled with intron splicing and/or exon recognition. There is an obvious correlation between the number and total length of introns on the one hand and the developmental complexity of organisms on the other, although the reasons for the abundance of intron sequences and their functions in higher organisms are not fully understood. The notion that introns are involved in complex regulation and development in higher eukaryotes is supported by several lines of evidence. For example, there is a negative correlation between the size of introns and the level of transcription of protein-coding genes. Furthermore, introns in highly expressed genes are substantially shorter than those in genes that are expressed at lower levels. This difference is greater in humans, where introns are, on average, 14 times shorter in highly expressed genes than in genes with low expression [5,56].
The intron sequences of mammalian protein-coding genes have also been shown to harbor independent transcriptional units, such as small RNA genes [57] and repetitive elements [3]. Repeats constitute about 45% of the human and the mouse genomes (Table 1) and can be found in both transcribed (introns and UTRs) and non-transcribed intergenic sequences. It is not obvious whether the proliferation of transposable repetitive elements in mammalian genomes is associated with some biological advantage. There are notable similarities in the genomic distribution of the major repetitive elements, LINEs (long interspersed nucleotide elements) and SINEs (short interspersed nucleotide elements), in the human and mouse genomes. Genome-wide profiling of human gene expression has revealed that SINE elements are mostly associated with highly expressed short-intron genes, while LINE elements are associated with weakly expressed long-intron genes [5]. Furthermore, similar repeats accumulate in orthologous locations in the human and mouse genomes [3,58].
The expanding world of non-coding RNA genes
Transcripts from non-coding RNA (ncRNA) genes are not translated into proteins and function directly as structural, regulatory or catalytic molecules. It is not clear how many ncRNA genes are present in the mammalian genome. The existing catalog of mammalian genes is strongly biased towards protein-coding genes, because most efforts were made in cloning and sequencing polyadenylated mRNAs, which tend not to be ncRNAs. Analysis of 33,409 full-length mouse cDNAs showed that ncRNA constitutes more than one third of all the identified transcripts [4]. Recently Ota et al. [6] reported the sequencing and characterization of 10,897 novel human full-length cDNA clones, and ncRNAs represent about half of these newly identified transcripts. Nevertheless, it is not known how many real RNA genes have been cloned, and how many clones in fact represent transcriptional artifacts. Surprisingly, a large proportion of ncRNA transcripts have introns, and many ncRNAs demonstrate distinct patterns of splicing [6]. The presence of introns in ncRNAs adds possibilities for regulation, given that the primary transcript might be functionally inactive, with subsequent cleavage and splicing being required to produce an active RNA molecule. Novel ncRNA genes are difficult to recognize and identify on the basis of sequence, and their discovery still depends largely on experimental approaches. The nature of ncRNA genes, which are often small and multicopy, lacking open reading frames and immune to point mutations, makes them difficult targets for genetic screens. Current estimates of the number of independent transcriptional units (around 70,000) and protein-coding genes (around 30,000) in the mouse transcriptome suggest that ncRNA genes may be highly abundant in the mouse genome [4].
Our understanding of the cellular function of ncRNAs has expanded far beyond the initial notion of their being intermediates and accessories in protein biosynthesis. The size of ncRNA molecules ranges from 20 nucleotides (microRNAs) to thousands of nucleotides (ncRNAs involved in gene silencing) [59]. Furthermore, ncRNAs are involved in many processes, including transcriptional and posttranscriptional regulation, chromosome replication, genomic imprinting, RNA processing, modification and alternative splicing, mRNA stability and translation, and even protein degradation and translocation [59-62]. Within the genome, ncRNA genes are found in extended stretches of conservation within orthologous regions of related genomes in intergenic and intronic sequences that have elevated GC content. Important noncanonical RNA species include families of translational repressors, such as microRNAs and small temporary RNAs (stRNAs) that inhibit translation of target mRNAs, small nuclear RNAs (snRNAs) that function as components of spliceosomes, and small nucleolar RNAs (snoRNAs) that are involved in the chemical modification of structural RNAs. Another important class of ncRNA molecules comprises those with catalytic activity, such as ribonuclease P. The functional importance of ncRNA genes is emphasized by the recent discoveries that link human genetic disorders with non-protein-coding genes [63,64].
The inhibition and silencing of genes by RNA molecules exploits the highly specific complementarity of nucleic acid interactions. There are two types of naturally occurring regulatory ncRNAs. First, cis-antisense transcripts originate from the same genomic region as the target gene, but have the opposite orientation, and can form long perfect duplexes with their targets; such cis-antisense transcripts may be expressed from imprinted regions of vertebrate chromosomes and play roles in chromatin structure. Second, trans-antisense RNAs are short molecules that are transcribed from loci distinct from their mRNA targets and form imperfect duplexes with complementary regions within their targets; examples of trans-antisense RNAs are microRNAs and small interfering (si) RNAs [59,62,65]. It seems that the increased complexity of gene expression and regulation in higher organisms has promoted the increased use (during evolution) of modular systems, whereby substrate recognition is delegated to diverse small RNA molecules that share a common protein catalytic subunit to exert their effects. An example of such a mechanism is the site-specific methylation of structural RNAs (rRNAs, tRNAs and snRNAs), in which numerous different snoRNAs provide specificity for methylation and pseudouridylation of target bases on the structural RNAs by complementarity, while catalytic activity is conferred by a protein methylase or pseudo-U synthetase associated with the snoRNA [61].
Another class of sequence elements that contributes to the mammalian transcriptome comprises Alu and other repeats. The observed evolutionary selection against change in Alu repeat sequences in the human genome has led to the hypothesis that they are functionally important. In a few cases, Alu elements have been shown to serve as regulators of transcription of adjacent genes [66] and in nucleosome positioning within chromatin [67]. More recent studies indicate that Alu repeats serve as templates for non-coding RNAs that can be involved in the regulation of gene activity and posttranscriptional gene silencing through repression of expression of other genes that contain similar repeats. A strong increase in the level of Alu transcripts in the cell is observed under stress conditions and after viral infection [61].
Non-coding sequences and the complexity of organisms
The function of non-coding DNA remains poorly studied, and interspecies comparison is often the only way to demonstrate that a conserved DNA sequence, which has evolved slowly as a result of negative selection, is functionally important. In general, non-coding regions are less conserved than the protein-coding parts of genes. Comparative analysis of orthologous non-coding regions in the genomes of higher eukaryotes has revealed a mosaic structure of alternating highly conserved and dissimilar segments. Conserved elements, the so-called phylogenetic footprints, constitute a significant proportion of non-coding DNA. Comparative analysis of the human and mouse genomes has demonstrated that about 5% of the genomic sequence consists of highly conserved segments of 50-100 base-pairs (bp); this proportion is much higher than can be explained by protein-coding sequences alone [3]. But this analysis of relatively long conserved segments did not take into account numerous shorter and weaker homologous elements of genomic DNA. The average selective constraint within mouse and human intergenic regions (15-19%) does not differ substantionally from the number of constrained nucleotides in the introns and intergenic regions of nematodes (18%) [44,68]. The average number of constrained nucleotides within a mammalian intergenic region is at least 2,000, which is twice as many as in an average protein-coding region. Some of the short conserved sequences in mammalian intergenic regions represent binding sites for known transcription factors and regulatory proteins, while others have no known biological function [69]. Needless to say, comparative inter-species analysis is not helpful in the detection of species-specific functional sequence elements. Some authors estimate that as much as one third of the human genome (about a billion base pairs) could be involved in cis-regulatory functions, such as the regulation of gene expression and the control of chromosomal replication, condensation, pairing and segregation [70].
The fraction of protein-coding DNA in the genome decreases with increasing organismal complexity. In bacteria, about 90% of the genome codes for proteins. This number drops off to 68% in yeast, to 23-24% in nematodes and to 1.5-2% in mammals (Table 1). Among the different mechanisms for increasing protein diversity (such as the use of multiple transcription start sites, alternative pre-mRNA splicing and polyadenylation, pre-mRNA editing, and posttranslational protein modification) alternative splicing is considered to be the most important source of protein diversity in mammals [71]. But this view was challenged when no significant difference in the level of alternative splicing was found in mammals as compared to other phyla, such as insects and nematodes [55]. Also, only a fraction of alternatively spliced human genes (10-30%) shows evidence of tissue-specific splice forms, mostly within the brain, testis and a few other tissues [72]. A relatively modest increase in the number of protein-coding genes from bacteria to unicellular eukaryotes to mammals does not account for the dramatic rise in the complexity of the organisms. The relatively small number of identified mammalian genes poses a question: what other factors contribute to the complexity of higher organisms?
We cannot rationally quantify the structural, physiological and behavioral complexity of organisms from different phyla. It is evident, however, that increased organismal complexity correlates less with the number of the protein-coding genes than with the length and diversity of non-protein-coding sequences. Generally, the complexity of organisms correlates with increases in the following parameters: first, the transcribed, but nontranslated, part of the genome; second, the length and number of introns in protein-coding genes; third, the number and complexity of cis-control elements and the increased use of complex and multiple promoters for a single gene; fourth, gene numbers, for both protein-coding and ncRNA genes; fifth, the complexity of UTRs and the length of 3' UTRs; and sixth, the ratio and the absolute number of transcription factors per genome [3,23,70,73]. In other words, the structural and physiological complexity of an organism is highly dependent on the complex regulation of gene expression and on the size and diversity of the transcriptome. Why is this so? Single-stranded RNA has some unique properties that make it suitable for regulatory roles. These include its ability to specifically recognize DNA sequences through complementary interactions; its conformational flexibility, which allows quick structural changes in a cooperative manner, and the ability to serve as a scaffold for protein molecules. The widespread use of RNA molecules in cell regulation and in the transient modulation of gene expression is also due to the quick and easy production of RNA (as no protein synthesis is required), and quick degradation by nucleases.
As discussed in this article, the non-coding transcribed part of the genome increases dramatically in size with the complexity of organisms, culminating in an estimated 1.2 billion nucleotides in humans. The function of these sequences still poses a challenge, some 30 years after their discovery. With the completion of the first three mammalian genome sequences, and more in view, the era of comparative mammalian genomics is coming to the fore, and efforts are increasingly focusing on genome annotation and the determination of functions for uncharacterized sequences. No genome annotation can be complete without characterization of the non-coding part of the transcriptome, however. This may become a priority for the future large-scale mammalian genome sequencing and annotation projects. We can hope that the scope and the complexity of the mammalian transcriptome will emerge in more detail with the discovery of orthologs of ncRNA genes, transcripts, and conserved functional sequence elements for closely and distantly related mammalian species.
References
- Scherrer K, Imaizumi-Scherrer MT, Reynaud CA, Therwath A. On pre-messenger RNA and transcriptons. A review. Mol Biol Rep. 1979;5:5–28. doi: 10.1007/BF00777484. [DOI] [PubMed] [Google Scholar]
- International Human Genome Sequencing Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- The FANTOM Consortium and the RIKEN Genome Exploration Research Group Phase I & II Team Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Versteeg R, van Schaik BDC, van Batenburg MF, Roos M, Monajemi R, Caron H, Bussemaker HJ, van Kampen AHC. The human transcriptome map reveals extremes in gene density, intron length, GC content, and repeat pattern for domains of highly and weakly expressed genes. Genome Res. 2003;13:1998–2004. doi: 10.1101/gr.1649303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, Kimura K, et al. Complete sequencing and characterization of 21,234 full-lemgth human cDNAs. Nat Genet . 2004;36:40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- Rat Genome Project http://www.hgsc.bcm.tmc.edu/projects/rat
- Evtushenko VI, Hanson KP, Barabitskaya OV, Emelyanov AV, Reshetnikov VL, Kozlov AP. An attempt to determine the maximal expression of the rat genome. Mol Biol (Mosk) 1989;23:663–675. [PubMed] [Google Scholar]
- Bantle JA, Hahn WE. Complexity and characterization of polyadenylated RNA in the mouse brain. Cell. 1976;8:139–150. doi: 10.1016/0092-8674(76)90195-1. [DOI] [PubMed] [Google Scholar]
- Hahn WE, Pettijohn DE, Van Ness J. One strand equivalent of the Escherichia coli genome is transcribed: complexity and abundance classes of mRNA. Science. 1977;197:582–585. doi: 10.1126/science.327551. [DOI] [PubMed] [Google Scholar]
- Hereford LM, Rosbash M. Number and distribution of polyadenylated RNA sequences in yeast. Cell. 1977;10:453–462. doi: 10.1016/0092-8674(77)90032-0. [DOI] [PubMed] [Google Scholar]
- Blattner FR, Plunkett G, III, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasnr JD, Rode CK, Mayhew GF, et al. The complete genome sequence of Escherichia coli K-12. Science. 1997;277:1453–1474. doi: 10.1126/science.277.5331.1453. [DOI] [PubMed] [Google Scholar]
- Dujon B. The yeast genome project: what did we learn? Trends Genet. 1996;12:263–270. doi: 10.1016/0168-9525(96)10027-5. [DOI] [PubMed] [Google Scholar]
- Mungall AJ, Palmer SA, Sims SK, Edwards CA, Ashurst JL, Wilming L, Jones MC, Horton R, Hunt SE, Scott CE, et al. The DNA sequence and analysis of human chromosome 6. Nature. 2003;425:805–811. doi: 10.1038/nature02055. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002;296:916–919. doi: 10.1126/science.1068597. [DOI] [PubMed] [Google Scholar]
- Davidson EH, Klein WH, Britten RJ. Sequence organization in animal DNA and a speculation on hnRNA as a coordinate regulatory transcript. Dev Biol. 1977;55:69–84. doi: 10.1016/0012-1606(77)90320-7. [DOI] [PubMed] [Google Scholar]
- Mattick JS, Gagen MJ. The evolution of controlled multitasked gene network: the role of introns and other non-coding RNAs in the development of complex organisms. Mol Biol Evol. 2001;18:1611–1630. doi: 10.1093/oxfordjournals.molbev.a003951. [DOI] [PubMed] [Google Scholar]
- Fields C, Adams MD, White O, Venter JC. How many genes in the human genome? Nat Genet. 1994;7:345–346. doi: 10.1038/ng0794-345. [DOI] [PubMed] [Google Scholar]
- Ewing B, Green P. Analysis of expressed sequence tags indicates 35,000 human genes. Nat Genet. 2000;25:232–234. doi: 10.1038/76115. [DOI] [PubMed] [Google Scholar]
- Liang F, Holt I, Pertea G, Karamysheva S, Salzberg SL, Quackenbush J. Gene index analysis of the human genome estimates approximately 120,000 genes. Nat Genet. 2000;25:239–240. doi: 10.1038/76126. [DOI] [PubMed] [Google Scholar]
- Zhuo D, Zhao WD, Wright FA, Yang HY, Wang JP, Sears R, Baer T, Kwon DH, Gordon D, Gibbs S, et al. Assembly, annotation, and integration of UNIGENE clusters into the human genome. Genome Res . 2001;11:904–918. doi: 10.1101/gr.GR-1645R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniGene http://www.ncbi.nlm.nih.gov/UniGene
- Pesole G, Mignone F, Gissi C, Grillo G, Licciulli F, Liuni S. Structural and functional features of eukaryotic mRNA untranslated regions. Gene. 2001;276:73–81. doi: 10.1016/S0378-1119(01)00674-6. [DOI] [PubMed] [Google Scholar]
- Larizza A, Makalowski W, Pesole G, Saccone C. Evolutionary dynamics of mammalian mRNA untranslated regions by comparative analysis of orthologous human, artiodactyl and rodent gene pairs. Comput Chem . 2002;26:479–490. doi: 10.1016/S0097-8485(02)00009-8. [DOI] [PubMed] [Google Scholar]
- Duret L, Dorkeld F, Gautier C. Strong conservation of noncoding sequences during vertebrate evolution: potential involvement in post-transcriptional regulation of gene expression. Nucleic Acids Res. 1993;21:2315–2322. doi: 10.1093/nar/21.10.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Bucher P. Searching for regulatory elements in human noncoding sequences. Curr Opin Struct Biol. 1997;7:399–406. doi: 10.1016/S0959-440X(97)80058-9. [DOI] [PubMed] [Google Scholar]
- Spicher A, Guicherit OM, Duret L, Aslanian A, Sanjines EM, Denko NC, Giaccia AJ, Blau HM. Highly conserved RNA sequences that are sensors of environmental stress. Mol Cell Biol. 1998;18:7371–7382. doi: 10.1128/mcb.18.12.7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignone F, Gissi C, Liuni S, Pesole G. Untranslated regions of mRNAs. Genome Biol. 2002;3:reviews0004–0004.10. doi: 10.1186/gb-2002-3-3-reviews0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzybowska EA, Wilczynska A, Siedlecki JA. Regulatory functions of 3' UTRs. Biochem Biophys Res Commun. 2001;288:291–295. doi: 10.1006/bbrc.2001.5738. [DOI] [PubMed] [Google Scholar]
- Bashirullah A, Cooperstock RL, Lipshitz HD. RNA localization in development. Annu Rev Biochem. 1998;67:335–394. doi: 10.1146/annurev.biochem.67.1.335. [DOI] [PubMed] [Google Scholar]
- Lipman DJ. Making (anti)sense of non-coding sequence conservation. Nucleic Acids Res. 1997;25:3580–3583. doi: 10.1093/nar/25.18.3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochetov AV, Sarai A, Vorob'ev DG, Kolchanov NA. The context organization of functional regions in yeast genes with high-level expression. Mol Biol (Mosk) 2002;36:1026–1034. [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Lipman DJ, Kondrashov AS. Patterns in interspecies similarity correlate with nucleotide composition in mammalian 3' UTRs. Nucleic Acids Res. 2003;31:5433–5439. doi: 10.1093/nar/gkg751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak M. The scanning model for translation: an update. J Cell Biol. 1989;108:229–241. doi: 10.1083/jcb.108.2.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestova TV, Kolupaeva VG, Lomakin IB, Pilipenko EV, Shatsky IN, Agol VI, Hellen CU. Molecular mechanisms of translation initiation in eukaryotes. Proc Natl Acad Sci USA. 2001;98:7029–7036. doi: 10.1073/pnas.111145798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin IB, Kochetov AV, Kondrashov FA, Koonin EV, Milanesi L. Presence of ATG triplets in 5' untranslated regions of eukaryotic cDNAs correlates with a 'weak' context of the start codon. Bioinformatics. 2001;17:890–900. doi: 10.1093/bioinformatics/17.10.890. [DOI] [PubMed] [Google Scholar]
- Gray NK, Wickens M. Control of translation initiation in animals. Annu Rev Cell Dev Biol. 1998;14:399–458. doi: 10.1146/annurev.cellbio.14.1.399. [DOI] [PubMed] [Google Scholar]
- Zubiaga AM, Belasco JG, Greenberg M. The nonamer UUAUUUAUU is the key AU-rich sequence motif that mediates mRNA degradation. Mol Cell Biol. 1995;15:2219–2230. doi: 10.1128/mcb.15.4.2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram G, Innes S, Minella O, Richardson J, Stansfield I. Endless possibilities: translation termination and stop codon recognition. Microbiology. 2001;147:255–269. doi: 10.1099/00221287-147-2-255. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Gladyshev VN. Mammalian selenoprotein gene signature: identification and functional analysis of selenoprotein genes using bioinformatics methods. Methods Enzymol. 2002;347:84–100. doi: 10.1016/s0076-6879(02)47010-3. [DOI] [PubMed] [Google Scholar]
- Kuersten S, Goodwin EB. The power of the 3' UTR: translational control and development. Nat Rev Genet. 2003;4:626–637. doi: 10.1038/nrg1125. [DOI] [PubMed] [Google Scholar]
- Lynch M, Richardson AO. The evolution of spliceosomal introns. Curr Opin Genet Dev. 2002;12:701–710. doi: 10.1016/S0959-437X(02)00360-X. [DOI] [PubMed] [Google Scholar]
- Lynch M, Kewalramani A. Messenger RNA surveillance and the evolutionary proliferation of introns. Mol Biol Evol. 2003;20:563–571. doi: 10.1093/molbev/msg068. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Kondrashov AS. Pattern of selective constraint in C. elegans and C. briggsae genomes. Genet Res. 1999;74:23–30. doi: 10.1017/S0016672399003821. [DOI] [PubMed] [Google Scholar]
- Bergman CM, Kreitman M. Analysis of conserved noncoding DNA in Drosophila reveals similar constraints in intergenic and intronic sequences. Genome Res. 2001;11:1335–1345. doi: 10.1101/gr.178701. [DOI] [PubMed] [Google Scholar]
- Wasserman WW, Palumbo M, Thompson M, Fickett JW, Lawrence CE. Human-mouse genome comparisons to locate regulatory sites. Nat Genet. 2000;26:225–228. doi: 10.1038/79965. [DOI] [PubMed] [Google Scholar]
- Jareborg N, Birney E, Durbin R. Comparative analysis of noncoding regions of 77 orthologous mouse and human gene pairs. Genome Res. 1999;9:815–824. doi: 10.1101/gr.9.9.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy S, Hannenhalli S, Workman C. Enrichment of regulatory signals in conserved non-coding genomic sequences. Bioinformatics . 2001;17:871–877. doi: 10.1093/bioinformatics/17.10.871. [DOI] [PubMed] [Google Scholar]
- Dermitzakis ET, Reymond A, Lyle R, Scamuffa N, Ucla C, Deutsch S, Stevenson BJ, Flegel V, Bucher P, Jongeneel CV, Antonarakis SE. Numerous potentionally functional but non-genic conserved sequences on human chromosome 21. Nature. 2002;420:578–582. doi: 10.1038/nature01251. [DOI] [PubMed] [Google Scholar]
- Hare MP, Palumbi SR. High intron sequence conservation across three mammalian orders suggests functional constraints. Mol Biol Evol. 2003;20:969–978. doi: 10.1093/molbev/msg111. [DOI] [PubMed] [Google Scholar]
- Cooper GM, Brudno M, Stone EA, Dubchak I, Batzoglou S, Sidow A. Characterization of evolutionary rates and constraints in three mammalian genomes. Genome Res. 2004. [DOI] [PMC free article] [PubMed]
- Levitsky VG, Podkolodnaya OA, Kolchanov NA, Podkolodny NL. Nucleosome formation potential of exons, introns and Alu repeats. Bioinformatics. 2001;17:1062–1064. doi: 10.1093/bioinformatics/17.11.1062. [DOI] [PubMed] [Google Scholar]
- Chernov IP, Akopov SB, Nikolaev LG, Sverdlov ED. Identification and mapping of nuclear matrix attachment regions in a one megabase locus of human chromosome 19q13.12: long-range correlation of S/MARs and gene positions. J Cell Biochem. 2002;84:590–600. doi: 10.1002/jcb.10043.abs. [DOI] [PubMed] [Google Scholar]
- Glazko GV, Koonin EV, Rogozin IB, Shabalina SA. A significant fraction of conserved noncoding DNA in human and mouse consists of predicted matrix attachment regions. Trends Genet. 2003;19:119–124. doi: 10.1016/S0168-9525(03)00016-7. [DOI] [PubMed] [Google Scholar]
- Brett D, Pospisil H, Valcarcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nature Genet. 2002;30:29–30. doi: 10.1038/ng803. [DOI] [PubMed] [Google Scholar]
- Castillo-Davis CI, Mekhedov SL, Hartl DL, Koonin EV, Kondrashov FA. Selection for short introns in highly expressed genes. Nat Genet. 2002;31:415–418. doi: 10.1038/ng940. [DOI] [PubMed] [Google Scholar]
- Vitali P, Royo H, Seitz H, Bachellerie JP, Huttenhofer A, Cavaille J. Identification of 13 human modification guide RNAs. Nucleic Acids Res. 2003;31:6543–6551. doi: 10.1093/nar/gkg849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Shabalina SA, Harris DG, Spouge JL, Kondrashov AS. Conserved fragments of transposable elements in intergenic regions: evidence for widespread recruitment of MIR- and L2-derived sequences within the mouse and human genomes. Genet Res. 2003;82:1–18. doi: 10.1017/S0016672303006268. [DOI] [PubMed] [Google Scholar]
- Hutvagner G, Zamore PD. RNAi: nature abhors a double strand. Curr Opin Genet Dev. 2002;12:225–232. doi: 10.1016/S0959-437X(02)00290-3. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Noncoding RNA genes. Curr Opin Genet Dev . 1999;9:695–699. doi: 10.1016/S0959-437X(99)00022-2. [DOI] [PubMed] [Google Scholar]
- Eddy SR. Non-coding RNA genes and the modern RNA world. Nat Rev Genet. 2001;2:919–929. doi: 10.1038/35103511. [DOI] [PubMed] [Google Scholar]
- Brandl S. Antisense-RNA regulation and RNA interference. Biochim Biophys Acta. 2002;1575:15–25. doi: 10.1016/s0167-4781(02)00280-4. [DOI] [PubMed] [Google Scholar]
- Ridanpaa M, van Eenennaam H, Pelin K, Chadwick R, Johnson C, Yuan B, vanVenrooij W, Pruijn G, Salmela R, Rockas S, et al. Mutations in the RNA component of RNase MRP gene cause a pleiotropic human disease, cartilage-hair displasia. Cell. 2001;104:195–203. doi: 10.1016/s0092-8674(01)00205-7. [DOI] [PubMed] [Google Scholar]
- Vulliamy T, Marrone A, Goldman F, Dearlove A, Bessler M, Mason PJ, Dokal I. The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature. 2001;413:432–435. doi: 10.1038/35096585. [DOI] [PubMed] [Google Scholar]
- Szymanski M, Barciszewska MZ, Zywicki M, Barciszewski J. Noncoding RNA transcripts. J Appl Genet. 2003;44:1–19. [PubMed] [Google Scholar]
- Britten RJ. Evolutionary selection against change in many Alu repeat sequences interspersed through primate genomes. Proc Natl Acad Sci USA. 1994;91:5992–5996. doi: 10.1073/pnas.91.13.5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englander EW, Howard BH. Nucleotide positioning by human Alu elements in chromatin. J Biol Chem. 1995;270:10091–10096. doi: 10.1074/jbc.270.17.10091. [DOI] [PubMed] [Google Scholar]
- Shabalina SA, Ogurtsov AY, Kondrashov VA, Kondrashov AS. Selective constraint in intergenic regions of human and mouse genomes. Trends Genet. 2001;17:373–376. doi: 10.1016/S0168-9525(01)02344-7. [DOI] [PubMed] [Google Scholar]
- Kondrashov AS, Shabalina SA. Classification of common conserved sequences in mammalian intergenic regions. Hum Mol Genet. 2002;11:669–674. doi: 10.1093/hmg/11.6.669. [DOI] [PubMed] [Google Scholar]
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003;424:147–151. doi: 10.1038/nature01763. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Tasic B. Alternative pre-mRNA splicing and proteome expansion in metazoans. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- Xu Q, Modrek B, Lee C. Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res . 2002;30:3754–3766. doi: 10.1093/nar/gkf492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein LD, Bao G, Blasiar D, Blumenthal T, Brent MR, Chen N, Chinwala A, Clarke L, Clee C, Coghlan A, et al. The genome sequence of Caenorhabditis briggsae: A platform for comparative genetics. PLoS Biology. 2003;1:E45. doi: 10.1371/journal.pbio.0000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tjaden B, Saxena RM, Stolyar S, Haynor DR, Kolker E, Rosenow C. Transcriptome analysis of Escherichia coli using high-density oligonucleotide probe arrays. Nucleic Acids Res. 2002;30:3732–3738. doi: 10.1093/nar/gkf505. [DOI] [PMC free article] [PubMed] [Google Scholar]